The Role of Hypoxia in Glioblastoma Invasion


Abstract
:1. Introduction
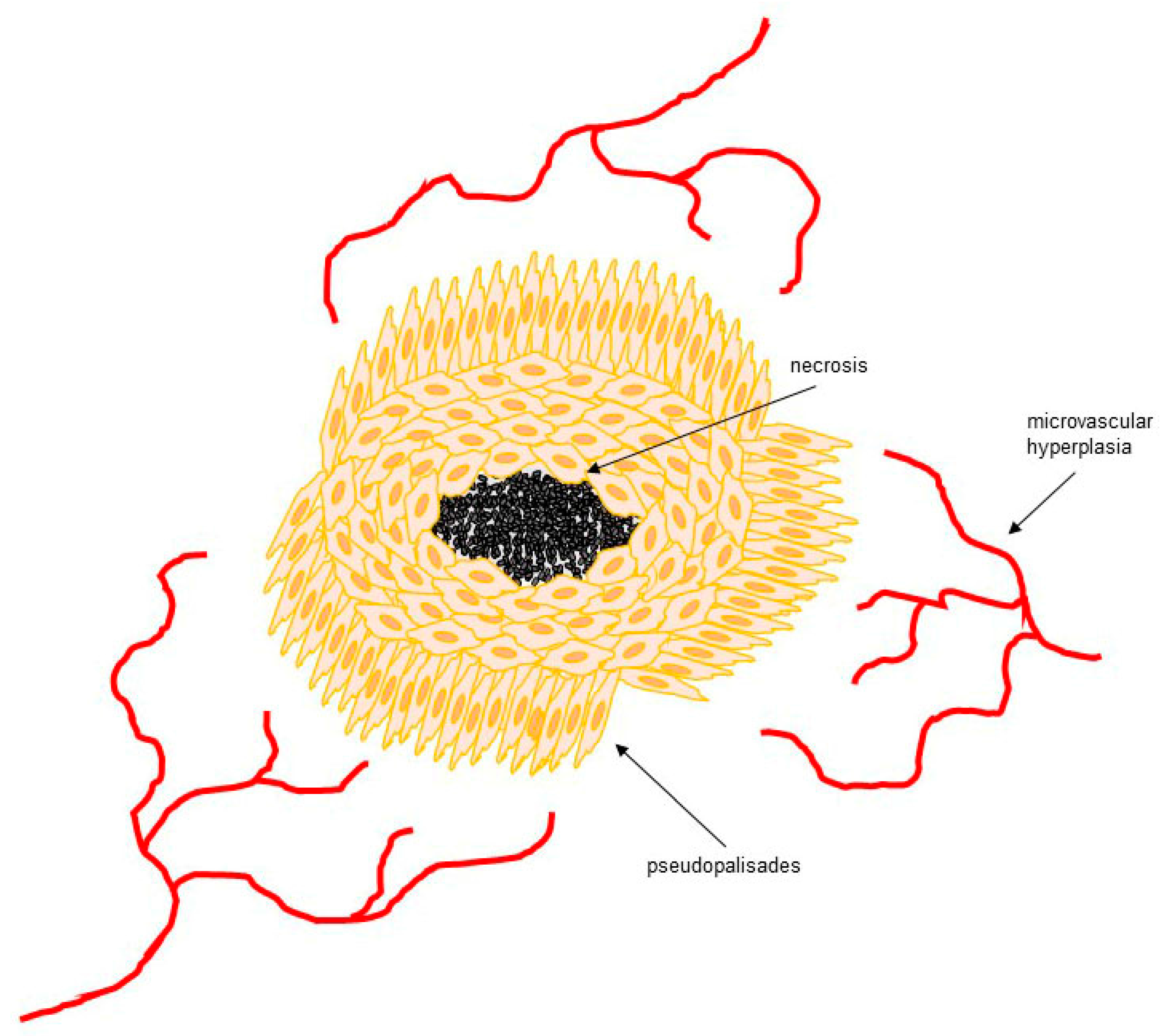
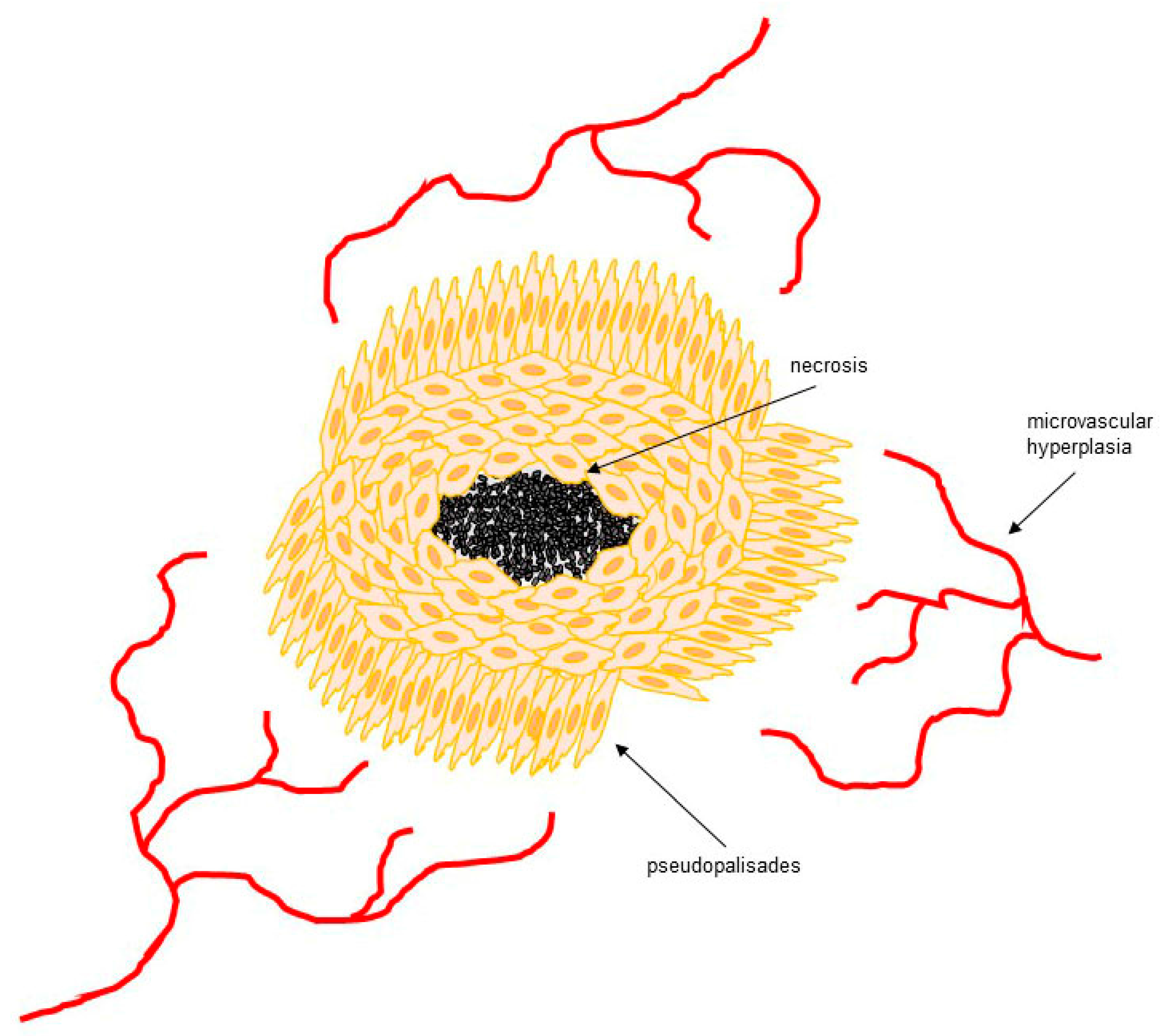
2. Hypoxia in GBM
3. HIF Transcription Factors
4. Hypoxia Driven Up-Regulation of Invasion Proteins in GBM
4.1. Extracellular Matrix Degradation and Remodeling
4.2. Epithelial to Mesenchymal Transition (EMT)
4.3. Chemokine Receptors
4.4. Cytoskeleton Dynamics
4.5. Hemostasis
4.6. Others
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Wang, M.; Dignam, J.J.; Won, M.; Curran, W.; Mehta, M.; Gilbert, M.R. Variation over time and interdependence between disease progression and death among patients with glioblastoma on RTOG 0525. Neuro-Oncology 2015, 17, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Löffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; Alfred Yung, W.K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef] [PubMed]
- Mahase, S.; Rattenni, R.N.; Wesseling, P.; Leenders, W.; Baldotto, C.; Jain, R.; Zagzag, D. Hypoxia-Mediated Mechanisms Associated with Antiangiogenic Treatment Resistance in Glioblastomas. Am. J. Pathol. 2017, 187, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Investig. 2004, 84, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Castellano-Sanchez, A.A.; Hunter, S.B.; Pecot, M.; Cohen, C.; Hammond, E.H.; Devi, S.N.; Kaur, B.; Van Meir, E.G. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004, 64, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Zhong, H.; Scalzitti, J.M.; Laughner, E.; Simons, J.W.; Semenza, G.L. Expression of hypoxia-inducible factor 1alpha in brain tumors: Association with angiogenesis, invasion, and progression. Cancer 2000, 88, 2606–2618. [Google Scholar] [CrossRef]
- Brat, D.J.; Castellano-Sanchez, A.; Kaur, B.; Van Meir, E.G. Genetic and biologic progression in astrocytomas and their relation to angiogenic dysregulation. Adv. Anat. Pathol. 2002, 9, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: A new world of angiogenesis research. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro-Oncology 2005, 7, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L.; Mumert, M.L.; Gillespie, D.L.; Kinney, A.Y.; Schabel, M.C.; Salzman, K.L. Preoperative dynamic contrast-enhanced MRI correlates with molecular markers of hypoxia and vascularity in specific areas of intratumoral microenvironment and is predictive of patient outcome. Neuro-Oncology 2014, 16, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. “Pseudopalisading” necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Clara, C.A.; Marie, S.K.N.; de Almeida, J.R.W.; Wakamatsu, A.; Oba-Shinjo, S.M.; Uno, M.; Neville, M.; Rosemberg, S. Angiogenesis and expression of PDGF-C, VEGF, CD105 and HIF-1α in human glioblastoma. Neuropathology 2014, 34, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Lin, A.; Mahairaki, V.; Matsui, W.; Eberhart, C.G. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am. J. Pathol. 2010, 177, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.-S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Salehi, H.A.; Boucher, Y.; Vasthare, U.S.; Tuma, R.F.; Jain, R.K. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994, 54, 4564–4568. [Google Scholar] [PubMed]
- Plate, K.H.; Mennel, H.D. Vascular morphology and angiogenesis in glial tumors. Exp. Toxicol. Pathol. 1995, 47, 89–94. [Google Scholar] [CrossRef]
- Bullitt, E.; Zeng, D.; Gerig, G.; Aylward, S.; Joshi, S.; Smith, J.K.; Lin, W.; Ewend, M.G. Vessel tortuosity and brain tumor malignancy: A blinded study. Acad. Radiol. 2005, 12, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.L.D.S.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.-J.; Duan, C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am. J. Physiol. Cell Physiol. 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Bunn, H.F. Hypoxia-inducible factor and its biomedical relevance. J. Biol. Chem. 2003, 278, 19575–19578. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia Inducible Factor-α Binding and Ubiquitylation by the von Hippel-Lindau Tumor Suppressor Protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, V.; Zhang, L.P.; Zhu, X.H.; Caro, J. Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor alpha (HIF-alpha) proteins. Biochem. Biophys. Res. Commun. 1999, 260, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Ebert, B.L.; Bunn, H.F. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol. Cell. Biol. 1998, 18, 4089–4096. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Kallio, P.J.; Pongratz, I.; Gradin, K.; McGuire, J.; Poellinger, L. Activation of hypoxia-inducible factor 1alpha: Posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc. Natl. Acad. Sci. USA 1997, 94, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Frederick, L.; Wang, X.Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar] [PubMed]
- Holland, E.C.; Hively, W.P.; DePinho, R.A.; Varmus, H.E. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998, 12, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
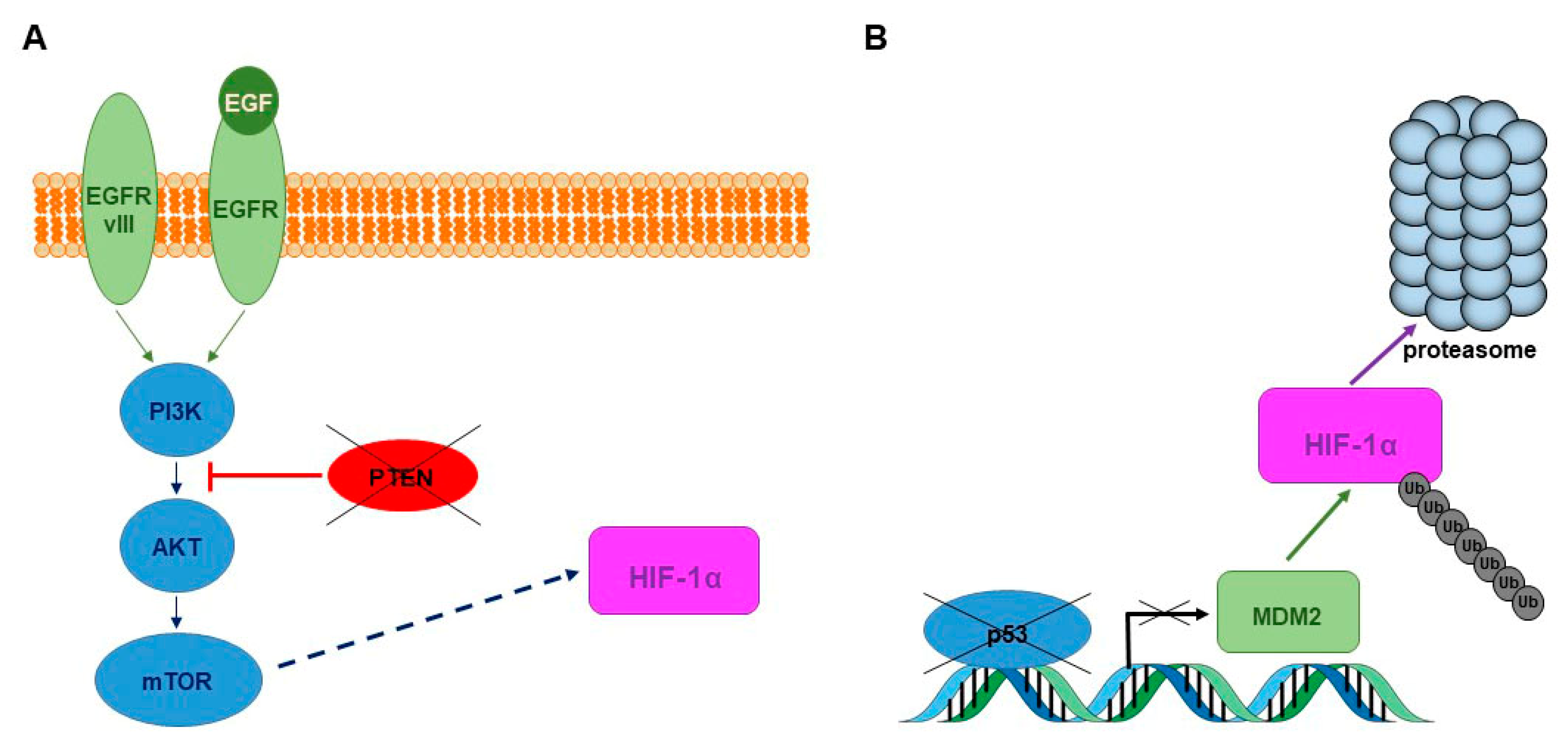
- Clarke, K.; Smith, K.; Gullick, W.J.; Harris, A.L. Mutant epidermal growth factor receptor enhances induction of vascular endothelial growth factor by hypoxia and insulin-like growth factor-1 via a PI3 kinase dependent pathway. Br. J. Cancer 2001, 84, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
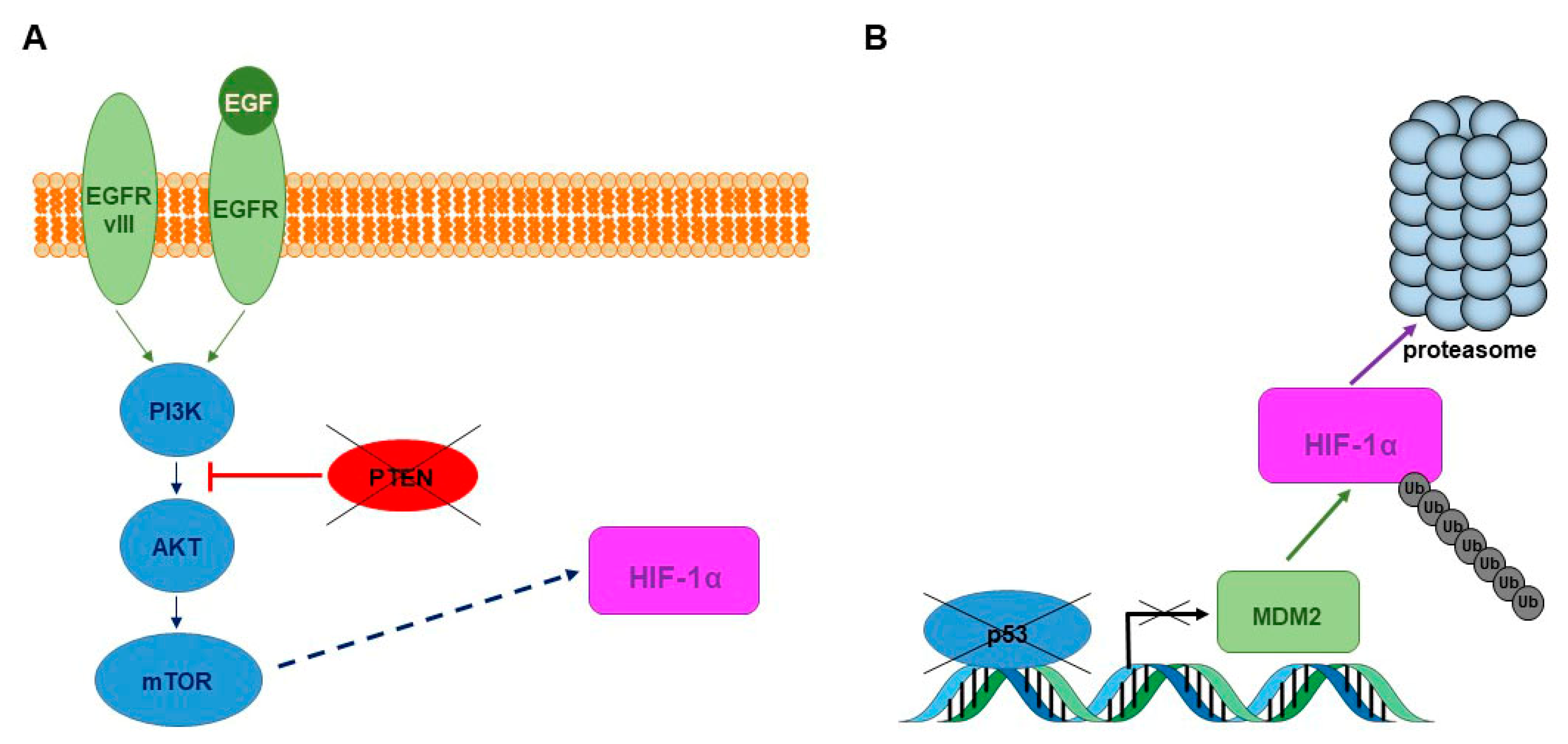
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Sansal, I.; Sellers, W.R. The biology and clinical relevance of the PTEN tumor suppressor pathway. J. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef] [PubMed]
- Ravi, R.; Mookerjee, B.; Bhujwalla, Z.M.; Sutter, C.H.; Artemov, D.; Zeng, Q.; Dillehay, L.E.; Madan, A.; Semenza, G.L.; Bedi, A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000, 14, 34–44. [Google Scholar] [PubMed]
- Madan, E.; Dikshit, B.; Gowda, S.H.; Srivastava, C.; Sarkar, C.; Chattopadhyay, P.; Sinha, S.; Chosdol, K. FAT1 is a novel upstream regulator of HIF1α and invasion of high grade glioma. Int. J. Cancer 2016, 139, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Z.; Shi, Q.; Jiao, J.; Bian, W.; Song, X.; Mo, J.; Sang, B.; Xu, Y.; Qian, J.; et al. Geranylgeranyltransferase I regulates HIF-1α promoting glioblastoma cell migration and invasion. J. Neurooncol. 2013, 112, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Cohen-Jonathan Moyal, E. Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-G.; Wang, C.; Liu, D.-Y.; Zhang, X.; Wang, L.; Yan, M.; Zhang, W.; Zhu, J.; Li, Z.-C.; Mi, C.; et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget 2016, 7, 47808–47820. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Koul, D.; Fu, J.; Shen, R.; LaFortune, T.A.; Wang, S.; Tiao, N.; Kim, Y.-W.; Liu, J.-L.; Ramnarian, D.; Yuan, Y.; et al. Antitumor activity of NVP-BKM120—A selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin. Cancer Res. 2012, 18, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.-M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Lee, E.Q.; Reardon, D.A.; Ligon, K.L.; Alfred Yung, W.K. Current clinical development of PI3K pathway inhibitors in glioblastoma. Neuro-Oncology 2012, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-J.; Koul, D.; LaFortune, T.; Tiao, N.; Shen, R.J.; Maira, S.-M.; Garcia-Echevrria, C.; Yung, W.K.A. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol. Cancer Ther. 2009, 8, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; de Gooijer, M.C.; Hanekamp, D.; Chandrasekaran, G.; Buil, L.C.M.; Thota, N.; Sparidans, R.W.; Beijnen, J.H.; Würdinger, T.; van Tellingen, O. PI3K-mTOR Pathway Inhibition Exhibits Efficacy Against High-grade Glioma in Clinically Relevant Mouse Models. Clin. Cancer Res. 2017, 23, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Netland, I.A.; Førde, H.E.; Sleire, L.; Leiss, L.; Rahman, M.A.; Skeie, B.S.; Gjerde, C.H.; Enger, P.Ø.; Goplen, D. Dactolisib (NVP-BEZ235) toxicity in murine brain tumour models. BMC Cancer 2016, 16, 657. [Google Scholar] [CrossRef] [PubMed]
- Huss, W.J.; Hanrahan, C.F.; Barrios, R.J.; Simons, J.W.; Greenberg, N.M. Angiogenesis and prostate cancer: Identification of a molecular progression switch. Cancer Res. 2001, 61, 2736–2743. [Google Scholar] [PubMed]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The Tumorigenic Roles of the Cellular REDOX Regulatory Systems. Oxid. Med. Cell. Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [PubMed]
- Klimova, T.; Chandel, N.S. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 2008, 15, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Brösicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Bache, M.; Rot, S.; Kessler, J.; Güttler, A.; Wichmann, H.; Greither, T.; Wach, S.; Taubert, H.; Söling, A.; Bilkenroth, U.; et al. mRNA expression levels of hypoxia-induced and stem cell-associated genes in human glioblastoma. Oncol. Rep. 2015, 33, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Polat, B.; Staab, A.; Hagemann, C.; Stein, S.; Flentje, M.; Theobald, M.; Katzer, A.; Vordermark, D. Rapid detection of the hypoxia-regulated CA-IX and NDRG1 gene expression in different glioblastoma cells in vitro. Oncol. Rep. 2008, 20, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Hagemann, C.; Staab, A.; Stojic, J.; Kühnel, S.; Vince, G.H.; Flentje, M.; Roosen, K.; Vordermark, D. Expression patterns of the hypoxia-related genes osteopontin, CA9, erythropoietin, VEGF and HIF-1α in human glioma in vitro and in vivo. Radiother. Oncol. 2007, 83, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Marotta, D.; Karar, J.; Jenkins, W.T.; Kumanova, M.; Jenkins, K.W.; Tobias, J.W.; Baldwin, D.; Hatzigeorgiou, A.; Alexiou, P.; Evans, S.M.; et al. In vivo profiling of hypoxic gene expression in gliomas using the hypoxia marker EF5 and laser-capture microdissection. Cancer Res. 2011, 71, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Proescholdt, M.A.; Merrill, M.J.; Stoerr, E.-M.; Lohmeier, A.; Pohl, F.; Brawanski, A. Function of carbonic anhydrase IX in glioblastoma multiforme. Neuro-Oncology 2012, 14, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Kobliakov, V.A. Role of Proton Pumps in Tumorigenesis. Biochemistry 2017, 82, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Kivelä, A.J.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivelä, J.; Parkkila, A.K.; Pastoreková, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. Expression of transmembrane carbonic anhydrase isoenzymes IX and XII in normal human pancreas and pancreatic tumours. Histochem. Cell Biol. 2000, 114, 197–204. [Google Scholar] [PubMed]
- Hilvo, M.; Baranauskiene, L.; Salzano, A.M.; Scaloni, A.; Matulis, D.; Innocenti, A.; Scozzafava, A.; Monti, S.M.; Di Fiore, A.; De Simone, G.; et al. Biochemical Characterization of CA IX, One of the Most Active Carbonic Anhydrase Isozymes. J. Biol. Chem. 2008, 283, 27799–27809. [Google Scholar] [CrossRef] [PubMed]
- Amiri, A.; Le, P.U.; Moquin, A.; Machkalyan, G.; Petrecca, K.; Gillard, J.W.; Yoganathan, N.; Maysinger, D. Inhibition of carbonic anhydrase IX in glioblastoma multiforme. Eur. J. Pharm. Biopharm. 2016, 109, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Burden-Gulley, S.M.; Qutaish, M.Q.; Sullivant, K.E.; Lu, H.; Wang, J.; Craig, S.E.L.; Basilion, J.P.; Wilson, D.L.; Brady-Kalnay, S.M. Novel cryo-imaging of the glioma tumor microenvironment reveals migration and dispersal pathways in vivid three-dimensional detail. Cancer Res. 2011, 71, 5932–5940. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, L.; Dong, Y.; Tan, Y.; Li, Y.; Zhao, M.; Xie, H.; Ju, H.; Wang, H.; Zhao, Y.; et al. EGFRvIII/integrin β3 interaction in hypoxic and vitronectinenriching microenvironment promote GBM progression and metastasis. Oncotarget 2016, 7, 4680–4694. [Google Scholar] [CrossRef] [PubMed]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Malden, L.T.; Novak, U.; Kaye, A.H.; Burgess, A.W. Selective amplification of the cytoplasmic domain of the epidermal growth factor receptor gene in glioblastoma multiforme. Cancer Res. 1988, 48, 2711–2714. [Google Scholar] [PubMed]
- Aldape, K.D.; Ballman, K.; Furth, A.; Buckner, J.C.; Giannini, C.; Burger, P.C.; Scheithauer, B.W.; Jenkins, R.B.; James, C.D. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J. Neuropathol. Exp. Neurol. 2004, 63, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.K.; van den Bent, M.; Lassman, A.B.; Reardon, D.A.; Scott, A.M. Antibody-drug conjugates in glioblastoma therapy: The right drugs to the right cells. Nat. Rev. Clin. Oncol. 2017, 14, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting EGFRvIII for glioblastoma multiforme. Cancer Lett. 2017, 403, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Flügel, D.; Görlach, A.; Michiels, C.; Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Mikkelsen, T.; Rosenfeld, S.S.; Hochberg, F.; Akella, N.S.; Fisher, J.D.; Cloud, G.A.; Zhang, Y.; Carson, K.; Wittemer, S.M.; et al. Phase I and correlative biology study of cilengitide in patients with recurrent malignant glioma. J. Clin. Oncol. 2007, 25, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Stewart, C.F.; Kocak, M.; Goldman, S.; Ellenbogen, R.G.; Phillips, P.; Lafond, D.; Poussaint, T.Y.; Kieran, M.W.; Boyett, J.M.; et al. Phase I clinical trial of cilengitide in children with refractory brain tumors: Pediatric Brain Tumor Consortium Study PBTC-012. J. Clin. Oncol. 2008, 26, 919–924. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.J.; Vezina, G.; Stewart, C.F.; Turner, D.; Pierson, C.R.; Chen, L.; Pollack, I.F.; Gajjar, A.; Kieran, M.W. Phase II study of cilengitide in the treatment of refractory or relapsed high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro-Oncology 2013, 15, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O’Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 2008, 26, 5610–5617. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Wei, Y.; Zhang, X.; Xu, R.; Han, M.; Huang, B.; Chen, A.; Li, W.; Zhang, Q.; et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes hypoxia-induced glioma migration and invasion. Oncotarget 2017, 8, 23401–23413. [Google Scholar] [CrossRef] [PubMed]
- Stracke, M.L.; Liotta, L.A. Multi-step cascade of tumor cell metastasis. In Vivo 1992, 6, 309–316. [Google Scholar] [PubMed]
- Schlaepfer, D.D.; Mitra, S.K. Multiple connections link FAK to cell motility and invasion. Curr. Opin. Genet. Dev. 2004, 14, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Mañes, S.; Mira, E.; Gómez-Moutón, C.; Lacalle, R.A.; Martínez, C. Cells on the move: A dialogue between polarization and motility. IUBMB Life 2000, 49, 89–96. [Google Scholar] [CrossRef] [PubMed]
- van der Slot, A.J.; Zuurmond, A.-M.; Bardoel, A.F.J.; Wijmenga, C.; Pruijs, H.E.H.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Stiff Collagen Matrices Increase Tumorigenic Prolactin Signaling in Breast Cancer Cells. J. Biol. Chem. 2013, 288, 12722–12732. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Pang, M.; Hou, X.; Yuan, S.; Sun, L. PLOD2 in cancer research. Biomed. Pharmacother. 2017, 90, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.K.; Zhang, M.; Qiu, Q.; Skuli, N.; Nakazawa, M.S.; Karakasheva, T.; Mucaj, V.; Shay, J.E.S.; Stangenberg, L.; Sadri, N.; et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013, 3, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Zuurmond, A.-M.; van der Slot-Verhoeven, A.J.; van Dura, E.A.; De Groot, J.; Bank, R.A. Minoxidil exerts different inhibitory effects on gene expression of lysyl hydroxylase 1, 2, and 3: Implications for collagen cross-linking and treatment of fibrosis. Matrix Biol. 2005, 24, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Hamsa, T.P.; Kuttan, G. Berberine inhibits pulmonary metastasis through down-regulation of MMP in metastatic B16F-10 melanoma cells. Phytother. Res. 2012, 26, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Guruvayoorappan, C.; Kuttan, G. Amentoflavone inhibits experimental tumor metastasis through a regulatory mechanism involving MMP-2, MMP-9, prolyl hydroxylase, lysyl oxidase, VEGF, ERK-1, ERK-2, STAT-1, NM23 and cytokines in lung tissues of C57BL/6 mice. Immunopharmacol. Immunotoxicol. 2008, 30, 711–727. [Google Scholar] [CrossRef] [PubMed]
- Guruvayoorappan, C.; Kuttan, G. Beta-carotene inhibits tumor-specific angiogenesis by altering the cytokine profile and inhibits the nuclear translocation of transcription factors in B16F-10 melanoma cells. Integr. Cancer Ther. 2007, 6, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Kang, Y. Multilayer control of the EMT master regulators. Oncogene 2014, 33, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertrán, E.; Pérez-Pomares, J.M.; Díez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisúa-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wu, Q.; Kim, L.; Miller, T.E.; Liau, B.B.; Mack, S.C.; Yang, K.; Factor, D.C.; Fang, X.; Huang, Z.; et al. RBPJ maintains brain tumor-initiating cells through CDK9-mediated transcriptional elongation. J. Clin. Investig. 2016, 126, 2757–2772. [Google Scholar] [CrossRef] [PubMed]
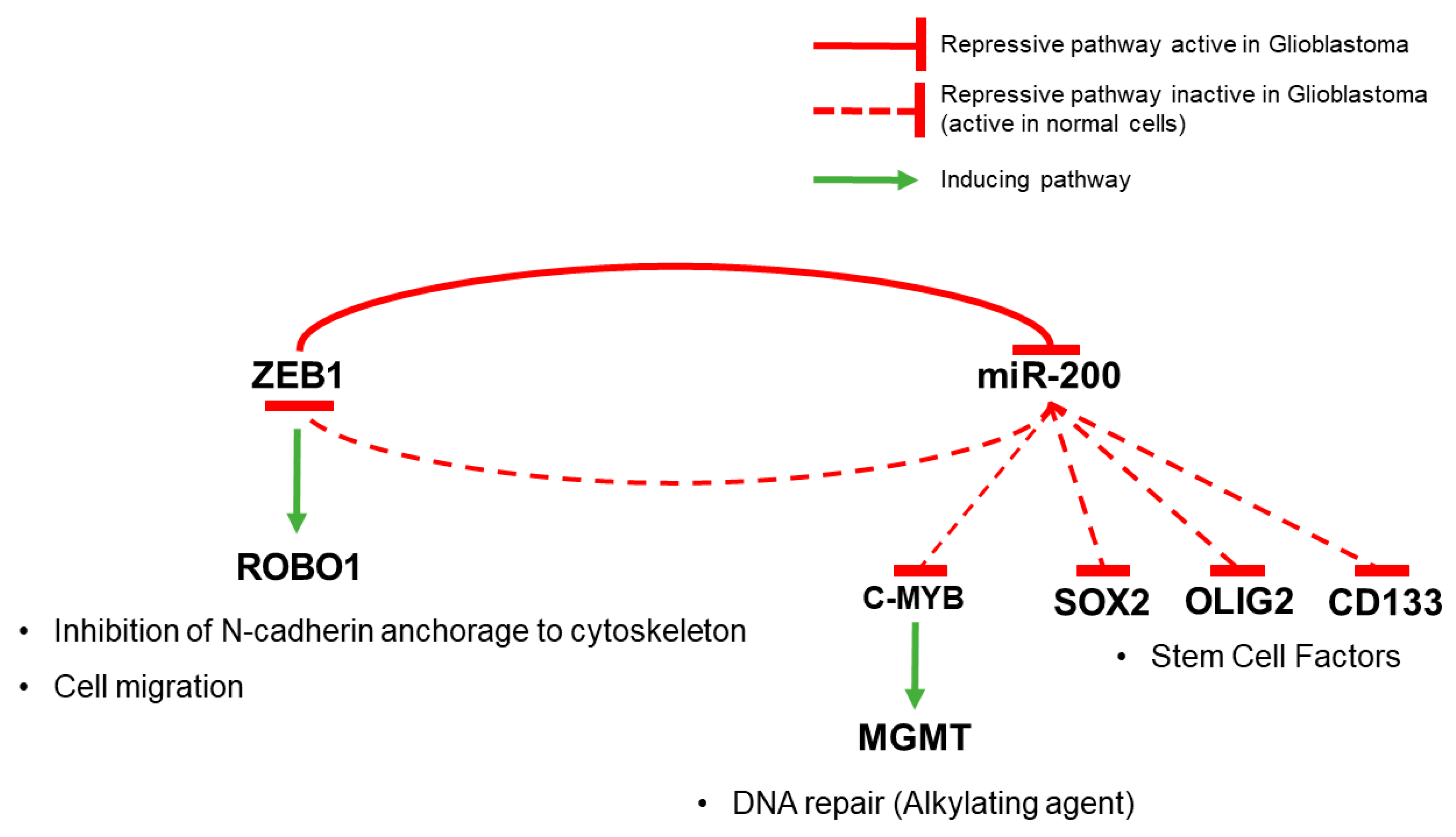
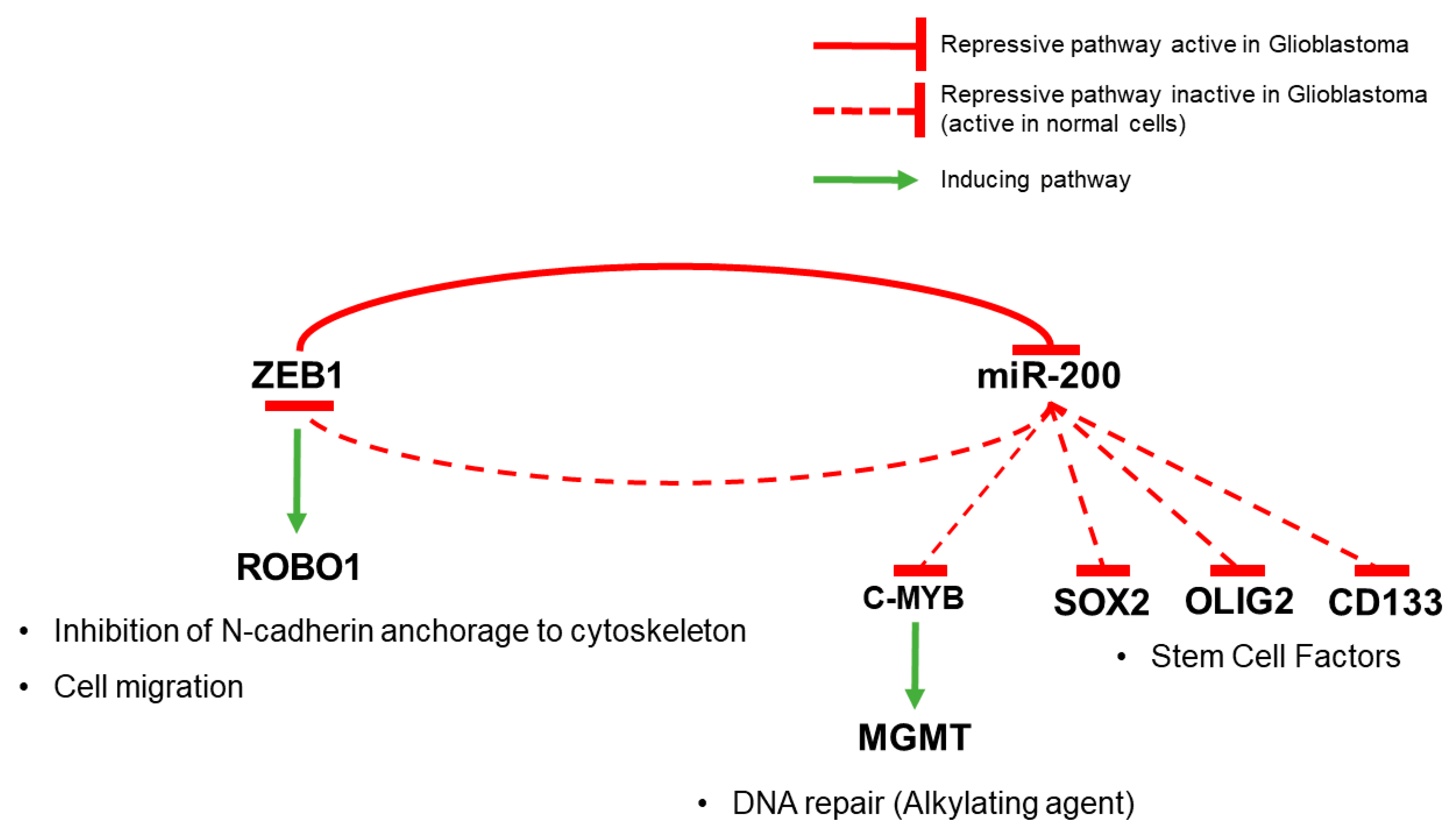
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.A.; Kruyt, F.A.E. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Kahlert, U.D.; Suwala, A.K.; Raabe, E.H.; Siebzehnrubl, F.A.; Suarez, M.J.; Orr, B.A.; Bar, E.E.; Maciaczyk, J.; Eberhart, C.G. ZEB1 Promotes Invasion in Human Fetal Neural Stem Cells and Hypoxic Glioma Neurospheres. Brain Pathol. 2015, 25, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Mahfooz, N.S.; Arregui, C.; Lilien, J.; Balsamo, J.; VanBerkum, M.F.A. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat. Cell Biol. 2002, 4, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-H.; Wu, K.-J. TWIST activation by hypoxia inducible factor-1 (HIF-1): Implications in metastasis and development. Cell Cycle 2008, 7, 2090–2096. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Morel, A.-P.; Hinkal, G.; Bastid, J.; Puisieux, A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 2010, 29, 3173–3184. [Google Scholar] [CrossRef] [PubMed]
- Castanon, I.; Baylies, M.K. A Twist in fate: Evolutionary comparison of Twist structure and function. Gene 2002, 287, 11–22. [Google Scholar] [CrossRef]
- Malek, R.; Gajula, R.P.; Williams, R.D.; Nghiem, B.; Simons, B.W.; Nugent, K.; Wang, H.; Taparra, K.; Lemtiri-Chlieh, G.; Yoon, A.R.; et al. TWIST1-WDR5-Hottip Regulates Hoxa9 Chromatin to Facilitate Prostate Cancer Metastasis. Cancer Res. 2017, 77, 3181–3193. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Nikhil, K.; Viccaro, K.; Chang, L.; Jacobsen, M.; Sandusky, G.; Shah, K. Aurora A-Twist1 axis promotes highly aggressive phenotypes in pancreatic carcinoma. J. Cell Sci. 2017, 130, 1078–1093. [Google Scholar] [CrossRef] [PubMed]
- Nordfors, K.; Haapasalo, J.; Mäkelä, K.; Granberg, K.J.; Nykter, M.; Korja, M.; Paavonen, T.; Haapasalo, H.; Soini, Y. Twist predicts poor outcome of patients with astrocytic glioma. J. Clin. Pathol. 2015, 68, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-H.; Wu, M.-Z.; Chiou, S.-H.; Chen, P.-M.; Chang, S.-Y.; Liu, C.-J.; Teng, S.-C.; Wu, K.-J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.-P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, M.C.; Tozer, K.R.; Silber, J.R.; Mikheeva, S.; Deng, M.; Morrison, R.S.; Manning, T.C.; Silbergeld, D.L.; Glackin, C.A.; Reh, T.A.; et al. TWIST is expressed in human gliomas and promotes invasion. Neoplasia 2005, 7, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2014, 33, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Kunigal, S.; Gondi, C.S.; Gujrati, M.; Lakka, S.S.; Dinh, D.H.; Olivero, W.C.; Rao, J.S. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. Int. J. Oncol. 2006, 29, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Menon, L.G.; Black, P.M.; Carroll, R.S.; Johnson, M.D. SNAI2/Slug promotes growth and invasion in human gliomas. BMC Cancer 2010, 10, 301. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011, 71, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, N.; Zhang, Y.; Yuan, F.; Pahuski, M.; Gibert, M.; Abounader, R. Role and Therapeutic Targeting of the HGF/MET Pathway in Glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.; Uno, M.; Marie, S.K.N.; Oba-Shinjo, S.M. LOX expression and functional analysis in astrocytomas and impact of IDH1 mutation. PLoS ONE 2015, 10, e0119781. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Phillips-Mason, P.J.; Burden-Gulley, S.M.; Kerstetter-Fogle, A.E.; Basilion, J.P.; Sloan, A.E.; Brady-Kalnay, S.M. Cadherin-11, a marker of the mesenchymal phenotype, regulates glioblastoma cell migration and survival in vivo. Mol. Cancer Res. 2012, 10, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef] [PubMed]
- Zagzag, D.; Lukyanov, Y.; Lan, L.; Ali, M.A.; Esencay, M.; Mendez, O.; Yee, H.; Voura, E.B.; Newcomb, E.W. Hypoxia-inducible factor 1 and VEGF upregulate CXCR4 in glioblastoma: Implications for angiogenesis and glioma cell invasion. Lab. Investig. 2006, 86, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Rempel, S.A.; Dudas, S.; Ge, S.; Gutiérrez, J.A. Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin. Cancer Res. 2000, 6, 102–111. [Google Scholar] [PubMed]
- Ehtesham, M.; Winston, J.A.; Kabos, P.; Thompson, R.C. CXCR4 expression mediates glioma cell invasiveness. Oncogene 2006, 25, 2801–2806. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Jiang, F.; Kalkanis, S.N.; Zhang, Z.G.; Zhang, X.-P.; DeCarvalho, A.C.; Katakowski, M.; Bobbitt, K.; Mikkelsen, T.; Chopp, M. SDF-1 and CXCR4 are up-regulated by VEGF and contribute to glioma cell invasion. Cancer Lett. 2006, 236, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, L.O.; Skerlj, R.T.; Bridger, G.J.; Schwartz, T.W. Molecular Interactions of Cyclam and Bicyclam Non-peptide Antagonists with the CXCR4 Chemokine Receptor. J. Biol. Chem. 2001, 276, 14153–14160. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Investig. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Mercurio, L.; Ajmone-Cat, M.A.; Cecchetti, S.; Ricci, A.; Bozzuto, G.; Molinari, A.; Manni, I.; Pollo, B.; Scala, S.; Carpinelli, G.; et al. Targeting CXCR4 by a selective peptide antagonist modulates tumor microenvironment and microglia reactivity in a human glioblastoma model. J. Exp. Clin. Cancer Res. 2016, 35, 55. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, Y.; Xue, Y.; Lv, W.; Zhang, Y.; He, S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim. Biophys. Sin. 2015, 47, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Schols, D.; Struyf, S.; Van Damme, J.; Esté, J.A.; Henson, G.; De Clercq, E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J. Exp. Med. 1997, 186, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; van der Ryst, E. CCR5 Antagonists: Host-Targeted Antivirals for the Treatment of HIV Infection. Antivir. Chem. Chemother. 2005, 16, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Blum, S.S.; Fung, H.B.; Bandres, J.C. Maraviroc: A CCR5-receptor antagonist for the treatment of HIV-1 infection. Clin. Ther. 2008, 30, 1228–1250. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.; Xolalpa, W.; Pestell, R.G. The potential to target CCL5/CCR5 in breast cancer. Expert Opin. Ther. Targets 2014, 18, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, Y.; Sasaki, S.; Mukaida, N.; Baba, T. Blockade of the chemokine receptor, CCR5, reduces the growth of orthotopically injected colon cancer cells via limiting cancer-associated fibroblast accumulation. Oncotarget 2016, 7, 48335–48345. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, E.C.; Hamilton, M.J.; Young, A.; Wadsworth, B.J.; LePard, N.E.; Lee, H.N.; Firmino, N.; Collier, J.L.; Bennewith, K.L. Maraviroc decreases CCL8-mediated migration of CCR5(+) regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology 2016, 5, e1150398. [Google Scholar] [CrossRef] [PubMed]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Ramiro, S.; Lladó, S.; Toro, S.; Coveñas, R.; Muñoz, M. Antitumor action of temozolomide, ritonavir and aprepitant against human glioma cells. J. Neurooncol. 2016, 126, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Ragel, B.T.; Couldwell, W.T.; Gillespie, D.L.; Jensen, R.L. Identification of hypoxia-induced genes in a malignant glioma cell line (U-251) by cDNA microarray analysis. Neurosurg. Rev. 2007, 30, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Méndez, O.; Zavadil, J.; Esencay, M.; Lukyanov, Y.; Santovasi, D.; Wang, S.-C.; Newcomb, E.W.; Zagzag, D. Knock down of HIF-1α in glioma cells reduces migration in vitro and invasion in vivo and impairs their ability to form tumor spheres. Mol. Cancer 2010, 9, 133. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, A.; Michiue, H.; Cheng, Y.; Uneda, A.; Tani, Y.; Nishiki, T.; Ichikawa, T.; Wei, F.-Y.; Tomizawa, K.; Matsui, H. Cyclin G2 promotes hypoxia-driven local invasion of glioblastoma by orchestrating cytoskeletal dynamics. Neoplasia 2013, 15, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Bennin, D.A.; Don, A.S.A.; Brake, T.; McKenzie, J.L.; Rosenbaum, H.; Ortiz, L.; DePaoli-Roach, A.A.; Horne, M.C. Cyclin G2 associates with protein phosphatase 2A catalytic and regulatory B’ subunits in active complexes and induces nuclear aberrations and a G1/S phase cell cycle arrest. J. Biol. Chem. 2002, 277, 27449–27467. [Google Scholar] [CrossRef] [PubMed]
- Arachchige Don, A.S.; Dallapiazza, R.F.; Bennin, D.A.; Brake, T.; Cowan, C.E.; Horne, M.C. Cyclin G2 is a centrosome-associated nucleocytoplasmic shuttling protein that influences microtubule stability and induces a p53-dependent cell cycle arrest. Exp. Cell Res. 2006, 312, 4181–4204. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Post, D.E.; Pieper, R.O.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005, 65, 1406–1413. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Hu, F.; Huang, R.; Mackman, N.; Horowitz, J.M.; Jensen, R.L.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006, 66, 7067–7074. [Google Scholar] [CrossRef] [PubMed]
- Belting, M.; Ahamed, J.; Ruf, W. Signaling of the tissue factor coagulation pathway in angiogenesis and cancer. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1545–1550. [Google Scholar] [CrossRef] [PubMed]
- Hamada, K.; Kuratsu, J.; Saitoh, Y.; Takeshima, H.; Nishi, T.; Ushio, Y. Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer 1996, 77, 1877–1883. [Google Scholar] [CrossRef]
- Harter, P.N.; Dützmann, S.; Drott, U.; Zachskorn, C.; Hattingen, E.; Capper, D.; Gessler, F.; Senft, C.; Seifert, V.; Plate, K.H.; et al. Anti-tissue factor (TF9–10H10) treatment reduces tumor cell invasiveness in a novel migratory glioma model. Neuropathology 2013, 33. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Ruf, W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Schaffner, F.; Ruf, W. Tissue factor and protease-activated receptor signaling in cancer. Semin. Thromb. Hemost. 2008, 34, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Gessler, F.; Voss, V.; Dützmann, S.; Seifert, V.; Gerlach, R.; Kögel, D. Inhibition of tissue factor/protease-activated receptor-2 signaling limits proliferation, migration and invasion of malignant glioma cells. Neuroscience 2010, 165, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. Interleukin-13 receptor alpha 2, EphA2, and Fos-related antigen 1 as molecular denominators of high-grade astrocytomas and specific targets for combinatorial therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol. Cancer Res. 2005, 3, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Fokas, E.; Bieker, M.; Rose, F.; Rexin, P.; Zhu, Y.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.-X. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol. Rep. 2008, 19, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, B.; Liu, X.; Gopal, U.; Isaacs, J.S. Hsp90 is an essential regulator of EphA2 receptor stability and signaling: Implications for cancer cell migration and metastasis. Mol. Cancer Res. 2009, 7, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Li, D.-Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Burnett, E.; Kinch, M.; Simon, E.; Wang, B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2000, 2, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Macrae, M.; Neve, R.M.; Rodriguez-Viciana, P.; Haqq, C.; Yeh, J.; Chen, C.; Gray, J.W.; McCormick, F. A conditional feedback loop regulates Ras activity through EphA2. Cancer Cell 2005, 8, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS ONE 2011, 6, e17649. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Inhibitors of the heat shock response: Biology and pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Chigurupati, S.; Venkataraman, R.; Barrera, D.; Naganathan, A.; Madan, M.; Paul, L.; Pattisapu, J.V.; Kyriazis, G.A.; Sugaya, K.; Bushnev, S.; et al. Receptor channel TRPC6 is a key mediator of Notch-driven glioblastoma growth and invasiveness. Cancer Res. 2010, 70, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Alptekin, M.; Eroglu, S.; Tutar, E.; Sencan, S.; Geyik, M.A.; Ulasli, M.; Demiryurek, A.T.; Camci, C. Gene expressions of TRP channels in glioblastoma multiforme and relation with survival. Tumor Biol. 2015, 36, 9209–9213. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Protein/Pathway | Role |
|---|---|
| Periostin | Recruitment of M2 tumor-associated macrophages which are tumor-supportive and immunosuppressive [130]. |
| Fibronectin 1 | Promotes cell cohesion and invasion of basement membrane [131]. |
| SPARC | Upholds ECM degradation through the uPA-uPAR pathway and stimulates survival, proliferation and invasion via the PI3K pathway [132]. |
| SNAI2 | Inhibits miR-34 activity, a regulatory microRNA for differentiation, promoting stemness [133,134]. |
| ID1 | Activates tumor progression pathways, such as Snail, ERK1/2 and AKT, and promotes stem cell self-renewal transcription factors Sox2, Oct3/4 and Nanog [135]. |
| HGF | Promotes survival, proliferation, transformation and invasion through activation of the PI3K/AKT, STAT3/JNK, SOS/RAS/ERK/MAPK pathways [136]. |
| LOX | Activates HIF-1α (via AKT pathway in a positive feed-back loop), FAK and VEGF gene expression [137]. |
| Cadherin 11 | Regulates cell-cell interactions and survival, and promotes cell migration [138]. |
| BMI1/EZH2 | Part of the Polycomb repressive complex 1 and 2, in that order, promotes gene silencing of the P16 and P14 tumor suppressors, and inhibits cancer stem cells differentiation [139]. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteiro, A.R.; Hill, R.; Pilkington, G.J.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. https://doi.org/10.3390/cells6040045
Monteiro AR, Hill R, Pilkington GJ, Madureira PA. The Role of Hypoxia in Glioblastoma Invasion. Cells. 2017; 6(4):45. https://doi.org/10.3390/cells6040045
Chicago/Turabian StyleMonteiro, Ana Rita, Richard Hill, Geoffrey J. Pilkington, and Patrícia A. Madureira. 2017. "The Role of Hypoxia in Glioblastoma Invasion" Cells 6, no. 4: 45. https://doi.org/10.3390/cells6040045
APA StyleMonteiro, A. R., Hill, R., Pilkington, G. J., & Madureira, P. A. (2017). The Role of Hypoxia in Glioblastoma Invasion. Cells, 6(4), 45. https://doi.org/10.3390/cells6040045