

IF1 Promotes Cellular Proliferation and Inhibits Oxidative Phosphorylation in Mouse Embryonic Fibroblasts under Normoxia and Hypoxia

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Isolation of Mouse Embryonic Fibroblasts
2.3. Cell Culture
2.4. Determination of Proliferation during Normoxia and Hypoxia
2.5. Cellular Bioenergetics
2.6. Lactate Determination
2.7. Statistical Analysis
3. Results
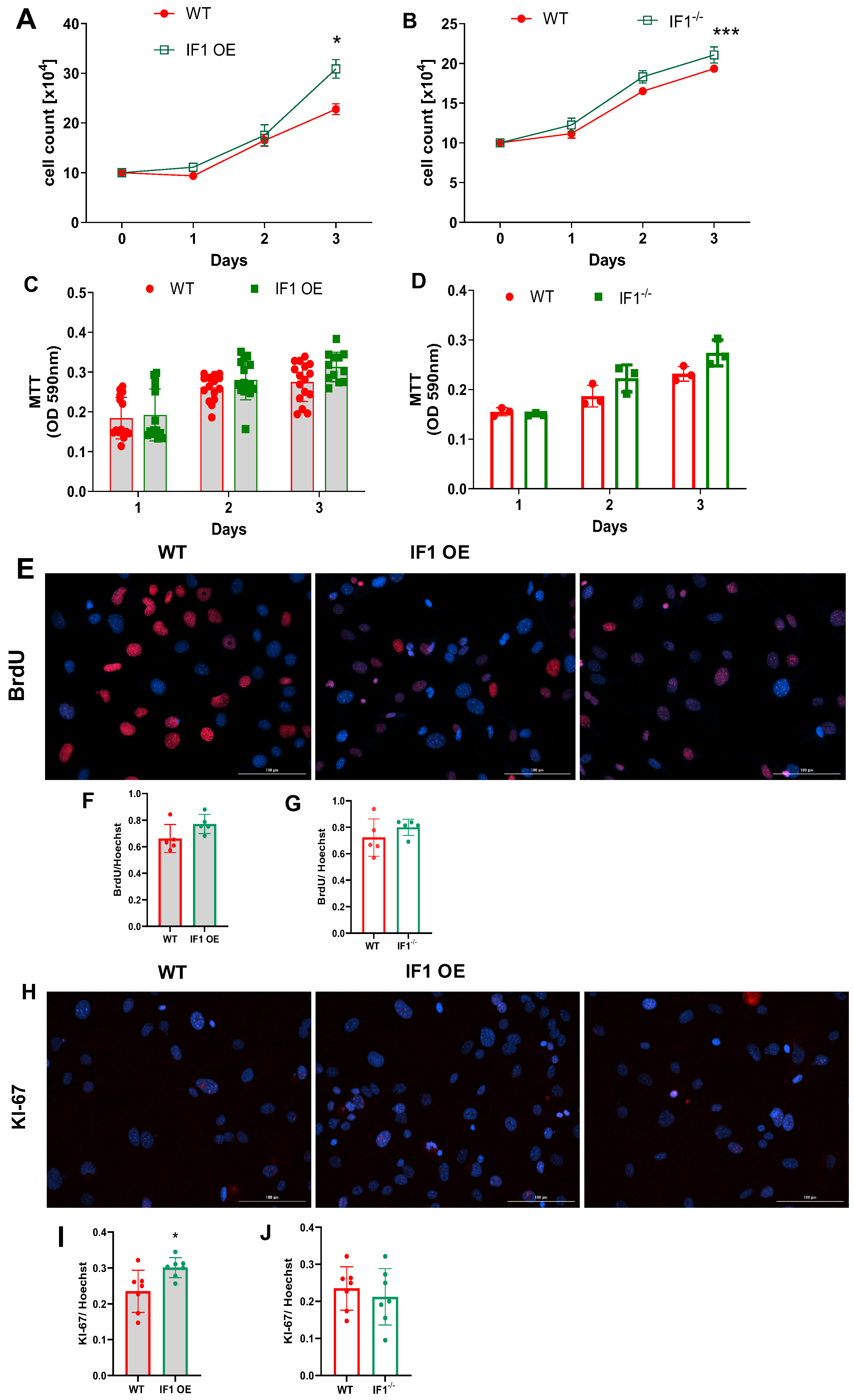
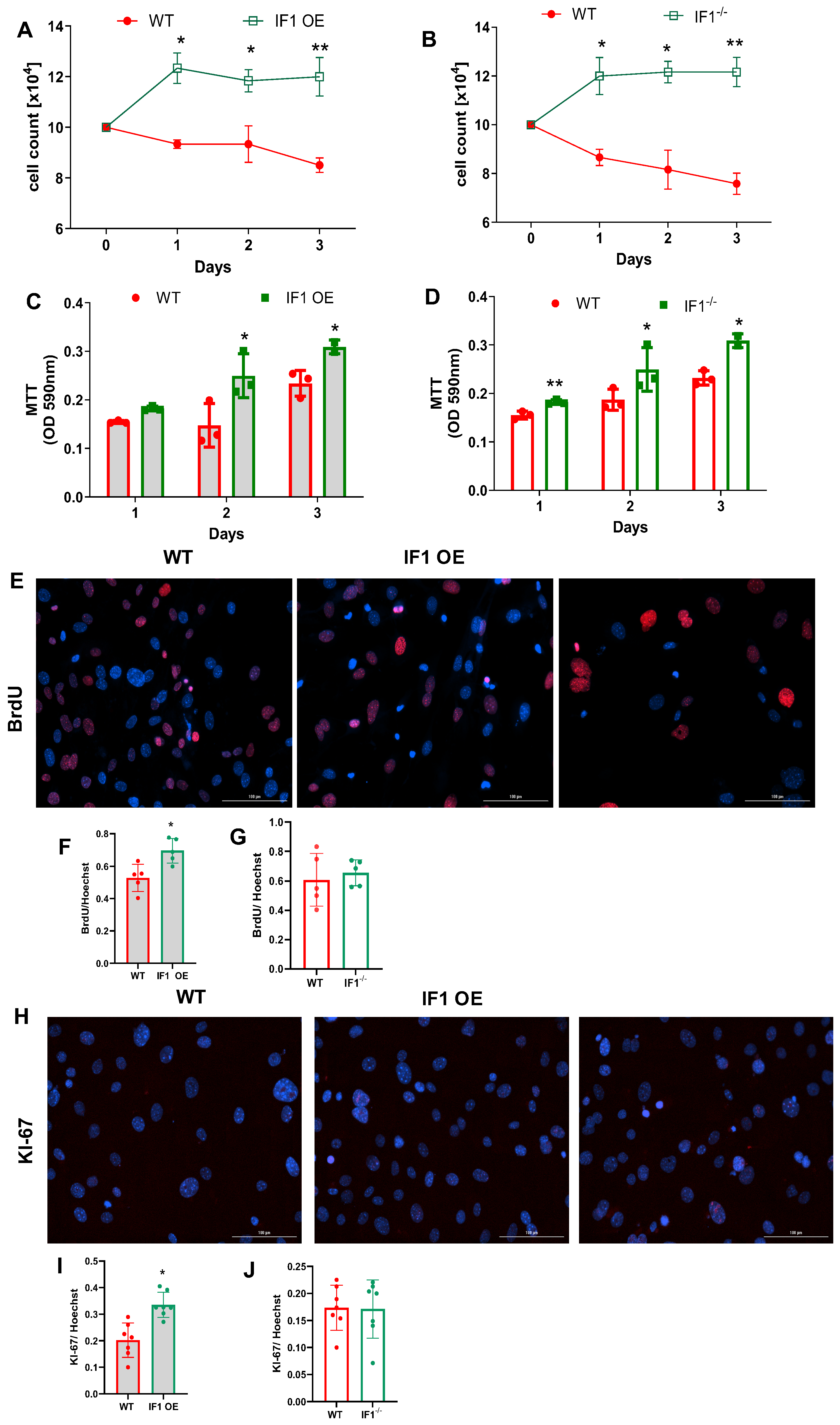
3.1. IF1 Plays a Role in Regulating Cell Survival and Proliferation, Especially under Hypoxia
3.2. Effects of IF1 on Mitochondrial ATP Content under Normoxia and Hypoxia
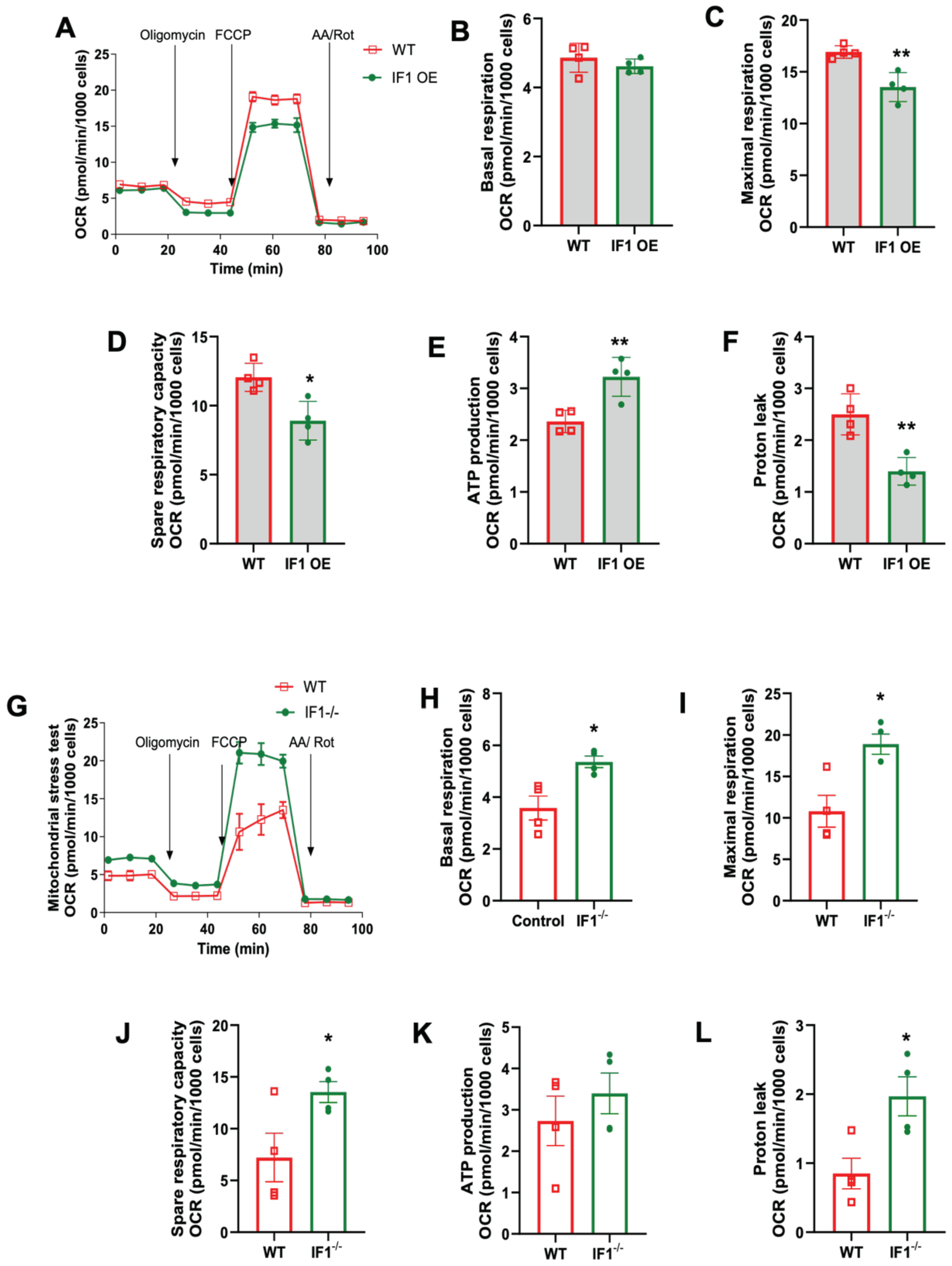
3.3. Effects of IF1 on Mitochondrial Respiration in MEFs Subjected to Normoxia and Hypoxia/Reoxygenation
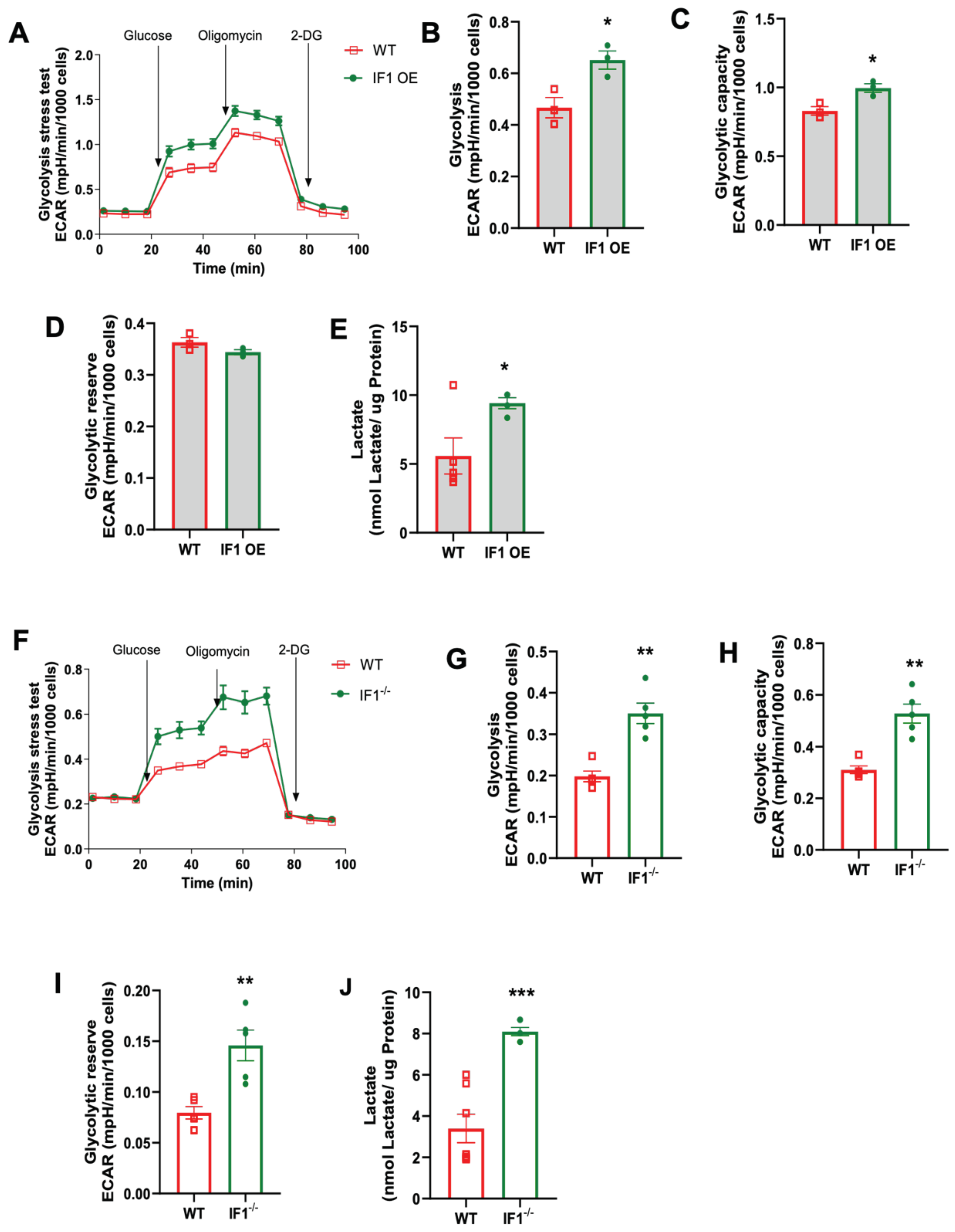
3.4. Effects of IF1 on Cellular Glycolysis in MEFs Subjected to Normoxia and Hypoxia/Reoxygenation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Antimycin A |
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| 2-DG | 2-Deoxy-D-glucose |
| ECAR | Extracellular acidification rate |
| FCCP | Carbonyl-cyanide-p-trifluoromethoxy phenylhydrazone |
| Glu | Glucose |
| PBS | Phosphate-buffered saline |
| FBS | Fetal bovine serum |
| IF1 | ATPase inhibitory factor subunit 1 |
| OCR | Oxygen consumption rate |
| Oligo | Oligomycin |
| OXPHOS | Oxidative phosphorylation |
| Rot | Rotenone |
| TMRM | Tetramethylrhodamine methyl ester perchlorate |
| WT | Wild-type |
References
- Pullman, M.E.; Monroy, G.C. A Naturally Occurring Inhibitor of Mitochondrial Adenosine Triphosphatase. J. Biol. Chem. 1963, 238, 3762–3769. [Google Scholar] [CrossRef]
- Cabezon, E.; Butler, P.J.; Runswick, M.J.; Walker, J.E. Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH. J. Biol. Chem. 2000, 275, 25460–25464. [Google Scholar] [CrossRef]
- Cabezon, E.; Montgomery, M.G.; Leslie, A.G.; Walker, J.E. The structure of bovine F1-ATPase in complex with its regulatory protein IF1. Nat. Struct. Biol. 2003, 10, 744–750. [Google Scholar] [CrossRef]
- Gu, J.; Zhang, L.; Zong, S.; Guo, R.; Liu, T.; Yi, J.; Wang, P.; Zhuo, W.; Yang, M. Cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science 2019, 364, 1068–1075. [Google Scholar] [CrossRef]
- Pinke, G.; Zhou, L.; Sazanov, L.A. Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat. Struct. Mol. Biol. 2020, 27, 1077–1085. [Google Scholar] [CrossRef]
- Asami, K.; Juniti, K.; Ernster, L. Possible regulatory function of a mitochondrial ATPase inhibitor in respiratory chain-linked energy transfer. Biochim. Biophys. Acta 1970, 205, 307–311. [Google Scholar] [CrossRef]
- Guerrieri, F.; Scarfo, R.; Zanotti, F.; Che, Y.W.; Papa, S. Regulatory role of the ATPase inhibitor protein on proton conduction by mitochondrial H+-ATPase complex. FEBS Lett. 1987, 213, 67–72. [Google Scholar] [CrossRef]
- Formentini, L.; Pereira, M.P.; Sánchez-Cenizo, L.; Santacatterina, F.; Lucas, J.J.; Navarro, C.; Martínez-Serrano, A.; Cuezva, J.M. In vivo inhibition of the mitochondrial H+-ATP synthase in neurons promotes metabolic preconditioning. EMBO J. 2014, 33, 762–778. [Google Scholar] [CrossRef]
- Kahancova, A.; Sklenar, F.; Jezek, P.; Dlaskova, A. Regulation of glucose-stimulated insulin secretion by ATPase Inhibitory Factor 1 (IF1). FEBS Lett. 2018, 592, 999–1009. [Google Scholar] [CrossRef]
- Zhou, B.; Caudal, A.; Tang, X.; Chavez, J.D.; McMillen, T.S.; Keller, A.; Villet, O.; Zhao, M.; Liu, Y.; Ritterhoff, J.; et al. Upregulation of mitochondrial ATPase inhibitory factor 1 (ATPIF1) mediates increased glycolysis in mouse hearts. J. Clin. Investig. 2022, 132, e155333. [Google Scholar] [CrossRef]
- Zhang, K.; Bao, R.; Huang, F.; Yang, K.; Ding, Y.; Lauterboeck, L.; Yoshida, M.; Long, Q.; Yang, Q. ATP synthase inhibitory factor subunit 1 regulates islet beta-cell function via repression of mitochondrial homeostasis. Lab. Investig. 2021, 102, 69–79. [Google Scholar] [CrossRef]
- Bravo, C.; Minauro-Sanmiguel, F.; Morales-Rios, E.; Rodriguez-Zavala, J.S.; Garcia, J.J. Overexpression of the inhibitor protein IF(1) in AS-30D hepatoma produces a higher association with mitochondrial F(1)F(0) ATP synthase compared to normal rat liver: Functional and cross-linking studies. J. Bioenerg. Biomembr. 2004, 36, 257–264. [Google Scholar] [CrossRef]
- Galber, C.; Fabbian, S.; Gatto, C.; Grandi, M.; Carissimi, S.; Acosta, M.J.; Sgarbi, G.; Tiso, N.; Argenton, F.; Solaini, G.; et al. The mitochondrial inhibitor IF1 binds to the ATP synthase OSCP subunit and protects cancer cells from apoptosis. Cell Death Dis. 2023, 14, 54. [Google Scholar] [CrossRef]
- Righetti, R.; Grillini, S.; Del Dotto, V.; Costanzini, A.; Liuzzi, F.; Zanna, C.; Sgarbi, G.; Solaini, G.; Baracca, A. The Pro-Oncogenic Protein IF(1) Promotes Proliferation of Anoxic Cancer Cells during Re-Oxygenation. Int. J. Mol. Sci. 2023, 24, 14624. [Google Scholar] [CrossRef]
- Formentini, L.; Sanchez-Arago, M.; Sanchez-Cenizo, L.; Cuezva, J.M. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol. Cell 2012, 45, 731–742. [Google Scholar] [CrossRef]
- Tanton, H.; Voronina, S.; Evans, A.; Armstrong, J.; Sutton, R.; Criddle, D.N.; Haynes, L.; Schmid, M.C.; Campbell, F.; Costello, E.; et al. F1F0-ATP Synthase Inhibitory Factor 1 in the Normal Pancreas and in Pancreatic Ductal Adenocarcinoma: Effects on Bioenergetics, Invasion and Proliferation. Front. Physiol. 2018, 9, 833. [Google Scholar] [CrossRef]
- Wei, S.; Fukuhara, H.; Kawada, C.; Kurabayashi, A.; Furihata, M.; Ogura, S.-I.; Inoue, K.; Shuin, T. Silencing of ATPase Inhibitory Factor 1 Inhibits Cell Growth via Cell Cycle Arrest in Bladder Cancer. Pathobiology 2015, 82, 224–232. [Google Scholar] [CrossRef]
- Faccenda, D.; Nakamura, J.; Gorini, G.; Dhoot, G.K.; Piacentini, M.; Yoshida, M.; Campanella, M. Control of Mitochondrial Remodeling by the ATPase Inhibitory Factor 1 Unveils a Pro-survival Relay via OPA1. Cell Rep. 2017, 18, 1869–1883. [Google Scholar] [CrossRef]
- Yang, K.; Long, Q.; Saja, K.; Huang, F.; Pogwizd, S.M.; Zhou, L.; Yoshida, M.; Yang, Q. Knockout of the ATPase inhibitory factor 1 protects the heart from pressure overload-induced cardiac hypertrophy. Sci. Rep. 2017, 7, 10501. [Google Scholar] [CrossRef]
- Nakamura, J.; Fujikawa, M.; Yoshida, M. IF1, a natural inhibitor of mitochondrial ATP synthase, is not essential for the normal growth and breeding of mice. Biosci. Rep. 2013, 33, e00067. [Google Scholar] [CrossRef]
- Liu, J.; Wang, P.; Luo, J.; Huang, Y.; He, L.; Yang, H.; Li, Q.; Wu, S.; Zhelyabovska, O.; Yang, Q. Peroxisome Proliferator-Activated Receptor β/δ Activation in Adult Hearts Facilitates Mitochondrial Function and Cardiac Performance Under Pressure-Overload Condition. Hypertension 2011, 57, 223–230. [Google Scholar] [CrossRef]
- Kim, T.; Zhelyabovska, O.; Liu, J.; Yang, Q. Generation of an inducible, cardiomyocyte-specific transgenic mouse model with PPAR beta/delta overexpression. Methods Mol. Biol. 2013, 952, 57–65. [Google Scholar]
- Luo, J.; Wu, S.; Liu, J.; Li, Y.; Yang, H.; Kim, T.; Zhelyabovska, O.; Ding, G.; Zhou, Y.; Yang, Y.; et al. Conditional PPARgamma knockout from cardiomyocytes of adult mice impairs myocardial fatty acid utilization and cardiac function. Am. J. Transl. Res. 2010, 3, 61–72. [Google Scholar]
- Durkin, M.E.; Qian, X.; Popescu, N.C.; Lowy, D.R. Isolation of Mouse Embryo Fibroblasts. Bio Protoc. 2013, 3, e908. [Google Scholar] [CrossRef]
- Arai, S.; Kriszt, R.; Harada, K.; Looi, L.; Matsuda, S.; Wongso, D.; Suo, S.; Ishiura, S.; Tseng, Y.; Raghunath, M.; et al. RGB-Color Intensiometric Indicators to Visualize Spatiotemporal Dynamics of ATP in Single Cells. Angew. Chem. Int. Ed. Engl. 2018, 57, 10873–10878. [Google Scholar] [CrossRef]
- White, D.; Lauterboeck, L.; Mobasheran, P.; Kitaguchi, T.; Chaanine, A.H.; Yang, Q. Real-Time Visualization of Cytosolic and Mitochondrial ATP Dynamics in Response to Metabolic Stress in Cultured Cells. Cells 2023, 12, 695. [Google Scholar] [CrossRef]
- Wang, K.; Chen, H.; Zhou, Z.; Zhang, H.; Zhou, H.J.; Min, W. ATPIF1 maintains normal mitochondrial structure which is impaired by CCM3 deficiency in endothelial cells. Cell Biosci. 2021, 11, 11. [Google Scholar] [CrossRef]
- Mendoza-Hoffmann, F.; Zarco-Zavala, M.; Ortega, R.; Garcia-Trejo, J.J. Control of rotation of the F(1)F(O)-ATP synthase nanomotor by an inhibitory alpha-helix from unfolded epsilon or intrinsically disordered zeta and IF(1) proteins. J. Bioenerg. Biomembr. 2018, 50, 403–424. [Google Scholar] [CrossRef]
- Garcia, J.J.; Morales-Rios, E.; Cortes-Hernandez, P.; Rodriguez-Zavala, J.S. The inhibitor protein (IF1) promotes dimerization of the mitochondrial F1F0-ATP synthase. Biochemistry 2006, 45, 12695–12703. [Google Scholar] [CrossRef]
- Minauro-Sanmiguel, F.; Wilkens, S.; Garcia, J.J. Structure of dimeric mitochondrial ATP synthase: Novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 12356–12358. [Google Scholar] [CrossRef]
- Guo, L. Mitochondrial ATP synthase inhibitory factor 1 interacts with the p53-cyclophilin D complex and promotes opening of the permeability transition pore. J. Biol. Chem. 2022, 298, 101858. [Google Scholar] [CrossRef] [PubMed]
- Wyant, G.A.; Yu, W.; Doulamis, I.P.; Nomoto, R.S.; Saeed, M.Y.; Duignan, T.; McCully, J.D.; Kaelin, W.G. Mitochondrial remodeling and ischemic protection by G protein-coupled receptor 35 agonists. Science 2022, 377, 621–629. [Google Scholar] [CrossRef]
- Formentini, L.; Santacatterina, F.; de Arenas, C.N.; Stamatakis, K.; López-Martínez, D.; Logan, A.; Cuezva, J.M. Mitochondrial ROS Production Protects the Intestine from Inflammation through Functional M2 Macrophage Polarization. Cell Rep. 2017, 19, 1202–1213. [Google Scholar] [CrossRef]
- Pavez-Giani, M.G.; Sánchez-Aguilera, P.I.; Bomer, N.; Miyamoto, S.; Booij, H.G.; Giraldo, P.; Oberdorf-Maass, S.U.; Nijholt, K.T.; Yurista, S.R.; Milting, H.; et al. ATPase Inhibitory Factor-1 Disrupts Mitochondrial Ca2+ Handling and Promotes Pathological Cardiac Hypertrophy through CaMKIIdelta. Int. J. Mol. Sci. 2021, 22, 4427. [Google Scholar] [CrossRef] [PubMed]
- Kahancova, A.; Sklenar, F.; Jezek, P.; Dlaskova, A. Overexpression of native IF1 downregulates glucose-stimulated insulin secretion by pancreatic INS-1E cells. Sci. Rep. 2020, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Romero-Carraminana, I.; Esparza-Molto, P.B.; Dominguez-Zorita, S.; Nuevo-Tapioles, C.; Cuezva, J.M. IF1 promotes oligomeric assemblies of sluggish ATP synthase and outlines the heterogeneity of the mitochondrial membrane potential. Commun. Biol. 2023, 6, 836. [Google Scholar] [CrossRef]
- Maldonado, E.N.; DeHart, D.N.; Patnaik, J.; Klatt, S.C.; Gooz, M.B.; Lemasters, J.J. ATP/ADP Turnover and Import of Glycolytic ATP into Mitochondria in Cancer Cells Is Independent of the Adenine Nucleotide Translocator. J. Biol. Chem. 2016, 291, 19642–19650. [Google Scholar] [CrossRef]
- St-Pierre, J.; Brand, M.D.; Boutilier, R.G. Mitochondria as ATP consumers: Cellular treason in anoxia. Proc. Natl. Acad. Sci. USA 2000, 97, 8670–8674. [Google Scholar] [CrossRef]
- Chinopoulos, C.; Adam-Vizi, V. Mitochondria as ATP consumers in cellular pathology. Biochim. Biophys. Acta 2010, 1802, 221–227. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauterboeck, L.; Kang, S.W.; White, D., III; Bao, R.; Mobasheran, P.; Yang, Q. IF1 Promotes Cellular Proliferation and Inhibits Oxidative Phosphorylation in Mouse Embryonic Fibroblasts under Normoxia and Hypoxia. Cells 2024, 13, 551. https://doi.org/10.3390/cells13060551
Lauterboeck L, Kang SW, White D III, Bao R, Mobasheran P, Yang Q. IF1 Promotes Cellular Proliferation and Inhibits Oxidative Phosphorylation in Mouse Embryonic Fibroblasts under Normoxia and Hypoxia. Cells. 2024; 13(6):551. https://doi.org/10.3390/cells13060551
Chicago/Turabian StyleLauterboeck, Lothar, Sung Wook Kang, Donnell White, III, Rong Bao, Parnia Mobasheran, and Qinglin Yang. 2024. "IF1 Promotes Cellular Proliferation and Inhibits Oxidative Phosphorylation in Mouse Embryonic Fibroblasts under Normoxia and Hypoxia" Cells 13, no. 6: 551. https://doi.org/10.3390/cells13060551
APA StyleLauterboeck, L., Kang, S. W., White, D., III, Bao, R., Mobasheran, P., & Yang, Q. (2024). IF1 Promotes Cellular Proliferation and Inhibits Oxidative Phosphorylation in Mouse Embryonic Fibroblasts under Normoxia and Hypoxia. Cells, 13(6), 551. https://doi.org/10.3390/cells13060551