
Protein Stability Regulation in Osteosarcoma: The Ubiquitin-like Modifications and Glycosylation as Mediators of Tumor Growth and as Targets for Therapy
 , , , and
, , , and
Abstract
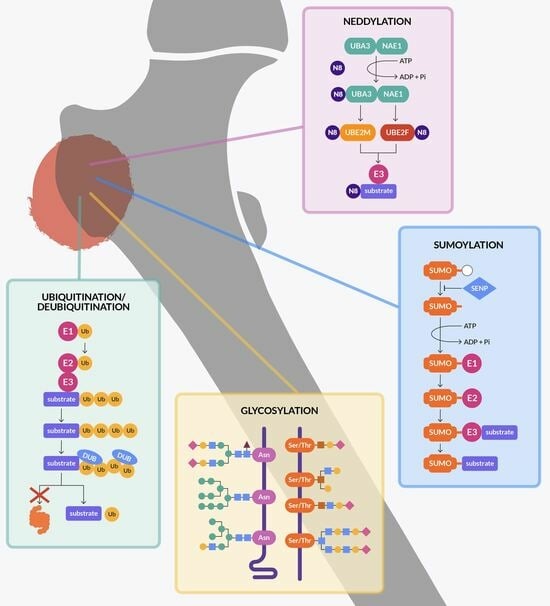
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Ubiquitination
2.1. p53 Ubiquitination
2.2. PI3K/AKT/mTOR Pathway and Ubiquitination
2.3. NRF2 Pathway and Ubiquitination
2.4. HIF Pathway and Ubiquitination
2.5. Other E3 Ligases and Ubiquitination
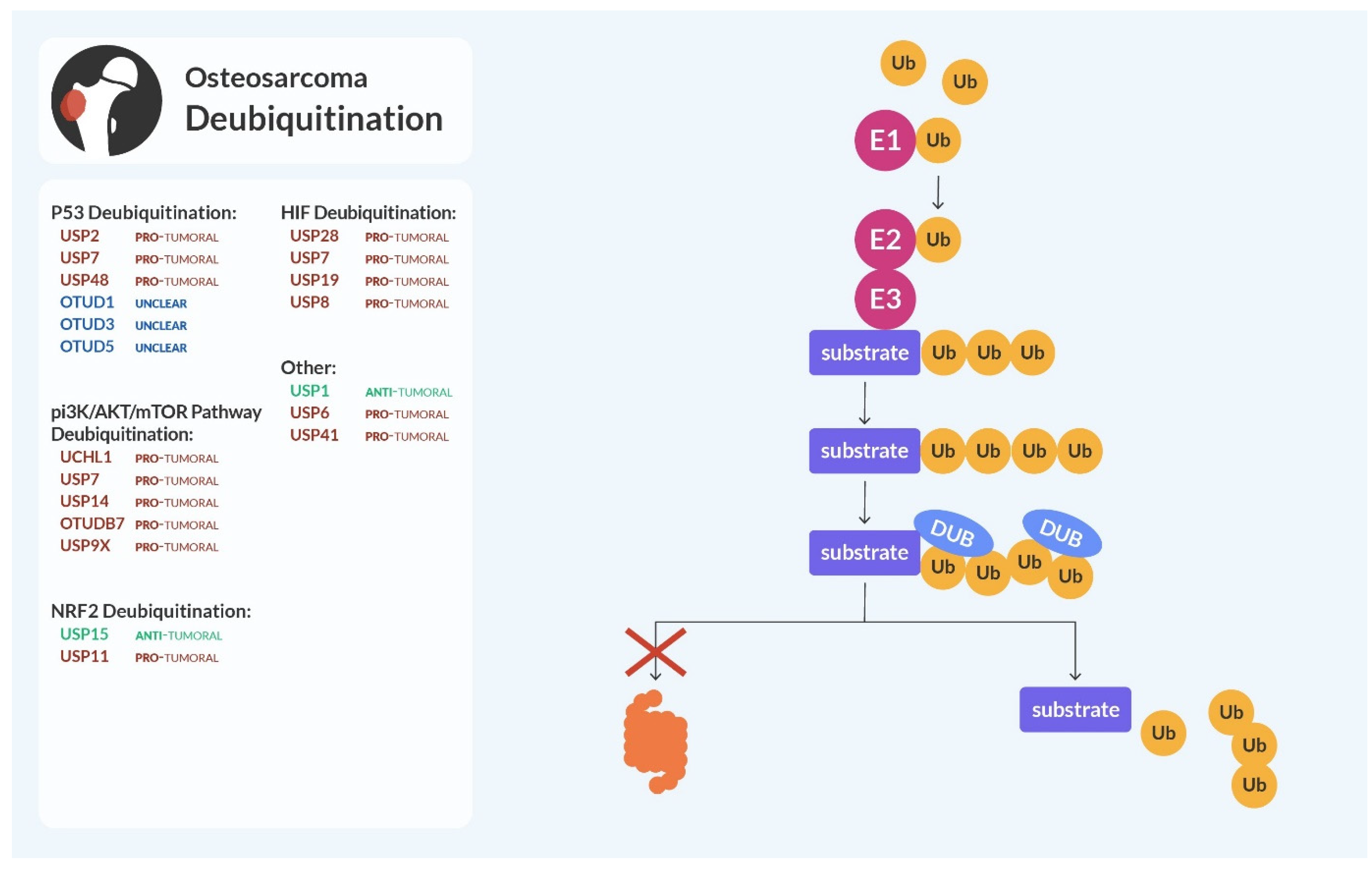
3. Deubiquitination
3.1. p53 and MDM2 Deubiquitination
3.2. PI3K/AKT/mTOR and Deubiquitination
3.3. NRF2 Deubiquitination
3.4. HIF Pathway and Deubiquitination
3.5. Other Deubiquitinating Enzymes of the USP Family
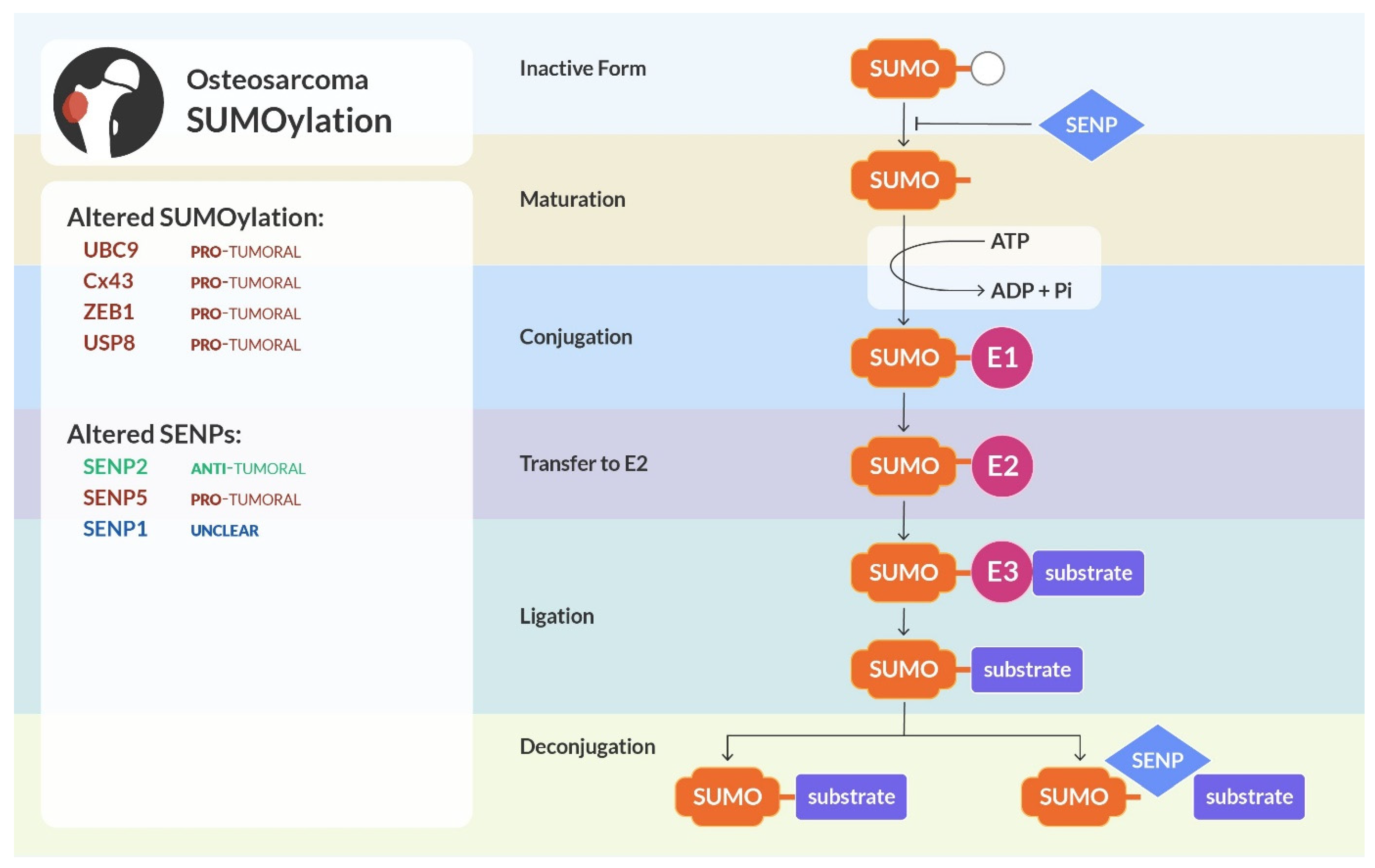
4. SUMOylation
4.1. Altered Pathways and Processes by SUMOylation in Osteosarcoma
4.2. SENPs Alterations in Osteosarcoma
5. NEDDylation
5.1. Derangements in NEDD8 Ligases
5.2. Substrates and Inhibitors of NEDDylation
6. Glycosylation
6.1. Glycosylation and Cell–Cell/Cell–Matrix Interaction
6.2. Glycosylation and Oncogenic Signalling in Cancer
7. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sadykova, L.R.; Ntekim, A.I.; Muyangwa-Semenova, M.; Rutland, C.S.; Jeyapalan, J.N.; Blatt, N.; Rizvanov, A.A. Epidemiology and Risk Factors of Osteosarcoma. Cancer Investig. 2020, 38, 259–269. [Google Scholar] [CrossRef]
- Kansara, M.; Teng, M.W.; Smyth, M.J.; Thomas, D.M. Translational biology of osteosarcoma. Nat. Rev. Cancer 2014, 14, 722–735. [Google Scholar] [CrossRef]
- Eccles, S.A.; Welch, D.R. Metastasis: Recent discoveries and novel treatment strategies. Lancet 2007, 369, 1742–1757. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammaren, H.M.; Savitski, M.M.; Baek, S.H. Control of protein stability by post-translational modifications. Nat. Commun. 2023, 14, 201. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Menezo, Y.; Clement, P.; Clement, A.; Elder, K. Methylation: An Ineluctable Biochemical and Physiological Process Essential to the Transmission of Life. Int. J. Mol. Sci. 2020, 21, 9311. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.; Bialek, W. Beyond K48 and K63: Non-canonical protein ubiquitination. Cell Mol. Biol. Lett. 2021, 26, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 ubiquitin ligases: Styles, structures and functions. Mol. Biomed. 2021, 2, 23. [Google Scholar] [CrossRef]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef]
- Yoshida, A.; Ushiku, T.; Motoi, T.; Beppu, Y.; Fukayama, M.; Tsuda, H.; Shibata, T. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: Correlation with a dedifferentiated subtype. Am. J. Surg. Pathol. 2012, 36, 423–431. [Google Scholar] [CrossRef]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53-MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 91. [Google Scholar] [CrossRef]
- Sane, S.; Rezvani, K. Essential Roles of E3 Ubiquitin Ligases in p53 Regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef]
- Xu, G.S.; Lin, Y.N.; Zeng, Q.; Li, Z.P.; Xiao, T.; Ye, Y.S.; Li, Z.Y.; Gao, X. HSP90-regulated CHIP/TRIM21/p21 Axis Involves in the Senescence of Osteosarcoma Cells. Protein Pept. Lett. 2023, 30, 513–519. [Google Scholar] [CrossRef]
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.; Feng, M.; Yu, Q.; Hu, R.; Wang, Z. The E3 Ligase RING1 Targets p53 for Degradation and Promotes Cancer Cell Proliferation and Survival. Cancer Res. 2018, 78, 359–371. [Google Scholar] [CrossRef]
- Hammer, E.; Heilbronn, R.; Weger, S. The E3 ligase Topors induces the accumulation of polysumoylated forms of DNA topoisomerase I in vitro and in vivo. FEBS Lett. 2007, 581, 5418–5424. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Allton, K.; Duncan, A.D.; Barton, M.C. TRIM24 is a p53-induced E3-ubiquitin ligase that undergoes ATM-mediated phosphorylation and autodegradation during DNA damage. Mol. Cell Biol. 2014, 34, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- de Groot, R.E.; Ganji, R.S.; Bernatik, O.; Lloyd-Lewis, B.; Seipel, K.; Sedova, K.; Zdrahal, Z.; Dhople, V.M.; Dale, T.C.; Korswagen, H.C.; et al. Huwe1-mediated ubiquitylation of dishevelled defines a negative feedback loop in the Wnt signaling pathway. Sci. Signal 2014, 7, ra26. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes. Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, J.; Hu, K.; Wang, G.; Li, M.; Zhang, J.; Cheng, G. FBXW7 acts as an independent prognostic marker and inhibits tumor growth in human osteosarcoma. Int. J. Mol. Sci. 2015, 16, 2294–2306. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Panwar, V.; Singh, A.; Bhatt, M.; Tonk, R.K.; Azizov, S.; Raza, A.S.; Sengupta, S.; Kumar, D.; Garg, M. Multifaceted role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal Transduct. Target. Ther. 2023, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Congwei, L.; Bei, Q.; Tao, Y.; Ruiguo, W.; Heze, Y.; Bo, D.; Zhihong, L. mTOR: An attractive therapeutic target for osteosarcoma? Oncotarget 2016, 7, 50805–50813. [Google Scholar] [CrossRef] [PubMed]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Zhang, W.; Xu, X.; Li, J.; Mu, H.; Liu, X.; Qin, L.; Zhu, X.; Zheng, M. The effects of TRAF6 on proliferation, apoptosis and invasion in osteosarcoma are regulated by miR-124. Int. J. Mol. Med. 2018, 41, 2968–2976. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.C.; Wang, W.; Xiong, Y. Cullin-RING E3 Ubiquitin Ligases: Bridges to Destruction. Subcell. Biochem. 2017, 83, 323–347. [Google Scholar] [CrossRef]
- Ghosh, P.; Wu, M.; Zhang, H.; Sun, H. mTORC1 signaling requires proteasomal function and the involvement of CUL4-DDB1 ubiquitin E3 ligase. Cell Cycle 2008, 7, 373–381. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, K.; Hou, C.; Jiang, K.; Chen, B.; Chen, J.; Lao, L.; Qian, L.; Zhong, G.; Liu, Z.; et al. CRL4B(DCAF11) E3 ligase targets p21 for degradation to control cell cycle progression in human osteosarcoma cells. Sci. Rep. 2017, 7, 1175. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, J.; Shao, J. Knockdown of CUL4A inhibits invasion and induces apoptosis in osteosarcoma cells. Int. J. Immunopathol. Pharmacol. 2015, 28, 263–269. [Google Scholar] [CrossRef]
- Chen, B.; Feng, Y.; Zhang, M.; Cheng, G.; Chen, B.; Wang, H. Small molecule TSC01682 inhibits osteosarcoma cell growth by specifically disrupting the CUL4B-DDB1 interaction and decreasing the ubiquitination of CRL4B E3 ligase substrates. Am. J. Cancer Res. 2019, 9, 1857–1870. [Google Scholar]
- Catena, V.; Fanciulli, M. Deptor: Not only a mTOR inhibitor. J. Exp. Clin. Cancer Res. 2017, 36, 12. [Google Scholar] [CrossRef]
- Bano, I.; Soomro, A.S.; Abbas, S.Q.; Ahmadi, A.; Hassan, S.S.U.; Behl, T.; Bungau, S. A Comprehensive Review of Biological Roles and Interactions of Cullin-5 Protein. ACS Omega 2022, 7, 5615–5624. [Google Scholar] [CrossRef]
- Sun, Y.; Li, H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell 2013, 4, 103–116. [Google Scholar] [CrossRef]
- Antonioli, M.; Albiero, F.; Nazio, F.; Vescovo, T.; Perdomo, A.B.; Corazzari, M.; Marsella, C.; Piselli, P.; Gretzmeier, C.; Dengjel, J.; et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev. Cell 2014, 31, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Popelka, H.; Lei, Y.; Yang, Y.; Klionsky, D.J. The Roles of Ubiquitin in Mediating Autophagy. Cells 2020, 9, 2025. [Google Scholar] [CrossRef]
- Hu, B.; Lv, X.; Gao, F.; Chen, S.; Wang, S.; Qing, X.; Liu, J.; Wang, B.; Shao, Z. Downregulation of DEPTOR inhibits the proliferation, migration, and survival of osteosarcoma through PI3K/Akt/mTOR pathway. Onco Targets Ther. 2017, 10, 4379–4391. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.J.; Kim, H.J.; Kim, C.H. Non-Thermal Plasma Induces Antileukemic Effect Through mTOR Ubiquitination. Cells 2020, 9, 595. [Google Scholar] [CrossRef]
- Jiang, J.; Xu, Y.; Ren, H.; Wudu, M.; Wang, Q.; Song, X.; Su, H.; Jiang, X.; Jiang, L.; Qiu, X. MKRN2 inhibits migration and invasion of non-small-cell lung cancer by negatively regulating the PI3K/Akt pathway. J. Exp. Clin. Cancer Res. 2018, 37, 189. [Google Scholar] [CrossRef]
- Paccosi, E.; Balzerano, A.; Proietti-De-Santis, L. Interfering with the Ubiquitin-Mediated Regulation of Akt as a Strategy for Cancer Treatment. Int. J. Mol. Sci. 2023, 24, 2809. [Google Scholar] [CrossRef]
- Wang, J.; Aldahamsheh, O.; Ferrena, A.; Borjihan, H.; Singla, A.; Yaguare, S.; Singh, S.; Viscarret, V.; Tingling, J.; Zi, X.; et al. The interaction of SKP2 with p27 enhances the progression and stemness of osteosarcoma. Ann. N. Y. Acad. Sci. 2021, 1490, 90–104. [Google Scholar] [CrossRef]
- Yao, W.; Wang, X.; Cai, Q.; Gao, S.; Wang, J.; Zhang, P. TRAF4 enhances osteosarcoma cell proliferation and invasion by Akt signaling pathway. Oncol. Res. 2014, 22, 21–28. [Google Scholar] [CrossRef]
- Strepkos, D.; Markouli, M.; Klonou, A.; Papavassiliou, A.G.; Piperi, C. Histone Methyltransferase SETDB1: A Common Denominator of Tumorigenesis with Therapeutic Potential. Cancer Res. 2021, 81, 525–534. [Google Scholar] [CrossRef]
- Jin, J.; He, J.; Li, X.; Ni, X.; Jin, X. The role of ubiquitination and deubiquitination in PI3K/AKT/mTOR pathway: A potential target for cancer therapy. Gene 2023, 889, 147807. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Tang, F.; Min, L.; Hornicek, F.; Duan, Z.; Tu, C. PTEN in osteosarcoma: Recent advances and the therapeutic potential. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188405. [Google Scholar] [CrossRef]
- Xi, X.; Bao, Y.; Zhou, Y.; Chen, Y.; Zhong, X.; Liao, J.; Zhou, J.; Xu, S.; Cao, Z.; Hu, K.; et al. Oncogenic gene TRIM10 confers resistance to cisplatin in osteosarcoma cells and activates the NF-kappaB signaling pathway. Cell Biol. Int. 2021, 45, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Guo, Y.; Xu, D.; Wang, Y.; Shen, Y.; Wang, F.; Lv, Y.; Song, F.; Jiang, D.; Zhang, Y.; et al. TRIM14 regulates cell proliferation and invasion in osteosarcoma via promotion of the AKT signaling pathway. Sci. Rep. 2017, 7, 42411. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Ma, Z.; Yang, F.; Bai, Y.; Li, J.; Luo, W.; Zhong, J. TRIM59 promotes osteosarcoma progression via activation of STAT3. Hum. Cell 2022, 35, 250–259. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.X.; Zhou, C.Y.; Xiao, X.; Tian, C.; Li, H.H.; Yin, C.L.; Wang, H.X. Tripartite motif 10 regulates cardiac hypertrophy by targeting the PTEN/AKT pathway. J. Cell Mol. Med. 2020, 24, 6233–6241. [Google Scholar] [CrossRef]
- Chen, J.; Huang, L.; Quan, J.; Xiang, D. TRIM14 regulates melanoma malignancy via PTEN/PI3K/AKT and STAT3 pathways. Aging 2021, 13, 13225–13238. [Google Scholar] [CrossRef]
- He, R.; Liu, H. TRIM59 knockdown blocks cisplatin resistance in A549/DDP cells through regulating PTEN/AKT/HK2. Gene 2020, 747, 144553. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, Y.W.; Park, Y.K. Nrf2 expression is associated with poor outcome in osteosarcoma. Pathology 2012, 44, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Je, E.M.; An, C.H.; Yoo, N.J.; Lee, S.H. Mutational and expressional analyses of NRF2 and KEAP1 in sarcomas. Tumori 2012, 98, 510–515. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, T.; Yang, X.; Cao, X.; Jin, G.; Zhang, P.; Guo, J.; Rong, K.; Li, B.; Hu, Y.; et al. DDRGK1 Enhances Osteosarcoma Chemoresistance via Inhibiting KEAP1-Mediated NRF2 Ubiquitination. Adv. Sci. 2023, 10, e2204438. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Z.; Yu, N.; Zhang, L.; Li, H.; Chen, Y.; Gong, F.; Lin, W.; He, X.; Wang, S.; et al. Bone-targeting exosome nanoparticles activate Keap1 / Nrf2 / GPX4 signaling pathway to induce ferroptosis in osteosarcoma cells. J. Nanobiotechnol. 2023, 21, 355. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhao, Y.; Wang, G.; Feng, S.; Ge, X.; Ye, W.; Wang, Z.; Zhu, Y.; Cai, W.; Bai, J.; et al. TRIM22 inhibits osteosarcoma progression through destabilizing NRF2 and thus activation of ROS/AMPK/mTOR/autophagy signaling. Redox Biol. 2022, 53, 102344. [Google Scholar] [CrossRef] [PubMed]
- Groulx, I.; Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell Biol. 2002, 22, 5319–5336. [Google Scholar] [CrossRef]
- Ren, H.Y.; Zhang, Y.H.; Li, H.Y.; Xie, T.; Sun, L.L.; Zhu, T.; Wang, S.D.; Ye, Z.M. Prognostic role of hypoxia-inducible factor-1 alpha expression in osteosarcoma: A meta-analysis. Onco Targets Ther. 2016, 9, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Du, R.; Shang, W.; Suo, S.; Yu, D.; Zhang, J. HIF-1alpha Silencing Inhibits the Growth of Osteosarcoma Cells by Inducing Apoptosis. Ann. Clin. Lab. Sci. 2016, 46, 140–146. [Google Scholar] [PubMed]
- Cassavaugh, J.M.; Hale, S.A.; Wellman, T.L.; Howe, A.K.; Wong, C.; Lounsbury, K.M. Negative regulation of HIF-1alpha by an FBW7-mediated degradation pathway during hypoxia. J. Cell Biochem. 2011, 112, 3882–3890. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.J.; Chang, B.Y.; Wang, X.F.; Zang, Y.F.; Zheng, Z.X.; Zhao, H.J.; Cui, Q.D. FBW7 regulates HIF-1alpha/VEGF pathway in the IL-1beta induced chondrocytes degeneration. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 5914–5924. [Google Scholar] [CrossRef] [PubMed]
- Sun, L. F-box and WD repeat domain-containing 7 (FBXW7) mediates the hypoxia inducible factor-1alpha (HIF-1alpha)/vascular endothelial growth factor (VEGF) signaling pathway to affect hypoxic-ischemic brain damage in neonatal rats. Bioengineered 2022, 13, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.C.; Gu, Y.; Yang, J.Y.; Xi, K.; Tang, J.C.; Bian, J.; Cai, F.; Chen, L. RACK1 mediates the advanced glycation end product-induced degradation of HIF-1alpha in nucleus pulposus cells via competing with HSP90 for HIF-1alpha binding. Cell Biol. Int. 2021, 45, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Zheng, D.; Wei, Z.; Liu, W.; Guo, W. TRIM26 inhibited osteosarcoma progression through destabilizing RACK1 and thus inactivation of MEK/ERK signaling. Cell Death Dis. 2023, 14, 529. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zheng, B.; Wu, Y.; Tang, Y.; Wang, L.; Gao, Y.; Gong, H.; Du, J.; Yu, R. Ubiquitin ligase Siah1 promotes the migration and invasion of human glioma cells by regulating HIF-1alpha signaling under hypoxia. Oncol. Rep. 2015, 33, 1185–1190. [Google Scholar] [CrossRef]
- Matsui-Hasumi, A.; Sato, Y.; Uto-Konomi, A.; Yamashita, S.; Uehori, J.; Yoshimura, A.; Yamashita, M.; Asahara, H.; Suzuki, S.; Kubo, M. E3 ubiquitin ligases SIAH1/2 regulate hypoxia-inducible factor-1 (HIF-1)-mediated Th17 cell differentiation. Int. Immunol. 2017, 29, 133–143. [Google Scholar] [CrossRef]
- Han, X.; Liu, F.; Zhang, C.; Ren, Z.; Li, L.; Wang, G. SIAH1/ZEB1/IL-6 axis is involved in doxorubicin (Dox) resistance of osteosarcoma cells. Biol. Chem. 2019, 400, 545–553. [Google Scholar] [CrossRef]
- Zheng, J.; You, W.; Zheng, C.; Wan, P.; Chen, J.; Jiang, X.; Zhu, Z.; Zhang, Z.; Gong, A.; Li, W.; et al. Knockdown of FBXO39 inhibits proliferation and promotes apoptosis of human osteosarcoma U-2OS cells. Oncol. Lett. 2018, 16, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, X.; Tang, W.; Zhu, Y.; Hu, J.; Zhang, X. Tripartite Motif Containing 11 Interacts with DUSP6 to Promote the Growth of Human Osteosarcoma Cells through Regulating ERK1/2 Pathway. BioMed Res. Int. 2019, 2019, 9612125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guo, Y.; Yang, H.; Shi, G.; Xu, G.; Shi, J.; Yin, N.; Chen, D. TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 2015, 6, 23708–23719. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Yuan, X.; Piao, L.; Wang, J.; Wang, P.; Zhuang, M.; Liu, J.; Liu, Z. Cellular functions and molecular mechanisms of ubiquitination in osteosarcoma. Front. Oncol. 2022, 12, 1072701. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Zhou, Y.; Wang, R.; Chen, S.; Wang, Q.; Xu, Z.; Liu, Y.; Yang, H. TRIM58 Interacts with Pyruvate Kinase M2 to Inhibit Tumorigenicity in Human Osteosarcoma Cells. BioMed Res. Int. 2020, 2020, 8450606. [Google Scholar] [CrossRef]
- Fu, L.; Cui, C.P.; Zhang, X.; Zhang, L. The functions and regulation of Smurfs in cancers. Semin. Cancer Biol. 2020, 67, 102–116. [Google Scholar] [CrossRef]
- Zhang, W.; Zhuang, Y.; Zhang, Y.; Yang, X.; Zhang, H.; Wang, G.; Yin, W.; Wang, R.; Zhang, Z.; Xiao, W. Uev1A facilitates osteosarcoma differentiation by promoting Smurf1-mediated Smad1 ubiquitination and degradation. Cell Death Dis. 2017, 8, e2974. [Google Scholar] [CrossRef]
- Shah, P.A.; Boutros-Suleiman, S.; Emanuelli, A.; Paolini, B.; Levy-Cohen, G.; Blank, M. The Emerging Role of E3 Ubiquitin Ligase SMURF2 in the Regulation of Transcriptional Co-Repressor KAP1 in Untransformed and Cancer Cells and Tissues. Cancers 2022, 14, 1607. [Google Scholar] [CrossRef] [PubMed]
- Van Stiphout, C.M.; Luu, A.K.; Viloria-Petit, A.M. Proteasome Inhibitors and Their Potential Applicability in Osteosarcoma Treatment. Cancers 2022, 14, 4544. [Google Scholar] [CrossRef] [PubMed]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta 2004, 1695, 189–207. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Han, S.; Wang, R.; Zhang, Y.; Li, X.; Gan, Y.; Gao, F.; Rong, P.; Wang, W.; Li, W. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int. J. Biol. Sci. 2022, 18, 2292–2303. [Google Scholar] [CrossRef]
- Piao, S.; Pei, H.Z.; Huang, B.; Baek, S.H. Ovarian tumor domain-containing protein 1 deubiquitinates and stabilizes p53. Cell Signal 2017, 33, 22–29. [Google Scholar] [CrossRef]
- Zeng, Q.; Li, Z.; Zhao, X.; Guo, L.; Yu, C.; Qin, J.; Zhang, S.; Zhang, Y.; Yang, X. Ubiquitin-specific protease 7 promotes osteosarcoma cell metastasis by inducing epithelial-mesenchymal transition. Oncol. Rep. 2019, 41, 543–551. [Google Scholar] [CrossRef]
- Kitamura, H.; Hashimoto, M. USP2-Related Cellular Signaling and Consequent Pathophysiological Outcomes. Int. J. Mol. Sci. 2021, 22, 1209. [Google Scholar] [CrossRef] [PubMed]
- Cetkovska, K.; Sustova, H.; Uldrijan, S. Ubiquitin-specific peptidase 48 regulates Mdm2 protein levels independent of its deubiquitinase activity. Sci. Rep. 2017, 7, 43180. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hou, B.; Huang, Y.; Wang, X.; Qian, Y.; Liang, Y.; Gu, X.; Ma, Z.; Sun, Y. USP48 alleviates bone cancer pain and regulates MrgC stabilization in spinal cord neurons of male mice. Eur. J. Pain. 2023, 27, 723–734. [Google Scholar] [CrossRef]
- Luo, Y.; He, J.; Yang, C.; Orange, M.; Ren, X.; Blair, N.; Tan, T.; Yang, J.M.; Zhu, H. UCH-L1 promotes invasion of breast cancer cells through activating Akt signaling pathway. J. Cell Biochem. 2018, 119, 691–700. [Google Scholar] [CrossRef]
- Zheng, S.; Qiao, G.; Min, D.; Zhang, Z.; Lin, F.; Yang, Q.; Feng, T.; Tang, L.; Sun, Y.; Zhao, H.; et al. Heterogeneous expression and biological function of ubiquitin carboxy-terminal hydrolase-L1 in osteosarcoma. Cancer Lett. 2015, 359, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, N.; Li, M.; Hong, T.; Meng, W.; Ouyang, T. Ubiquitin C-terminal hydrolase-L1: A new cancer marker and therapeutic target with dual effects (Review). Oncol. Lett. 2023, 25, 123. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.T.; Kuo, Y.C.; Hung, J.J.; Huang, C.H.; Chen, W.Y.; Chou, T.Y.; Chen, Y.; Chen, Y.J.; Chen, Y.J.; Cheng, W.C.; et al. K63-polyubiquitinated HAUSP deubiquitinates HIF-1alpha and dictates H3K56 acetylation promoting hypoxia-induced tumour progression. Nat. Commun. 2016, 7, 13644. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Hong, J.; Zhang, J.H.; Wang, X.F.; Ma, Y.S.; Xiong, Z.X.; Sun, H.R.; Cheng, C.; Xie, B.Z.; Liu, J.B.; et al. Treatment with b-AP15 to Inhibit UCHL5 and USP14 Deubiquitinating Activity and Enhance p27 and Cyclin E1 for Tumors with p53 Deficiency. Technol. Cancer Res. Treat. 2022, 21, 15330338221119745. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jie, Z.; Joo, D.; Ordureau, A.; Liu, P.; Gan, W.; Guo, J.; Zhang, J.; North, B.J.; Dai, X.; et al. TRAF2 and OTUD7B govern a ubiquitin-dependent switch that regulates mTORC2 signalling. Nature 2017, 545, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, W.; Ma, X.; Hao, Y.; Liu, G.; Hu, X.; Shang, H.; Wu, P.; Zhao, Z.; Liu, W. Logistic regression analysis for the identification of the metastasis-associated signaling pathways of osteosarcoma. Int. J. Mol. Med. 2018, 41, 1233–1244. [Google Scholar] [CrossRef]
- Zheng, W.; Li, S.; Huang, J.; Dong, Y.; Zhang, H.; Zheng, J. Down-Regulation of Ubiquitin-Specific Peptidase 9X Inhibited Proliferation, Migration and Invasion of Osteosarcoma via ERK1/2 and PI3K/Akt Signaling Pathways. Biol. Pharm. Bull. 2022, 45, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhan, J.; Chen, D.; Shao, G.; Zhang, H.; Gu, W.; Luo, J. The deubiquitinase USP11 regulates cell proliferation and ferroptotic cell death via stabilization of NRF2 USP11 deubiquitinates and stabilizes NRF2. Oncogene 2021, 40, 1706–1720. [Google Scholar] [CrossRef] [PubMed]
- Stockum, A.; Snijders, A.P.; Maertens, G.N. USP11 deubiquitinates RAE1 and plays a key role in bipolar spindle formation. PLoS ONE 2018, 13, e0190513. [Google Scholar] [CrossRef]
- Rothzerg, E.; Xu, J.; Wood, D.; Kõks, S. 12 Survival-related differentially expressed genes based on the TARGET-osteosarcoma database. Exp. Biol. Med. 2021, 246, 2072–2081. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Tian, W.; Wu, T.; Sun, Z.; Lau, A.; Chapman, E.; Fang, D.; Zhang, D.D. USP15 negatively regulates Nrf2 through deubiquitination of Keap1. Mol. Cell 2013, 51, 68–79. [Google Scholar] [CrossRef]
- Ren, Y.; Song, Z.; Rieser, J.; Ackermann, J.; Koch, I.; Lv, X.; Ji, T.; Cai, X. USP15 Represses Hepatocellular Carcinoma Progression by Regulation of Pathways of Cell Proliferation and Cell Migration: A System Biology Analysis. Cancers 2023, 15, 1371. [Google Scholar] [CrossRef]
- Zhou, L.; Jiang, H.; Du, J.; Li, L.; Li, R.; Lu, J.; Fu, W.; Hou, J. USP15 inhibits multiple myeloma cell apoptosis through activating a feedback loop with the transcription factor NF-kappaBp65. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Ren, X.; Jiang, M.; Ding, P.; Zhang, X.; Zhou, X.; Shen, J.; Liu, D.; Yan, X.; Ma, Z. Ubiquitin-specific protease 28: The decipherment of its dual roles in cancer development. Exp. Hematol. Oncol. 2023, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Garcia, C.; Tomaskovic, I.; Shah, V.J.; Dikic, I.; Diefenbacher, M. USP28: Oncogene or Tumor Suppressor? A Unifying Paradigm for Squamous Cell Carcinoma. Cells 2021, 10, 2652. [Google Scholar] [CrossRef] [PubMed]
- Altun, M.; Zhao, B.; Velasco, K.; Liu, H.; Hassink, G.; Paschke, J.; Pereira, T.; Lindsten, K. Ubiquitin-specific protease 19 (USP19) regulates hypoxia-inducible factor 1alpha (HIF-1alpha) during hypoxia. J. Biol. Chem. 2012, 287, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Hinton, K.; Kirk, A.; Paul, P.; Persad, S. Regulation of the Epithelial to Mesenchymal Transition in Osteosarcoma. Biomolecules 2023, 13, 398. [Google Scholar] [CrossRef]
- Zhang, J.; van Dinther, M.; Thorikay, M.; Gourabi, B.M.; Kruithof, B.P.T.; Ten Dijke, P. Opposing USP19 splice variants in TGF-beta signaling and TGF-beta-induced epithelial-mesenchymal transition of breast cancer cells. Cell Mol. Life Sci. 2023, 80, 43. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Su, Y.; Fei, X.; Wang, X.; Zhang, G.; Su, C.; Du, T.; Yang, T.; Wang, G.; Tang, Z.; et al. Ubiquitin specific peptidase 19 is a prognostic biomarker and affect the proliferation and migration of clear cell renal cell carcinoma. Oncol. Rep. 2020, 43, 1964–1974. [Google Scholar] [CrossRef]
- Troilo, A.; Alexander, I.; Muehl, S.; Jaramillo, D.; Knobeloch, K.P.; Krek, W. HIF1alpha deubiquitination by USP8 is essential for ciliogenesis in normoxia. EMBO Rep. 2014, 15, 77–85. [Google Scholar] [CrossRef]
- Xie, F.; Zhou, X.; Li, H.; Su, P.; Liu, S.; Li, R.; Zou, J.; Wei, X.; Pan, C.; Zhang, Z.; et al. USP8 promotes cancer progression and extracellular vesicle-mediated CD8+ T cell exhaustion by deubiquitinating the TGF-beta receptor TbetaRII. EMBO J. 2022, 41, e108791. [Google Scholar] [CrossRef]
- Zhao, Y.; Peng, D.; Liu, Y.; Zhang, Q.; Liu, B.; Deng, Y.; Ding, W.; Zhou, Z.; Liu, Q. Usp8 promotes tumor cell migration through activating the JNK pathway. Cell Death Dis. 2022, 13, 286. [Google Scholar] [CrossRef]
- Williams, S.A.; Maecker, H.L.; French, D.M.; Liu, J.; Gregg, A.; Silverstein, L.B.; Cao, T.C.; Carano, R.A.; Dixit, V.M. USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 2011, 146, 918–930. [Google Scholar] [CrossRef]
- Yuan, P.; Feng, Z.; Huang, H.; Wang, G.; Chen, Z.; Xu, G.; Xie, Z.; Jie, Z.; Zhao, X.; Ma, Q.; et al. USP1 inhibition suppresses the progression of osteosarcoma via destabilizing TAZ. Int. J. Biol. Sci. 2022, 18, 3122–3136. [Google Scholar] [CrossRef] [PubMed]
- Lavaud, M.; Mullard, M.; Tesfaye, R.; Amiaud, J.; Legrand, M.; Danieau, G.; Brion, R.; Morice, S.; Regnier, L.; Dupuy, M.; et al. Overexpression of the Ubiquitin Specific Proteases USP43, USP41, USP27x and USP6 in Osteosarcoma Cell Lines: Inhibition of Osteosarcoma Tumor Growth and Lung Metastasis Development by the USP Antagonist PR619. Cells 2021, 10, 2268. [Google Scholar] [CrossRef] [PubMed]
- Gomarasca, M.; Lombardi, G.; Maroni, P. SUMOylation and NEDDylation in Primary and Metastatic Cancers to Bone. Front. Cell Dev. Biol. 2022, 10, 889002. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, D.; Wang, W.; Yang, W.C.; He, S.; Garcia, A.; Matunis, M.J. SUMO paralogue-specific functions revealed through systematic analysis of human knockout cell lines and gene expression data. Mol. Biol. Cell 2021, 32, 1849–1866. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Lu, Y.; Chan, Y.T.; Zhang, C.; Wang, N.; Feng, Y. The Role of Protein SUMOylation in Human Hepatocellular Carcinoma: A Potential Target of New Drug Discovery and Development. Cancers 2021, 13, 5700. [Google Scholar] [CrossRef] [PubMed]
- Knipscheer, P.; Flotho, A.; Klug, H.; Olsen, J.V.; van Dijk, W.J.; Fish, A.; Johnson, E.S.; Mann, M.; Sixma, T.K.; Pichler, A. Ubc9 sumoylation regulates SUMO target discrimination. Mol. Cell 2008, 31, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Kukkula, A.; Ojala, V.K.; Mendez, L.M.; Sistonen, L.; Elenius, K.; Sundvall, M. Therapeutic Potential of Targeting the SUMO Pathway in Cancer. Cancers 2021, 13, 4402. [Google Scholar] [CrossRef]
- Tokarz, P.; Wozniak, K. SENP Proteases as Potential Targets for Cancer Therapy. Cancers 2021, 13, 2059. [Google Scholar] [CrossRef]
- Sahin, U.; de The, H.; Lallemand-Breitenbach, V. Sumoylation in Physiology, Pathology and Therapy. Cells 2022, 11, 814. [Google Scholar] [CrossRef]
- Zhang, D.; Yu, K.; Yang, Z.; Li, Y.; Ma, X.; Bian, X.; Liu, F.; Li, L.; Liu, X.; Wu, W. Silencing Ubc9 expression suppresses osteosarcoma tumorigenesis and enhances chemosensitivity to HSV-TK/GCV by regulating connexin 43 SUMOylation. Int. J. Oncol. 2018, 53, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Zheng, G.Z.; Xie, P.; Zhang, Q.H.; Lin, F.X.; Chang, B.; Hu, Q.X.; Du, S.X.; Li, X.D. Antitumor activity of resveratrol against human osteosarcoma cells: A key role of Cx43 and Wnt/beta-catenin signaling pathway. Oncotarget 2017, 8, 111419–111432. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Barker, D.; Gibbins, J.M.; Dash, P.R. Talin is a substrate for SUMOylation in migrating cancer cells. Exp. Cell Res. 2018, 370, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Ruh, M.; Stemmler, M.P.; Frisch, I.; Fuchs, K.; van Roey, R.; Kleemann, J.; Roas, M.; Schuhwerk, H.; Eccles, R.L.; Agaimy, A.; et al. The EMT transcription factor ZEB1 blocks osteoblastic differentiation in bone development and osteosarcoma. J. Pathol. 2021, 254, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Perez-Oquendo, M.; Gibbons, D.L. Regulation of ZEB1 Function and Molecular Associations in Tumor Progression and Metastasis. Cancers 2022, 14, 1864. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, Y.; Zhou, F.; Jie, L.; Pu, L.; Ju, J.; Li, F.; Dai, Z.; Wang, X.; Zhou, S. Sox9 regulates hyperexpression of Wnt1 and Fzd1 in human osteosarcoma tissues and cells. Int. J. Clin. Exp. Pathol. 2014, 7, 4795–4805. [Google Scholar]
- Pei, H.; Chen, L.; Liao, Q.M.; Wang, K.J.; Chen, S.G.; Liu, Z.J.; Zhang, Z.C. SUMO-specific protease 2 (SENP2) functions as a tumor suppressor in osteosarcoma via SOX9 degradation. Exp. Ther. Med. 2018, 16, 5359–5365. [Google Scholar] [CrossRef]
- Wang, X.; Liang, X.; Liang, H.; Wang, B. SENP1/HIF-1alpha feedback loop modulates hypoxia-induced cell proliferation, invasion, and EMT in human osteosarcoma cells. J. Cell Biochem. 2018, 119, 1819–1826. [Google Scholar] [CrossRef]
- Wang, L.; Wu, J.; Song, S.; Chen, H.; Hu, Y.; Xu, B.; Liu, J. Plasma Exosome-Derived Sentrin SUMO-Specific Protease 1: A Prognostic Biomarker in Patients With Osteosarcoma. Front. Oncol. 2021, 11, 625109. [Google Scholar] [CrossRef]
- Liu, F.; Li, L.; Li, Y.; Ma, X.; Bian, X.; Liu, X.; Wang, G.; Zhang, D. Overexpression of SENP1 reduces the stemness capacity of osteosarcoma stem cells and increases their sensitivity to HSVtk/GCV. Int. J. Oncol. 2018, 53, 2010–2020. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, X.C. Inhibition of SENP5 suppresses cell growth and promotes apoptosis in osteosarcoma cells. Exp. Ther. Med. 2014, 7, 1691–1695. [Google Scholar] [CrossRef]
- Liu, T.; Wang, H.; Chen, Y.; Wan, Z.; Du, Z.; Shen, H.; Yu, Y.; Ma, S.; Xu, Y.; Li, Z.; et al. SENP5 promotes homologous recombination-mediated DNA damage repair in colorectal cancer cells through H2AZ deSUMOylation. J. Exp. Clin. Cancer Res. 2023, 42, 234. [Google Scholar] [CrossRef] [PubMed]
- Saggu, G.; Stroopinsky, D.; Dudek, A.Z.; Olszanski, A.J.; Juric, D.; Dowlati, A.; Vaishampayan, U.; Assad, H.; Rodón, J.; Gibbs, J.; et al. 352 (PB132)—Subasumstat, a first-in-class inhibitor of SUMO-activating enzyme, demonstrates dose-dependent target engagement and SUMOylation inhibition, leading to rapid activation of innate and adaptive immune responses in the dose escalation portion of a phase 1/2 clinical study. Eur. J. Cancer 2022, 174, S125–S126. [Google Scholar] [CrossRef]
- Gabellier, L.; De Toledo, M.; Chakraborty, M.; Akl, D.; Hallal, R.; Aqrouq, M.; Buonocore, G.; Recasens-Zorzo, C.; Cartron, G.; Delort, A.; et al. SUMOylation inhibitor TAK-981 (subasumstat) synergizes with 5-azacytidine in preclinical models of acute myeloid leukemia. Haematologica 2024, 109, 98–114. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Roleder, C.; Liu, T.; Bruss, N.; Best, S.; Wang, X.; Phillips, T.; Shouse, G.; Berger, A.J.; Alinari, L.; et al. T Cell-intrinsic Immunomodulatory Effects of TAK-981 (Subasumstat), a SUMO-activating Enzyme Inhibitor, in Chronic Lymphocytic Leukemia. Mol. Cancer Ther. 2023, 22, 1040–1051. [Google Scholar] [CrossRef]
- Nakamura, A.; Grossman, S.; Song, K.; Xega, K.; Zhang, Y.; Cvet, D.; Berger, A.; Shapiro, G.; Huszar, D. The SUMOylation inhibitor subasumstat potentiates rituximab activity by IFN1-dependent macrophage and NK cell stimulation. Blood 2022, 139, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jiang, Y.; Luo, Q.; Li, L.; Jia, L. Neddylation: A novel modulator of the tumor microenvironment. Mol. Cancer 2019, 18, 77. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef]
- Rabut, G.; Peter, M. Function and regulation of protein neddylation. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Jia, L. Targeting Protein Neddylation for Cancer Therapy. Adv. Exp. Med. Biol. 2020, 1217, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Laigle, V.; Dingli, F.; Amhaz, S.; Perron, T.; Chouchene, M.; Colasse, S.; Petit, I.; Poullet, P.; Loew, D.; Prunier, C.; et al. Quantitative Ubiquitylome Analysis Reveals the Specificity of RNF111/Arkadia E3 Ubiquitin Ligase for its Degradative Substrates SKI and SKIL/SnoN in TGF-beta Signaling Pathway. Mol. Cell Proteom. 2021, 20, 100173. [Google Scholar] [CrossRef] [PubMed]
- Broemer, M.; Tenev, T.; Rigbolt, K.T.; Hempel, S.; Blagoev, B.; Silke, J.; Ditzel, M.; Meier, P. Systematic in vivo RNAi analysis identifies IAPs as NEDD8-E3 ligases. Mol. Cell 2010, 40, 810–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, B.; Jiang, K.; Lao, L.; Shen, H.; Chen, Z. Activation of TNF-alpha/NF-kappaB axis enhances CRL4B(DCAF)(11) E3 ligase activity and regulates cell cycle progression in human osteosarcoma cells. Mol. Oncol. 2018, 12, 476–494. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Du, D.; Peng, Y.; Liu, J.; Long, J. The functional analysis of Cullin 7 E3 ubiquitin ligases in cancer. Oncogenesis 2020, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, W.; Gao, X.; Lu, X.; Xie, F.; Um, S.W.; Kang, M.W.; Yang, H.; Shang, Y.; Wang, Z.; et al. Cullin7 induces docetaxel resistance by regulating the protein level of the antiapoptotic protein Survivin in lung adenocarcinoma cells. J. Thorac. Dis. 2023, 15, 5006–5019. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Z.; Qian, M.; Wang, S.; Qiu, W.; Chen, Z.; Sun, Z.; Xiong, Y.; Wang, C.; Sun, X.; et al. Cullin-7 (CUL7) is overexpressed in glioma cells and promotes tumorigenesis via NF-kappaB activation. J. Exp. Clin. Cancer Res. 2020, 39, 59. [Google Scholar] [CrossRef]
- Bravo-Navas, S.; Yanez, L.; Romon, I.; Briz, M.; Dominguez-Garcia, J.J.; Pipaon, C. Map of ubiquitin-like post-translational modifications in chronic lymphocytic leukemia. Role of p53 lysine 120 NEDDylation. Leukemia 2021, 35, 3568–3572. [Google Scholar] [CrossRef]
- Xie, P.; Peng, Z.; Chen, Y.; Li, H.; Du, M.; Tan, Y.; Zhang, X.; Lu, Z.; Cui, C.P.; Liu, C.H.; et al. Neddylation of PTEN regulates its nuclear import and promotes tumor development. Cell Res. 2021, 31, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Zhang, M.; He, S.; Lu, K.; Chen, Y.; Xing, G.; Lu, Y.; Liu, P.; Li, Y.; Wang, S.; et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat. Commun. 2014, 5, 3733. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Si, Y.; Yu, H.; Zhang, L.; Xie, P.; Jiang, W. MLN4924 (Pevonedistat), a protein neddylation inhibitor, suppresses proliferation and migration of human clear cell renal cell carcinoma. Sci. Rep. 2017, 7, 5599. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kuo, K.L.; Lin, W.C.; Chen, M.S.; Liu, S.H.; Liao, S.M.; Hsu, C.H.; Chang, Y.W.; Chang, H.C.; Huang, K.H. Neddylation inhibitor, MLN4924 suppresses angiogenesis in huvecs and solid cancers: In vitro and in vivo study. Am. J. Cancer Res. 2020, 10, 953–964. [Google Scholar] [PubMed]
- Liu, H.; Bei, Q.; Luo, X. MLN4924 inhibits cell proliferation by targeting the activated neddylation pathway in endometrial carcinoma. J. Int. Med. Res. 2021, 49, 3000605211018592. [Google Scholar] [CrossRef]
- Ferris, J.; Espona-Fiedler, M.; Hamilton, C.; Holohan, C.; Crawford, N.; McIntyre, A.J.; Roberts, J.Z.; Wappett, M.; McDade, S.S.; Longley, D.B.; et al. Pevonedistat (MLN4924): Mechanism of cell death induction and therapeutic potential in colorectal cancer. Cell Death Discov. 2020, 6, 61. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, C.C.; Zhang, H.P.; Li, G.Q.; Li, S.S. MLN4924 suppresses neddylation and induces cell cycle arrest, senescence, and apoptosis in human osteosarcoma. Oncotarget 2016, 7, 45263–45274. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Esmail, S.; Manolson, M.F. Advances in understanding N-glycosylation structure, function, and regulation in health and disease. Eur. J. Cell Biol. 2021, 100, 151186. [Google Scholar] [CrossRef]
- Wandall, H.H.; Nielsen, M.A.I.; King-Smith, S.; de Haan, N.; Bagdonaite, I. Global functions of O-glycosylation: Promises and challenges in O-glycobiology. FEBS J. 2021, 288, 7183–7212. [Google Scholar] [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188464. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Teng, H.; Zhang, Y.; Wang, F.; Tang, L.; Zhang, C.; Hu, Z.; Chen, Y.; Ge, Y.; Wang, Z.; et al. A glycosylation-related gene signature predicts prognosis, immune microenvironment infiltration, and drug sensitivity in glioma. Front. Pharmacol. 2024, 14, 1259051. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.K.; Bouchie, M.P.; Kukuruzinska, M.A. N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J. Biol. Chem. 2010, 285, 31164–31173. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.K.; Bouchie, M.P.; Nita-Lazar, M.; Yang, H.Y.; Kukuruzinska, M.A. Coordinate regulation of N-glycosylation gene DPAGT1, canonical Wnt signaling and E-cadherin adhesion. J. Cell Sci. 2013, 126, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Liu, L.; Zeng, T.; Liang, J.Z.; Song, Y.; Chen, K.; Li, Y.; Chen, L.; Zhu, Y.H.; Li, J.; et al. Overexpression of MUC13, a Poor Prognostic Predictor, Promotes Cell Growth by Activating Wnt Signaling in Hepatocellular Carcinoma. Am. J. Pathol. 2018, 188, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Nita-Lazar, M.; Rebustini, I.; Walker, J.; Kukuruzinska, M.A. Hypoglycosylated E-cadherin promotes the assembly of tight junctions through the recruitment of PP2A to adherens junctions. Exp. Cell Res. 2010, 316, 1871–1884. [Google Scholar] [CrossRef]
- Meng, Q.; Ren, C.; Wang, L.; Zhao, Y.; Wang, S. Knockdown of ST6Gal-I inhibits the growth and invasion of osteosarcoma MG-63 cells. Biomed. Pharmacother. 2015, 72, 172–178. [Google Scholar] [CrossRef]
- Harosh-Davidovich, S.B.; Khalaila, I. O-GlcNAcylation affects beta-catenin and E-cadherin expression, cell motility and tumorigenicity of colorectal cancer. Exp. Cell Res. 2018, 364, 42–49. [Google Scholar] [CrossRef]
- Hamester, F.; Legler, K.; Wichert, B.; Kelle, N.; Eylmann, K.; Rossberg, M.; Ding, Y.; Kurti, S.; Schmalfeldt, B.; Milde-Langosch, K.; et al. Prognostic relevance of the Golgi mannosidase MAN1A1 in ovarian cancer: Impact of N-glycosylation on tumour cell aggregation. Br. J. Cancer 2019, 121, 944–953. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Bachvarova, M.; Morin, C.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Bachvarov, D. Role of the polypeptide N-acetylgalactosaminyltransferase 3 in ovarian cancer progression: Possible implications in abnormal mucin O-glycosylation. Oncotarget 2014, 5, 544–560. [Google Scholar] [CrossRef]
- Schroeder, J.A.; Adriance, M.C.; Thompson, M.C.; Camenisch, T.D.; Gendler, S.J. MUC1 alters beta-catenin-dependent tumor formation and promotes cellular invasion. Oncogene 2003, 22, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Baldari, S.; Di Modugno, F.; Nistico, P.; Toietta, G. Strategies for Efficient Targeting of Tumor Collagen for Cancer Therapy. Cancers 2022, 14, 4706. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhong, L.; Li, Y.; Xiao, D.; Zhang, R.; Liao, D.; Lv, D.; Wang, X.; Wang, J.; Xie, X.; et al. Up-regulation of PCOLCE by TWIST1 promotes metastasis in Osteosarcoma. Theranostics 2019, 9, 4342–4353. [Google Scholar] [CrossRef] [PubMed]
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [CrossRef]
- Singla, A.; Wang, J.; Yang, R.; Geller, D.S.; Loeb, D.M.; Hoang, B.H. Wnt Signaling in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1258, 125–139. [Google Scholar] [CrossRef]
- Kariya, Y.; Oyama, M.; Hashimoto, Y.; Gu, J.; Kariya, Y. beta4-Integrin/PI3K Signaling Promotes Tumor Progression through the Galectin-3-N-Glycan Complex. Mol. Cancer Res. 2018, 16, 1024–1034. [Google Scholar] [CrossRef]
- Wang, Y.; Fukuda, T.; Isaji, T.; Lu, J.; Im, S.; Hang, Q.; Gu, W.; Hou, S.; Ohtsubo, K.; Gu, J. Loss of alpha1,6-fucosyltransferase inhibits chemical-induced hepatocellular carcinoma and tumorigenesis by down-regulating several cell signaling pathways. FASEB J. 2015, 29, 3217–3227. [Google Scholar] [CrossRef]
- Agrawal, P.; Fontanals-Cirera, B.; Sokolova, E.; Jacob, S.; Vaiana, C.A.; Argibay, D.; Davalos, V.; McDermott, M.; Nayak, S.; Darvishian, F.; et al. A Systems Biology Approach Identifies FUT8 as a Driver of Melanoma Metastasis. Cancer Cell 2017, 31, 804–819. [Google Scholar] [CrossRef]
- Tu, C.F.; Wu, M.Y.; Lin, Y.C.; Kannagi, R.; Yang, R.B. FUT8 promotes breast cancer cell invasiveness by remodeling TGF-beta receptor core fucosylation. Breast Cancer Res. 2017, 19, 111. [Google Scholar] [CrossRef]
- Zhao, Y.P.; Xu, X.Y.; Fang, M.; Wang, H.; You, Q.; Yi, C.H.; Ji, J.; Gu, X.; Zhou, P.T.; Cheng, C.; et al. Decreased core-fucosylation contributes to malignancy in gastric cancer. PLoS ONE 2014, 9, e94536. [Google Scholar] [CrossRef]
- Lin, S.; Zhou, L.; Dong, Y.; Yang, Q.; Yang, Q.; Jin, H.; Yuan, T.; Zhou, S. Alpha-(1,6)-fucosyltransferase (FUT8) affects the survival strategy of osteosarcoma by remodeling TNF/NF-kappaB2 signaling. Cell Death Dis. 2021, 12, 1124. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Cegang, F.; Zhao, H.; Wang, B.; Jia, S.; Chen, H.; Cai, H. Up-regulation of Core 1 Beta 1, 3-Galactosyltransferase Suppresses Osteosarcoma Growth with Induction of IFN-gamma Secretion and Proliferation of CD8(+) T Cells. Curr. Cancer Drug Targets 2023, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Manara, M.C.; Baldini, N.; Serra, M.; Lollini, P.L.; De Giovanni, C.; Vaccari, M.; Argnani, A.; Benini, S.; Maurici, D.; Picci, P.; et al. Reversal of malignant phenotype in human osteosarcoma cells transduced with the alkaline phosphatase gene. Bone 2000, 26, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757–766. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Gregorio, J.; Di Giuseppe, L.; Terreri, S.; Rossi, M.; Battafarano, G.; Pagliarosi, O.; Flati, V.; Del Fattore, A. Protein Stability Regulation in Osteosarcoma: The Ubiquitin-like Modifications and Glycosylation as Mediators of Tumor Growth and as Targets for Therapy. Cells 2024, 13, 537. https://doi.org/10.3390/cells13060537
Di Gregorio J, Di Giuseppe L, Terreri S, Rossi M, Battafarano G, Pagliarosi O, Flati V, Del Fattore A. Protein Stability Regulation in Osteosarcoma: The Ubiquitin-like Modifications and Glycosylation as Mediators of Tumor Growth and as Targets for Therapy. Cells. 2024; 13(6):537. https://doi.org/10.3390/cells13060537
Chicago/Turabian StyleDi Gregorio, Jacopo, Laura Di Giuseppe, Sara Terreri, Michela Rossi, Giulia Battafarano, Olivia Pagliarosi, Vincenzo Flati, and Andrea Del Fattore. 2024. "Protein Stability Regulation in Osteosarcoma: The Ubiquitin-like Modifications and Glycosylation as Mediators of Tumor Growth and as Targets for Therapy" Cells 13, no. 6: 537. https://doi.org/10.3390/cells13060537