Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Viability Assay
2.3. Apoptosis Assay
2.3.1. Annexin V-FITC-PI Staining
2.3.2. TUNEL Assay for Late Apoptosis Detection
2.4. Estimation of ROS Generation
2.5. Measurement of Antioxidants
2.6. Western Blot Analysis
2.7. Determination of Mitochondrial Membrane Potential
2.8. Chemicals and Media
2.9. Statistical Analysis
3. Results
3.1. Effects of Lipid Emulsion on Doxorubicin-Induced Reduced H9c2 Cell Viability
3.2. Doxorubicin-Induced Late Apoptosis Was Reduced by Lipid Emulsion Pretreatment in H9c2 Rat Cardiomyoblasts
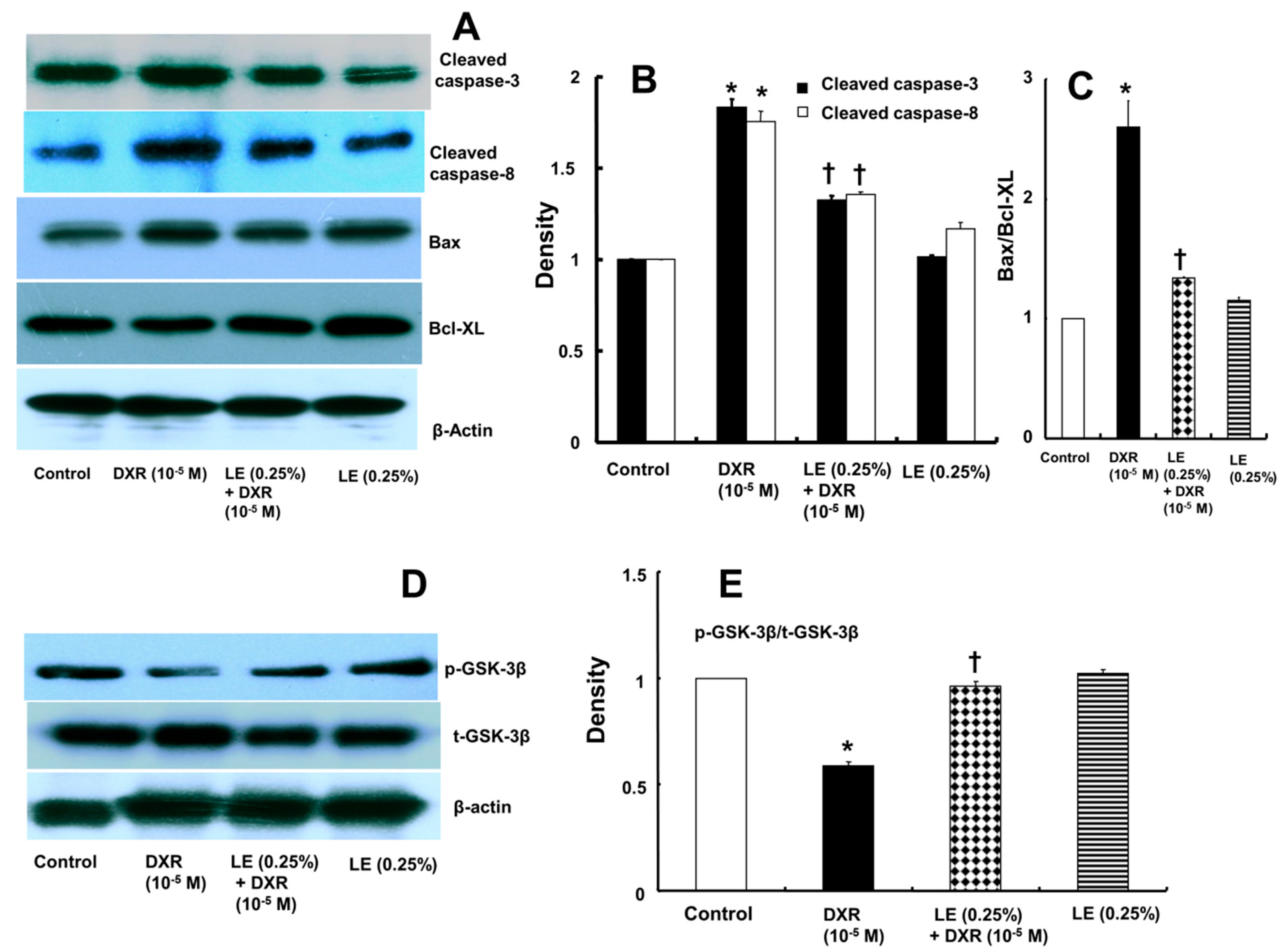
3.3. Lipid Emulsion Mediated Downregulation of the Apoptotic Signaling Cascade Activated by Doxorubicin in H9c2 Rat Cardiomyoblasts
3.4. Role of Lipid Emulsion in Modulating GSK-3β Phosphorylation in H9c2 Rat Cardiomyoblasts
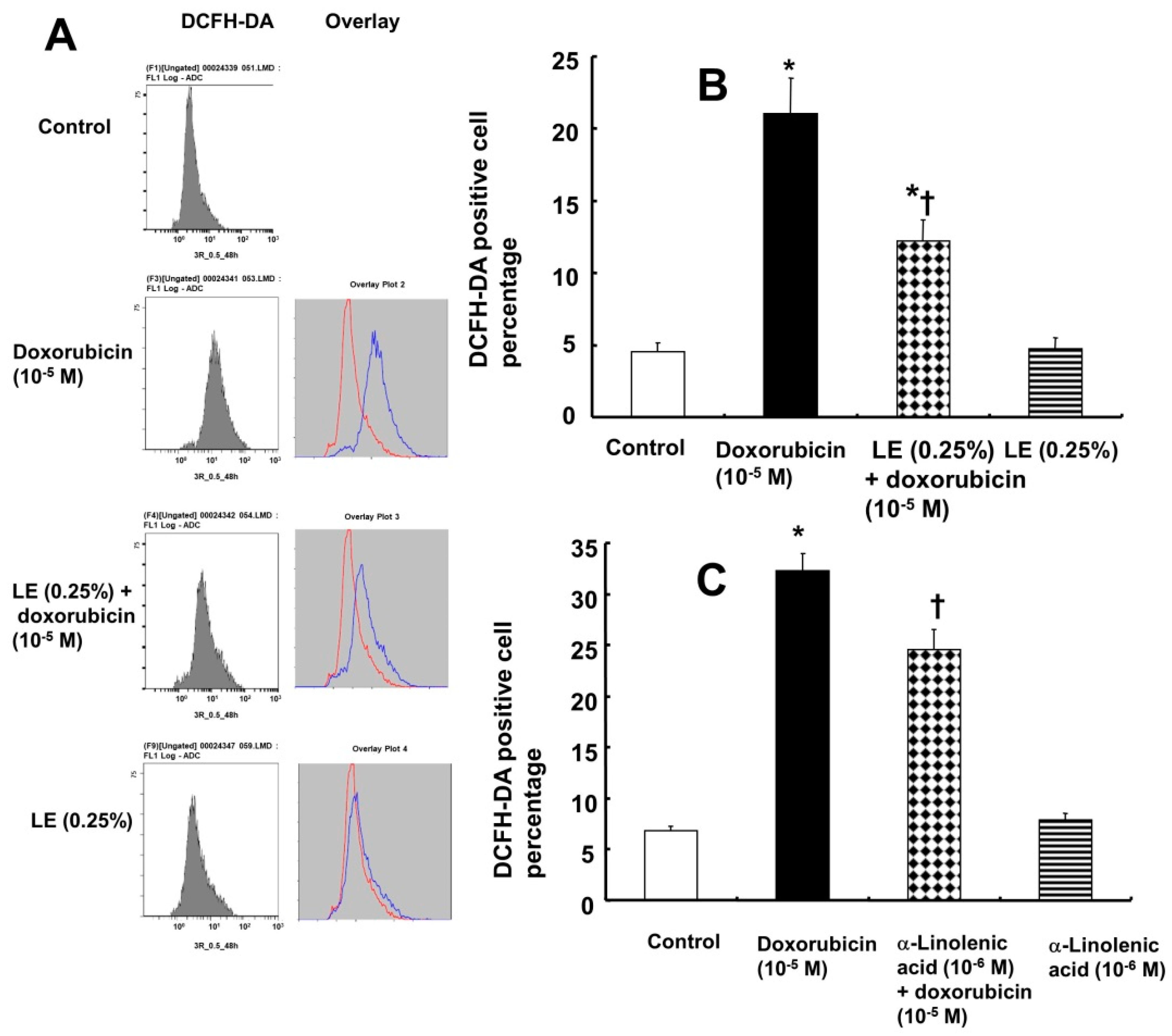
3.5. Doxorubicin-Induced Oxidative Stress Was Attenuated by Lipid Emulsion Pretreatment in H9c2 Rat Cardiomyoblasts
3.6. Role of Lipid Emulsion in Regulating the MMP
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Simůnek, T.; Stérba, M.; Popelová, O.; Adamcová, M.; Hrdina, R.; Gersl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Xu, M.F.; Tang, P.L.; Qian, Z.M.; Ashraf, M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sci. 2001, 68, 889–901. [Google Scholar] [CrossRef]
- Anderson, A.B.; Arriaga, E.A. Subcellular metabolite profiles of the parent CCRF-CEM and the derived CEM/C2 cell lines after treatment with doxorubicin. J. Chromatogr. B 2004, 808, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Sarvazyan, N. Visualization of doxorubicin-induced oxidative stress in isolated cardiac myocytes. Am. J. Physiol. 1996, 271, H2079–H2085. [Google Scholar] [CrossRef] [PubMed]
- Wanten, G.J.; Calder, P.C. Immune modulation by parenteral lipid emulsions. Am. J. Clin. Nutr. 2007, 85, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Ok, S.H.; Hong, J.M.; Lee, S.H.; Sohn, J.T. Lipid emulsion for treating local anesthetic systemic toxicity. Int. J. Med. Sci. 2018, 15, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, X.; Li, J.; Zhang, X.; Gong, T.; Zhang, Z. Lipid nanoemulsions loaded with doxorubicin-oleic acid ionic complex: Characterization, in vitro and in vivo studies. Pharmazie 2011, 66, 496–505. [Google Scholar] [PubMed]
- Yu, X.; Cui, L.; Zhang, Z.; Zhao, Q.; Li, S. α-Linolenic acid attenuates doxorubicin-induced cardiotoxicity in rats through suppression of oxidative stress and apoptosis. Acta Biochim. Biophys. Sin. 2013, 45, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Almutairdi, A.; Mulesa, L.; Alberda, C.; Beattie, C.; Gramlich, L. Parenteral nutrition and lipids. Nutrients 2017, 9, E388. [Google Scholar] [CrossRef] [PubMed]
- Uysal, M.; Karaman, S. In vivo effects of intravenous lipid emulsion on lung tissue in an experimental model of acute malathion intoxication. Toxicol. Ind. Health 2018, 34, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Ok, S.H.; Choi, M.H.; Shin, I.W.; Lee, S.H.; Kang, S.; Oh, J.; Han, J.Y.; Sohn, J.T. Lipid Emulsion Inhibits Apoptosis Induced by a Toxic Dose of Verapamil via the Delta-Opioid Receptor in H9c2 Rat Cardiomyoblasts. Cardiovasc. Toxicol. 2017, 17, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Ok, S.H.; Yu, J.; Lee, Y.; Cho, H.; Shin, I.W.; Sohn, J.T. Lipid emulsion attenuates apoptosis induced by a toxic dose of bupivacaine in H9c2 rat cardiomyoblast cells. Hum. Exp. Toxicol. 2016, 35, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Intralipid® [Package Insert] Uppsala, Sweden. Fresenius Kabi. 2015. Available online: https://www.fresenius-kabi.com/in/products/intralipid (accessed on 9 September 2018).
- Arola, O.J.; Saraste, A.; Pulkki, K.; Kallajoki, M.; Parvinen, M.; Voipio-Pulkki, L.M. Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res. 2000, 60, 1789–1792. [Google Scholar] [PubMed]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar] [PubMed]
- Biancaniello, T.; Meyer, R.A.; Wong, K.Y.; Sager, C.; Kaplan, S. Doxorubicin cardiotoxicity in children. J. Pediatr. 1980, 97, 45–50. [Google Scholar] [CrossRef]
- Singal, P.K.; Iliskovic, N. Doxorubicin-induced cardiomyopathy. N. Engl. J. Med. 1998, 339, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Pein, F.; Sakiroglu, O.; Dahan, M.; Lebidois, J.; Merlet, P.; Shamsaldin, A.; Villain, E.; de Vathaire, F.; Sidi, D.; Hartmann, O. Cardiac abnormalities 15 years and more after adriamycin therapy in 229 childhood survivors of a solid tumour at the Institut Gustave Roussy. Br. J. Cancer 2004, 91, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, M.L.; Lange, R.A.; Parsons, H.; Andrews, T.; Aune, G.J. The tell-tale heart: Molecular and cellular responses to childhood anthracycline exposure. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1379–H1389. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, J.H. Effect of anthracycline antibiotics on oxygen radical formation in rat heart. Cancer Res. 1983, 43, 460–472. [Google Scholar] [PubMed]
- Zhang, Y.W.; Shi, J.; Li, Y.J.; Wei, L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch. Immunol. Ther. Exp. 2009, 57, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Kostin, S.; Pool, L.; Elsässer, A.; Hein, S.; Drexler, H.C.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.P.; et al. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Wencker, D.; Chandra, M.; Nguyen, K.; Miao, W.; Garantziotis, S.; Factor, S.M.; Shirani, J.; Armstrong, R.C.; Kitsis, R.N. A mechanistic role for cardiac myocyte apoptosis in heart failure. J. Clin. Investig. 2003, 111, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kotamraju, S.; Konorev, E.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: The role of hydrogen peroxide. Biochem. J. 2002, 367, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Hrelia, S.; Bordoni, A.; Biagi, P.L. Role of gamma-linolenic acid in counteracting doxorubicin-induced damage in cultured rat cardiomyocytes. Prostag. Leuk. Essent. Fat. Acids 2001, 64, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Bai, Z.; Yang, L.; Li, X.; Chen, X. Lipid emulsion reverses bupivacaine-induced apoptosis of h9c2 cardiomyocytes: PI3K/Akt/GSK-3β signaling pathway. Environ. Toxicol. Pharmacol. 2016, 42, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Bai, Z.; Lv, D.; Liu, H.; Li, X.; Chen, X. Rescue effect of lipid emulsion on bupivacaine-induced cardiac toxicity in cardiomyocytes. Mol. Med. Rep. 2015, 12, 3739–3747. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M. Cell death pathways—Potential therapeutic targets. Xenobiotica 2009, 39, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3β: A bifunctional role in cell death pathways. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Yi, M.; Huang, Y.P. Oxymatrine ameliorates doxorubicin-induced cardiotoxicity in rats. Cell. Physiol. Biochem. 2017, 43, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef] [PubMed]
- Wattanapitayakul, S.K.; Bauer, J.A. Oxidative pathways in cardiovascular disease: Roles, mechanisms, and therapeutic implications. Pharmacol. Ther. 2001, 89, 187–206. [Google Scholar] [CrossRef]
- Kahl, R.; Kampkötter, A.; Wätjen, W.; Chovolou, Y. Antioxidant enzymes and apoptosis. Drug Metab. Rev. 2004, 36, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.E.; Jansen, E.S.; Sanchez, W.; Waterhouse, N.J. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarization and cytochrome c release. Methods 2013, 61, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, J.A.; Schramko, A.A.; Skrifvars, M.B.; Litonius, E.; Backman, J.T.; Mervaala, E.; Rosenberg, P.H. The effects of intravenous lipid emulsion on hemodynamic recovery and myocardial cell mitochondrial function after bupivacaine toxicity in anesthetized pigs. Hum. Exp. Toxicol. 2017, 36, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.M.; Eksborg, S.; Björk, O.; Abrahamsson, J.; Behrendtz, M.; Castor, A.; Forestier, E.; Lönnerholm, G. Pharmacokinetics of doxorubicin in children with acute lymphoblastic leukemia: Multi-institutional collaborative study. Med. Pediatr. Oncol. 2002, 38, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Volkova, M.; Russell, R., III. Anthracycline cardiotoxicity: Prevalence, pathogenesis and treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subbarao, R.B.; Ok, S.-H.; Lee, S.H.; Kang, D.; Kim, E.-J.; Kim, J.-Y.; Sohn, J.-T. Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts. Cells 2018, 7, 144. https://doi.org/10.3390/cells7100144
Subbarao RB, Ok S-H, Lee SH, Kang D, Kim E-J, Kim J-Y, Sohn J-T. Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts. Cells. 2018; 7(10):144. https://doi.org/10.3390/cells7100144
Chicago/Turabian StyleSubbarao, Raghavendra Baregundi, Seong-Ho Ok, Soo Hee Lee, Dawon Kang, Eun-Jin Kim, Ji-Yoon Kim, and Ju-Tae Sohn. 2018. "Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts" Cells 7, no. 10: 144. https://doi.org/10.3390/cells7100144
APA StyleSubbarao, R. B., Ok, S.-H., Lee, S. H., Kang, D., Kim, E.-J., Kim, J.-Y., & Sohn, J.-T. (2018). Lipid Emulsion Inhibits the Late Apoptosis/Cardiotoxicity Induced by Doxorubicin in Rat Cardiomyoblasts. Cells, 7(10), 144. https://doi.org/10.3390/cells7100144