
Abnormal Morphology and Synaptogenic Signaling in Astrocytes Following Prenatal Opioid Exposure

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Prenatal Drug Treatment
2.3. Cortical Neuron Purification, Culture, and Treatment
2.4. Cortical Astrocyte Purification, Culture, Transfection, and Lysis
2.5. Immunocytochemistry (ICC)
2.6. Cell Viability Assay
2.7. Western Blotting
2.8. Image Analysis and Statistics
3. Results
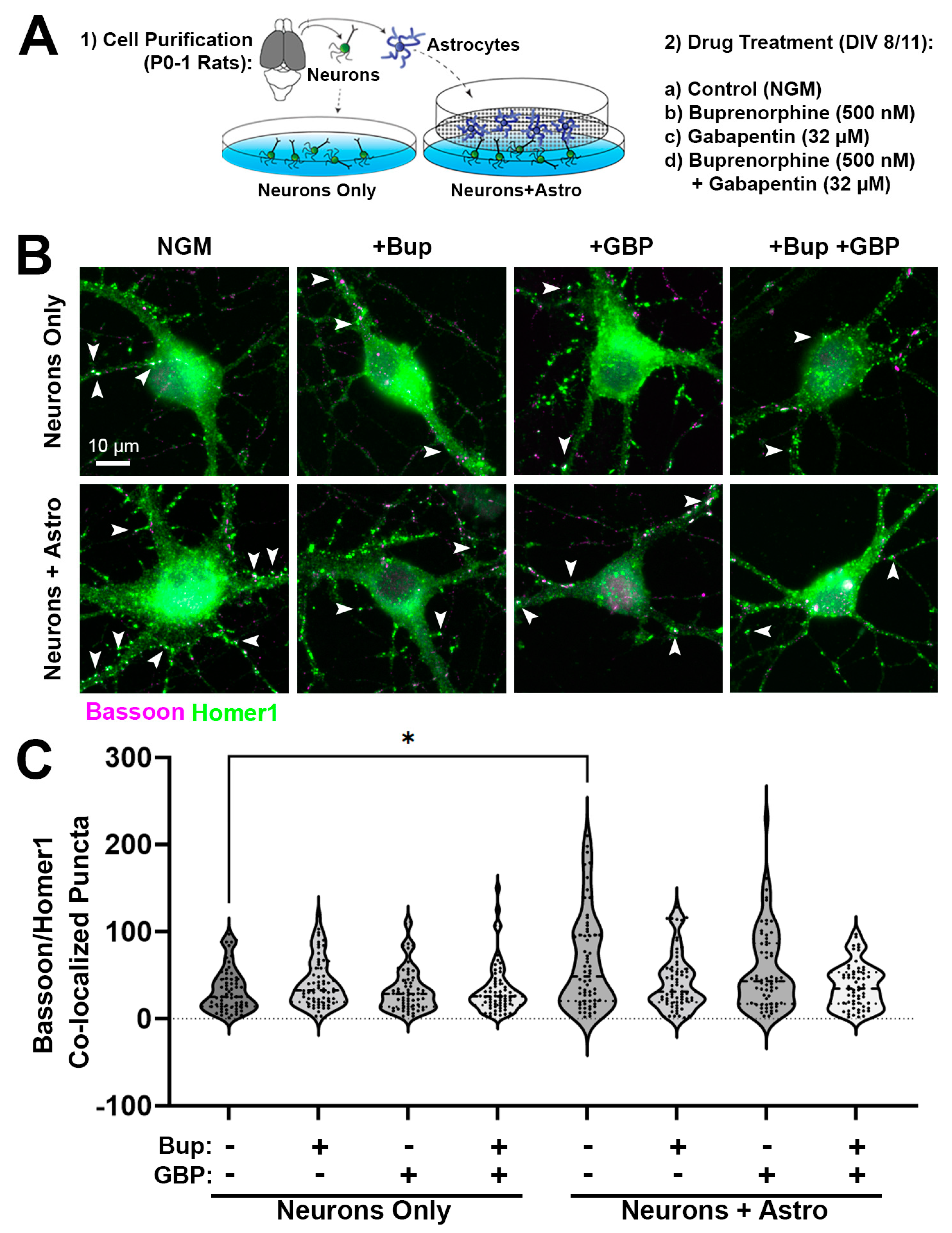
3.1. Acute Buprenorphine Exposure Inhibits Astrocyte-Mediated Synaptogenesis
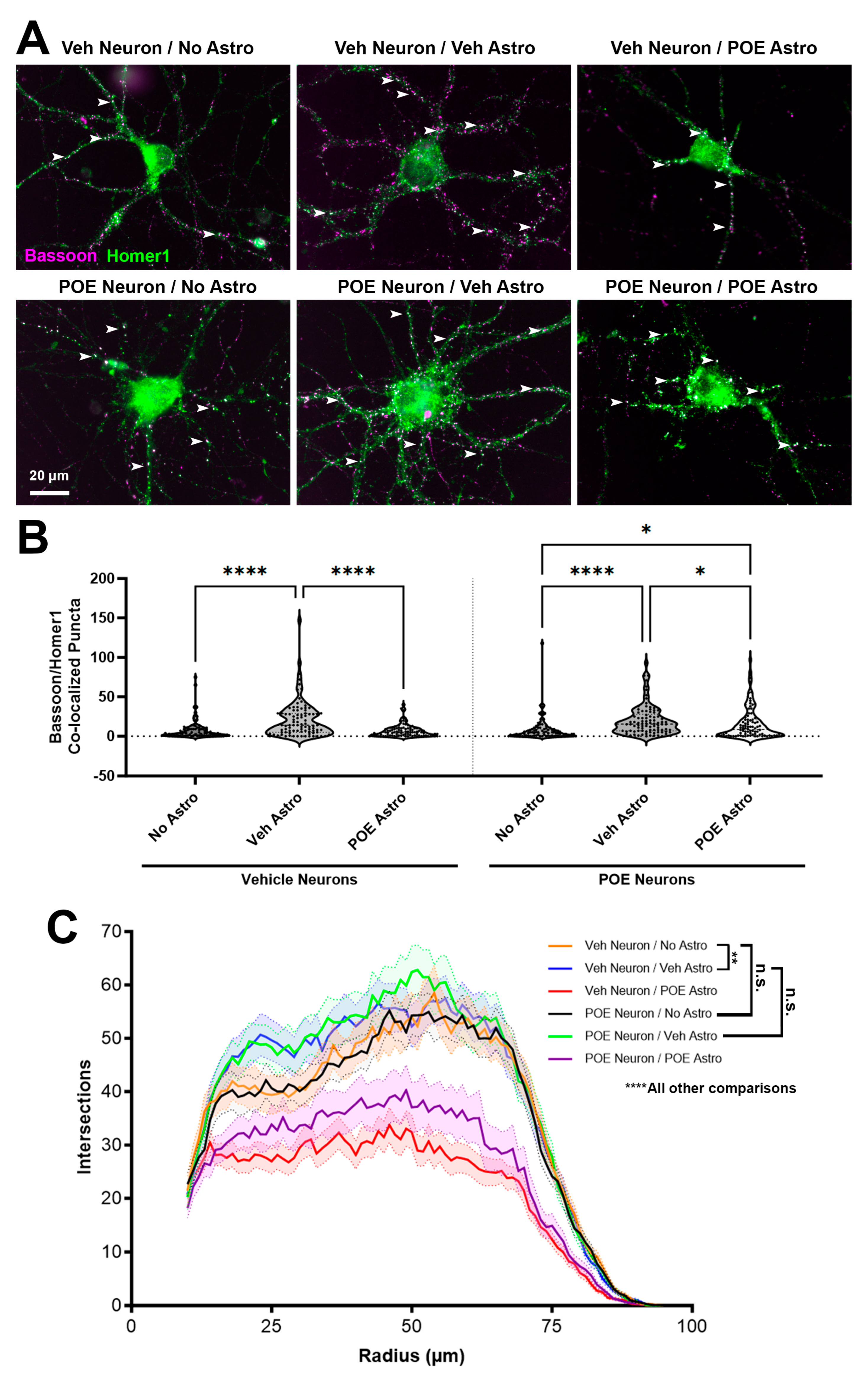
3.2. Prenatal Buprenorphine Promotes Neuroadaptive Changes in Response to Astrocyte-Secreted Factors
3.3. POE Does Not Affect the Short-Term Survival of Cultured Neurons and Astrocytes
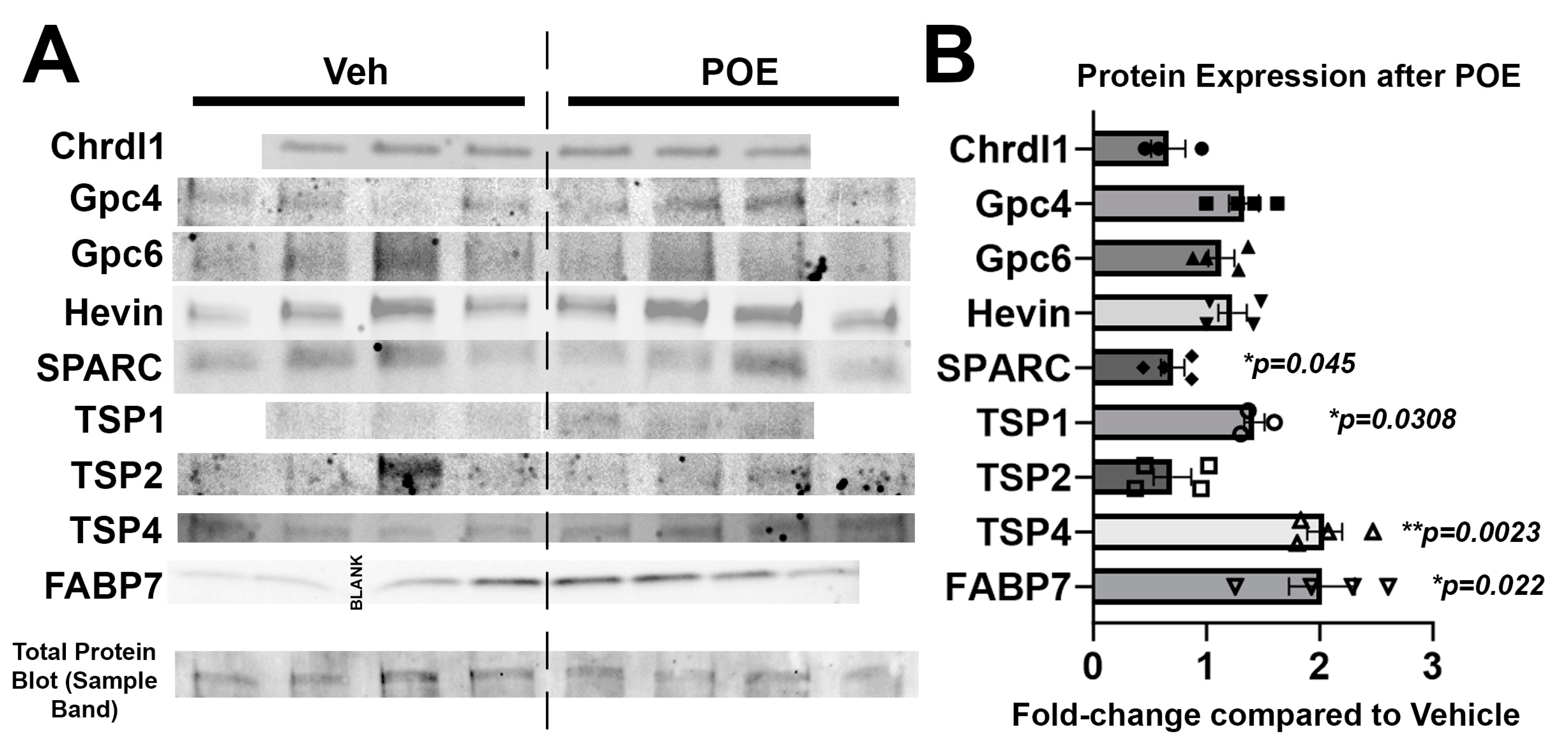
3.4. Buprenorphine Alters the Expression of Astrocytic Proteins Known to Regulate Synaptic and Neuronal Maturation
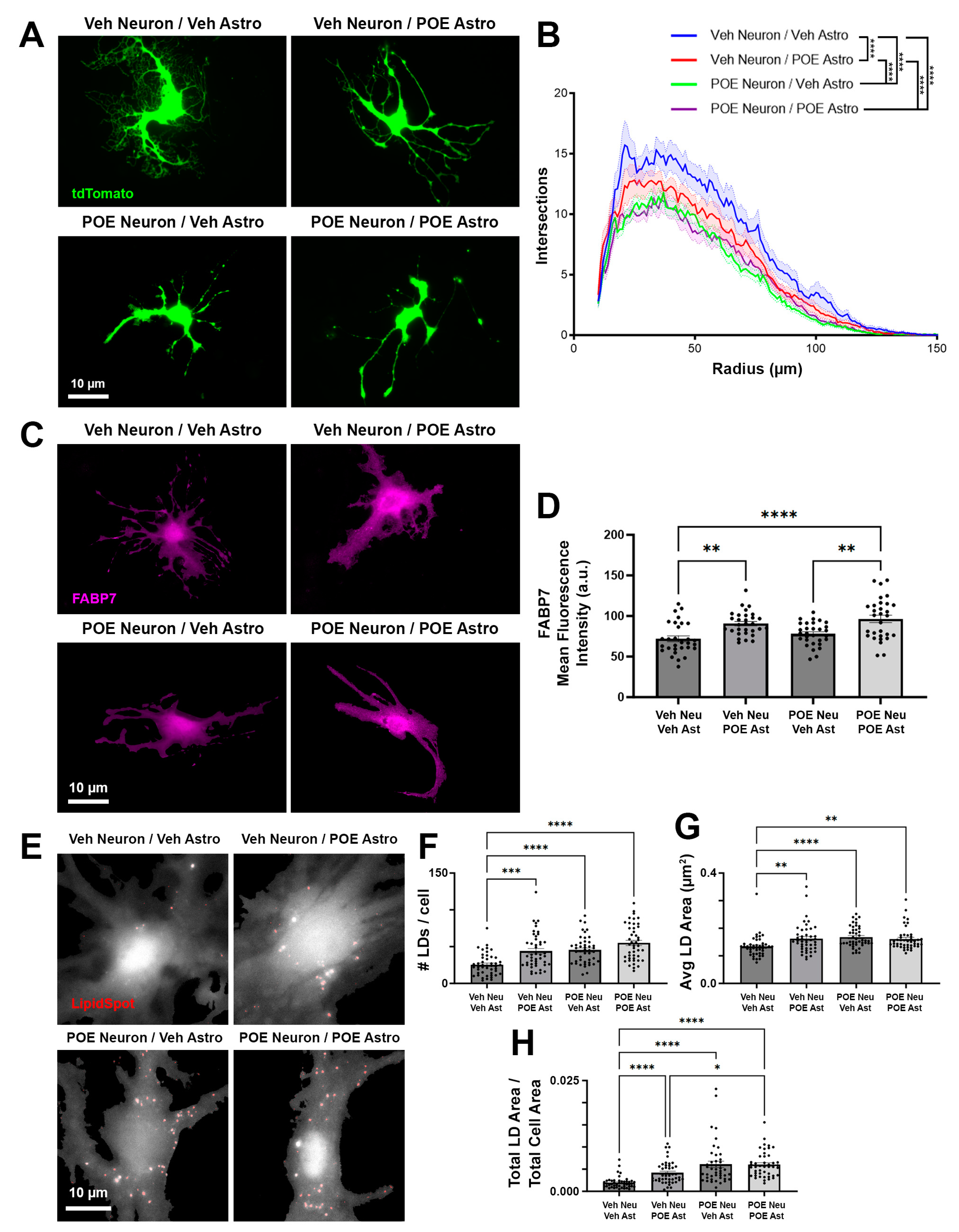
3.5. Disruptions in Astrocyte Morphology and Lipid Droplets Associated with POE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
| Abbreviation/Acronym | Definition |
| AGM | Astrocyte growth medium |
| ANCOVA | Analysis of co-variance |
| ANOVA | Analysis of variance |
| AraC | Cytosine arabinoside |
| Astro | Astrocyte |
| BDNF | Brain-derived neurotrophic factor |
| Bup | Buprenorphine |
| Chrdl1 | Chordin-like 1 |
| CNS | Central nervous system |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DIV# | Day in vitro # |
| DPBS | Dulbecco’s phosphate-buffered saline |
| E# | Embryonic day # |
| ECM | Extracellular matrix |
| FABP7 | Fatty acid binding protein 7 |
| GBP | Gabapentin |
| Gpc | Glypican |
| ICC | Immunocytochemistry |
| LD | Lipid droplet |
| MAT | Medication-assisted treatment |
| MOR | µ-opioid receptor |
| NAS | Neonatal abstinence syndrome |
| NGM | Neuronal growth medium |
| NGS | Normal goat serum |
| P# | Postnatal day # |
| PBS | Phosphate-buffered saline |
| PDL | Poly-D-lysine |
| PFA | Paraformaldehyde |
| POE | Prenatal opioid exposure |
| PUFA | Polyunsaturated fatty acid |
| SPARC | Secreted protein acidic and rich in cysteine |
| TSP | Thrombospondin |
| Veh | Vehicle |
References
- Kocherlakota, P. Neonatal abstinence syndrome. Pediatrics 2014, 134, e547–e561. [Google Scholar] [CrossRef]
- Wachman, E.M.; Warden, A.H.; Thomas, Z.; Thomas-Lewis, J.A.; Shrestha, H.; Nikita, F.N.U.; Shaw, D.; Saia, K.; Schiff, D.M. Impact of psychiatric medication co-exposure on Neonatal Abstinence Syndrome severity. Drug Alcohol Depend. 2018, 192, 45–50. [Google Scholar] [CrossRef]
- Patrick, S.W.; Davis, M.M.; Lehmann, C.U.; Cooper, W.O. Increasing incidence and geographic distribution of neonatal abstinence syndrome: United States 2009 to 2012. J. Perinatol. 2015, 35, 650–655. [Google Scholar] [CrossRef]
- Batra, K.; Cruz, P.; Cross, C.L.; Bhandari, N.; Abdulla, F.; Pharr, J.R.; Buttner, M.P. Incidence of neonatal abstinence syndrome epidemic and associated predictors in Nevada: A statewide audit, 2016–2018. Int. J. Environ. Res. Public Health 2020, 18, 232. [Google Scholar] [CrossRef]
- Boggess, T.; Risher, W.C. Clinical and basic research investigations into the long-term effects of prenatal opioid exposure on brain development. J. Neurosci. Res. 2022, 100, 396–409. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.C.; Grecco, G.G.; Atwood, B.K.; Nugent, F.S. Effects of prenatal opioid exposure on synaptic adaptations and behaviors across development. Neuropharmacology 2023, 222, 109312. [Google Scholar] [CrossRef]
- Yeoh, S.L.; Eastwood, J.; Wright, I.M.; Morton, R.; Melhuish, E.; Ward, M.; Oei, J.L. Cognitive and motor outcomes of children with prenatal opioid exposure: A systematic review and meta-analysis. JAMA Netw. Open 2019, 2, e197025. [Google Scholar] [CrossRef]
- Nygaard, E.; Moe, V.; Slinning, K.; Walhovd, K.B. Longitudinal cognitive development of children born to mothers with opioid and polysubstance use. Pediatr. Res. 2015, 78, 330–335. [Google Scholar] [CrossRef]
- Boggess, T.; Williamson, J.C.; Niebergall, E.B.; Sexton, H.; Mazur, A.; Egleton, R.D.; Grover, L.M.; Risher, W.C. Alterations in excitatory and inhibitory synaptic development within the mesolimbic dopamine pathway in a mouse model of prenatal drug exposure. Front. Pediatr. 2021, 9, 794544. [Google Scholar] [CrossRef]
- Chung, W.S.; Allen, N.J.; Eroglu, C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.X.; Eroglu, C. Cell adhesion molecules regulating astrocyte-neuron interactions. Curr. Opin. Neurobiol. 2021, 69, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Kucukdereli, H.; Allen, N.J.; Lee, A.T.; Feng, A.; Ozlu, M.I.; Conatser, L.M.; Chakraborty, C.; Workman, G.; Weaver, M.; Sage, E.H.; et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proc. Natl. Acad. Sci. USA 2011, 108, E440–E449. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Suarez, E.; Liu, T.F.; Kopelevich, A.; Allen, N.J. Astrocyte-secreted chordin-like 1 drives synapse maturation and limits plasticity by increasing synaptic GluA2 AMPA receptors. Neuron 2018, 100, 1116–1132.e13. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, J.A.; Ramirez, J.; Liu, D.; Kim, Y.H.; Baldwin, K.T.; Enustun, E.; Ejikeme, T.; Ji, R.R.; Eroglu, C. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 2017, 551, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.T.; Tan, C.X.; Strader, S.T.; Jiang, C.; Savage, J.T.; Elorza-Vidal, X.; Contreras, X.; Rulicke, T.; Hippenmeyer, S.; Estevez, R.; et al. HepaCAM controls astrocyte self-organization and coupling. Neuron 2021, 109, 2427–2442.e10. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.X.; Bindu, D.S.; Hardin, E.J.; Sakers, K.; Baumert, R.; Ramirez, J.J.; Savage, J.T.; Eroglu, C. delta-Catenin controls astrocyte morphogenesis via layer-specific astrocyte-neuron cadherin interactions. J. Cell Biol. 2023, 222, e202303138. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.L.; Bisagno, V. Glial-neuronal ensembles: Partners in drug addiction-associated synaptic plasticity. Front. Pharmacol. 2014, 5, 204. [Google Scholar] [CrossRef]
- Kruyer, A.; Kalivas, P.W. Astrocytes as cellular mediators of cue reactivity in addiction. Curr. Opin. Pharmacol. 2021, 56, 1–6. [Google Scholar] [CrossRef]
- Ikeda, H.; Miyatake, M.; Koshikawa, N.; Ochiai, K.; Yamada, K.; Kiss, A.; Donlin, M.J.; Panneton, W.M.; Churchill, J.D.; Green, M.; et al. Morphine modulation of thrombospondin levels in astrocytes and its implications for neurite outgrowth and synapse formation. J. Biol. Chem. 2010, 285, 38415–38427. [Google Scholar] [CrossRef] [PubMed]
- Phamduong, E.; Rathore, M.K.; Crews, N.R.; D’Angelo, A.S.; Leinweber, A.L.; Kappera, P.; Krenning, T.M.; Rendell, V.R.; Belcheva, M.M.; Coscia, C.J. Acute and chronic mu opioids differentially regulate thrombospondins 1 and 2 isoforms in astrocytes. ACS Chem. Neurosci. 2014, 5, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Corkrum, M.; Rothwell, P.E.; Thomas, M.J.; Kofuji, P.; Araque, A. Opioid-mediated astrocyte-neuron signaling in the nucleus accumbens. Cells 2019, 8, 586. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Knapp, P.E. Opiate drugs with abuse liability hijack the endogenous opioid system to disrupt neuronal and glial maturation in the central nervous system. Front. Pediatr. 2017, 5, 294. [Google Scholar] [CrossRef]
- Martin, L.B.; Thompson, A.C.; Martin, T.; Kristal, M.B. Analgesic efficacy of orally administered buprenorphine in rats. Comp. Med. 2001, 51, 43–48. [Google Scholar] [PubMed]
- Lindemalm, S.; Nydert, P.; Svensson, J.O.; Stahle, L.; Sarman, I. Transfer of buprenorphine into breast milk and calculation of infant drug dose. J. Hum. Lact. 2009, 25, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Risher, W.C.; Kim, N.; Koh, S.; Choi, J.E.; Mitev, P.; Spence, E.F.; Pilaz, L.J.; Wang, D.; Feng, G.; Silver, D.L.; et al. Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J. Cell Biol. 2018, 217, 3747–3765. [Google Scholar] [CrossRef]
- Mazur, A.; Bills, E.H.; DeSchepper, K.M.; Williamson, J.C.; Henderson, B.J.; Risher, W.C. Astrocyte-derived thrombospondin induces cortical synaptogenesis in a sex-specific manner. eNeuro 2021, 8, 1–15. [Google Scholar] [CrossRef]
- Ippolito, D.M.; Eroglu, C. Quantifying synapses: An immunocytochemistry-based assay to quantify synapse number. J. Vis. Exp. 2010, 45, e2270. [Google Scholar] [CrossRef]
- Ferreira, T.A.; Blackman, A.V.; Oyrer, J.; Jayabal, S.; Chung, A.J.; Watt, A.J.; Sjostrom, P.J.; van Meyel, D.J. Neuronal morphometry directly from bitmap images. Nat. Methods 2014, 11, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Arshadi, C.; Gunther, U.; Eddison, M.; Harrington, K.I.S.; Ferreira, T.A. SNT: A unifying toolbox for quantification of neuronal anatomy. Nat. Methods 2021, 18, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Kutra, D.; Kroeger, T.; Straehle, C.N.; Kausler, B.X.; Haubold, C.; Schiegg, M.; Ales, J.; Beier, T.; Rudy, M.; et al. ilastik: Interactive machine learning for (bio)image analysis. Nat. Methods 2019, 16, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Farhy-Tselnicker, I.; Allen, N.J. Astrocytes, neurons, synapses: A tripartite view on cortical circuit development. Neural Dev. 2018, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Risher, W.C.; Eroglu, C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biol. 2012, 31, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, C.; Allen, N.J.; Susman, M.W.; O’Rourke, N.A.; Park, C.Y.; Ozkan, E.; Chakraborty, C.; Mulinyawe, S.B.; Annis, D.S.; Huberman, A.D.; et al. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 2009, 139, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Kraft, W.K. Buprenorphine in neonatal abstinence syndrome. Clin. Pharmacol. Ther. 2018, 103, 112–119. [Google Scholar] [CrossRef]
- Jha, M.K.; Kim, J.H.; Song, G.J.; Lee, W.H.; Lee, I.K.; Lee, H.W.; An, S.S.A.; Kim, S.; Suk, K. Functional dissection of astrocyte-secreted proteins: Implications in brain health and diseases. Prog. Neurobiol. 2018, 162, 37–69. [Google Scholar] [CrossRef]
- Jones, E.V.; Bouvier, D.S. Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast. 2014, 2014, 321209. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Yamamoto, Y.; Sharifi, K.; Kida, H.; Kagawa, Y.; Yasumoto, Y.; Islam, A.; Miyazaki, H.; Shimamoto, C.; Maekawa, M.; et al. Astrocyte-expressed FABP7 regulates dendritic morphology and excitatory synaptic function of cortical neurons. Glia 2016, 64, 48–62. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Islam, A.; Kagawa, Y.; Miyazaki, H.; Shil, S.K.; Umaru, B.A.; Yasumoto, Y.; Yamamoto, Y.; Owada, Y. FABP7 protects astrocytes against ROS toxicity via lipid droplet formation. Mol. Neurobiol. 2019, 56, 5763–5779. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, Y.; Kuniyoshi, Y. A translational model to determine rodent’s age from human foetal age. Sci. Rep. 2017, 7, 17248. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.-H.; Han, K.-S.; Lee, J.; Bae, J.Y.; An, H.; Park, S.; Oh, S.-J.; Kim, E.; Hwang, E.; Bae, Y.C.; et al. Expression of µ-opioid receptor in CA1 hippocampal astrocytes. Exp. Neurobiol. 2018, 27, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Corkrum, M.; Araque, A. Astrocyte-neuron signaling in the mesolimbic dopamine system: The hidden stars of dopamine signaling. Neuropsychopharmacology 2021, 46, 1864–1872. [Google Scholar] [CrossRef] [PubMed]
- Hauser, K.F.; Stiene-Martin, A.; Mattson, M.P.; Elde, R.P.; Ryan, S.E.; Godleske, C.C. mu-Opioid receptor-induced Ca2+ mobilization and astroglial development: Morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca(2+)-dependent mechanism. Brain Res. 1996, 720, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Seney, M.L.; Kim, S.M.; Glausier, J.R.; Hildebrand, M.A.; Xue, X.; Zong, W.; Wang, J.; Shelton, M.A.; Phan, B.N.; Srinivasan, C.; et al. Transcriptional alterations in dorsolateral prefrontal cortex and nucleus accumbens implicate neuroinflammation and synaptic remodeling in opioid use disorder. Biol. Psychiatry 2021, 90, 550–562. [Google Scholar] [CrossRef]
- Narita, M.; Miyatake, M.; Narita, M.; Shibasaki, M.; Shindo, K.; Nakamura, A.; Kuzumaki, N.; Nagumo, Y.; Suzuki, T. Direct evidence of astrocytic modulation in the development of rewarding effects induced by drugs of abuse. Neuropsychopharmacology 2006, 31, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Hung, C.J.; Shen, C.H.; Chen, W.Y.; Chang, C.Y.; Pan, H.C.; Liao, S.L.; Chen, C.J. Prenatal buprenorphine exposure decreases neurogenesis in rats. Toxicol. Lett. 2014, 225, 92–101. [Google Scholar] [CrossRef]
- Linker, K.E.; Cross, S.J.; Leslie, F.M. Glial mechanisms underlying substance use disorders. Eur. J. Neurosci. 2019, 50, 2574–2589. [Google Scholar] [CrossRef]
- Hughes, E.G.; Elmariah, S.B.; Balice-Gordon, R.J. Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Mol. Cell. Neurosci. 2010, 43, 136–145. [Google Scholar] [CrossRef]
- Irala, D.; Wang, S.; Sakers, K.; Nagendren, L.; Ulloa Severino, F.P.; Bindu, D.S.; Savage, J.T.; Eroglu, C. Astrocyte-secreted neurocan controls inhibitory synapse formation and function. Neuron 2024, 112, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Li, Q.; Dong, Y.; Yan, W.; Song, K.; Lin, Y.Q.; Sun, Y.G. Mu-opioid receptors expressed in glutamatergic neurons are essential for morphine withdrawal. Neurosci. Bull. 2020, 36, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Grecco, G.G.; Mork, B.E.; Huang, J.Y.; Metzger, C.E.; Haggerty, D.L.; Reeves, K.C.; Gao, Y.; Hoffman, H.; Katner, S.N.; Masters, A.R.; et al. Prenatal methadone exposure disrupts behavioral development and alters motor neuron intrinsic properties and local circuitry. eLife 2021, 10, e66230. [Google Scholar] [CrossRef]
- Wang, Y.; Han, T.Z. Prenatal exposure to heroin in mice elicits memory deficits that can be attributed to neuronal apoptosis. Neuroscience 2009, 160, 330–338. [Google Scholar] [CrossRef]
- Mei, B.; Niu, L.; Cao, B.; Huang, D.; Zhou, Y. Prenatal morphine exposure alters the layer II/III pyramidal neurons morphology in lateral secondary visual cortex of juvenile rats. Synapse 2009, 63, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, K.; Morihiro, Y.; Maekawa, M.; Yasumoto, Y.; Hoshi, H.; Adachi, Y.; Sawada, T.; Tokuda, N.; Kondo, H.; Yoshikawa, T.; et al. FABP7 expression in normal and stab-injured brain cortex and its role in astrocyte proliferation. Histochem. Cell Biol. 2011, 136, 501–513. [Google Scholar] [CrossRef]
- Kagawa, Y.; Yasumoto, Y.; Sharifi, K.; Ebrahimi, M.; Islam, A.; Miyazaki, H.; Yamamoto, Y.; Sawada, T.; Kishi, H.; Kobayashi, S.; et al. Fatty acid-binding protein 7 regulates function of caveolae in astrocytes through expression of caveolin-1. Glia 2015, 63, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef]
- Bantle, C.M.; Hirst, W.D.; Weihofen, A.; Shlevkov, E. Mitochondrial dysfunction in astrocytes: A role in parkinson’s disease? Front. Cell Dev. Biol. 2020, 8, 608026. [Google Scholar] [CrossRef]
- Lee, J.A.; Hall, B.; Allsop, J.; Alqarni, R.; Allen, S.P. Lipid metabolism in astrocytic structure and function. Semin. Cell Dev. Biol. 2021, 112, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletic-Savatic, M.; Bellen, H.J. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metab. 2017, 26, 719–737.e6. [Google Scholar] [CrossRef]
- Smolic, T.; Tavcar, P.; Horvat, A.; Cerne, U.; Haluzan Vasle, A.; Tratnjek, L.; Kreft, M.E.; Scholz, N.; Matis, M.; Petan, T.; et al. Astrocytes in stress accumulate lipid droplets. Glia 2021, 69, 1540–1562. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M. Prevention of neonatal abstinence syndrome in an outpatient prenatal buprenorphine tapering program. South. Med. J. 2020, 113, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, E.M.; Vassoler, F.M. Modeling prenatal opioid exposure in animals: Current findings and future directions. Front. Neuroendocrinol. 2018, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Roth, D.; Loudin, S.; Andrews, L.; Evans, J.; Davies, T.H. Inclusion of positive self-reporting by mothers of substance exposed neonates increases the predictability of NAS severity over toxicology alone. Matern. Child Health J. 2020, 24, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, S.; Bruni, T.; Bishop, L.; Turuba, R.; Olibris, B.; Jumah, N.A. Differences in hospital length of stay between neonates exposed to buprenorphine versus methadone in utero: A retrospective chart review. Paediatr. Child Health 2019, 24, e104–e110. [Google Scholar] [CrossRef] [PubMed]
- Kongstorp, M.; Bogen, I.L.; Stiris, T.; Andersen, J.M. Prenatal exposure to methadone or buprenorphine impairs cognitive performance in young adult rats. Drug Alcohol Depend. 2020, 212, 108008. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Chiang, Y.C.; Yuan, Z.F.; Kuo, C.C.; Lai, M.D.; Hung, T.W.; Ho, I.K.; Chen, S.T. Buprenorphine, methadone, and morphine treatment during pregnancy: Behavioral effects on the offspring in rats. Neuropsychiatr. Dis. Treat. 2015, 11, 609–618. [Google Scholar] [CrossRef]
- Smith, B.L.; Hassler, A.; Lloyd, K.R.; Reyes, T.M. Perinatal morphine but not buprenorphine affects gestational and offspring neurobehavioral outcomes in mice. Neurotoxicology 2023, 99, 292–304. [Google Scholar] [CrossRef]
- Elam, H.B.; Donegan, J.J.; Hsieh, J.; Lodge, D.J. Gestational buprenorphine exposure disrupts dopamine neuron activity and related behaviors in adulthood. eNeuro 2022, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sundelin Wahlsten, V.; Sarman, I. Neurobehavioural development of preschool-age children born to addicted mothers given opiate maintenance treatment with buprenorphine during pregnancy. Acta Paediatr. 2013, 102, 544–549. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niebergall, E.B.; Weekley, D.; Mazur, A.; Olszewski, N.A.; DeSchepper, K.M.; Radant, N.; Vijay, A.S.; Risher, W.C. Abnormal Morphology and Synaptogenic Signaling in Astrocytes Following Prenatal Opioid Exposure. Cells 2024, 13, 837. https://doi.org/10.3390/cells13100837
Niebergall EB, Weekley D, Mazur A, Olszewski NA, DeSchepper KM, Radant N, Vijay AS, Risher WC. Abnormal Morphology and Synaptogenic Signaling in Astrocytes Following Prenatal Opioid Exposure. Cells. 2024; 13(10):837. https://doi.org/10.3390/cells13100837
Chicago/Turabian StyleNiebergall, Ethan B., Daron Weekley, Anna Mazur, Nathan A. Olszewski, Kayla M. DeSchepper, N. Radant, Aishwarya S. Vijay, and W. Christopher Risher. 2024. "Abnormal Morphology and Synaptogenic Signaling in Astrocytes Following Prenatal Opioid Exposure" Cells 13, no. 10: 837. https://doi.org/10.3390/cells13100837