A Phospho-SIM in the Antiviral Protein PML is Required for Its Recruitment to HSV-1 Genomes

Abstract
:1. Introduction
2. Experimental Section
2.1. Cells
2.2. Plasmids
| S8A forward | 5'-ggagcctgcacccgcccgaGctccgaggccccagc-3' |
| S8E forward | 5'-ggagcctgcacccgcTcgaGAAccgaggccccagcag-3' |
| S8 reverse | 5'-atggcgttgtcatcgtcatccttg-3' |
| T28;S36/38/40A forward | 5'-atgcctccccccgagGcGccctctgaaggccgccagcccGCTcccG-CGccGGCccctacagagcgagc-3' |
| T28;S36/38/40E forward | 5'-atgcctccccccgagGAAccctctgaaggccgGcagcccGAAccc-GAAcccGAAcctacagagcgagcc-3' |
| T28;S36/38/40 reverse | 5'-ggtgggctcctggggccgggcggg-3' |
| S117A | 5'-agtctgcagcggcgcTtAGcggtgtaccggcaga-3' |
| S117E forward | 5'-tctgcagcggcgcctCGAggtgtaccggcagat-3' |
| S117E reverse | 5'-ctctcgaaaaagacgttatccagggcggg-3' |
| S399/403;T409A forward | 5'-aaGgaCgcGgcGgtGGCTaaAaaGgccGCcccagaggctgcca-gcGctcccagggacccta-3' |
| S399/403;T409E forward | 5'-aaGgaCgcGgcGgtGGAGaaAaaGgccGAAccTgaggctgcc-agcGAAcccagggaccctatt-3' |
| S399/403;T409 reverse | 5'-cccctgggtgatgcaagagctgag-3' |
| S480;T482;S493A forward | 5'-cagaagaggaagtgcGCGcagGcccagtgccccaggaaggtcatca-agatggagGctgaggaggggaagg-3' |
| S480;T482;S493E forward | 5'-cagaagaggaagtgcGAGcagGAAcagtgcccTaggaaggtcatc-aagatggagGAAgaggaggggaaggag-3' |
| S480;T482;S493 | 5'-ggctgtcgttgtattggagacatc-3' |
| S504/505A forward | 5'-ggcaaggttggctcgAGCcGccccggagcagccca-3' |
| S504/505A reverse | 5'-tccttcccctcctcagactccatc-3' |
| S504/505E forward | 5'-gcaaggttggctcggGAAGAGccggagcagcccagg-3' |
| S505/505E reverse | 5'-ctccttcccctcctcagactccat-3' |
| S518A forward | 5'-cagcacctccaaggcagtcGcaccTccTcacctggatggaccg-3' |
| S518E forward | 5'-cagcacctccaaggcCgtGGAaccaccccacctgga-3' |
| S518 reverse | 5'-ggcctgggctgctccgg-3' |
| S527/530A | 5'-ctggatggaccgcctGCccccaggGCccccgtcataggaag-3' |
| S527/530E forward | 5'-ctggatggaccgcctGAAccTaggGAAcccgtcataggaagt-3' |
| S527/530E reverse | 5'-gtggggtggtgagactgccttggag-3' |
| S560/561/562/565A forward | 5′-cgcgttgtggtgatcGCGGCcGcggaagacGcagatgccgaaaact-3′ |
| S560/561/562/565A reverse | 5'-ttcctctgcctccccggcgccact-3' |
| S560/561/562/565E forward | 5'-ggaggcagaggaacgTgttgtggtgatcGAAGAAGAggaagacGA-Agatgccgaaaactcg-3' |
| S560/561/562/565E reverse | 5'-ccggcgccactggccacgtggttg-3' |
| S565A forward | 5'-agcagctcggaagacGcagatgccgaaaact-3' |
| S565A reverse | 5'-gatcaccacaacgcgttcctctgc-3' |
| S565E forward | 5'-gttgtggtgatcagcTCTtcggaagacGAagatgccgaaaactc-3' |
| S565E reverse | 5'-gcgttcctctgcctccccggcgcc-3' |
| V556/557/558A;I559S forward | 5'-aggcagaggaacgcgCtgCggCTaGcagcagctcggaaga-3' |
| Δ476–490 forward | 5'-gaggcaaggttggctcgga-3' |
| Δ476–490 reverse | 5'-ctgggctgtcgttgtattggaga-3' |
| K65R | 5'-atgccaggcggaagcGCGCtgcccgaagctgctg-3' |
| K160R forward | 5'-acaccagtggttcctACGTcaTgaAgcccggcccctagca-3' |
| K160R reverse | 5'-gcctcgaagcacttggcgcag-3' |
| K490R | 5'-cccaggaaggtcatcCGgatggagtctgagga-3' |
| K616R forward | 5'-gttttctttgacctcCGgattgacaatgaaa-3' |
| K616R reverse | 5'-cagaggtctgtcttctgcttggg-3' |
2.3. Viruses
2.4. PML Immunoprecipitation
2.5. Mass Spectrometry (MS)
2.6. Western Blots
2.7. Immunofluorescence/Fluorescence Microscopy
3. Results and Discussion
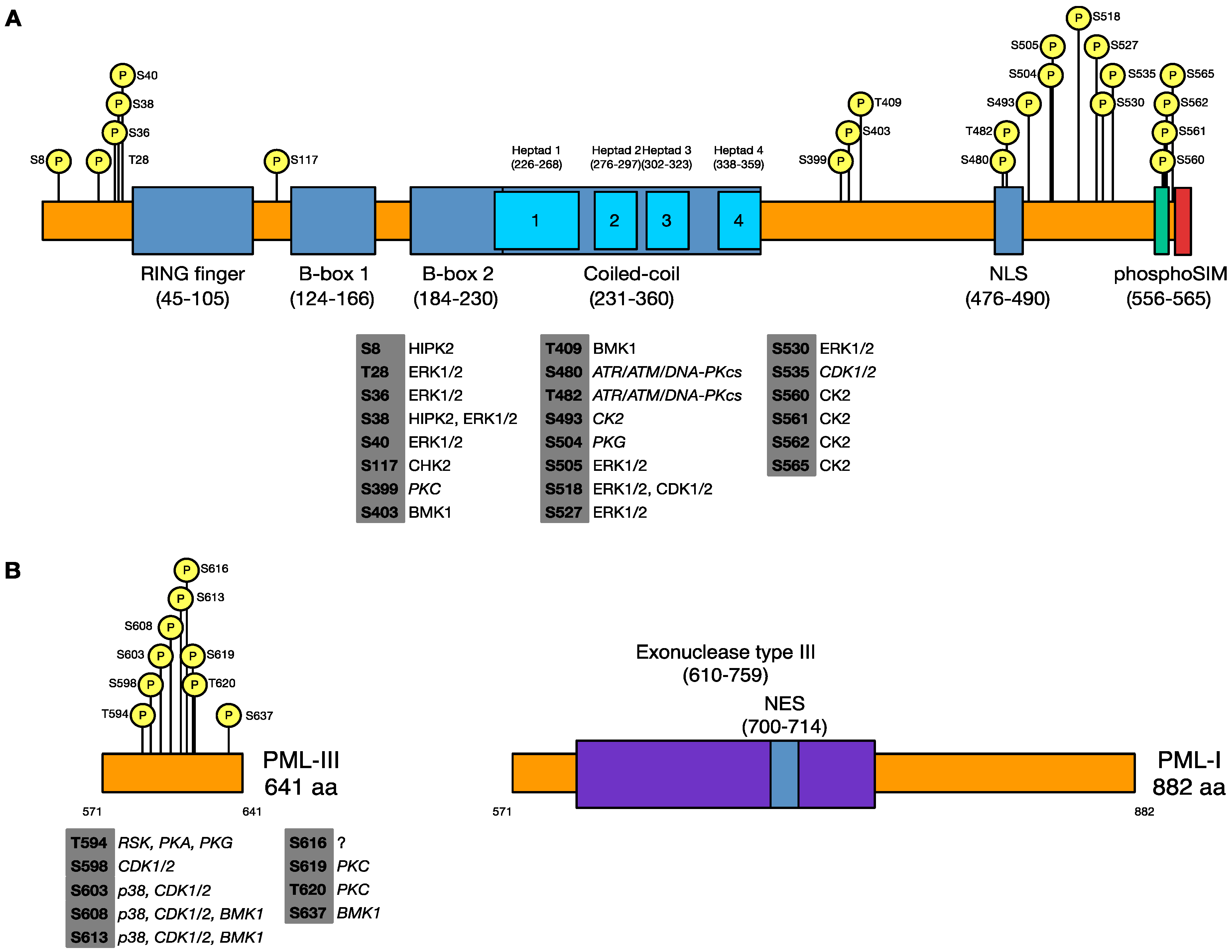
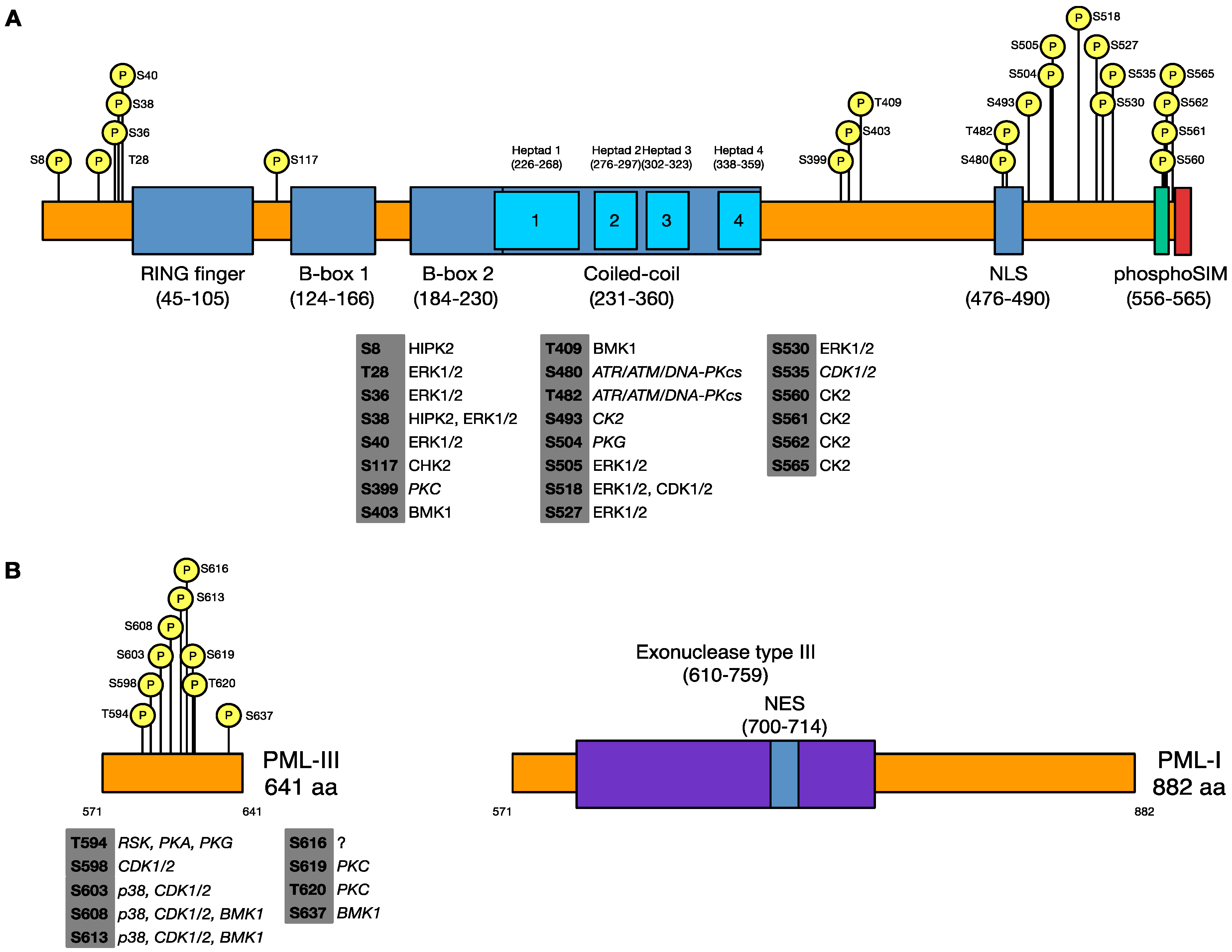
3.1. Identification of Phosphorylated Residues of PML in Uninfected and HSV-1 Infected Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uninfected | HSV-1 Infected | ||
|---|---|---|---|
| Coverage gaps | Coverage gaps | ||
| S399 | 1–44 | S403 | 1–7 |
| S403 | 57–86 | T409 | 18–44 |
| T409 | 147–149 | S505 | 57–97 |
| S480 | 212–216 | S518 | 132–153 |
| T482 | S527 | 206–216 | |
| S493 | S565 | 336–337 | |
| S504 | T594* | 395–399 | |
| S505 | S598* | 478–486 | |
| S518 | S603* | ||
| S527 | S613* | ||
| T594* | S637* | ||
| S598* | |||
| S603* | |||
| S608* | |||
| S613* | |||
| S616* | |||
| S619* | |||
| T620* | |||
| S637* | |||
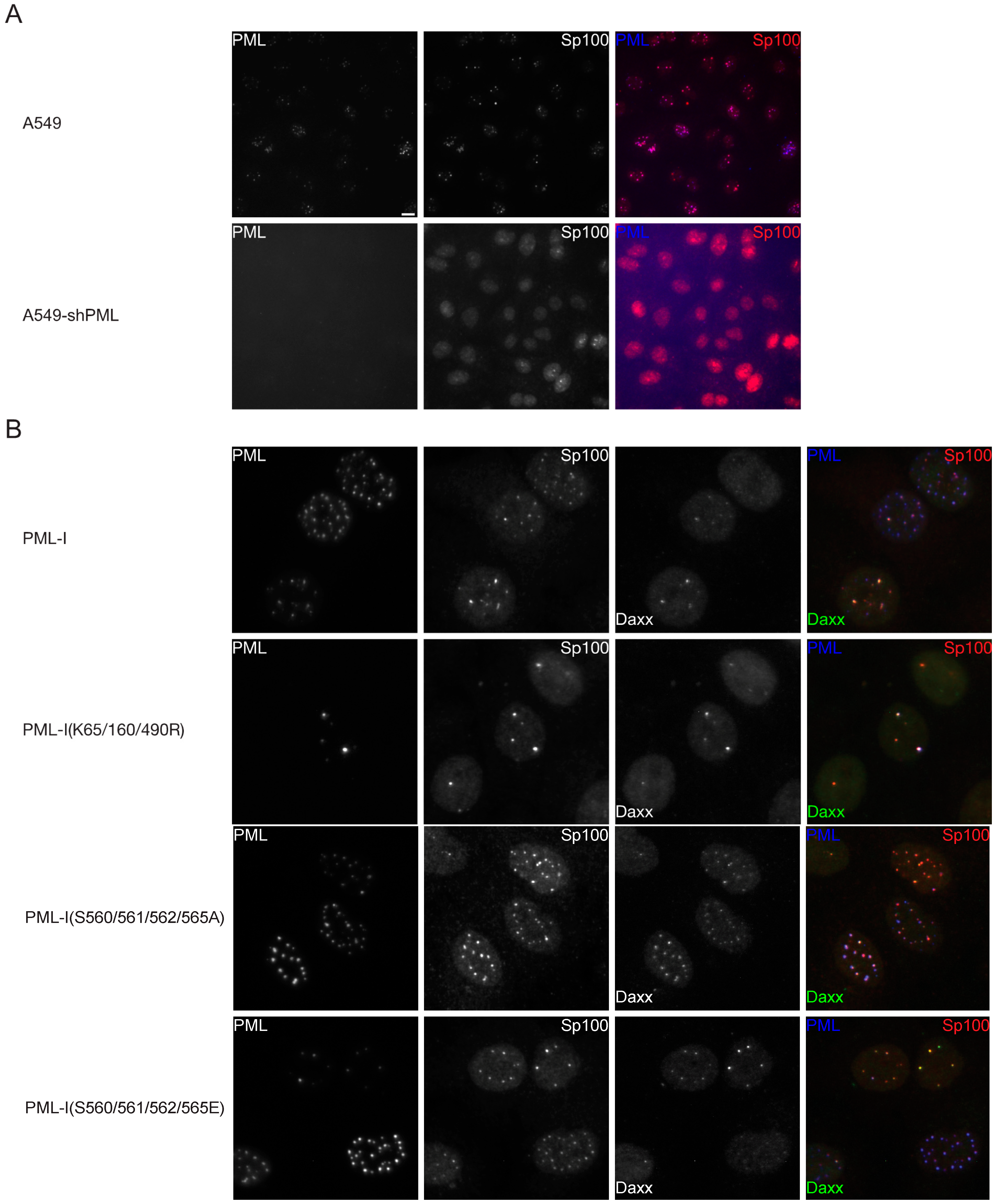
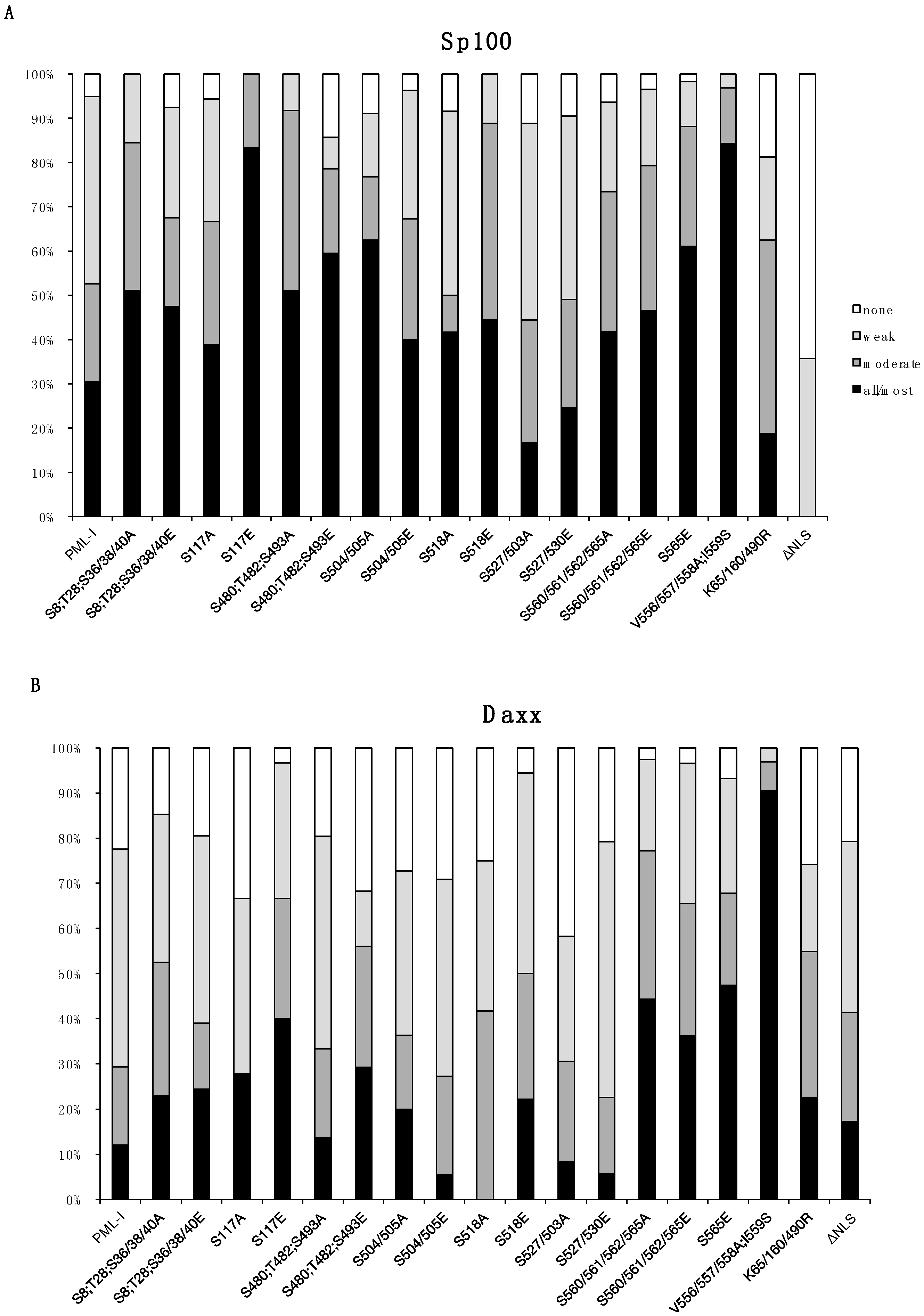
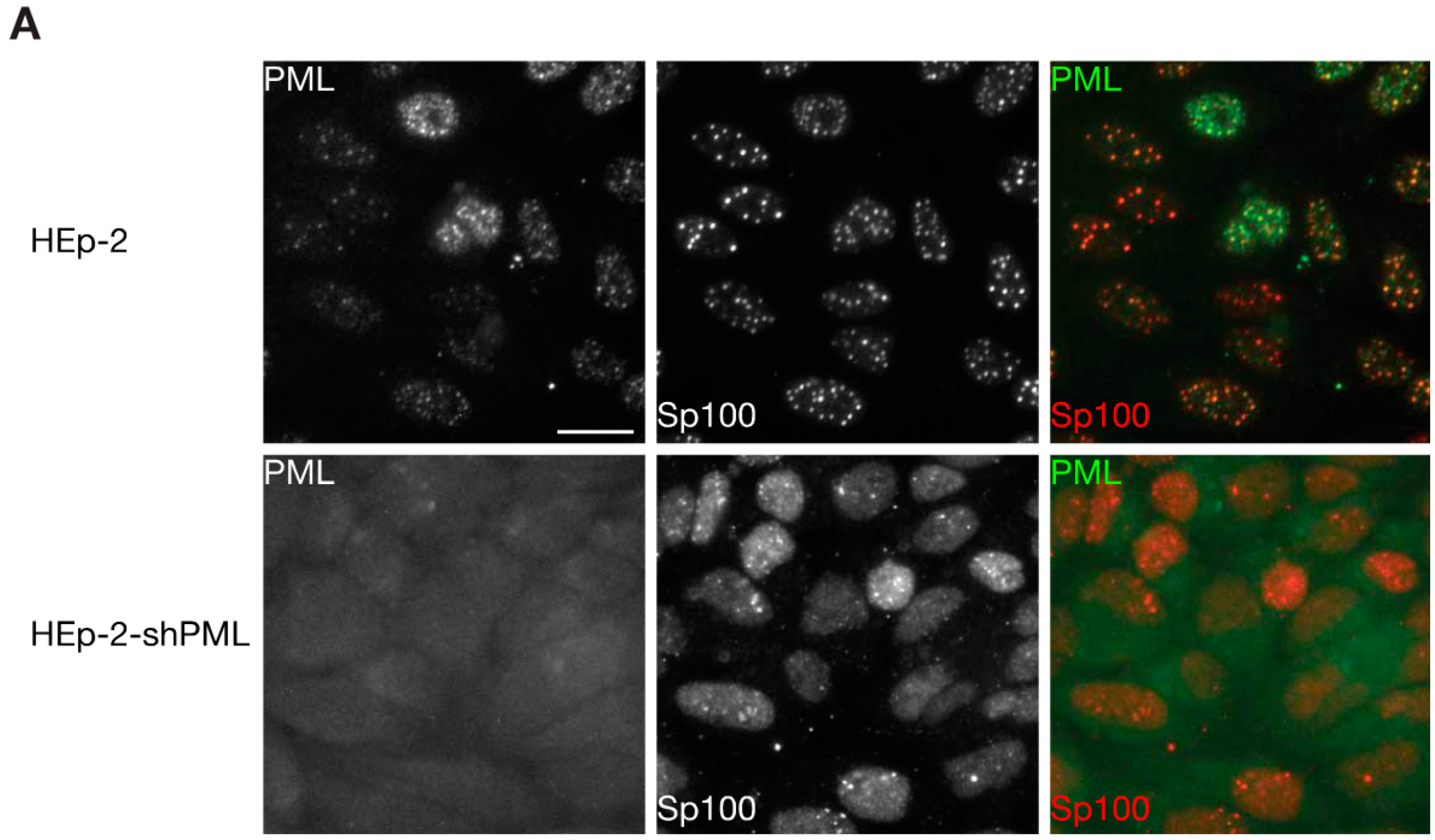
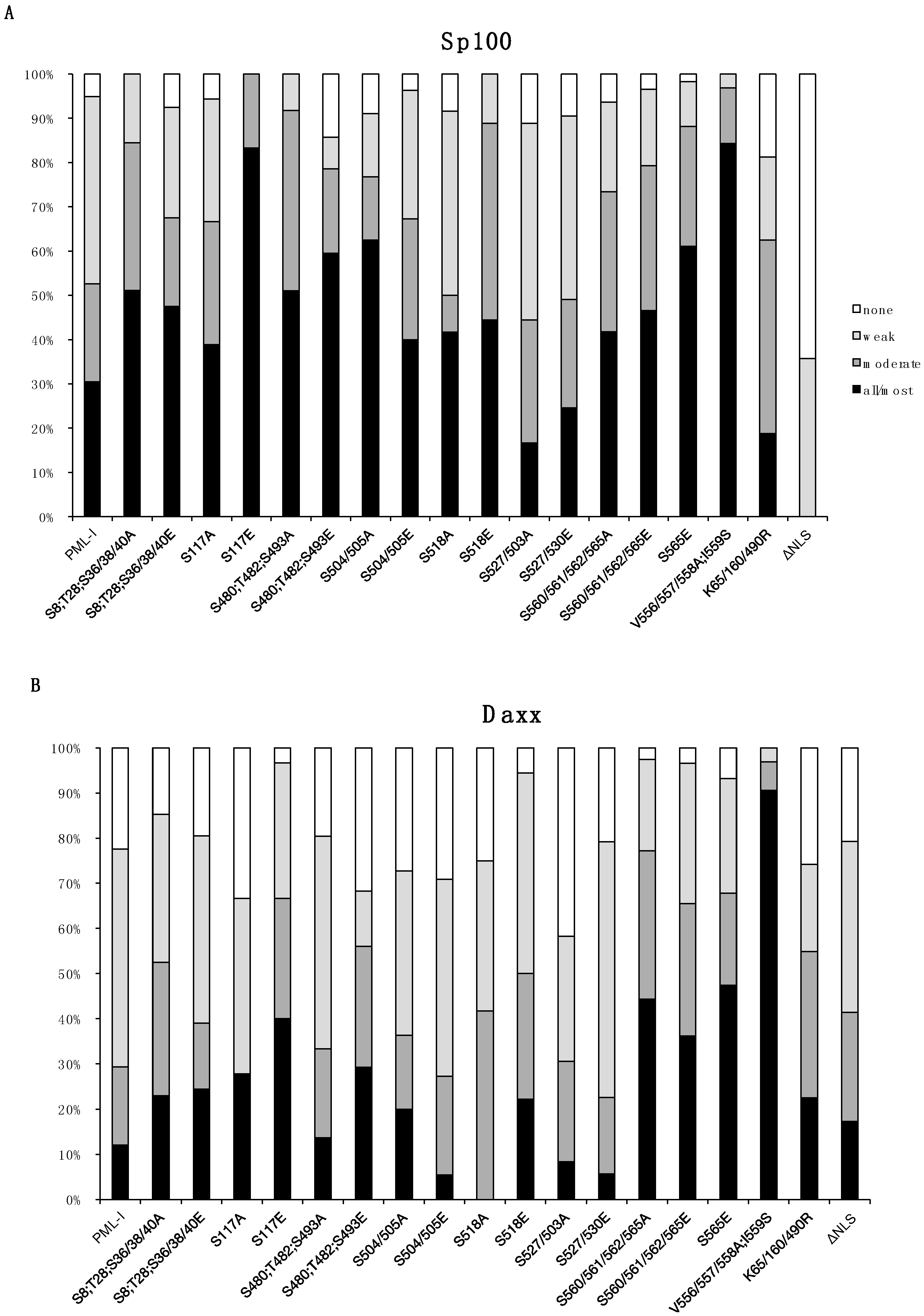
3.2. Phosphorylation at Sites near the SIM Alter ND10 Morphology and Influences Sp100 and Daxx Recruitment to ND10s

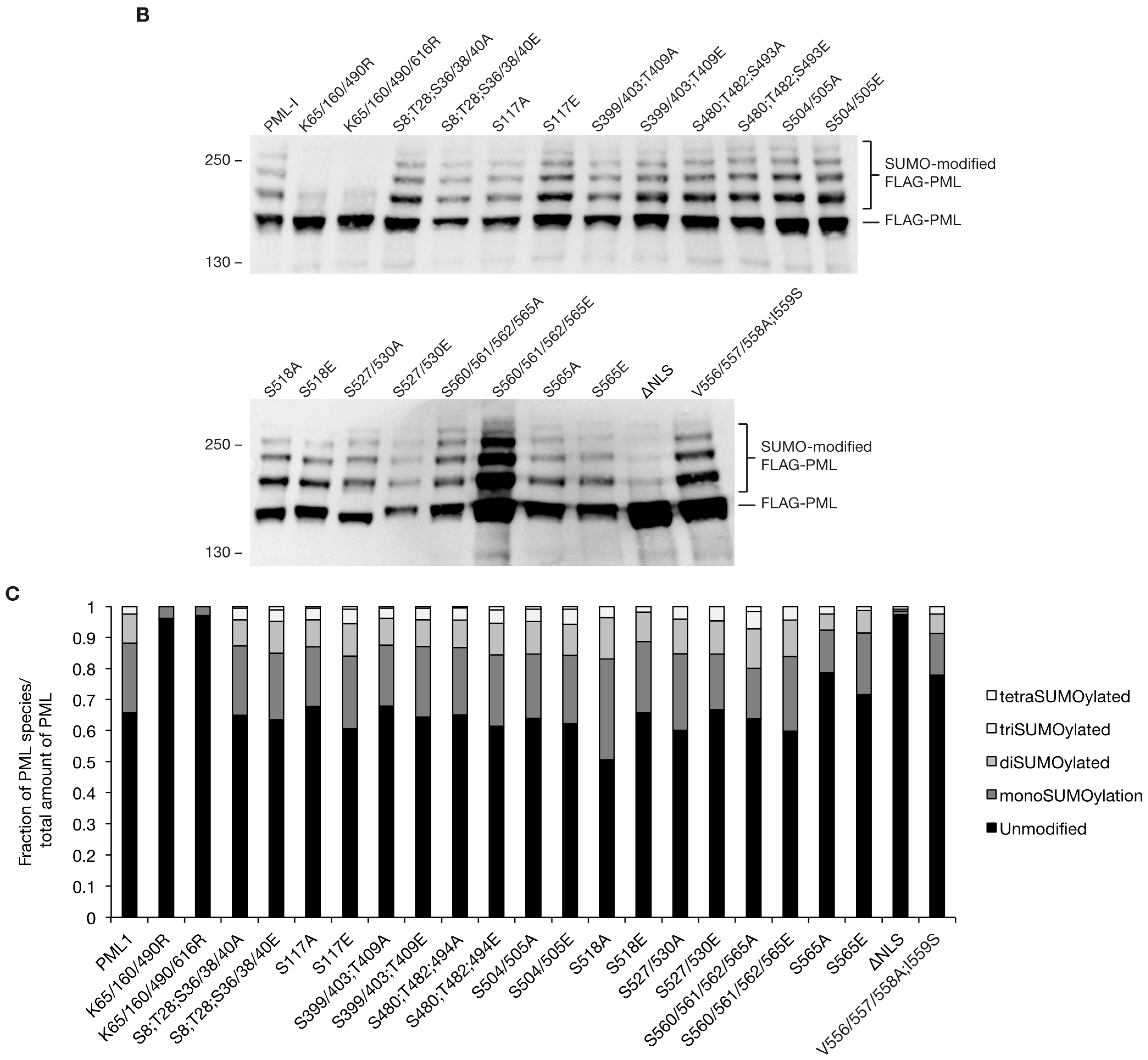
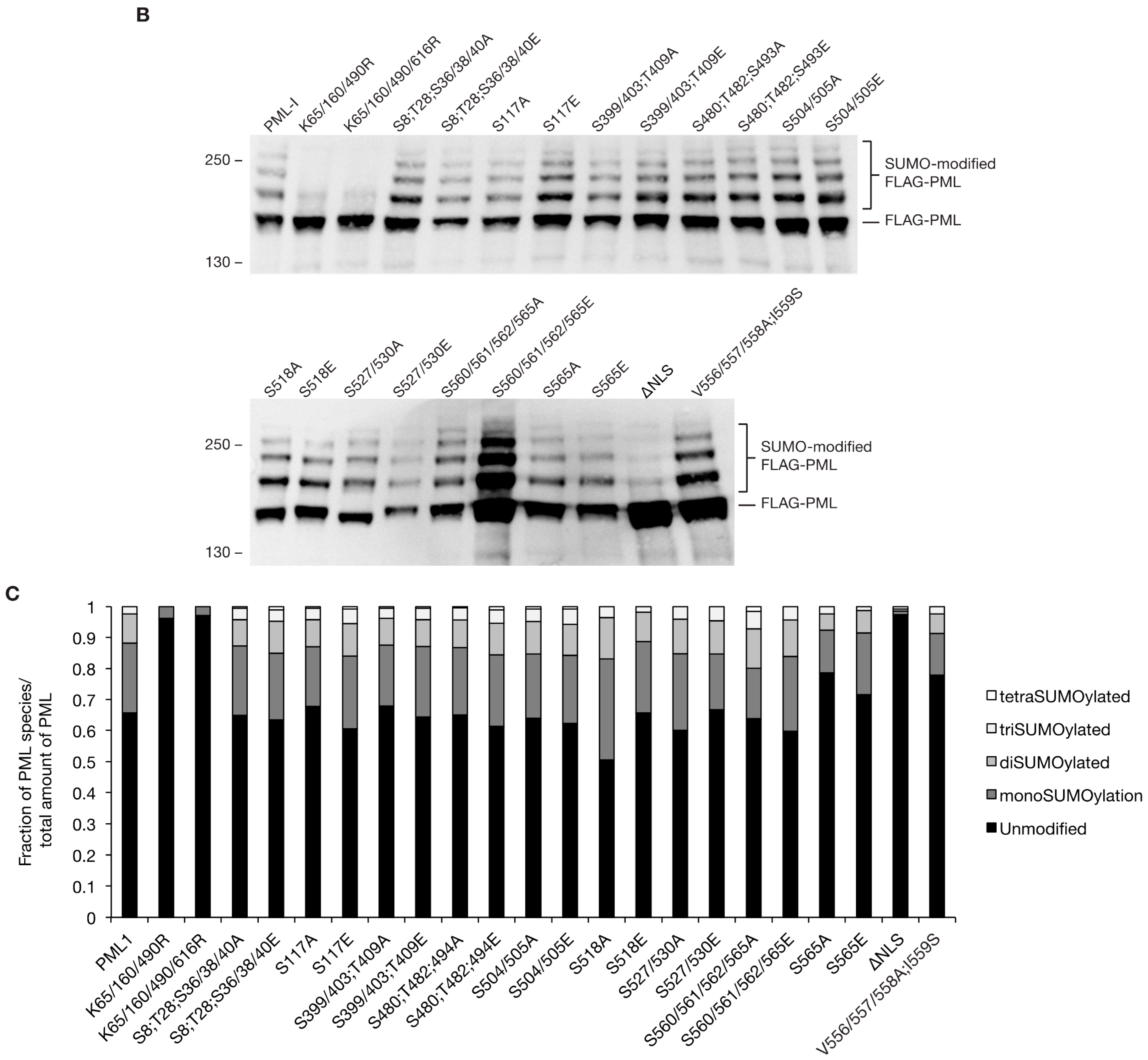
3.3. Phosphorylation does not Largely Impact SUMOylation Levels

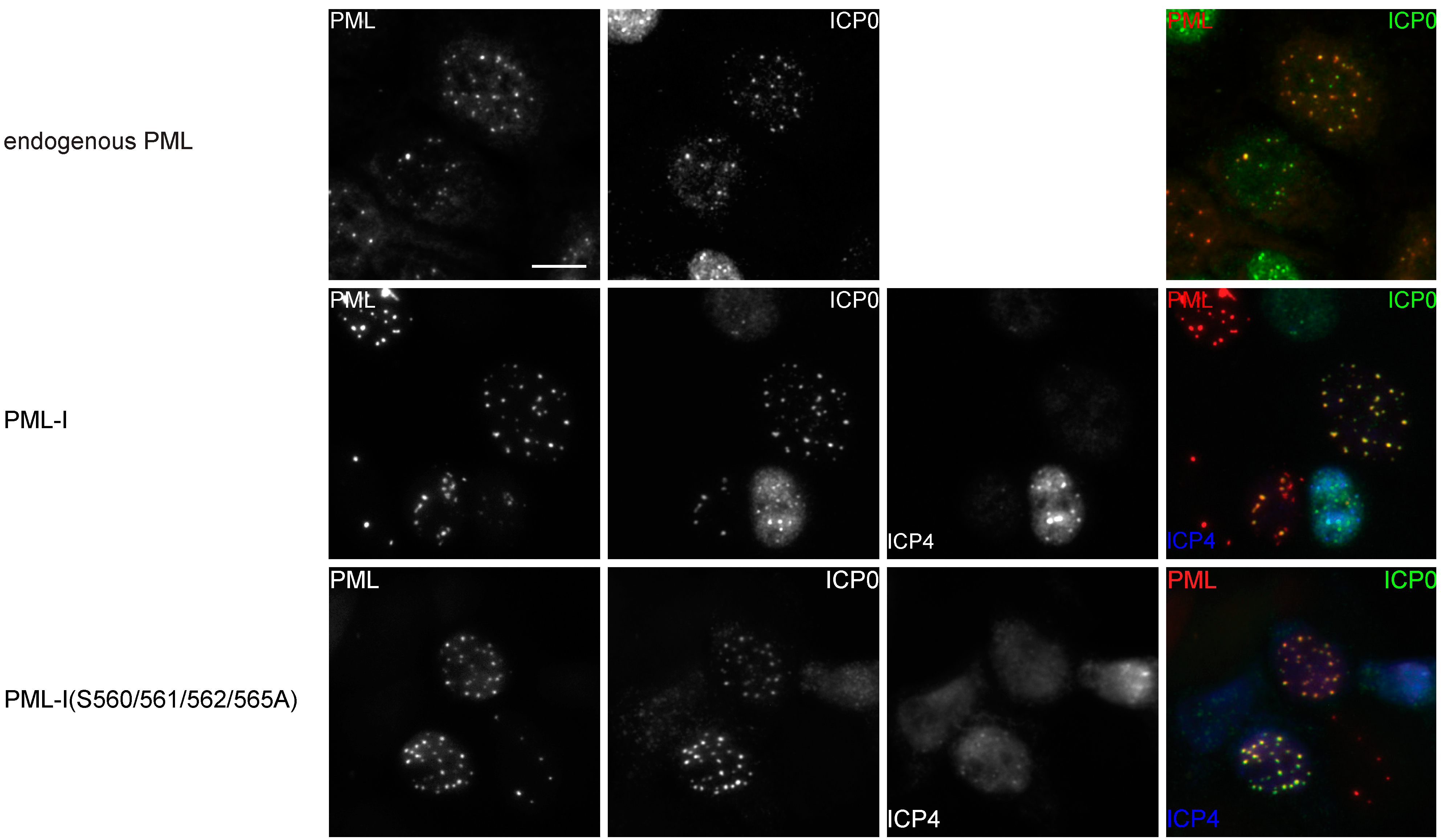
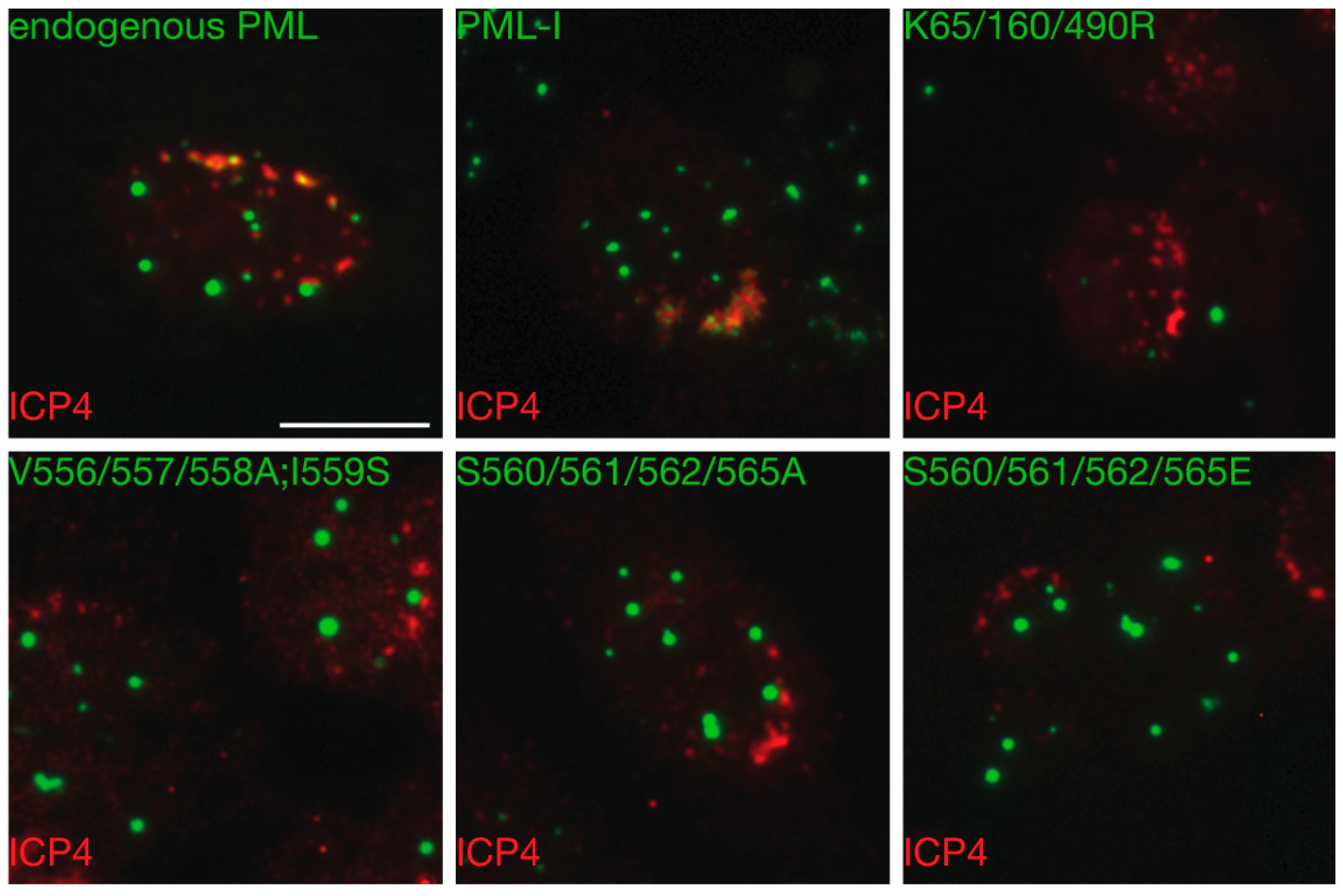
3.4. Phosphorylation is not Required for the Colocalization of PML-I and ICP0
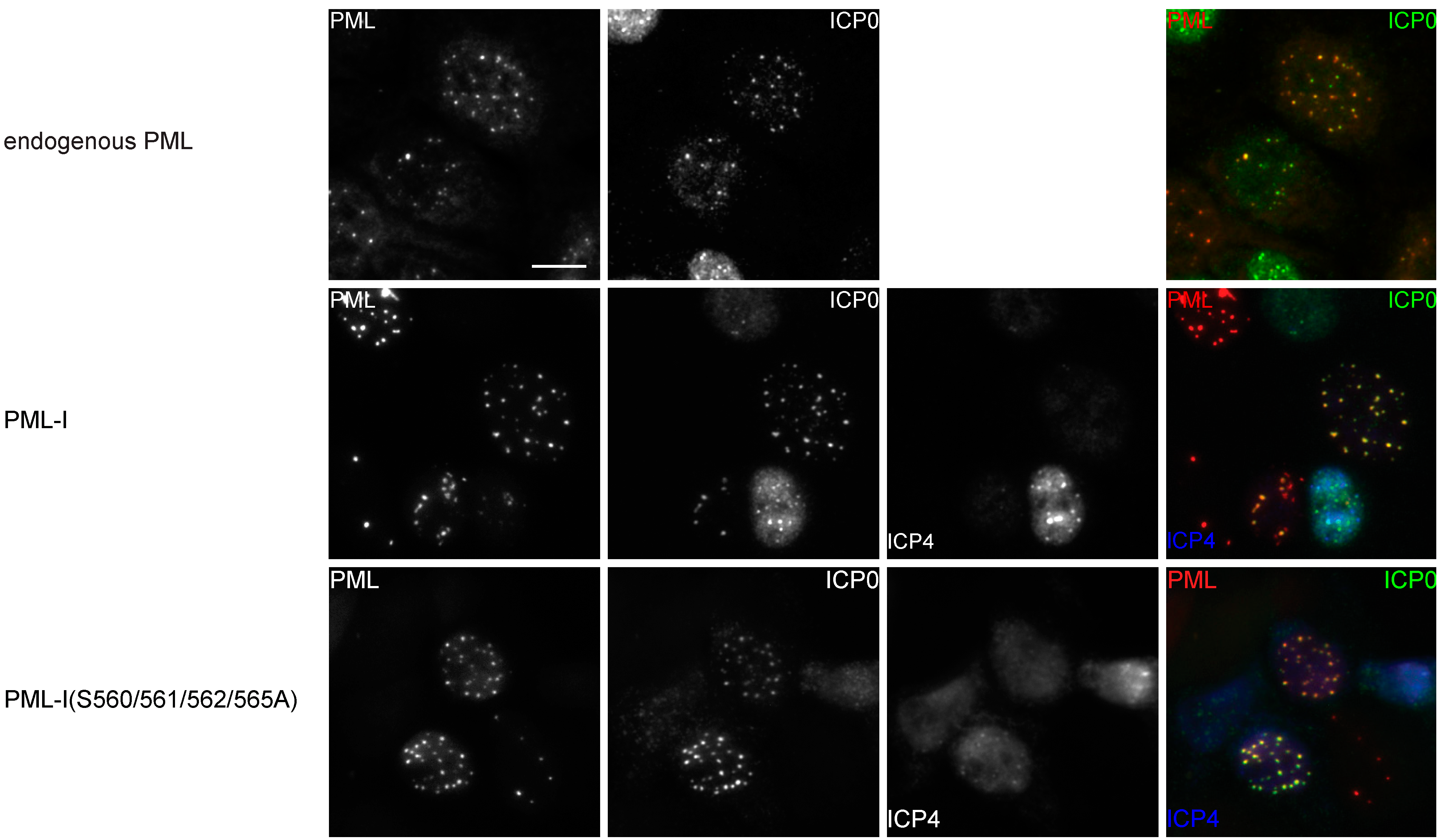
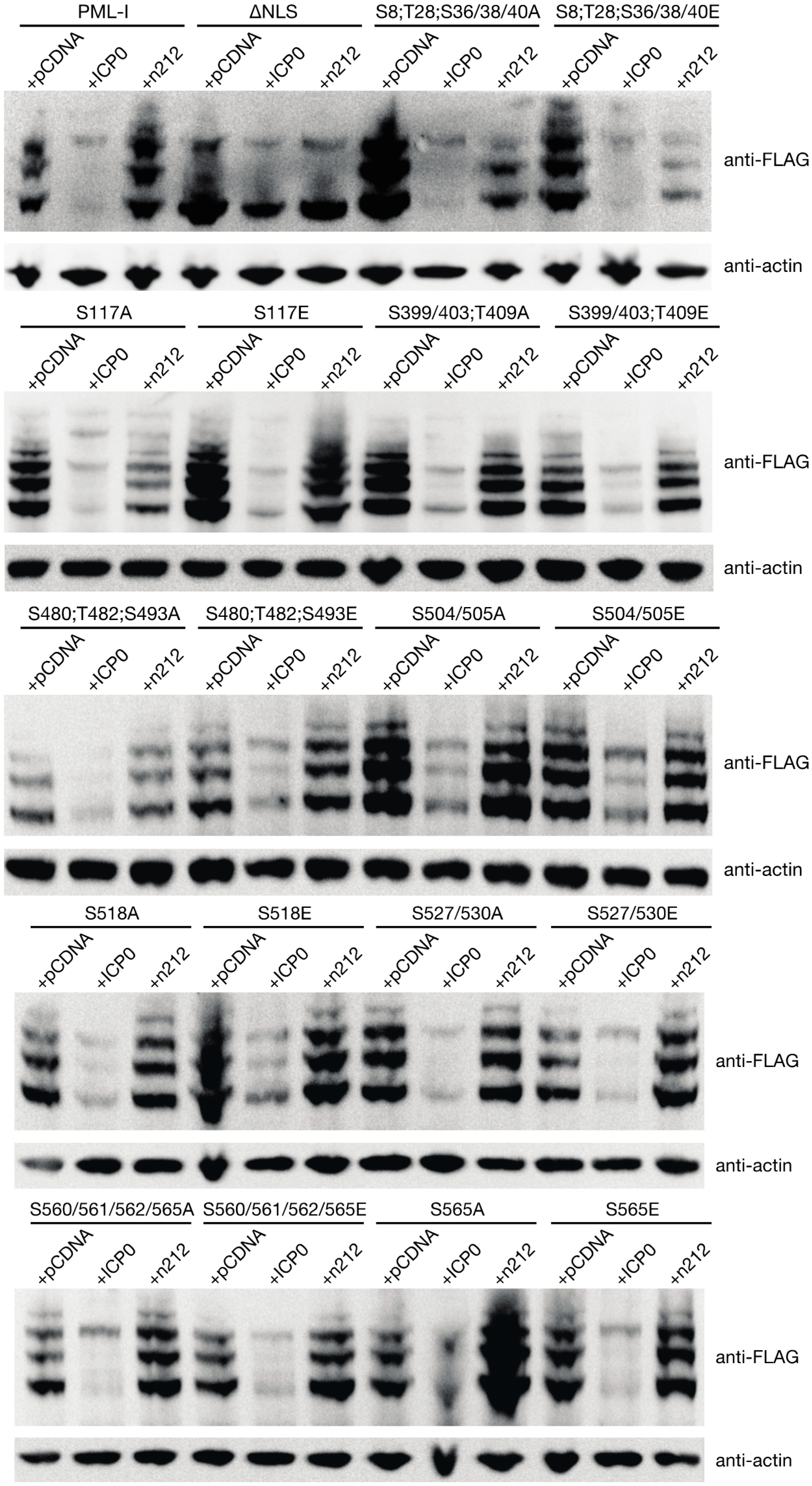
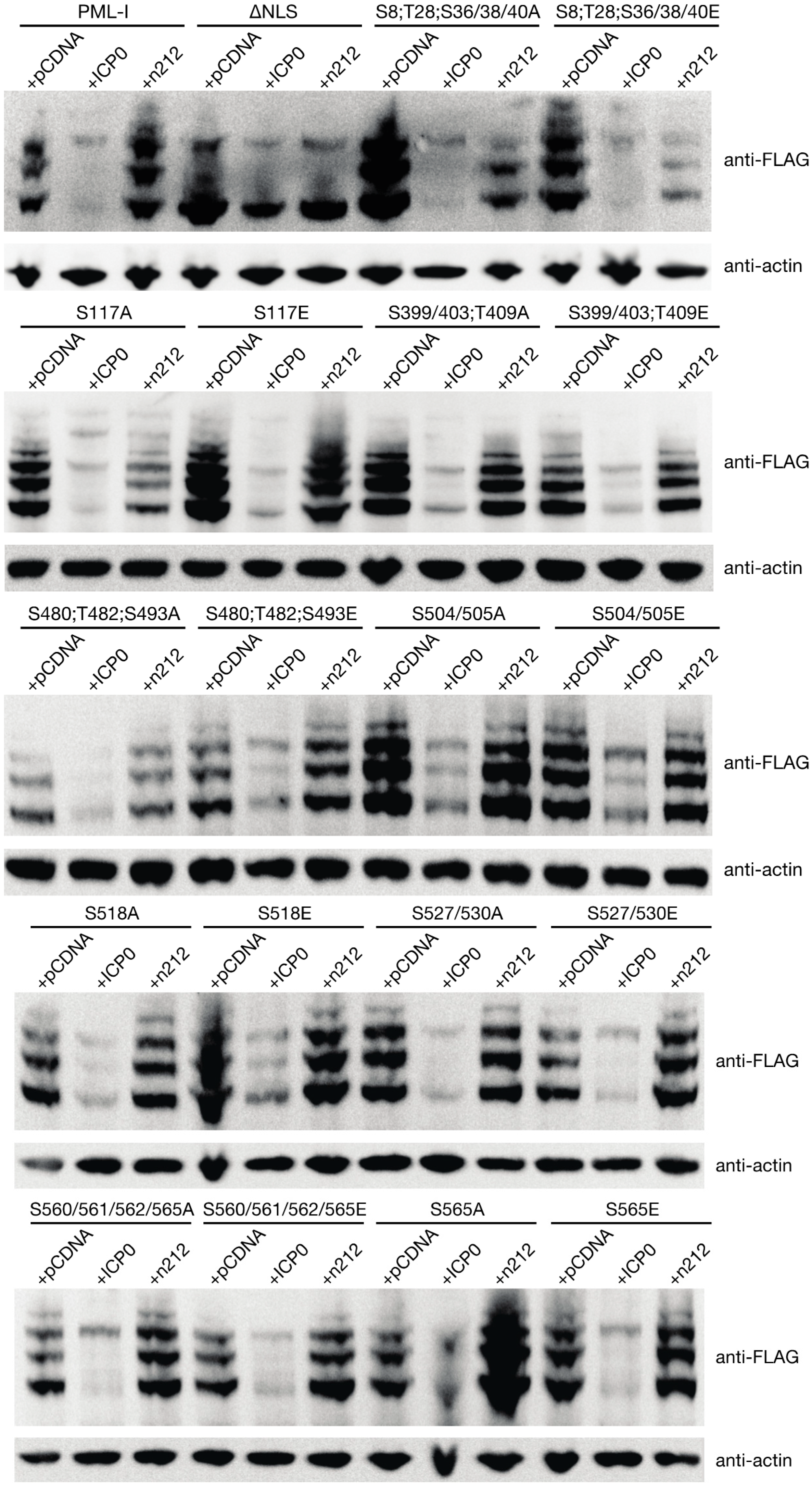
3.5. PML Phosphorylation has Minor Effects on ICP0-Induced Degradation
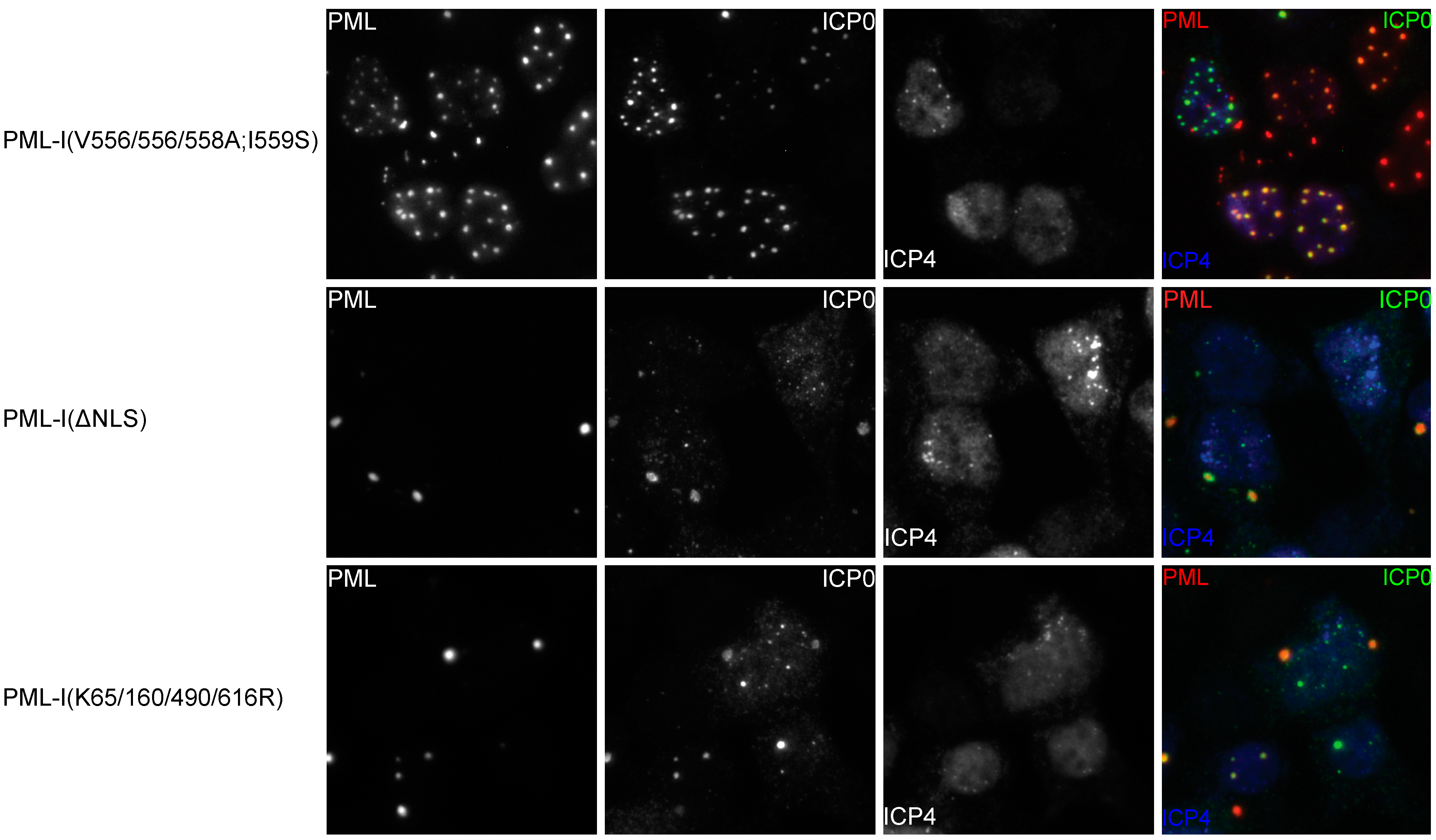
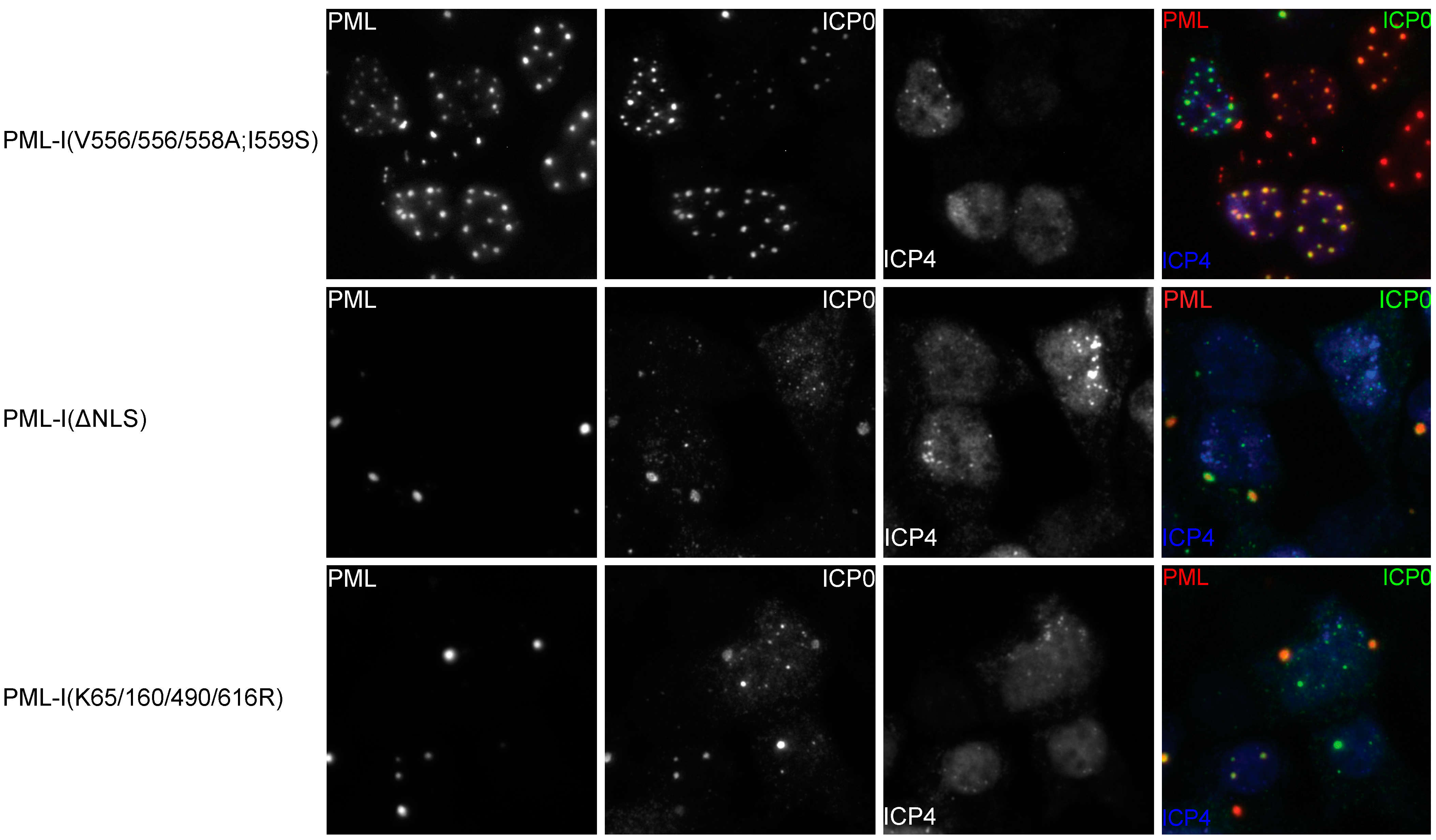
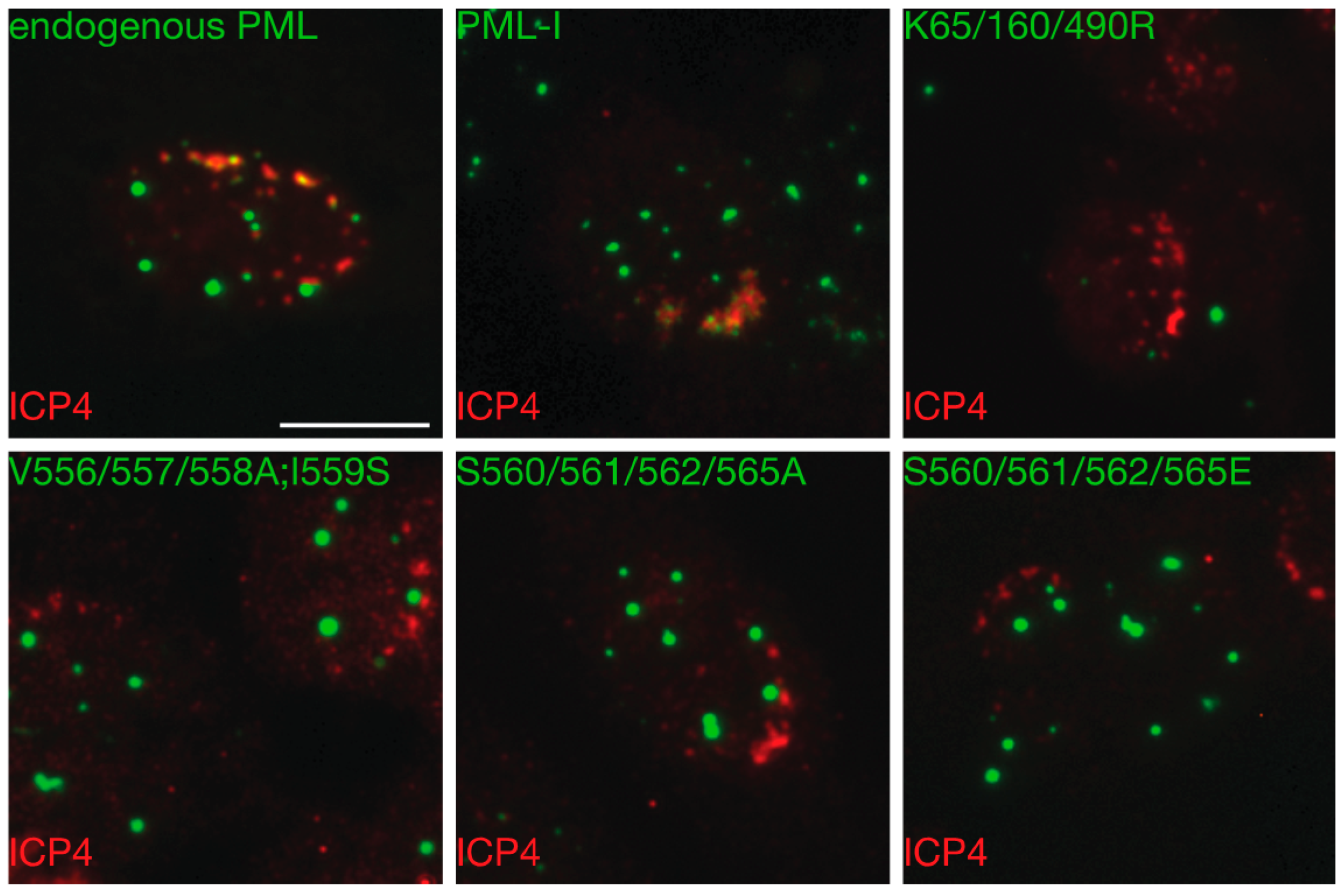
3.6. Mutation of the Phosphoacceptor Sites in the Phospho-SIM of PML-I Prevents its Recruitment to Incoming Viral Genomes
4. Conclusions
| Incoming Genome Recruitment | ||||||||
|---|---|---|---|---|---|---|---|---|
| ++ | +++ | ND | ++ | ND | +++ | +++ | ND | +++ |
| + | ++ | - | - | ++ | +++ | +++ | - | +++ |
| ++ | - | + | - | - | +++ | +++ | - | +++ |
| ++ | ++ | + | + | ++ | +++ | + | - | +++ |
| ++ | ND | + | ND | ++ | +++ | ND | - | +++ |
| ++ | ++ | + | - | ++ | +++ | +++ | - | +++ |
| +++ | ND | ++ | ND | ++ | +++ | ND | - | +++ |
| ++ | ++ | - | - | ++ | +++ | +++ | - | +++ |
| ++ | ND | + | ND | ++ | +++ | ND | + | +++ |
| ++ | ++ | + | + | ++ | +++ | +++ | + | +++ |
| ++ | ND | - | ND | ++ | +++ | ND | + | +++ |
| ++ | ++ | - | + | +++ | +++ | +++ | - | +++ |
| ++ | ND | + | ND | ++ | +++ | ND | + | +++ |
| + | ++ | - | ++ | ++ | +++ | +++ | - | +++ |
| + | ND | - | ND | ++ | +++ | ND | - | +++ |
| ++ | +++ | ++ | +++ | + | +++ | +++ | - | - |
| ++ | ND | ++ | ND | ++ | +++ | ND | - | - |
| ND | +++ | ND | +++ | + | +++ | +++ | - | +++ |
| ++ | ND | ++ | ND | ++ | +++ | ND | - | +++ |
| +++ | +++ | +++ | +++ | + | +++ | +++ | ND | - |
| - | ND | + | ND | - | +++ | ++ | ++ | - |
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Whitley, R.J.; Kimberlin, D.W.; Roizman, B. Herpes Simplex Viruses. Clin. Infect. Dis. 1998, 26, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Pfister, J.R.; Spear, S.J. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex. Transm. Dis. 2003, 30, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Li, D.; Mikhail, F.M.; Sassano, A.; Platanias, L.C.; Colamonici, O.; Anastasi, J.; Nucifora, G. EVI1 Abrogates Interferon-alpha Response by Selectively Blocking PML Induction. J. Biol. Chem. 2005, 280, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Shiels, C.; Freemont, P.S. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001, 20, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, E.; Laukens, K.; Dang, T.H.; Van Ostade, X. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int. J. Biol. Sci. 2010, 6, 51–67. [Google Scholar]
- Shen, T.H.; Lin, H.K.; Scaglioni, P.P.; Yung, T.M.; Pandolfi, P.P. The Mechanisms of PML-Nuclear Body Formation. Mol. Cell 2006, 24, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML Nuclear Bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Quimby, B.B.; Yong-Gonzalez, V.; Anan, T.; Strunnikov, A.V.; Dasso, M. The promyelocytic leukemia protein stimulates SUMO conjugation in yeast. Oncogene 2006, 25, 2999–3005. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2010, 30, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 2007, 8, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Chee, A.V.; Lopez, P.; Pandolfi, P.P.; Roizman, B. Promyelocytic Leukemia Protein Mediates Interferon-Based Anti-Herpes Simplex Virus 1 Effects. J. Virol. 2003, 77, 7101–7105. [Google Scholar] [CrossRef] [PubMed]
- Cuchet, D.; Sykes, A.; Nicolas, A.; Orr, A.; Murray, J.; Sirma, H.; Heeren, J.; Bartelt, A.; Everett, R.D. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 2011, 124, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parada, C.; Gripon, P.; Sirma, H.; Orr, A. Replication of ICP0-Null Mutant Herpes Simplex Virus Type 1 Is Restricted by both PML and Sp100. J. Virol. 2008, 82, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML Contributes to a Cellular Mechanism of Repression of Herpes Simplex Virus Type 1 Infection That Is Inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Sourvinos, G.; Leiper, C.; Clements, J.B.; Orr, A. Formation of Nuclear Foci of the Herpes Simplex Virus Type 1 Regulatory Protein ICP4 at Early Times of Infection: Localization, Dynamics, Recruitment of ICP27, and Evidence for the De Novo Induction of ND10-Like Complexes. J. Virol. 2004, 78, 1903–1917. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J. ND10 Components Relocate to Sites Associated with Herpes Simplex Virus Type 1 Nucleoprotein Complexes during Virus Infection. J. Virol. 2005, 79, 5078–5089. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J.; Orr, A.; Preston, C.M. Herpes Simplex Virus Type 1 Genomes Are Associated with ND10 Nuclear Substructures in Quiescently Infected Human Fibroblasts. J. Virol. 2007, 81, 10991–11004. [Google Scholar] [CrossRef]
- Cheng, X.; Kao, H.Y. Post-translational modifications of PML: Consequences and implications. Front. Oncol. 2013, 2. [Google Scholar] [CrossRef]
- Nisole, S.; Maroui, M.A.; Mascle, X.H.; Aubry, M.; Chelbi-Alix, M.K. Differential Roles of PML Isoforms. Front. Oncol. 2013, 3. [Google Scholar] [CrossRef]
- Sternsdorf, T.; Jensen, K.; Will, H. Evidence for Covalent Modification of the Nuclear Dot-associated Proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 1997, 139, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Wada, H.; Fukuda-Kamitani, T.; Yeh, E.T.H. Identification of Three Major Sentrinization Sites in PML. J. Biol. Chem. 1998, 273, 26675–26682. [Google Scholar] [CrossRef] [PubMed]
- Galisson, F.; Mahrouche, L.; Courcelles, M.; Bonneil, E.; Meloche, S.; Chelbi-Alix, M.K.; Thibault, P. A novel proteomics approach to identify SUMOylated proteins and their modification sites in human cells. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.O.; Andersen, J.S.; Ogg, S.C.; Hay, R.T.; Mann, M.; Lamond, A.I. Distinct and Overlapping Sets of SUMO-1 and SUMO-2 Target Proteins Revealed by Quantitative Proteomics. Mol. Cell. Proteomics 2006, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Weidtkamp-Peters, S.; Lenser, T.; Negorev, D.; Gerstner, N.; Hofmann, T.G.; Schwanitz, G.; Hoischen, C.; Maul, G.; Dittrich, P.; Hemmerich, P. Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 2008, 121, 2731–2743. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.Y.; Huang, Y.S.; Jeng, J.C.; Kuo, H.Y.; Chang, C.C.; Chao, T.T.; Ho, C.C.; Chen, Y.C.; Lin, T.P.; Fang, H.I.; et al. Role of SUMO-Interacting Motif in Daxx SUMO Modification, Subnuclear Localization, and Repression of Sumoylated Transcription Factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Rabellino, A.; Carter, B.; Konstantinidou, G.; Wu, S.Y.; Rimessi, A.; Byers, L.A.; Heymach, J.V.; Girard, L.; Chiang, C.M.; Teruya-Feldstein, J.; et al. The SUMO E3-ligase PIAS1 Regulates the Tumor Suppressor PML and Its Oncogenic Counterpart PML-RARA. Cancer Res. 2012, 72, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de Thé, H. Arsenic degrades PML or PML-RAR[alpha] through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Erker, Y.; Neyret-Kahn, H.; Seeler, J.S.; Dejean, A.; Atfi, A.; Levy, L. Arkadia, a novel SUMO-targeted ubiquitin ligase involved in PML degradation. Mol. Cell. Biol. 2013, 33, 2163–2177. [Google Scholar] [CrossRef] [PubMed]
- Reineke, E.L.; Lam, M.; Liu, Q.; Liu, Y.; Stanya, K.J.; Chang, K.S.; Means, A.R.; Kao, H.Y. Degradation of the Tumor Suppressor PML by Pin1 Contributes to the Cancer Phenotype of Breast Cancer MDA-MB-231 Cells. Mol. Cell. Biol. 2008, 28, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, F.; Privalsky, M.L. Phosphorylation of PML by mitogen-activated protein kinases plays a key role in arsenic trioxide-mediated apoptosis. Cancer Cell 2004, 5, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Scaglioni, P.P.; Yung, T.M.; Cai, L.F.; Erdjument-Bromage, H.; Kaufman, A.J.; Singh, B.; Teruya-Feldstein, J.; Tempst, P.; Pandolfi, P.P. A CK2-Dependent Mechanism for Degradation of the PML Tumor Suppressor. Cell 2006, 126, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates III, J.R.; Gray, N.S.; Lee, J.D. Pharmacological Inhibition of BMK1 Suppresses Tumor Growth through Promyelocytic Leukemia Protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Gammeltoft, S.; Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 1999, 294, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, C.J.A.; Cerutti, L.; Hulo, N.; Gattiker, A.; Falquet, L.; Pagni, M.; Bairoch, A.; Bucher, P. PROSITE: A documented database using patterns and profiles as motif descriptors. Brief. Bioinform. 2002, 3, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenço, D.; Boutell, C.; Lukashchuk, V.; Grant, K.; Sykes, A.; Murray, J.; Orr, A.; Everett, R.D. SUMO Pathway Dependent Recruitment of Cellular Repressors to Herpes Simplex Virus Type 1 Genomes. PLoS Pathog 2011, 7, e1002123. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenço, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes Simplex Virus 1 Ubiquitin Ligase ICP0 Interacts with PML Isoform I and Induces Its SUMO-Independent Degradation. J. Virol. 2012, 86, 11209–11222. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Orr, A.; Everett, R.D. PML Residue Lysine 160 Is Required for the Degradation of PML Induced by Herpes Simplex Virus Type 1 Regulatory Protein ICP0. J. Virol. 2003, 77, 8686–8694. [Google Scholar] [CrossRef] [PubMed]
- Gresko, E.; Ritterhoff, S.; Sevilla-Perez, J.; Roscic, A.; Frobius, K.; Kotevic, I.; Vichalkovski, A.; Hess, D.; Hemmings, B.A.; Schmitz, M.L. PML tumor suppressor is regulated by HIPK2-mediated phosphorylation in response to DNA damage. Oncogene 2008, 28, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Everett, R.D. Components of Promyelocytic Leukemia Nuclear Bodies (ND10) Act Cooperatively To Repress Herpesvirus Infection. J. Virol. 2013, 87, 2174–2185. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.O. Relationship Between the Envelope and the Infectivity of Herpes Simplex Virus. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. N. Y. N 1964, 115, 814–816. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.Z.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 1989, 63, 4579–4589. [Google Scholar] [PubMed]
- Everett, R. D.; Young, D.F.; Randall, R.E.; Orr, A. STAT-1- and IRF-3-Dependent Pathways Are Not Essential for Repression of ICP0-Null Mutant Herpes Simplex Virus Type 1 in Human Fibroblasts. J. Virol. 2008, 82, 8871–8881. [Google Scholar] [CrossRef] [PubMed]
- Strang, B.L.; Stow, N.D. Blocks to herpes simplex virus type 1 replication in a cell line, tsBN2, encoding a temperature-sensitive RCC1 protein. J. Gen. Virol. 2007, 88, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, P.A.; Aron, G.M.; Biswal, N.; Benyesh-Melnick, M. Temperature-sensitive mutants of herpes simplex virus type 1: isolation, complementation and partial characterization. Virology 1973, 52, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Davido, D.J.; Zagorski, W.F.; Lane, W.S.; Schaffer, P.A. Phosphorylation Site Mutations Affect Herpes Simplex Virus Type 1 ICP0 Function. J. Virol. 2005, 79, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Kane, S.; Sano, H.; Liu, S.C.H.; Asara, J.M.; Lane, W.S.; Garner, C.C.; Lienhard, G.E. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J. Biol. Chem. 2002, 277, 22115–22118. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R., III. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [PubMed]
- Sotnikov, A.G.; Negorev, D.; Ishov, A.M.; Maul, G.G. Monoclonal antibodies against protein Daxx and its localization in nuclear domains 10. Tsitologiia 2001, 43, 1123–1129. [Google Scholar] [PubMed]
- Le, X.F.; Yang, P.; Chang, K.S. Analysis of the Growth and Transformation Suppressor Domains of Promyelocytic Leukemia Gene, PML. J. Biol. Chem. 1996, 271, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Condemine, W.; Takahashi, Y.; Zhu, J.; Puvion-Dutilleul, F.; Guegan, S.; Janin, A.; de Thé, H. Characterization of Endogenous Human Promyelocytic Leukemia Isoforms. Cancer Res. 2006, 66, 6192–6198. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the Type I Interferon Response by the Nucleoprotein of the Prototypic Arenavirus Lymphocytic Choriomeningitis Virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, M.; Klingström, J. Alpha/Beta Interferon (IFN-α/β)-Independent Induction of IFN-λ1 (Interleukin-29) in Response to Hantaan Virus Infection. J. Virol. 2010, 84, 9140–9148. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Kotla, S.; Bumgarner, R.E.; Gustin, K.E. Human Rhinovirus Attenuates the Type I Interferon Response by Disrupting Activation of Interferon Regulatory Factor 3. J. Virol. 2006, 80, 5021–5031. [Google Scholar] [CrossRef] [PubMed]
- Umareddy, I.; Tang, K.F.; Vasudevan, S.G.; Devi, S.; Hibberd, M.L.; Gu, F. Dengue virus regulates type I interferon signalling in a strain-dependent manner in human cell lines. J. Gen. Virol. 2008, 89, 3052–3062. [Google Scholar] [CrossRef] [PubMed]
- Sutejo, R.; Yeo, D.S.; Myaing, M.Z.; Hui, C.; Xia, J.; Ko, D.; Cheung, P.C.F.; Tan, B.H.; Sugrue, R.J. Activation of Type I and III Interferon Signalling Pathways Occurs in Lung Epithelial Cells Infected with Low Pathogenic Avian Influenza Viruses. PLoS One 2012, 7, e33732. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.E. International symposium on the mitral valve. Biomed. Eng. 1976, 11, 59. [Google Scholar] [PubMed]
- Smith, C.D.; Craft, D.W.; Shiromoto, R.S.; Yan, P.O. Alternative cell line for virus isolation. J. Clin. Microbiol. 1986, 24, 265–268. [Google Scholar]
- Sanda, C.; Weitzel, P.; Tsukahara, T.; Schaley, J.; Edenberg, H.J.; Stephens, M.A.; McClintick, J.N.; Blatt, L.M.; Li, L.; Brodsky, L.; et al. Differential Gene Induction by Type I and Type II Interferons and Their Combination. J. Interferon Cytokine Res. 2006, 26, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Boonyaratanakornkit, J.B.; Bartlett, E.J.; Amaro-Carambot, E.; Collins, P.L.; Murphy, B.R.; Schmidt, A.C. The C Proteins of Human Parainfluenza Virus Type 1 (HPIV1) Control the Transcription of a Broad Array of Cellular Genes That Would Otherwise Respond to HPIV1 Infection. J. Virol. 2009, 83, 1892–1910. [Google Scholar] [PubMed]
- Dankar, S.K.; Miranda, E.; Forbes, N.E.; Pelchat, M.; Tavassoli, A.; Selman, M.; Ping, J.; Jia, J.; Brown, E.G. Influenza A/Hong Kong/156/1997(H5N1) virus NS1 gene mutations F103L and M106I both increase IFN antagonism, virulence and cytoplasmic localization but differ in binding to RIG-I and CPSF30. Virol. J. 2013, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parsy, M.L.; Orr, A. Analysis of the Functions of Herpes Simplex Virus Type 1 Regulatory Protein ICP0 That Are Critical for Lytic Infection and Derepression of Quiescent Viral Genomes. J. Virol. 2009, 83, 4963–4977. [Google Scholar] [CrossRef] [PubMed]
- Stehmeier, P.; Muller, S. Phospho-Regulated SUMO Interaction Modules Connect the SUMO System to CK2 Signaling. Mol. Cell 2009, 33, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Percherancier, Y.; Germain-Desprez, D.; Galisson, F.; Mascle, X.H.; Dianoux, L.; Estephan, P.; Chelbi-Alix, M.K.; Aubry, M. Role of SUMO in RNF4-mediated PML degradation: PML sumoylation and phospho-switch control of its SUMO binding domain dissected in living cells. J Biol Chem 2009, 284, 16595–16608. [Google Scholar] [CrossRef] [PubMed]
- Tagata, Y.; Yoshida, H.; Nguyen, L.A.; Kato, H.; Ichikawa, H.; Tashiro, F.; Kitabayashi, I. Phosphorylation of PML is essential for activation of C/EBPε and PU.1 to accelerate granulocytic differentiation. Leukemia 2008, 22, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kuo, C.; Bisi, J.E.; Kim, M.K. PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat. Cell. Biol. 2002, 4, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of Missing Proteins in the neXtProt Database and Unregistered Phosphopeptides in the PhosphoSitePlus Database As Part of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013. [Google Scholar]
- Christensen, G.L.; Kelstrup, C.D.; Lyngsø, C.; Sarwar, U.; Bøgebo, R.; Sheikh, S.P.; Gammeltoft, S.; Olsen, J.V.; Hansen, J.L. Quantitative Phosphoproteomics Dissection of Seven-transmembrane Receptor Signaling Using Full and Biased Agonists. Mol. Cell. Proteomics 2010, 9, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate Specificities and Identification of Putative Substrates of ATM Kinase Family Members. J. Biol. Chem. 1999, 274, 37538–37543. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Bayless, A.M.; Goddard, E.T.; Davido, D.J. CK2 inhibitors increase the sensitivity of HSV-1 to interferon-[beta]. Antiviral Res. 2011, 91, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; Kheddache-Atmane, S.; El Asmi, F.; Dianoux, L.; Aubry, M.; Chelbi-Alix, M.K. Requirement of PML SUMO Interacting Motif for RNF4- or Arsenic Trioxide-Induced Degradation of Nuclear PML Isoforms. PLoS One 2012, 7, e44949. [Google Scholar] [CrossRef] [PubMed]
- Condemine, W.; Takahashi, Y.; Le Bras, M.; de Thé, H. A nucleolar targeting signal in PML-I addresses PML to nucleolar caps in stressed or senescent cells. J. Cell Sci. 2007, 120, 3219–3227. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The Intrinsic Antiviral Defense to Incoming HSV-1 Genomes Includes Specific DNA Repair Proteins and Is Counteracted by the Viral Protein ICP0. PLoS Pathog 2011, 7, e1002084. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Cuchet-Lourenço, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A Viral Ubiquitin Ligase Has Substrate Preferential SUMO Targeted Ubiquitin Ligase Activity that Counteracts Intrinsic Antiviral Defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Dellaire, G.; Ching, R.W.; Ahmed, K.; Jalali, F.; Tse, K.C.K.; Bristow, R.G.; Bazett-Jones, D.P. Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J. Cell Biol. 2006, 175, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Naik, M.T.; Huang, Y.S.; Jeng, J.C.; Liao, P.H.; Kuo, H.Y.; Ho, C.C.; Hsieh, Y.L.; Lin, C.H.; Huang, N.J.; et al. Structural and Functional Roles of Daxx SIM Phosphorylation in SUMO Paralog-Selective Binding and Apoptosis Modulation. Mol. Cell 2011, 42, 62–74. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, M.C.; Box, A.C.; Haug, J.S.; Lane, W.S.; Davido, D.J. A Phospho-SIM in the Antiviral Protein PML is Required for Its Recruitment to HSV-1 Genomes. Cells 2014, 3, 1131-1158. https://doi.org/10.3390/cells3041131
Smith MC, Box AC, Haug JS, Lane WS, Davido DJ. A Phospho-SIM in the Antiviral Protein PML is Required for Its Recruitment to HSV-1 Genomes. Cells. 2014; 3(4):1131-1158. https://doi.org/10.3390/cells3041131
Chicago/Turabian StyleSmith, Miles C., Andrew C. Box, Jeffrey S. Haug, William S. Lane, and David J. Davido. 2014. "A Phospho-SIM in the Antiviral Protein PML is Required for Its Recruitment to HSV-1 Genomes" Cells 3, no. 4: 1131-1158. https://doi.org/10.3390/cells3041131