Mcl-1 Ubiquitination: Unique Regulation of an Essential Survival Protein

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
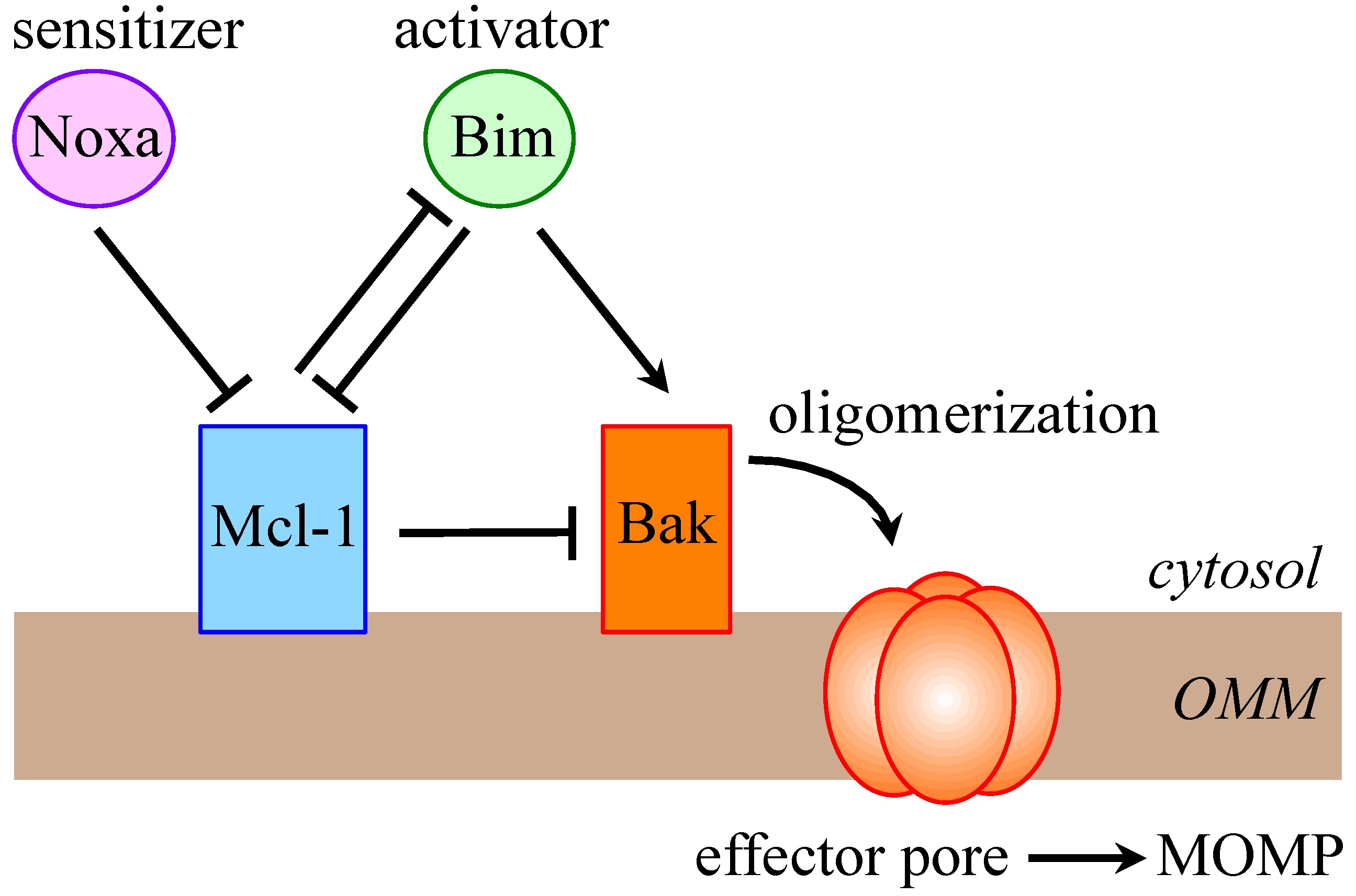
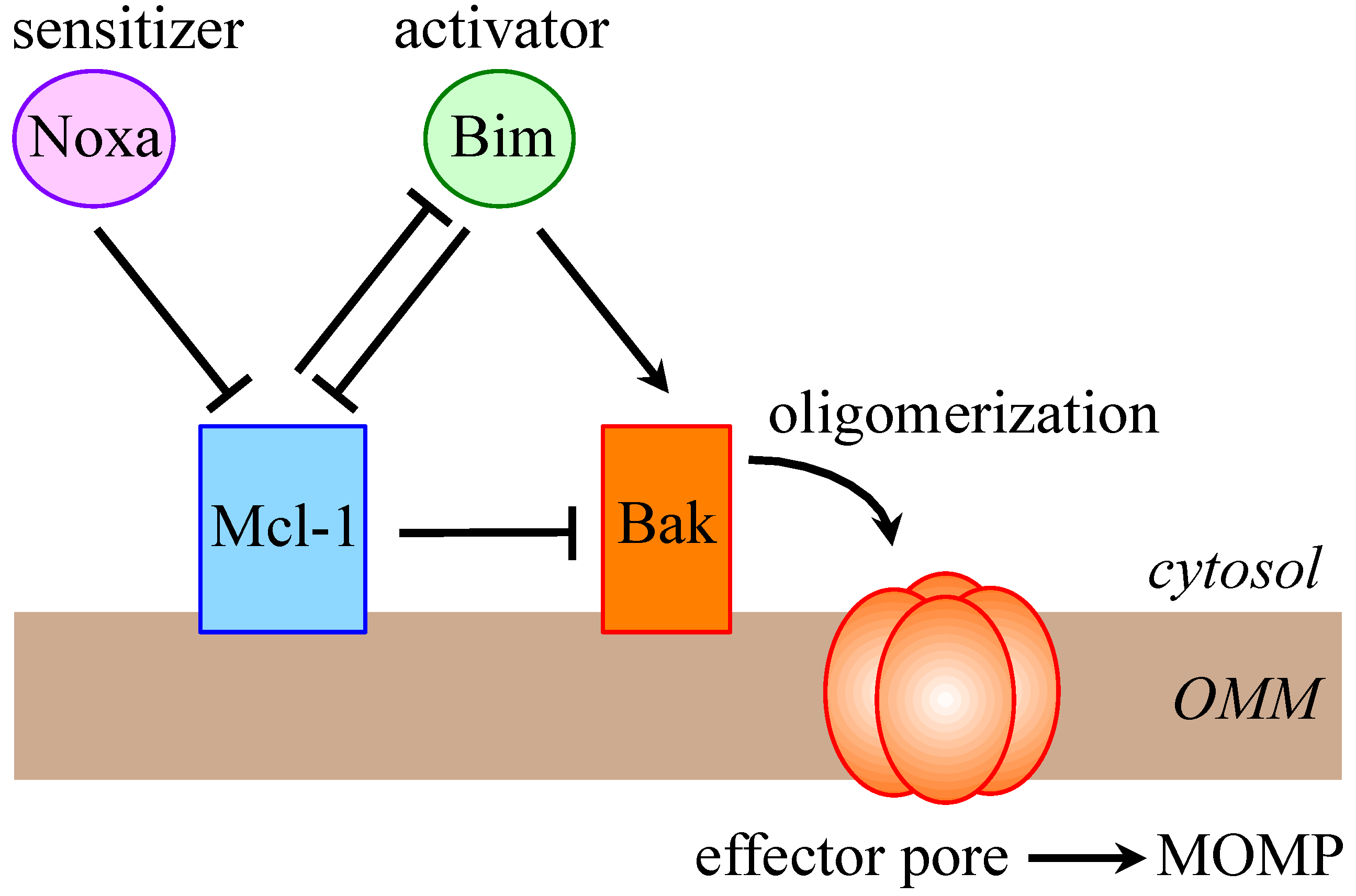
1.1. The Bcl-2 Family
1.2. The Ubiquitin-Proteasome Machinery
2. Mcl-1 is a Crucial Prosurvival Protein
3. Multi-Level Regulation of Mcl-1
3.1. Transcriptional, Post-Transcriptional and Translational Regulation of Mcl-1
3.2. Post-Translational Regulation of Mcl-1

4. Control of Mcl-1 Protein Level by the Ubiquitin-Proteasome System
4.1. The E3 Ubiquitin-Ligases of Mcl-1
4.1.1. Mule
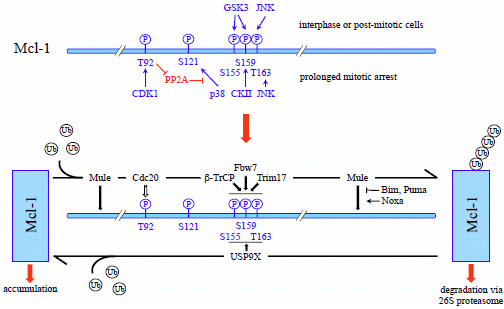
4.1.2. Phosphorylation-Dependent Degradation of Mcl-1
4.1.3. SCFβ-TrCP
4.1.4. SCFFbw7
4.1.5. APC/CCdc20
4.1.6. Trim17
4.2. Deubiquitination of Mcl-1 by USP9X

4.3. Ubiquitin-Independent Degradation of Mcl-1
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Programed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Desagher, S.; Martinou, J.-C. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000, 10, 369–377. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell. Biol. 2014, 15, 49–63. [Google Scholar]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar]
- Broemer, M.; Meier, P. Ubiquitin-mediated regulation of apoptosis. Trends Cell Biol. 2009, 19, 130–140. [Google Scholar] [CrossRef]
- Neutzner, A.; Li, S.; Xu, S.; Karbowski, M. The ubiquitin/proteasome system-dependent control of mitochondrial steps in apoptosis. Semin. Cell Dev. Biol. 2012, 23, 499–508. [Google Scholar] [CrossRef]
- Thompson, S.J.; Loftus, L.T.; Ashley, M.D.; Meller, R. Ubiquitin-proteasome system as a modulator of cell fate. Curr. Opin. Pharmacol. 2008, 8, 90–95. [Google Scholar] [CrossRef]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell. Biol. 2011, 12, 439–452. [Google Scholar] [CrossRef]
- Happo, L.; Strasser, A.; Cory, S. BH3-only proteins in apoptosis at a glance. J. Cell Sci. 2012, 125, 1081–1087. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar]
- Chen, Z.J.; Sun, L.J. Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell. Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef]
- Clague, M.J.; Coulson, J.M.; Urbe, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125, 277–286. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Suresh, B.; Baek, K.H. The role of deubiquitinating enzymes in apoptosis. Cell. Mol. Life Sci. 2011, 68, 15–26. [Google Scholar] [CrossRef]
- Kozopas, K.M.; Yang, T.; Buchan, H.L.; Zhou, P.; Craig, R.W. MCL1, a gene expressed in programed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 3516–3520. [Google Scholar] [CrossRef]
- Krajewski, S.; Bodrug, S.; Krajewska, M.; Shabaik, A.; Gascoyne, R.; Berean, K.; Reed, J.C. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programed cell death in vivo. Am. J. Pathol. 1995, 146, 1309–1319. [Google Scholar]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1's contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef]
- Rinkenberger, J.L.; Horning, S.; Klocke, B.; Roth, K.; Korsmeyer, S.J. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000, 14, 23–27. [Google Scholar]
- Opferman, J.T.; Letai, A.; Beard, C.; Sorcinelli, M.D.; Ong, C.C.; Korsmeyer, S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003, 426, 671–676. [Google Scholar]
- Vikstrom, I.; Carotta, S.; Luthje, K.; Peperzak, V.; Jost, P.J.; Glaser, S.; Busslinger, M.; Bouillet, P.; Strasser, A.; Nutt, S.L.; et al. Mcl-1 is essential for germinal center formation and B cell memory. Science 2010, 330, 1095–1099. [Google Scholar] [CrossRef]
- Dzhagalov, I.; Dunkle, A.; He, Y.W. The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J. Immunol. 2008, 181, 521–528. [Google Scholar] [CrossRef]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar]
- Dzhagalov, I.; St John, A.; He, Y.W. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007, 109, 1620–1626. [Google Scholar] [CrossRef]
- Steimer, D.A.; Boyd, K.; Takeuchi, O.; Fisher, J.K.; Zambetti, G.P.; Opferman, J.T. Selective roles for antiapoptotic MCL-1 during granulocyte development and macrophage effector function. Blood 2009, 113, 2805–2815. [Google Scholar] [CrossRef]
- Arbour, N.; Vanderluit, J.L.; Le Grand, J.N.; Jahani-Asl, A.; Ruzhynsky, V.A.; Cheung, E.C.; Kelly, M.A.; MacKenzie, A.E.; Park, D.S.; Opferman, J.T.; et al. Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 2008, 28, 6068–6078. [Google Scholar] [CrossRef] [Green Version]
- Malone, C.D.; Hasan, S.M.; Roome, R.B.; Xiong, J.; Furlong, M.; Opferman, J.T.; Vanderluit, J.L. Mcl-1 regulates the survival of adult neural precursor cells. Mol. Cell. Neurosci. 2012, 49, 439–447. [Google Scholar] [CrossRef]
- Vick, B.; Weber, A.; Urbanik, T.; Maass, T.; Teufel, A.; Krammer, P.H.; Opferman, J.T.; Schuchmann, M.; Galle, P.R.; Schulze-Bergkamen, H. Knockout of myeloid cell leukemia-1 induces liver damage and increases apoptosis susceptibility of murine hepatocytes. Hepatology 2009, 49, 627–636. [Google Scholar] [CrossRef]
- Wang, X.; Bathina, M.; Lynch, J.; Koss, B.; Calabrese, C.; Frase, S.; Schuetz, J.D.; Rehg, J.E.; Opferman, J.T. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 2013, 27, 1351–1364. [Google Scholar] [CrossRef]
- Peperzak, V.; Vikstrom, I.; Walker, J.; Glaser, S.P.; LePage, M.; Coquery, C.M.; Erickson, L.D.; Fairfax, K.; Mackay, F.; Strasser, A.; et al. Mcl-1 is essential for the survival of plasma cells. Nat. Immunol. 2013, 14, 290–297. [Google Scholar] [CrossRef]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef]
- Germain, M.; Nguyen, A.P.; Le Grand, J.N.; Arbour, N.; Vanderluit, J.L.; Park, D.S.; Opferman, J.T.; Slack, R.S. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011, 30, 395–407. [Google Scholar] [CrossRef]
- Thomas, R.L.; Roberts, D.J.; Kubli, D.A.; Lee, Y.; Quinsay, M.N.; Owens, J.B.; Fischer, K.M.; Sussman, M.A.; Miyamoto, S.; Gustafsson, A.B. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013, 27, 1365–1377. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Mazumder, S.; Hill, B.T.; Kalaycio, M.; Almasan, A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013, 4, e628. [Google Scholar] [CrossRef]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schafer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin. Invest. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [Green Version]
- Perciavalle, R.M.; Stewart, D.P.; Koss, B.; Lynch, J.; Milasta, S.; Bathina, M.; Temirov, J.; Cleland, M.M.; Pelletier, S.; Schuetz, J.D.; et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat. Cell Biol. 2012, 14, 575–583. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [Green Version]
- Glaser, S.P.; Lee, E.F.; Trounson, E.; Bouillet, P.; Wei, A.; Fairlie, W.D.; Izon, D.J.; Zuber, J.; Rappaport, A.R.; Herold, M.J.; et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012, 26, 120–125. [Google Scholar] [CrossRef]
- Xiang, Z.; Luo, H.; Payton, J.E.; Cain, J.; Ley, T.J.; Opferman, J.T.; Tomasson, M.H. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J. Clin. Invest. 2010, 120, 2109–2118. [Google Scholar] [CrossRef]
- Wei, G.; Twomey, D.; Lamb, J.; Schlis, K.; Agarwal, J.; Stam, R.W.; Opferman, J.T.; Sallan, S.E.; den Boer, M.L.; Pieters, R.; et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell 2006, 10, 331–342. [Google Scholar] [CrossRef]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.L.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef]
- Konopleva, M.; Contractor, R.; Tsao, T.; Samudio, I.; Ruvolo, P.P.; Kitada, S.; Deng, X.; Zhai, D.; Shi, Y.X.; Sneed, T.; et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006, 10, 375–388. [Google Scholar] [CrossRef]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar]
- Mazumder, S.; Choudhary, G.S.; Al-Harbi, S.; Almasan, A. Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012, 72, 3069–3079. [Google Scholar] [CrossRef]
- Ertel, F.; Nguyen, M.; Roulston, A.; Shore, G.C. Programming cancer cells for high expression levels of Mcl1. EMBO Rep. 2013, 14, 328–336. [Google Scholar] [CrossRef]
- Le Gouill, S.; Podar, K.; Harousseau, J.L.; Anderson, K.C. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004, 3, 1259–1262. [Google Scholar] [CrossRef]
- Thomas, L.W.; Lam, C.; Edwards, S.W. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010, 584, 2981–2989. [Google Scholar] [CrossRef]
- Croxton, R.; Ma, Y.; Song, L.; Haura, E.B.; Cress, W.D. Direct repression of the Mcl-1 promoter by E2F1. Oncogene 2002, 21, 1359–1369. [Google Scholar]
- Bingle, C.D.; Craig, R.W.; Swales, B.M.; Singleton, V.; Zhou, P.; Whyte, M.K. Exon skipping in Mcl-1 results in a bcl-2 homology domain 3 only gene product that promotes cell death. J. Biol. Chem. 2000, 275, 22136–22146. [Google Scholar]
- Kim, J.H.; Bae, J. MCL-1ES induces MCL-1L-dependent BAX- and BAK-independent mitochondrial apoptosis. PLoS One 2013, 8, e79626. [Google Scholar]
- Yang, T.; Buchan, H.L.; Townsend, K.J.; Craig, R.W. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J. Cell Physiol. 1996, 166, 523–536. [Google Scholar] [CrossRef]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef]
- Lam, L.T.; Lu, X.; Zhang, H.; Lesniewski, R.; Rosenberg, S.; Semizarov, D. A microRNA screen to identify modulators of sensitivity to BCL2 inhibitor ABT-263 (navitoclax). Mol. Cancer Ther. 2010, 9, 2943–2950. [Google Scholar] [CrossRef]
- Subramaniam, D.; Natarajan, G.; Ramalingam, S.; Ramachandran, I.; May, R.; Queimado, L.; Houchen, C.W.; Anant, S. Translation inhibition during cell cycle arrest and apoptosis: Mcl-1 is a novel target for RNA binding protein CUGBP2. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G1025–G1032. [Google Scholar]
- Mills, J.R.; Hippo, Y.; Robert, F.; Chen, S.M.; Malina, A.; Lin, C.J.; Trojahn, U.; Wendel, H.G.; Charest, A.; Bronson, R.T.; et al. mTORC1 promotes survival through translational control of Mcl-1. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10853–10858. [Google Scholar] [CrossRef]
- Fritsch, R.M.; Schneider, G.; Saur, D.; Scheibel, M.; Schmid, R.M. Translational repression of MCL-1 couples stress-induced eIF2 alpha phosphorylation to mitochondrial apoptosis initiation. J. Biol. Chem. 2007, 282, 22551–22562. [Google Scholar] [CrossRef]
- Han, J.; Goldstein, L.A.; Gastman, B.R.; Rabinovitz, A.; Rabinowich, H. Disruption of Mcl-1.Bim complex in granzyme B-mediated mitochondrial apoptosis. J. Biol. Chem. 2005, 280, 16383–16392. [Google Scholar] [CrossRef]
- Michels, J.; O'Neill, J.W.; Dallman, C.L.; Mouzakiti, A.; Habens, F.; Brimmell, M.; Zhang, K.Y.; Craig, R.W.; Marcusson, E.G.; Johnson, P.W.; et al. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene 2004, 23, 4818–4827. [Google Scholar] [CrossRef]
- Herrant, M.; Jacquel, A.; Marchetti, S.; Belhacene, N.; Colosetti, P.; Luciano, F.; Auberger, P. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene 2004, 23, 7863–7873. [Google Scholar]
- Clohessy, J.G.; Zhuang, J.; Brady, H.J. Characterisation of Mcl-1 cleavage during apoptosis of haematopoietic cells. Br. J. Haematol. 2004, 125, 655–665. [Google Scholar] [CrossRef]
- Weng, C.; Li, Y.; Xu, D.; Shi, Y.; Tang, H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 2005, 280, 10491–10500. [Google Scholar] [CrossRef]
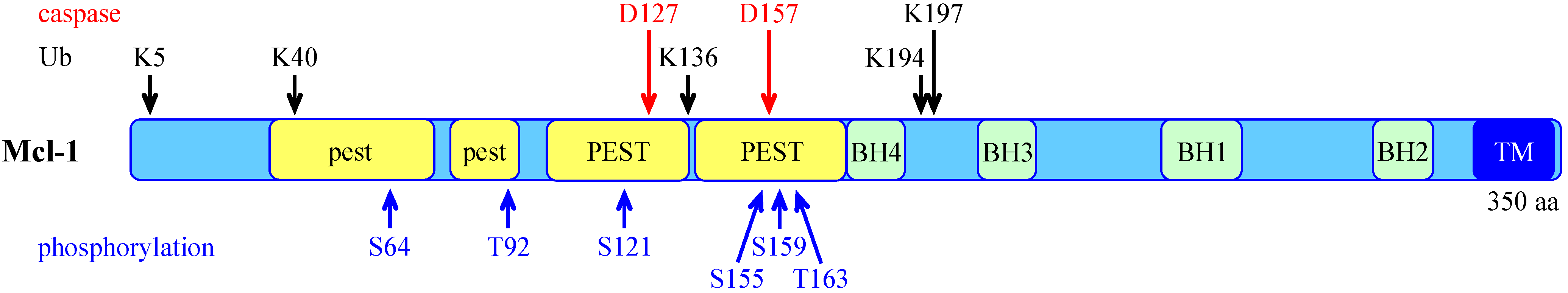
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar] [CrossRef]
- Kobayashi, S.; Lee, S.H.; Meng, X.W.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Craig, R.W.; Kaufmann, S.H.; Gores, G.J. Serine 64 phosphorylation enhances the antiapoptotic function of Mcl-1. J. Biol. Chem. 2007, 282, 18407–18417. [Google Scholar] [CrossRef]
- Domina, A.M.; Vrana, J.A.; Gregory, M.A.; Hann, S.R.; Craig, R.W. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004, 23, 5301–5315. [Google Scholar] [CrossRef]
- Ding, Q.; Huo, L.; Yang, J.Y.; Xia, W.; Wei, Y.; Liao, Y.; Chang, C.J.; Yang, Y.; Lai, C.C.; Lee, D.F.; et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 2008, 68, 6109–6117. [Google Scholar] [CrossRef]
- Nifoussi, S.K.; Vrana, J.A.; Domina, A.M.; De Biasio, A.; Gui, J.; Gregory, M.A.; Hann, S.R.; Craig, R.W. Thr 163 phosphorylation causes Mcl-1 stabilization when degradation is independent of the adjacent GSK3-targeted phosphodegron, promoting drug resistance in cancer. PloS one 2012, 7, e47060. [Google Scholar] [CrossRef]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezawa, A.; Ichijo, H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar]
- Kodama, Y.; Taura, K.; Miura, K.; Schnabl, B.; Osawa, Y.; Brenner, D.A. Antiapoptotic effect of c-Jun N-terminal Kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology 2009, 136, 1423–1434. [Google Scholar]
- Nijhawan, D.; Fang, M.; Traer, E.; Zhong, Q.; Gao, W.; Du, F.; Wang, X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003, 17, 1475–1486. [Google Scholar] [CrossRef]
- Cuconati, A.; Mukherjee, C.; Perez, D.; White, E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003, 17, 2922–2932. [Google Scholar] [CrossRef]
- Derouet, M.; Thomas, L.; Cross, A.; Moots, R.J.; Edwards, S.W. Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J. Biol. Chem. 2004, 279, 26915–26921. [Google Scholar]
- Nencioni, A.; Hua, F.; Dillon, C.P.; Yokoo, R.; Scheiermann, C.; Cardone, M.H.; Barbieri, E.; Rocco, I.; Garuti, A.; Wesselborg, S.; et al. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood 2005, 105, 3255–3262. [Google Scholar] [CrossRef]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef]
- Warr, M.R.; Acoca, S.; Liu, Z.; Germain, M.; Watson, M.; Blanchette, M.; Wing, S.S.; Shore, G.C. BH3-ligand regulates access of MCL-1 to its E3 ligase. FEBS Lett. 2005, 579, 5603–5608. [Google Scholar]
- Warr, M.R.; Mills, J.R.; Nguyen, M.; Lemaire-Ewing, S.; Baardsnes, J.; Sun, K.L.; Malina, A.; Young, J.C.; Jeyaraju, D.V.; O'Connor-McCourt, M.; et al. Mitochondrion-dependent N-terminal processing of outer membrane Mcl-1 protein removes an essential Mule/Lasu1 protein-binding site. J. Biol. Chem. 2011, 286, 25098–25107. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lee, E.F.; van Delft, M.F.; Day, C.L.; Smith, B.J.; Huang, D.C.; Fairlie, W.D.; Hinds, M.G.; Colman, P.M. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6217–6222. [Google Scholar] [CrossRef]
- Mei, Y.; Du, W.; Yang, Y.; Wu, M. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene 2005, 24, 7224–7237. [Google Scholar] [CrossRef]
- Wuilleme-Toumi, S.; Trichet, V.; Gomez-Bougie, P.; Gratas, C.; Bataille, R.; Amiot, M. Reciprocal protection of Mcl-1 and Bim from ubiquitin-proteasome degradation. Biochem. Biophys. Res. Commun. 2007, 361, 865–869. [Google Scholar] [CrossRef]
- Gomez-Bougie, P.; Menoret, E.; Juin, P.; Dousset, C.; Pellat-Deceunynck, C.; Amiot, M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/USP9X interaction. Biochem. Biophys. Res. Commun. 2011, 413, 460–464. [Google Scholar] [CrossRef]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef]
- Hao, Z.; Duncan, G.S.; Su, Y.W.; Li, W.Y.; Silvester, J.; Hong, C.; You, H.; Brenner, D.; Gorrini, C.; Haight, J.; et al. The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 2012, 209, 173–186. [Google Scholar] [CrossRef]
- Ding, Q.; He, X.; Hsu, J.M.; Xia, W.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Liu, J.C.; Zhong, Q.; Wang, X.; et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 2007, 27, 4006–4017. [Google Scholar] [CrossRef]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef]
- Maurer, U.; Charvet, C.; Wagman, A.S.; Dejardin, E.; Green, D.R. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 2006, 21, 749–760. [Google Scholar] [CrossRef]
- Morel, C.; Carlson, S.M.; White, F.M.; Davis, R.J. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol. Cell. Biol. 2009, 29, 3845–3852. [Google Scholar]
- Zhao, Y.; Altman, B.J.; Coloff, J.L.; Herman, C.E.; Jacobs, S.R.; Wieman, H.L.; Wofford, J.A.; Dimascio, L.N.; Ilkayeva, O.; Kelekar, A.; et al. Glycogen Synthase Kinase 3{alpha} and 3{beta} Mediate a Glucose-Sensitive Antiapoptotic Signaling Pathway To Stabilize Mcl-1. Mol. Cell. Biol. 2007, 27, 4328–4339. [Google Scholar] [CrossRef]
- Magiera, M.M.; Mora, S.; Mojsa, B.; Robbins, I.; Lassot, I.; Desagher, S. Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ. 2013, 20, 281–292. [Google Scholar] [CrossRef]
- Jope, R.S.; Johnson, G.V. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004, 29, 95–102. [Google Scholar] [CrossRef]
- Ding, Q.; He, X.; Xia, W.; Hsu, J.M.; Chen, C.T.; Li, L.Y.; Lee, D.F.; Yang, J.Y.; Xie, X.; Liu, J.C.; et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. 2007, 67, 4564–4571. [Google Scholar] [CrossRef]
- Ren, H.; Koo, J.; Guan, B.; Yue, P.; Deng, X.; Chen, M.; Khuri, F.R.; Sun, S.Y. The E3 ubiquitin ligases beta-TrCP and FBXW7 cooperatively mediates GSK3-dependent Mcl-1 degradation induced by the Akt inhibitor API-1, resulting in apoptosis. Mol Cancer 2013, 12, 146. [Google Scholar] [CrossRef]
- Dehan, E.; Bassermann, F.; Guardavaccaro, D.; Vasiliver-Shamis, G.; Cohen, M.; Lowes, K.N.; Dustin, M.; Huang, D.C.; Taunton, J.; Pagano, M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell 2009, 33, 109–116. [Google Scholar] [CrossRef]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef]
- Wang, Z.; Inuzuka, H.; Zhong, J.; Wan, L.; Fukushima, H.; Sarkar, F.H.; Wei, W. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 2012, 586, 1409–1418. [Google Scholar] [CrossRef]
- Millman, S.E.; Pagano, M. MCL1 meets its end during mitotic arrest. EMBO Rep. 2011, 12, 384–385. [Google Scholar] [CrossRef]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef]
- Meroni, G.; Diez-Roux, G. TRIM/RBCC, a novel class of 'single protein RING finger' E3 ubiquitin ligases. Bioessays 2005, 27, 1147–1157. [Google Scholar] [CrossRef]
- Napolitano, L.M.; Meroni, G. TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 2012, 64, 64–71. [Google Scholar] [CrossRef]
- Ogawa, S.; Goto, W.; Orimo, A.; Hosoi, T.; Ouchi, Y.; Muramatsu, M.; Inoue, S. Molecular cloning of a novel RING finger-B box-coiled coil (RBCC) protein, terf, expressed in the testis. Biochem. Biophys. Res. Commun. 1998, 251, 515–519. [Google Scholar] [CrossRef]
- Urano, T.; Usui, T.; Takeda, S.; Ikeda, K.; Okada, A.; Ishida, Y.; Iwayanagi, T.; Otomo, J.; Ouchi, Y.; Inoue, S. TRIM44 interacts with and stabilizes terf, a TRIM ubiquitin E3 ligase. Biochem. Biophys. Res. Commun. 2009, 383, 263–268. [Google Scholar] [CrossRef]
- Endo, H.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Inoue, S. Terf/TRIM17 stimulates degradation of kinetochore protein ZWINT and regulates cell proliferation. J. Biochem. (Tokyo). 2012, 151, 139–144. [Google Scholar] [CrossRef]
- Lassot, I.; Robbins, I.; Kristiansen, M.; Rahmeh, R.; Jaudon, F.; Magiera, M.M.; Mora, S.; Vanhille, L.; Lipkin, A.; Pettmann, B.; et al. Trim17, a novel E3 ubiquitin-ligase, initiates neuronal apoptosis. Cell Death Differ. 2010, 17, 1928–1941. [Google Scholar] [CrossRef]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O'Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Stewart, D.P.; Koss, B.; Bathina, M.; Perciavalle, R.M.; Bisanz, K.; Opferman, J.T. Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol. Cell. Biol. 2010, 30, 3099–3110. [Google Scholar] [CrossRef]
- Jariel-Encontre, I.; Bossis, G.; Piechaczyk, M. Ubiquitin-independent degradation of proteins by the proteasome. Biochim. Biophys. Acta 2008, 1786, 153–177. [Google Scholar]
- Baugh, J.M.; Viktorova, E.G.; Pilipenko, E.V. Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. 2009, 386, 814–827. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 Ubiquitination: Unique Regulation of an Essential Survival Protein. Cells 2014, 3, 418-437. https://doi.org/10.3390/cells3020418
Mojsa B, Lassot I, Desagher S. Mcl-1 Ubiquitination: Unique Regulation of an Essential Survival Protein. Cells. 2014; 3(2):418-437. https://doi.org/10.3390/cells3020418
Chicago/Turabian StyleMojsa, Barbara, Iréna Lassot, and Solange Desagher. 2014. "Mcl-1 Ubiquitination: Unique Regulation of an Essential Survival Protein" Cells 3, no. 2: 418-437. https://doi.org/10.3390/cells3020418