
Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies


 , ,
, ,
Abstract
1. Introduction
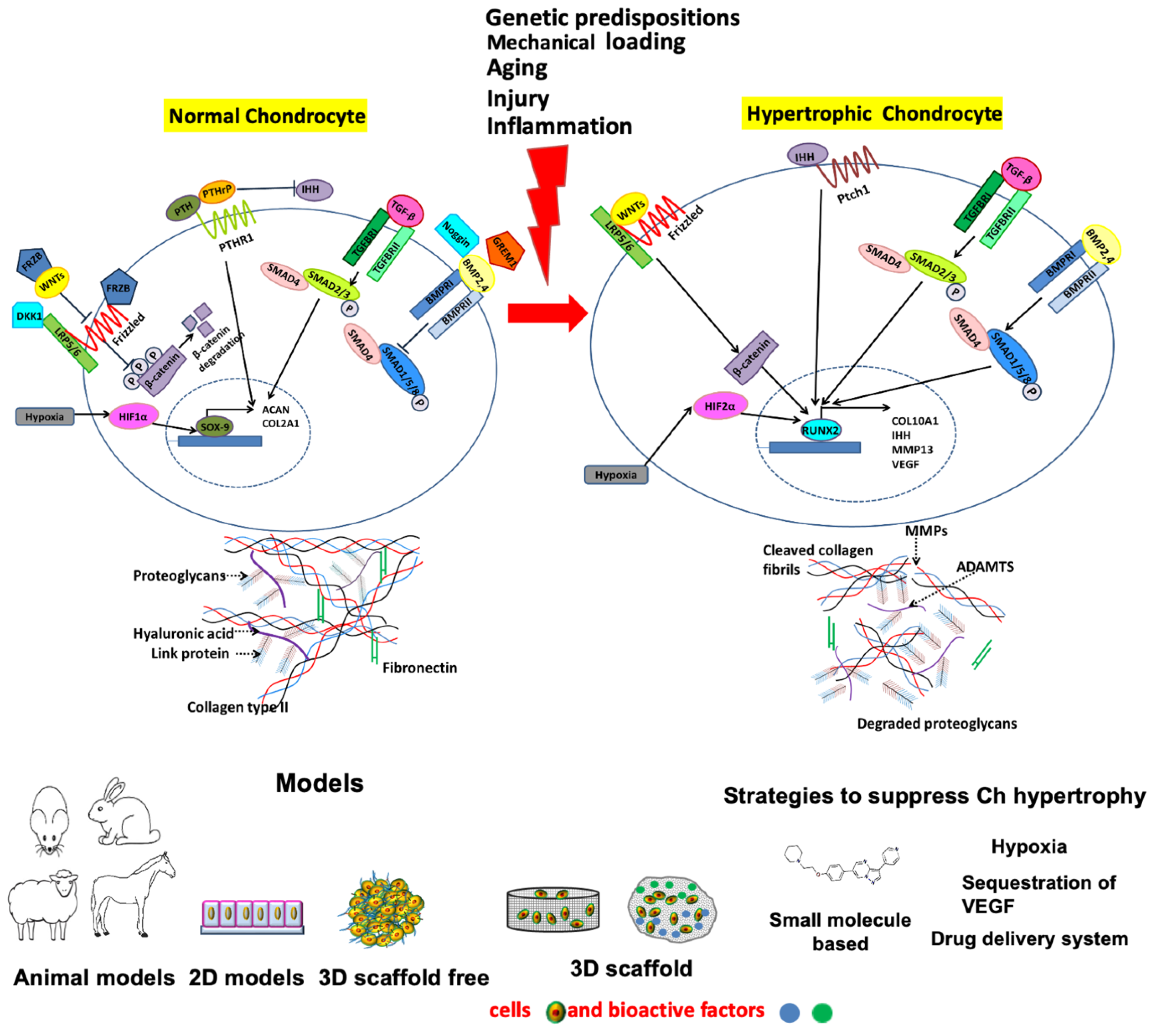
2. Chondrocyte Hypertrophy in OA
2.1. Major Cellular Signaling Pathways Involved in Chondrocyte Hypertrophy, Cartilage Mineralization/Calcification and Osteophyte Formation
2.1.1. IHH/PTHrP Pathway
2.1.2. Wnt Pathway
2.1.3. TGF Beta Pathway
2.1.4. BMP Pathway
2.1.5. FGF Pathway
3. Models to Study Chondrocyte Hypertrophy
3.1. In Vivo Models
3.1.1. Spontaneous Models
3.1.2. Surgically Induced Models
3.1.3. Chemically Induced Models
3.1.4. Ex Vivo Explant Models
3.2. In Vitro Models
3.2.1. Cell Sources
Articular Chondrocytes
Chondro-Progenitors (ChPs)
Pluripotent Stem Cells
Mesenchymal Stem/Stromal Cells (MSCs)
3.2.2. Two-Dimensional (2D) In Vitro Models
3.2.3. Three-Dimensional (3D) Scaffold-Free In Vitro Models
3.2.4. In Vitro Scaffold/Biomaterial-Based 3D Models
3.2.5. Bioreactor-Based Models
3.2.6. Considerations on the Relevance of OA Chondrocyte Hypertrophy Models
4. Strategies to Suppress Chondrocyte Hypertrophy and Cartilage Mineralization/Calcification
4.1. Small-Molecule-Based Modulation of Signaling Pathways
4.2. Role of VEGF Sequestration and Hypoxia
4.3. Biomaterial-Based Drug Delivery Systems to Mitigate Chondrocyte Hypertrophy
5. Conclusions and Future Perspectives
Funding
Conflicts of Interest
References
- Rim, Y.A.; Nam, Y.; Ju, J.H. The Role of Chondrocyte Hypertrophy and Senescence in Osteoarthritis Initiation and Progression. Int. J. Mol. Sci. 2020, 21, 2358. [Google Scholar] [CrossRef] [PubMed]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef] [PubMed]
- Laine, L.; White, W.B.; Rostom, A.; Hochberg, M. COX-2 Selective Inhibitors in the Treatment of Osteoarthritis. Semin. Arthritis Rheum. 2008, 38, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Vavken, P.; Samartzis, D. Effectiveness of autologous chondrocyte implantation in cartilage repair of the knee: A systematic review of controlled trials. Osteoarthr. Cartil. 2010, 18, 857–863. [Google Scholar] [CrossRef]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar] [CrossRef]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2018, 1442, 17–34. [Google Scholar] [CrossRef]
- Tigli, R.S.; Ghosh, S.; Laha, M.M.; Shevde, N.K.; Daheron, L.; Gimble, J.; Gümüşderelioglu, M.; Kaplan, D.L. Comparative chondrogenesis of human cell sources in 3D scaffolds. J. Tissue Eng. Regen. Med. 2009, 3, 348–360. [Google Scholar] [CrossRef]
- Jaswal, A.P.; Bandyopadhyay, A. Re-examining osteoarthritis therapy from a developmental biologist’s perspective. Biochem. Pharmacol. 2019, 165, 17–23. [Google Scholar] [CrossRef]
- Yasuda, T. Cartilage Destruction by Matrix Degradation Products. Mod. Rheumatol. 2006, 16, 197–205. [Google Scholar] [CrossRef]
- Enomoto, H.; Inoki, I.; Komiya, K.; Shiomi, T.; Ikeda, E.; Obata, K.-I.; Matsumoto, H.; Toyama, Y.; Okada, Y. Vascular Endothelial Growth Factor Isoforms and Their Receptors Are Expressed in Human Osteoarthritic Cartilage. Am. J. Pathol. 2003, 162, 171–181. [Google Scholar] [CrossRef]
- Yang, S.; Kim, J.; Ryu, J.-H.; Oh, H.; Chun, C.-H.; Kim, B.J.; Min, B.H.; Chun, J.-S. Hypoxia-inducible factor-2α is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B. Anatomy and physiology of the mineralized tissues: Role in the pathogenesis of osteoarthrosis. Osteoarthr. Cartil. 2004, 12, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M.; Plumb, D.A.; Dragomir, C.; Favero, M.; El Hachem, K.; Hashimoto, K.; Roach, H.I.; Olivotto, E.; Borzì, R.M.; et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: Signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur. Cells Mater. 2011, 21, 202–220. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage–bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Cho, Y.; Jeong, S.; Kim, H.; Kang, D.; Lee, J.; Kang, S.-B.; Kim, J.-H. Disease-modifying therapeutic strategies in osteoarthritis: Current status and future directions. Exp. Mol. Med. 2021, 53, 1689–1696. [Google Scholar] [CrossRef]
- Lories, R.J.; Corr, M.; Lane, N.E. To Wnt or not to Wnt: The bone and joint health dilemma. Nat. Rev. Rheumatol. 2013, 9, 328–339. [Google Scholar] [CrossRef]
- Lian, C.; Wang, X.; Qiu, X.; Wu, Z.; Gao, B.; Liu, L.; Liang, G.; Zhou, H.; Yang, X.; Peng, Y.; et al. Collagen type II suppresses articular chondrocyte hypertrophy and osteoarthritis progression by promoting integrin β1−SMAD1 interaction. Bone Res. 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Pitsillides, A.A.; Beier, F. Cartilage biology in osteoarthritis—Lessons from developmental biology. Nat. Rev. Rheumatol. 2011, 7, 654–663. [Google Scholar] [CrossRef]
- Kawaguchi, H. Endochondral ossification signals in cartilage degradation during osteoarthritis progression in experimental mouse models. Mol. Cells 2008, 25, 1–6. [Google Scholar] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.-L.A.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Billinghurst, R.C.; Dahlberg, L.; Ionescu, M.; Reiner, A.; Bourne, R.; Rorabeck, C.; Mitchell, P.; Hambor, J.; Diekmann, O.; Tschesche, H.; et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J. Clin. Investig. 1997, 99, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Nelson, F.; Dahlberg, L.; Laverty, S.; Reiner, A.; Pidoux, I.; Ionescu, M.; Fraser, G.L.; Brooks, E.; Tanzer, M.; Rosenberg, L.C.; et al. Evidence for altered synthesis of type II collagen in patients with osteoarthritis. J. Clin. Investig. 1998, 102, 2115–2125. [Google Scholar] [CrossRef]
- Hayami, T.; Funaki, H.; Yaoeda, K.; Mitui, K.; Yamagiwa, H.; Tokunaga, K.; Hatano, H.; Kondo, J.; Hiraki, Y.; Yamamoto, T.; et al. Expression of the cartilage derived anti-angiogenic factor chondromodulin-I decreases in the early stage of experimental osteoarthritis. J. Rheumatol. 2003, 30, 2207–2217. [Google Scholar]
- Zhang, X.; Prasadam, I.; Fang, W.; Crawford, R.; Xiao, Y. Chondromodulin-1 ameliorates osteoarthritis progression by inhibiting HIF-2α activity. Osteoarthr. Cartil. 2016, 24, 1970–1980. [Google Scholar] [CrossRef]
- Zhang, W.; Moskowitz, R.W.; Nuki, G.; Abramson, S.; Altman, R.D.; Arden, N.; Bierma-Zeinstra, S.; Brandt, K.D.; Croft, P.; Doherty, M.; et al. OARSI recommendations for the management of hip and knee osteoarthritis, Part II: OARSI evidence-based, expert consensus guidelines. Osteoarthr. Cartil. 2008, 16, 137–162. [Google Scholar] [CrossRef]
- Park, S.; Bello, A.; Arai, Y.; Ahn, J.; Kim, D.; Cha, K.-Y.; Baek, I.; Park, H.; Lee, S.-H. Functional Duality of Chondrocyte Hypertrophy and Biomedical Application Trends in Osteoarthritis. Pharmaceutics 2021, 13, 1139. [Google Scholar] [CrossRef]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the Contribution of the Changing Hypertrophic Chondrocyte Phenotype in the Development and Progression of Osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Pritzker, K.P.H.; Gay, S.; Jimenez, S.A.; Ostergaard, K.; Pelletier, J.-P.; Revell, P.A.; Salter, D.; van den Berg, W.B. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthr. Cartil. 2006, 14, 13–29. [Google Scholar] [CrossRef]
- Anderson-Baron, M.; Liang, Y.; Kunze, M.; Mulet-Sierra, A.; Osswald, M.; Ansari, K.; Seikaly, H.; Adesida, A.B. Suppression of Hypertrophy During in vitro Chondrogenesis of Cocultures of Human Mesenchymal Stem Cells and Nasal Chondrocytes Correlates With Lack of in vivo Calcification and Vascular Invasion. Front. Bioeng. Biotechnol. 2021, 8, 572356. [Google Scholar] [CrossRef] [PubMed]
- Von Der Mark, K.; Kirsch, T.; Nerlich, A.; Kuss, A.; Weseloh, G.; Glückert, K.; Stöss, H. Type x collagen synthesis in human osteoarthritic cartilage. indication of chondrocyte hypertrophy. Arthritis Care Res. 1992, 35, 806–811. [Google Scholar] [CrossRef]
- Kirsch, T.; von der Mark, K. Remodelling of collagen types I, II and X and calcification of human fetal cartilage. Bone Miner. 1992, 18, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Fosang, A.J.; Last, K.; Stanton, H.; Weeks, D.B.; Campbell, I.K.; Hardingham, T.; Hembry, R.M. Generation and Novel Distribution of Matrix Metalloproteinase-derived Aggrecan Fragments in Porcine Cartilage Explants. J. Biol. Chem. 2000, 275, 33027–33037. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Berkelaar, M.H.M.; Dasen, B.; Halleux, C.; Guth-Gundel, S.; Kramer, I.; Ghosh, S.; Martin, I.; Barbero, A.; Occhetta, P. Blockage of bone morphogenetic protein signalling counteracts hypertrophy in a human osteoarthritic micro-cartilage model. J. Cell Sci. 2020, 133, jcs249094. [Google Scholar] [CrossRef]
- Pavlou, E.; Zhang, X.; Wang, J.; Kourkoumelis, N. Raman spectroscopy for the assessment of osteoarthritis. Ann. Jt. 2018, 3, 83. [Google Scholar] [CrossRef]
- Kumar, R.; Grønhaug, K.M.; Afseth, N.K.; Isaksen, V.; Davies, C.D.L.; Drogset, J.O.; Lilledahl, M.B. Optical investigation of osteoarthritic human cartilage (ICRS grade) by confocal Raman spectroscopy: A pilot study. Anal. Bioanal. Chem. 2015, 407, 8067–8077. [Google Scholar] [CrossRef]
- Gottardi, R.; Hansen, U.; Raiteri, R.; Loparic, M.; Düggelin, M.; Mathys, D.; Friederich, N.F.; Bruckner, P.; Stolz, M. Supramolecular Organization of Collagen Fibrils in Healthy and Osteoarthritic Human Knee and Hip Joint Cartilage. PLoS ONE 2016, 11, e0163552. [Google Scholar] [CrossRef]
- Han, B.; Nia, H.T.; Wang, C.; Chandrasekaran, P.; Li, Q.; Chery, D.R.; Li, H.; Grodzinsky, A.J.; Han, L. AFM-Nanomechanical Test: An Interdisciplinary Tool That Links the Understanding of Cartilage and Meniscus Biomechanics, Osteoarthritis Degeneration, and Tissue Engineering. ACS Biomater. Sci. Eng. 2017, 3, 2033–2049. [Google Scholar] [CrossRef]
- Tchetina, E.V.; Squires, G.; Poole, A.R. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol. 2005, 32, 876–886. [Google Scholar]
- Zhong, L.; Huang, X.; Rodrigues, E.D.; Leijten, J.C.; Verrips, T.; El Khattabi, M.; Karperien, M.; Post, J.N. Endogenous DKK1 and FRZB Regulate Chondrogenesis and Hypertrophy in Three-Dimensional Cultures of Human Chondrocytes and Human Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 1808–1817. [Google Scholar] [CrossRef]
- Orfanidou, T.; Iliopoulos, D.; Malizos, K.N.; Tsezou, A. Involvement of SOX-9 and FGF-23 in RUNX-2 regulation in osteoarthritic chondrocytes. J. Cell. Mol. Med. 2010, 13, 3186–3194. [Google Scholar] [CrossRef]
- Hellingman, C.A.; Davidson, E.N.B.; Koevoet, W.; Vitters, E.L.; Berg, W.B.V.D.; Van Osch, G.J.; Van Der Kraan, P.M. Smad Signaling Determines Chondrogenic Differentiation of Bone-Marrow-Derived Mesenchymal Stem Cells: Inhibition of Smad1/5/8P Prevents Terminal Differentiation and Calcification. Tissue Eng. Part A 2011, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Rigueur, D.; Brugger, S.; Anbarchian, T.; Kil Kim, J.; Lee, Y.; Lyons, K.M. The Type I BMP Receptor ACVR1/ALK2 is Required for Chondrogenesis During Development. J. Bone Miner. Res. 2014, 30, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, U.; Ferlosio, A.; Arcuri, G.; Spagnoli, L.G.; Orlandi, A. Transglutaminase 2 as a biomarker of osteoarthritis: An update. Amino Acids 2011, 44, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Pirosa, A.; Tankus, E.B.; Mainardi, A.; Occhetta, P.; Dönges, L.; Baum, C.; Rasponi, M.; Martin, I.; Barbero, A. Modeling In Vitro Osteoarthritis Phenotypes in a Vascularized Bone Model Based on a Bone-Marrow Derived Mesenchymal Cell Line and Endothelial Cells. Int. J. Mol. Sci. 2021, 22, 9581. [Google Scholar] [CrossRef]
- Ravindra, K.C.; Ahrens, C.C.; Wang, Y.; Ramseier, J.Y.; Wishnok, J.S.; Griffith, L.G.; Grodzinsky, A.J.; Tannenbaum, S.R. Chemoproteomics of matrix metalloproteases in a model of cartilage degeneration suggests functional biomarkers associated with posttraumatic osteoarthritis. J. Biol. Chem. 2018, 293, 11459–11469. [Google Scholar] [CrossRef]
- Sumer, E.; Sondergaard, B.; Rousseau, J.; Delmas, P.; Fosang, A.; Karsdal, M.; Christiansen, C.; Qvist, P. MMP and non-MMP-mediated release of aggrecan and its fragments from articular cartilage: A comparative study of three different aggrecan and glycosaminoglycan assays. Osteoarthr. Cartil. 2007, 15, 212–221. [Google Scholar] [CrossRef]
- Chen, S.; Fu, P.; Cong, R.; Wu, H.; Pei, M. Strategies to minimize hypertrophy in cartilage engineering and regeneration. Genes Dis. 2015, 2, 76–95. [Google Scholar] [CrossRef]
- Studer, D.; Millan, C.; Öztürk, E.; Maniura-Weber, K.; Zenobi-Wong, M. Molecular and biophysical mechanisms regulating hypertrophic differentiation in chondrocytes and mesenchymal stem cells. Eur. Cells Mater. 2012, 24, 118–135. [Google Scholar] [CrossRef]
- Lin, N.-Y.; Distler, A.; Beyer, C.; Philipi-Schöbinger, A.; Breda, S.; Dees, C.; Stock, M.; Tomcik, M.; Niemeier, A.; Dell’Accio, F.; et al. Inhibition of Notch1 promotes hedgehog signalling in a HES1-dependent manner in chondrocytes and exacerbates experimental osteoarthritis. Ann. Rheum. Dis. 2016, 75, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-F.; Dong, Y.; Ionescu, A.M.; Rosier, R.N.; Zuscik, M.J.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Parathyroid hormone-related peptide (PTHrP) inhibits Runx2 expression through the PKA signaling pathway. Exp. Cell Res. 2004, 299, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Vega, R.B.; Matsuda, K.; Oh, J.; Barbosa, A.C.; Yang, X.; Meadows, E.; McAnally, J.; Pomajzl, C.; Shelton, J.M.; Richardson, J.A.; et al. Histone Deacetylase 4 Controls Chondrocyte Hypertrophy during Skeletogenesis. Cell 2004, 119, 555–566. [Google Scholar] [CrossRef]
- Guo, L.; Wei, X.; Zhang, Z.; Wang, X.; Wang, C.; Li, P.; Wei, L. Ipriflavone attenuates the degeneration of cartilage by blocking the Indian hedgehog pathway. Osteoarthr. Cartil. 2020, 28, S477. [Google Scholar] [CrossRef]
- Bradley, E.W.; Drissi, M.H. WNT5A Regulates Chondrocyte Differentiation through Differential Use of the CaN/NFAT and IKK/NF-κB Pathways. Mol. Endocrinol. 2010, 24, 1581–1593. [Google Scholar] [CrossRef]
- Van den Bosch, M.H.; Blom, A.B.; van Lent, P.L.; van Beuningen, H.M.; Davidson, E.N.B.; van der Kraan, P.M.; Berg, W.B.V.D. Canonical Wnt signaling skews TGF-β signaling in chondrocytes towards signaling via ALK1 and Smad 1/5/8. Cell. Signal. 2014, 26, 951–958. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Y.; Wu, Y.; Wang, Y.; Sun, L.; Li, F. The inhibition of EZH2 ameliorates osteoarthritis development through the Wnt/β-catenin pathway. Sci. Rep. 2016, 6, 29176. [Google Scholar] [CrossRef]
- Huang, X.; Zhong, L.; Hendriks, J.; Post, J.N.; Karperien, M. The Effects of the WNT-Signaling Modulators BIO and PKF118-310 on the Chondrogenic Differentiation of Human Mesenchymal Stem Cells. Int. J. Mol. Sci. 2018, 19, 561. [Google Scholar] [CrossRef]
- Held, A.; Glas, A.; Dietrich, L.; Bollmann, M.; Brandstädter, K.; Grossmann, T.; Lohmann, C.; Pap, T.; Bertrand, J. Targeting β-catenin dependent Wnt signaling via peptidomimetic inhibitors in murine chondrocytes and OA cartilage. Osteoarthr. Cartil. 2018, 26, 818–823. [Google Scholar] [CrossRef]
- Scharstuhl, A.; Glansbeek, H.L.; van Beuningen, H.M.; Vitters, E.L.; van der Kraan, P.M.; Berg, W.B.V.D. Inhibition of Endogenous TGF-β During Experimental Osteoarthritis Prevents Osteophyte Formation and Impairs Cartilage Repair. J. Immunol. 2002, 169, 507–514. [Google Scholar] [CrossRef]
- Van Der Kraan, P.M. The Changing Role of TGFβ in Healthy, Ageing and Osteoarthritic Joints. Nat. Rev. Rheumatol. 2017, 13, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Trehan, S.K.; Guan, Y.; Sun, C.; Moore, D.C.; Jayasuriya, C.T.; Chen, Q. Matrilin-3 Inhibits Chondrocyte Hypertrophy as a Bone Morphogenetic Protein-2 Antagonist. J. Biol. Chem. 2014, 289, 34768–34779. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-Y.; Tsai, C.-H.; Liu, S.-C.; Huang, C.-C.; Lin, T.-H.; Yang, Y.-Z.; Tang, C.-H. Noggin Inhibits IL-1β and BMP-2 Expression, and Attenuates Cartilage Degeneration and Subchondral Bone Destruction in Experimental Osteoarthritis. Cells 2020, 9, 927. [Google Scholar] [CrossRef] [PubMed]
- Iwai, T.; Murai, J.; Yoshikawa, H.; Tsumaki, N. Smad7 Inhibits Chondrocyte Differentiation at Multiple Steps during Endochondral Bone Formation and Down-regulates p38 MAPK Pathways. J. Biol. Chem. 2008, 283, 27154–27164. [Google Scholar] [CrossRef]
- Bonen, D.K.; Schmid, T.M. Elevated extracellular calcium concentrations induce type X collagen synthesis in chondrocyte cultures. J. Cell Biol. 1991, 115, 1171–1178. [Google Scholar] [CrossRef]
- Burton, D.; Foster, M.; Johnson, K.; Hiramoto, M.; Deftos, L.; Terkeltaub, R. Chondrocyte calcium-sensing receptor expression is up-regulated in early guinea pig knee osteoarthritis and modulates PTHrP, MMP-13, and TIMP-3 expression. Osteoarthr. Cartil. 2005, 13, 395–404. [Google Scholar] [CrossRef]
- Wu, S.; Palese, T.; Mishra, O.P.; Delivoria-Papadopoulos, M.; De Luca, F. Effects of Ca2-sensing receptor activation in the growth plate. FASEB J. 2003, 18, 143–145. [Google Scholar] [CrossRef]
- Luckman, S.P.; Rees, E.; Kwan, A.P.L. Partial characterization of cell-type X collagen interactions. Biochem. J. 2003, 372, 485–493. [Google Scholar] [CrossRef]
- Wang, G.; Woods, A.; Sabari, S.; Pagnotta, L.; Stanton, L.-A.; Beier, F. RhoA/ROCK Signaling Suppresses Hypertrophic Chondrocyte Differentiation. J. Biol. Chem. 2004, 279, 13205–13214. [Google Scholar] [CrossRef]
- Wang, G.; Beier, F. Rac1/Cdc42 and RhoA GTPases Antagonistically Regulate Chondrocyte Proliferation, Hypertrophy, and Apoptosis. J. Bone Miner. Res. 2005, 20, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Carlson, C.S.; McGee, M.P. Expression of β1 Integrins by Cultured Articular Chondrocytes and in Osteoarthritic Cartilage. Exp. Cell Res. 1995, 217, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Saito, T.; Sugita, S.; Hikata, T.; Kobayashi, H.; Fukai, A.; Taniguchi, Y.; Hirata, M.; Akiyama, H.; Chung, U.-I.; et al. Notch signaling in chondrocytes modulates endochondral ossification and osteoarthritis development. Proc. Natl. Acad. Sci. USA 2013, 110, 1875–1880. [Google Scholar] [CrossRef]
- Loeser, R.F.; Erickson, E.A.; Long, D.L. Mitogen-activated protein kinases as therapeutic targets in osteoarthritis. Curr. Opin. Rheumatol. 2008, 20, 581–586. [Google Scholar] [CrossRef]
- Li, T.-F.; Gao, L.; Sheu, T.-J.; Sampson, E.R.; Flick, L.M.; Konttinen, Y.T.; Chen, D.; Schwarz, E.M.; Zuscik, M.; Jonason, J.H.; et al. Aberrant hypertrophy in Smad3-deficient murine chondrocytes is rescued by restoring transforming growth factor β-activated kinase 1/activating transcription factor 2 signaling: A potential clinical implication for osteoarthritis. Arthritis Care Res. 2010, 62, 2359–2369. [Google Scholar] [CrossRef]
- Prasadam, I.; Mao, X.; Shi, W.; Crawford, R.; Xiao, Y. Combination of MEK-ERK inhibitor and hyaluronic acid has a synergistic effect on anti-hypertrophic and pro-chondrogenic activities in osteoarthritis treatment. J. Mol. Med. 2012, 91, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Arai, Y.; Bello, A.; Park, H.; Kim, D.; Park, K.-S.; Lee, S.-H. SPRY4 acts as an indicator of osteoarthritis severity and regulates chondrocyte hypertrophy and ECM protease expression. NPJ Regen. Med. 2021, 6, 1–9. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Yi, D.; Yu, H.; Lu, K.; Ruan, C.; Ding, C.; Tong, L.; Zhao, X.; Chen, D. AMPK Signaling in Energy Control, Cartilage Biology, and Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 1551. [Google Scholar] [CrossRef]
- Zhang, Q.; Lai, S.; Hou, X.; Cao, W.; Zhang, Y.; Zhang, Z. Protective effects of PI3K/Akt signal pathway induced cell autophagy in rat knee joint cartilage injury. Am. J. Transl. Res. 2018, 10, 762–770. [Google Scholar] [PubMed]
- Liu, N.; Fu, D.; Yang, J.; Liu, P.; Song, X.; Wang, X.; Li, R.; Fu, Z.; Chen, J.; Gong, X.; et al. Asiatic acid attenuates hypertrophic and fibrotic differentiation of articular chondrocytes via AMPK/PI3K/AKT signaling pathway. Arthritis Res. Ther. 2020, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Xie, Y.; Wang, Q.; Wang, X.; Luo, F.; Zhou, S.; Wang, Z.; Huang, J.; Tan, Q.; Jin, M.; et al. A novel fibroblast growth factor receptor 1 inhibitor protects against cartilage degradation in a murine model of osteoarthritis. Sci. Rep. 2016, 6, 24042. [Google Scholar] [CrossRef]
- Mannstadt, M.; Jüppner, H.; Gardella, T.J. Receptors for PTH and PTHrP: Their biological importance and functional properties. Am. J. Physiol.-Renal Physiol. 1999, 277, F665–F675. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhou, J.; Wei, X.; Zhang, J.; Fleming, B.; Terek, R.; Pei, M.; Chen, Q.; Liu, T.; Wei, L. Activation of Indian hedgehog promotes chondrocyte hypertrophy and upregulation of MMP-13 in human osteoarthritic cartilage. Osteoarthr. Cartil. 2012, 20, 755–763. [Google Scholar] [CrossRef]
- Sasagawa, S.; Takemori, H.; Uebi, T.; Ikegami, D.; Hiramatsu, K.; Ikegawa, S.; Yoshikawa, H.; Tsumaki, N. SIK3 is essential for chondrocyte hypertrophy during skeletal development in mice. Development 2012, 139, 1153–1163. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, Q.; Lanske, B.; Fleming, B.C.; Terek, R.; Wei, X.; Zhang, G.; Wang, S.; Li, K.; Wei, L. Disrupting the Indian hedgehog signaling pathway in vivo attenuates surgically induced osteoarthritis progression in Col2a1-CreERT2; Ihhfl/fl mice. Arthritis Res. Ther. 2014, 16, R11. [Google Scholar] [CrossRef]
- Yahara, Y.; Takemori, H.; Okada, M.; Kosai, A.; Yamashita, A.; Kobayashi, T.; Fujita, K.; Itoh, Y.; Nakamura, M.; Fuchino, H.; et al. Pterosin B prevents chondrocyte hypertrophy and osteoarthritis in mice by inhibiting Sik3. Nat. Commun. 2016, 7, 10959. [Google Scholar] [CrossRef]
- Karaplis, A.C.; Luz, A.; Glowacki, J.; Bronson, R.T.; Tybulewicz, V.L.; Kronenberg, H.M.; Mulligan, R.C. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994, 8, 277–289. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.K.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP Receptor in Early Development and Indian Hedgehog—Regulated Bone Growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Provot, S.; Kempf, H.; Murtaugh, L.C.; Chung, U.-I.; Kim, D.-W.; Chyung, J.; Kronenberg, H.M.; Lassar, A.B. Nkx3.2/Bapx1 acts as a negative regulator of chondrocyte maturation. Development 2006, 133, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Rainbow, R.S.; Kwon, H.; Zeng, L. The Role of Nkx3.2 in Chondrogenesis. Front. Biol. 2014, 9, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Simon, M.; Dheedene, A.; Alanay, Y.; Mihci, E.; Rifai, L.; Sefiani, A.; van Bever, Y.; Meradji, M.; Superti-Furga, A.; et al. Homozygous Inactivating Mutations in the NKX3-2 Gene Result in Spondylo-Megaepiphyseal-Metaphyseal Dysplasia. Am. J. Hum. Genet. 2009, 85, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fan, X.; Xing, L.; Tian, F. Wnt signaling: A promising target for osteoarthritis therapy. Cell Commun. Signal. 2019, 17, 1–14. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Soung, D.Y.; Schwarz, E.M.; O’Keefe, R.J.; Drissi, H. Wnt induction of chondrocyte hypertrophy through the Runx2 transcription factor. J. Cell. Physiol. 2006, 208, 77–86. [Google Scholar] [CrossRef]
- Yuasa, T.; Otani, T.; Koike, T.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt/β-catenin signaling stimulates matrix catabolic genes and activity in articular chondrocytes: Its possible role in joint degeneration. Lab. Investig. 2008, 88, 264–274. [Google Scholar] [CrossRef]
- Bertrand, J.; Kräft, T.; Gronau, T.; Sherwood, J.; Rutsch, F.; Lioté, F.; Dell’Accio, F.; Lohmann, C.H.; Bollmann, M.; Held, A.; et al. BCP crystals promote chondrocyte hypertrophic differentiation in OA cartilage by sequestering Wnt3a. Ann. Rheum. Dis. 2020, 79, 975–984. [Google Scholar] [CrossRef]
- Rosenthal, A.K. Crystals, inflammation, and osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 170–173. [Google Scholar] [CrossRef]
- Rosenthal, A.K. Basic calcium phosphate crystal-associated musculoskeletal syndromes: An update. Curr. Opin. Rheumatol. 2018, 30, 168–172. [Google Scholar] [CrossRef]
- Leijten, J.C.; Bos, S.D.; Landman, E.B.; Georgi, N.; Jahr, H.; Meulenbelt, I.; Post, J.N.; A van Blitterswijk, C.; Karperien, M. GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and are regulated by osteoarthritis-associated factors. Thromb. Haemost. 2013, 15, R126. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Chun, C.-H.; Chun, J.-S. Dkk-1 expression in chondrocytes inhibits experimental osteoarthritic cartilage destruction in mice. Arthritis Care Res. 2012, 64, 2568–2578. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Mardones, R.; Pritzker, K.; van Wijnen, A.J.; Galindo, M.A.; Heras, F.L. Dickkopf-1 reduces hypertrophic changes in human chondrocytes derived from bone marrow stem cells. Gene 2018, 687, 228–237. [Google Scholar] [CrossRef]
- Zhen, G.; Guo, Q.; Li, Y.; Wu, C.; Zhu, S.; Wang, R.; Guo, X.E.; Kim, B.C.; Huang, J.; Hu, Y.; et al. Mechanical stress determines the configuration of TGFβ activation in articular cartilage. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Qiu, Y.-T.; Chen, M.-J.; Zhang, Z.-Y.; Yang, C. Synovial TGF-β1 and MMP-3 levels and their correlation with the progression of temporomandibular joint osteoarthritis combined with disc displacement: A preliminary study. Biomed. Rep. 2012, 1, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Van Beuningen, H.M.; Van der Kraan, P.M.; Arntz, O.J.; Van den Berg, W.B. Transforming Growth Factor-Β1 Stimulates Articular Chondrocyte Proteoglycan Synthesis and Induces Osteophyte Formation in the Murine Knee Joint. Lab. Investig. 1994, 71, 279–290. [Google Scholar]
- Allas, L.; Rochoux, Q.; Leclercq, S.; Boumédiene, K.; Baugé, C. Development of a simple osteoarthritis model useful to predict in vitro the anti-hypertrophic action of drugs. Lab. Investig. 2019, 100, 64–71. [Google Scholar] [CrossRef]
- Chawla, S.; Kumar, A.; Admane, P.; Bandyopadhyay, A.; Ghosh, S. Elucidating role of silk-gelatin bioink to recapitulate articular cartilage differentiation in 3D bioprinted constructs. Bioprinting 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Futrega, K.; Robey, P.G.; Klein, T.J.; Crawford, R.W.; Doran, M.R. A single day of TGF-β1 exposure activates chondrogenic and hypertrophic differentiation pathways in bone marrow-derived stromal cells. Commun. Biol. 2021, 4, 1–12. [Google Scholar] [CrossRef]
- Narcisi, R.; Quarto, R.; Ulivi, V.; Muraglia, A.; Molfetta, L.; Giannoni, P. TGF β-1 administration during Ex vivo expansion of human articular chondrocytes in a serum-free medium redirects the cell phenotype toward hypertrophy. J. Cell. Physiol. 2011, 227, 3282–3290. [Google Scholar] [CrossRef]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, H.; Zhang, D.; Xie, J.; Zhou, X. The role of stromal cell-derived factor 1 on cartilage development and disease. Osteoarthr. Cartil. 2020, 29, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Zhang, D.; Lin, Y.; Yuan, Q.; Zhou, X. Anterior Cruciate Ligament Transection–Induced Cellular and Extracellular Events in Menisci: Implications for Osteoarthritis. Am. J. Sports Med. 2018, 46, 1185–1198. [Google Scholar] [CrossRef]
- Kawakami, Y.; Leon, J.M.R.; Belmonte, J.C.I. The role of TGFβs and Sox9 during limb chondrogenesis. Curr. Opin. Cell Biol. 2006, 18, 723–729. [Google Scholar] [CrossRef]
- Balooch, G.; Balooch, M.; Nalla, R.K.; Schilling, S.; Filvaroff, E.H.; Marshall, G.W.; Marshall, S.J.; Ritchie, R.O.; Derynck, R.; Alliston, T. TGF-β regulates the mechanical properties and composition of bone matrix. Proc. Natl. Acad. Sci. USA 2005, 102, 18813–18818. [Google Scholar] [CrossRef] [PubMed]
- Claus, S.; Aubert-Foucher, E.; Demoor, M.; Camuzeaux, B.; Paumier, A.; Piperno, M.; Damour, O.; Duterque-Coquillaud, M.; Galéra, P.; Mallein-Gerin, F. Chronic exposure of bone morphogenetic protein-2 favors chondrogenic expression in human articular chondrocytes amplified in monolayer cultures. J. Cell. Biochem. 2010, 111, 1642–1651. [Google Scholar] [CrossRef]
- Steinert, A.F.; Proffen, B.; Kunz, M.; Hendrich, C.; Ghivizzani, S.C.; Nöth, U.; Rethwilm, A.; Eulert, J.; Evans, C.H. Hypertrophy is induced during the in vitro chondrogenic differentiation of human mesenchymal stem cells by bone morphogenetic protein-2 and bone morphogenetic protein-4 gene transfer. Thromb. Haemost. 2009, 11, R148. [Google Scholar] [CrossRef]
- Kuroda, R.; Usas, A.; Kubo, S.; Corsi, K.; Peng, H.; Rose, T.; Cummins, J.; Fu, F.H.; Huard, J. Cartilage repair using bone morphogenetic protein 4 and muscle-derived stem cells. Arthritis Care Res. 2006, 54, 433–442. [Google Scholar] [CrossRef]
- Chubinskaya, S.; Hurtig, M.; Rueger, D.C. OP-1/BMP-7 in cartilage repair. Int. Orthop. 2007, 31, 773–781. [Google Scholar] [CrossRef]
- Sekiya, I.; Tang, T.; Hayashi, M.; Morito, T.; Ju, Y.-J.; Mochizuki, T.; Muneta, T. Periodic knee injections of BMP-7 delay cartilage degeneration induced by excessive running in rats. J. Orthop. Res. 2009, 27, 1088–1092. [Google Scholar] [CrossRef]
- Lowery, J.W.; Rosen, V. The BMP Pathway and Its Inhibitors in the Skeleton. Physiol. Rev. 2018, 98, 2431–2452. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.; A Parker, E.; Hegde, A.; Chau, M.; Barnes, K.M.; Baron, J. Gradients in bone morphogenetic protein-related gene expression across the growth plate. J. Endocrinol. 2007, 193, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kawakami, H.; Tahara, N.; Olmer, M.; Hayashi, S.; Akiyama, R.; Bagchi, A.; Lotz, M.; Kawakami, Y. Expression of Noggin and Gremlin1 and its implications in fine-tuning BMP activities in mouse cartilage tissues. J. Orthop. Res. 2016, 35, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Singh, P.N.; Sohaskey, M.L.; Harland, R.M.; Bandyopadhyay, A. Precise spatial restriction of BMP signaling is essential for articular cartilage differentiation. Development 2015, 142, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Daans, M.; Lories, R.J.; Luyten, F.P. Dynamic activation of bone morphogenetic protein signaling in collagen-induced arthritis supports their role in joint homeostasis and disease. Arthritis Res. Ther. 2008, 10, R115. [Google Scholar] [CrossRef]
- Leijten, J.C.H.; Emons, J.; Sticht, C.; van Gool, S.; Decker, E.; Uitterlinden, A.; Rappold, G.; Hofman, A.; Rivadeneira, F.; Scherjon, S.; et al. Gremlin 1, Frizzled-related protein, and Dkk-1 are key regulators of human articular cartilage homeostasis. Arthritis Rheum. 2012, 64, 3302–3312. [Google Scholar] [CrossRef]
- Zuniga, A.; Michos, O.; Spitz, F.; Haramis, A.-P.G.; Panman, L.; Galli, A.; Vintersten, K.; Klasen, C.; Mansfield, W.; Kuc, S.; et al. Mouse limb deformity mutations disrupt a global control region within the large regulatory landscape required for Gremlin expression. Genes Dev. 2004, 18, 1553–1564. [Google Scholar] [CrossRef]
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.-I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1–NF-κB pathway. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Davidson, E.N.B.; Vitters, E.L.; Berg, W.B.V.D.; Van Der Kraan, P.M. TGF β-induced cartilage repair is maintained but fibrosis is blocked in the presence of Smad7. Thromb. Haemost. 2006, 8, R65. [Google Scholar] [CrossRef]
- Feng, J.Q.; Xing, L.; Zhang, J.-H.; Zhao, M.; Horn, D.; Chan, J.; Boyce, B.F.; Harris, S.E.; Mundy, G.R.; Chen, D. NF-κB Specifically Activates BMP-2 Gene Expression in Growth Plate Chondrocytes in Vivo and in a Chondrocyte Cell Line in Vitro. J. Biol. Chem. 2003, 278, 29130–29135. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Raimann, A.; Ertl, D.A.; Helmreich, M.; Sagmeister, S.; Egerbacher, M.; Haeusler, G. Fibroblast Growth Factor 23 and Klotho Are Present in the Growth Plate. Connect. Tissue Res. 2013, 54, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Guibert, M.; Cailotto, F.; Gasser, A.; Presle, N.; Mainard, D.; Netter, P.; Kempf, H.; Jouzeau, J.-Y.; Reboul, P. Fibroblast Growth Factor 23 drives MMP13 expression in human osteoarthritic chondrocytes in a Klotho-independent manner. Osteoarthr. Cartil. 2016, 24, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xie, Y.; Li, W.; Huang, J.; Wang, Z.; Tang, J.; Xu, W.; Sun, X.; Tan, Q.; Huang, S.; et al. Conditional Deletion of Fgfr3 in Chondrocytes leads to Osteoarthritis-like Defects in Temporomandibular Joint of Adult Mice. Sci. Rep. 2016, 6, 24039. [Google Scholar] [CrossRef] [PubMed]
- Kuyinu, E.L.; Narayanan, G.; Nair, L.S.; Laurencin, C.T. Animal models of osteoarthritis: Classification, update, and measurement of outcomes. J. Orthop. Surg. Res. 2016, 11, 1–27. [Google Scholar] [CrossRef]
- McDougall, J.J.; Andruski, B.; Schuelert, N.; Hallgrímsson, B.; Matyas, J.R. Unravelling the relationship between age, nociception and joint destruction in naturally occurring osteoarthritis of Dunkin Hartley guinea pigs. Pain 2009, 141, 222–232. [Google Scholar] [CrossRef]
- Flahiff, C.M.; Kraus, V.B.; Huebner, J.L.; Setton, L.A. Cartilage mechanics in the guinea pig model of osteoarthritis studied with an osmotic loading method. Osteoarthr. Cartil. 2004, 12, 383–388. [Google Scholar] [CrossRef][Green Version]
- Liu, W.; Burton-Wurster, N.; Glant, T.T.; Tashman, S.; Sumner, D.R.; Kamath, R.V.; Lust, G.; Kimura, J.H.; Cs-Szabo, G. Spontaneous and experimental osteoarthritis in dog: Similarities and differences in proteoglycan levels. J. Orthop. Res. 2003, 21, 730–737. [Google Scholar] [CrossRef]
- Staines, K.; Poulet, B.; Wentworth, D.; Pitsillides, A. The STR/ort mouse model of spontaneous osteoarthritis–an update. Osteoarthr. Cartil. 2016, 25, 802–808. [Google Scholar] [CrossRef]
- A Jimenez, P.; Glasson, S.S.; Trubetskoy, O.V.; Haimes, H.B. Spontaneous osteoarthritis in Dunkin Hartley guinea pigs: Histologic, radiologic, and biochemical changes. Lab. Anim. Sci. 1997, 47, 598–601. [Google Scholar]
- Panula, H.E.; Helminen, H.J.; Kiviranta, I. Slowly Progressive Osteoarthritis After Tibial Valgus Osteotomy in Young Beagle Dogs. Clin. Orthop. Relat. Res. 1997, 343, 192–202. [Google Scholar] [CrossRef]
- McIlwraith, C.W.; Frisbie, D.D.; Kawcak, C.E. The horse as a model of naturally occurring osteoarthritis. Bone Jt. Res. 2012, 1, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Malda, J.; Benders, K.; Klein, T.; de Grauw, J.; Kik, M.; Hutmacher, D.W.; Saris, D.; van Weeren, P.; Dhert, W. Comparative study of depth-dependent characteristics of equine and human osteochondral tissue from the medial and lateral femoral condyles. Osteoarthr. Cartil. 2012, 20, 1147–1151. [Google Scholar] [CrossRef]
- Glasson, S.; Blanchet, T.; Morris, E. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 2007, 15, 1061–1069. [Google Scholar] [CrossRef]
- Marijnissen, A.; van Roermund, P.; TeKoppele, J.; Bijlsma, J.; Lafeber, F. The canine ‘groove’ model, compared with the ACLT model of osteoarthritis. Osteoarthr. Cartil. 2002, 10, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Kamekura, S.; Hoshi, K.; Shimoaka, T.; Chung, U.; Chikuda, H.; Yamada, T.; Uchida, M.; Ogata, N.; Seichi, A.; Nakamura, K.; et al. Osteoarthritis development in novel experimental mouse models induced by knee joint instability. Osteoarthr. Cartil. 2005, 13, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Bascuñán, A.L.; Biedrzycki, A.; Banks, S.A.; Lewis, D.D.; Kim, S.E. Large Animal Models for Anterior Cruciate Ligament Research. Front. Vet. Sci. 2019, 6, 292. [Google Scholar] [CrossRef]
- Kaur, G.; Patel, D.; Sawant, M.G. Evaluation of anti-osteoarthritic activity of Vigna mungo in papain induced osteoarthritis model. Indian J. Pharmacol. 2015, 47, 59–64. [Google Scholar] [CrossRef]
- Botter, S.; van Osch, G.; Waarsing, J.; van der Linden, J.; Verhaar, J.; Pols, H.; van Leeuwen, J.; Weinans, H. Cartilage damage pattern in relation to subchondral plate thickness in a collagenase-induced model of osteoarthritis. Osteoarthr. Cartil. 2008, 16, 506–514. [Google Scholar] [CrossRef]
- de Sousa Valente, J. The Pharmacology of Pain Associated With the Monoiodoacetate Model of Osteoarthritis. Front. Pharmacol. 2019, 10, 974. [Google Scholar] [CrossRef]
- McCoy, A.M. Animal Models of Osteoarthritis: Comparisons and Key Considerations. Vet. Pathol. 2015, 52, 803–818. [Google Scholar] [CrossRef]
- Cope, P.; Ourradi, K.; Li, Y.; Sharif, M. Models of osteoarthritis: The good, the bad and the promising. Osteoarthr. Cartil. 2018, 27, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, M.; Lesur, C.; Thomas, M.; Anract, P.; De Nanteuil, G.; Pastoureau, P. Effect of inhibition of matrix metalloproteinases on cartilage loss in vitro and in a guinea pig model of osteoarthritis. Arthritis Care Res. 2005, 52, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Tchetina, E.V.; Antoniou, J.; Tanzer, M.; Zukor, D.J.; Poole, A.R. Transforming Growth Factor-β2 Suppresses Collagen Cleavage in Cultured Human Osteoarthritic Cartilage, Reduces Expression of Genes Associated with Chondrocyte Hypertrophy and Degradation, and Increases Prostaglandin E2 Production. Am. J. Pathol. 2006, 168, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Wezel, A.; Madry, H.; Cucchiarini, M. rAAV-mediated overexpression of TGF-β stably restructures human osteoarthritic articular cartilage in situ. J. Transl. Med. 2013, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.I.; Argyle, D.J.; Clements, D.N. In vitro models for the study of osteoarthritis. Vet. J. 2016, 209, 40–49. [Google Scholar] [CrossRef]
- Tang, Q.; Zheng, G.; Feng, Z.; Tong, M.; Xu, J.; Hu, Z.; Shang, P.; Chen, Y.; Wang, C.; Lou, Y.; et al. Wogonoside inhibits IL-1β induced catabolism and hypertrophy in mouse chondrocyte and ameliorates murine osteoarthritis. Oncotarget 2017, 8, 61440–61456. [Google Scholar] [CrossRef]
- Matsumoto, T.; Cooper, G.M.; Gharaibeh, B.; Meszaros, L.B.; Li, G.; Usas, A.; Fu, F.H.; Huard, J. Cartilage repair in a rat model of osteoarthritis through intraarticular transplantation of muscle-derived stem cells expressing bone morphogenetic protein 4 and soluble flt-1. Arthritis Care Res. 2009, 60, 1390–1405. [Google Scholar] [CrossRef]
- Limat, A.; Hunziker, T.; Waelti, E.R.; Inaebnit, S.P.; Wiesmann, U.; Braathen, L.R. Soluble factors from human hair papilla cells and dermal fibroblasts dramatically increase the clonal growth of outer root sheath cells. Arch. Dermatol. Res. 1993, 285, 205–210. [Google Scholar] [CrossRef]
- Eger, W.; Schumacher, B.L.; Mollenhauer, J.; Kuettner, K.E.; Cole, A.A. Human knee and ankle cartilage explants: Catabolic differences. J. Orthop. Res. 2002, 20, 526–534. [Google Scholar] [CrossRef]
- Ji, Q.; Zheng, Y.; Zhang, G.; Hu, Y.; Fan, X.; Hou, Y.; Wen, L.; Li, L.; Xu, Y.; Wang, Y.; et al. Single-cell RNA-seq analysis reveals the progression of human osteoarthritis. Ann. Rheum. Dis. 2018, 78, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ning, Y.; Zhang, P.; Poulet, B.; Huang, R.; Gong, Y.; Hu, M.; Li, C.; Zhou, R.; Lammi, M.J.; et al. Comparison of the major cell populations among osteoarthritis, Kashin–Beck disease and healthy chondrocytes by single-cell RNA-seq analysis. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, M.; Coburn, J.; Centola, M.; Murab, S.; Barbero, A.; Kaplan, D.L.; Martin, I.; Ghosh, S. Tissue engineering strategies to study cartilage development, degeneration and regeneration. Adv. Drug Deliv. Rev. 2014, 84, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Barbero, A.; Ploegert, S.; Heberer, M.; Martin, I. Plasticity of clonal populations of dedifferentiated adult human articular chondrocytes. Arthritis Rheum. 2003, 48, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Darling, E.M.; Athanasiou, K.A. Retaining Zonal Chondrocyte Phenotype by Means of Novel Growth Environments. Tissue Eng. 2005, 11, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Dowthwaite, G.P.; Bishop, J.C.; Redman, S.N.; Khan, I.M.; Rooney, P.; Evans, D.J.R.; Haughton, L.; Bayram, Z.; Boyer, S.; Thomson, B.; et al. The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 2004, 117, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Pretzel, D.; Linss, S.; Rochler, S.; Endres, M.; Kaps, C.; Alsalameh, S.; Kinne, R.W. Relative percentage and zonal distribution of mesenchymal progenitor cells in human osteoarthritic and normal cartilage. Arthritis Res. Ther. 2011, 13, R64. [Google Scholar] [CrossRef]
- Su, X.; Zuo, W.; Wu, Z.; Chen, J.; Wu, N.; Ma, P.; Xia, Z.; Jiang, C.; Ye, Z.; Liu, S.; et al. CD146 as a new marker for an increased chondroprogenitor cell sub-population in the later stages of osteoarthritis. J. Orthop. Res. 2014, 33, 84–91. [Google Scholar] [CrossRef]
- Koelling, S.; Kruegel, J.; Irmer, M.; Path, J.R.; Sadowski, B.; Miro, X.; Miosge, N. Migratory Chondrogenic Progenitor Cells from Repair Tissue during the Later Stages of Human Osteoarthritis. Cell Stem Cell 2009, 4, 324–335. [Google Scholar] [CrossRef]
- Liu, W.; Feng, M.; Jayasuriya, C.T.; Peng, H.; Zhang, L.; Guan, Y.; Froehlich, J.A.; Terek, R.M.; Chen, Q. Human osteoarthritis cartilage-derived stromal cells activate joint degeneration through TGF-beta lateral signaling. FASEB J. 2020, 34, 16552–16566. [Google Scholar] [CrossRef]
- McCarthy, H.E.; Bara, J.J.; Brakspear, K.; Singhrao, S.K.; Archer, C.W. The comparison of equine articular cartilage progenitor cells and bone marrow-derived stromal cells as potential cell sources for cartilage repair in the horse. Vet. J. 2012, 192, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Zheng, H.; Seol, D.; Yu, Y.; Martin, J.A. Gene expression profiles reveal that chondrogenic progenitor cells and synovial cells are closely related. J. Orthop. Res. 2014, 32, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, C.T.; Twomey-Kozak, J.; Newberry, J.; Desai, S.; Feltman, P.; Franco, J.R.; Li, N.; Terek, R.; Ehrlich, M.G.; Owens, B.D. Human Cartilage-Derived Progenitors Resist Terminal Differentiation and Require CXCR4 Activation to Successfully Bridge Meniscus Tissue Tears. Stem Cells 2018, 37, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Hammerick, K.E.; Huang, Z.; Sun, N.; Lam, M.T.; Prinz, F.B.; Wu, J.C.; Commons, G.W.; Longaker, M.T. Elastic Properties of Induced Pluripotent Stem Cells. Tissue Eng. Part A 2011, 17, 495–502. [Google Scholar] [CrossRef]
- Jukes, J.M.; Both, S.K.; Leusink, A.; Sterk, L.M.T.; van Blitterswijk, C.A.; de Boer, J. Endochondral bone tissue engineering using embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6840–6845. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Csobonyeiova, M.; Polak, S.; Nicodemou, A.; Zamborsky, R.; Danisovic, L. iPSCs in Modeling and Therapy of Osteoarthritis. Biomedicines 2021, 9, 186. [Google Scholar] [CrossRef]
- Yamashita, A.; Morioka, M.; Yahara, Y.; Okada, M.; Kobayashi, T.; Kuriyama, S.; Matsuda, S.; Tsumaki, N. Generation of Scaffoldless Hyaline Cartilaginous Tissue from Human iPSCs. Stem Cell Rep. 2015, 4, 404–418. [Google Scholar] [CrossRef]
- Xu, M.; Stattin, E.-L.; Shaw, G.; Heinegård, D.; Sullivan, G.; Wilmut, I.; Colman, A.; Önnerfjord, P.; Khabut, A.; Aspberg, A.; et al. Chondrocytes Derived From Mesenchymal Stromal Cells and Induced Pluripotent Cells of Patients With Familial Osteochondritis Dissecans Exhibit an Endoplasmic Reticulum Stress Response and Defective Matrix Assembly. Stem Cells Transl. Med. 2016, 5, 1171–1181. [Google Scholar] [CrossRef]
- Urlić, I.; Ivković, A. Cell Sources for Cartilage Repair—Biological and Clinical Perspective. Cells 2021, 10, 2496. [Google Scholar] [CrossRef]
- Mueller, M.B.; Fischer, M.; Zellner, J.; Berner, A.; Dienstknecht, T.; Prantl, L.; Kujat, R.; Nerlich, M.; Tuan, R.S.; Angele, P. Hypertrophy in Mesenchymal Stem Cell Chondrogenesis: Effect of TGF-β Isoforms and Chondrogenic Conditioning. Cells Tissues Organs 2010, 192, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Occhetta, P.; Pigeot, S.; Rasponi, M.; Dasen, B.; Mehrkens, A.; Ullrich, T.; Kramer, I.; Guth-gundel, S.; Barbero, A.; Martin, I. Developmentally Inspired Programming of Adult Human Mesenchymal Stromal Cells towards Stable Chondrogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4625–4630. [Google Scholar] [CrossRef] [PubMed]
- Rennerfeldt, D.A.; Raminhos, J.S.; Leff, S.M.; Manning, P.; Van Vliet, K.J. Emergent heterogeneity in putative mesenchymal stem cell colonies: Single-cell time lapsed analysis. PLoS ONE 2019, 14, e0213452. [Google Scholar] [CrossRef] [PubMed]
- Mwale, F.; Rampersad, S.; Richard, H.; Guoying, Y.; Al Rowas, S.; Madiraju, P.; Antoniou, J.; Laverty, S. The Constitutive Expression of Type X Collagen in Mesenchymal Stem Cells from Osteoarthritis Patients Is Reproduced in a Rabbit Model of Osteoarthritis. J. Tissue Eng. 2011, 2011, 587547. [Google Scholar] [CrossRef]
- Cheng, N.-C.; Estes, B.T.; Awad, H.A.; Guilak, F. Chondrogenic Differentiation of Adipose-Derived Adult Stem Cells by a Porous Scaffold Derived from Native Articular Cartilage Extracellular Matrix. Tissue Eng. Part A 2009, 15, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Beane, O.S.; Fonseca, V.C.; Cooper, L.L.; Koren, G.; Darling, E.M. Impact of Aging on the Regenerative Properties of Bone Marrow-, Muscle-, and Adipose-Derived Mesenchymal Stem/Stromal Cells. PLoS ONE 2014, 9, e115963. [Google Scholar] [CrossRef] [PubMed]
- Mohamed-Ahmed, S.; Fristad, I.; Lie, S.A.; Suliman, S.; Mustafa, K.; Vindenes, H.; Idris, S.B. Adipose-derived and bone marrow mesenchymal stem cells: A donor-matched comparison. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Koga, H.; Muneta, T.; Ju, Y.-J.; Nagase, T.; Nimura, A.; Mochizuki, T.; Ichinose, S.; von der Mark, K.; Sekiya, I. Synovial Stem Cells Are Regionally Specified According to Local Microenvironments After Implantation for Cartilage Regeneration. Stem Cells 2006, 25, 689–696. [Google Scholar] [CrossRef]
- Parolini, O.; Alviano, F.; Bagnara, G.P.; Bilic, G.; Bühring, H.-J.; Evangelista, M.; Hennerbichler, S.; Liu, B.; Magatti, M.; Mao, N.; et al. Concise Review: Isolation and Characterization of Cells from Human Term Placenta: Outcome of the First International Workshop on Placenta Derived Stem Cells. Stem Cells 2007, 26, 300–311. [Google Scholar] [CrossRef]
- E Barlow, S.; Brooke, G.; Chatterjee, K.; Price, G.; Pelekanos, R.; Rossetti, T.; Doody, M.; Venter, D.; Pain, S.; Gilshenan, K.; et al. Comparison of Human Placenta- and Bone Marrow–Derived Multipotent Mesenchymal Stem Cells. Stem Cells Dev. 2008, 17, 1095–1108. [Google Scholar] [CrossRef]
- Anker, P.S.I.T.; Scherjon, S.A.; der Keur, C.K.; de Groot-Swings, G.M.J.S.; Claas, F.H.J.; Fibbe, W.E.; Kanhai, H.H.H. Isolation of Mesenchymal Stem Cells of Fetal or Maternal Origin from Human Placenta. Stem Cells 2004, 22, 1338–1345. [Google Scholar] [CrossRef]
- Baksh, D.; Yao, R.; Tuan, R.S. Comparison of Proliferative and Multilineage Differentiation Potential of Human Mesenchymal Stem Cells Derived from Umbilical Cord and Bone Marrow. Stem Cells 2007, 25, 1384–1392. [Google Scholar] [CrossRef] [PubMed]
- Can, A.; Karahuseyinoglu, S. Concise Review: Human Umbilical Cord Stroma with Regard to the Source of Fetus-Derived Stem Cells. Stem Cells 2007, 25, 2886–2895. [Google Scholar] [CrossRef]
- El Omar, R.; Beroud, J.; Stoltz, J.F.; Menu, P.; Velot, E.; Decot, V. Umbilical Cord Mesenchymal Stem Cells: The New Gold Standard for Mesenchymal Stem Cell-Based Therapies? Tissue Eng.-Part B Rev. 2014, 20, 523–544. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Kern, S.; Klüter, H.; Eichler, H. Critical Parameters for the Isolation of Mesenchymal Stem Cells from Umbilical Cord Blood. Stem Cells 2004, 22, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Deberg, M.; Piccardi, N.; Msika, P.; Reginster, J.-Y.; Henrotin, Y. Subchondral bone osteoblasts induce phenotypic changes in human osteoarthritic chondrocytes. Osteoarthr. Cartil. 2005, 13, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pati, F.; Choi, Y.-J.; Rijal, G.; Shim, J.-H.; Kim, S.W.; Ray, A.R.; Cho, D.-W.; Ghosh, S. Bioprintable, cell-laden silk fibroin–gelatin hydrogel supporting multilineage differentiation of stem cells for fabrication of three-dimensional tissue constructs. Acta Biomater. 2015, 11, 233–246. [Google Scholar] [CrossRef]
- Jeyaraman, N.; Prajwal, G.S.; Jeyaraman, M.; Muthu, S.; Khanna, M. Chondrogenic Potential of Dental-Derived Mesenchymal Stromal Cells. Osteology 2021, 1, 149–174. [Google Scholar] [CrossRef]
- Caron, M.M.J.; Emans, P.J.; Coolsen, M.M.E.; Voss, L.; Surtel, D.A.M.; Cremers, A.; van Rhijn, L.W.; Welting, T.J.M. Redifferentiation of dedifferentiated human articular chondrocytes: Comparison of 2D and 3D cultures. Osteoarthr. Cartil. 2012, 20, 1170–1178. [Google Scholar] [CrossRef]
- Chen, J.-L.; Zou, C.; Chen, Y.; Zhu, W.; Liu, W.; Huang, J.; Liu, Q.; Wang, D.; Duan, L.; Xiong, J.; et al. TGFβ1 induces hypertrophic change and expression of angiogenic factors in human chondrocytes. Oncotarget 2017, 8, 91316–91327. [Google Scholar] [CrossRef]
- Filip, A.; Bianchi, A.; Mainard, D.; Lacolley, P.; Magdalou, J.; Mercier, N. A simple two dimensional culture method to study the hypertrophic differentiation of rat articular chondrocytes. Bio-Medical Mater. Eng. 2015, 25, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-H.; Li, L.-Z.; Sun, J.-B.; Wu, F.-H.; Liang, J.-Y. A new antioxidant flavone glycoside from Scutellaria baicalensis Georgi. Nat. Prod. Res. 2014, 28, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.C.; Chawla, S.; Hegde, A.; Singh, D.; Bandyopadhyay, B.; Lakshmanan, C.C.; Kalsi, G.; Ghosh, S. Establishment of an in vitro organoid model of dermal papilla of human hair follicle. J. Cell. Physiol. 2018, 233, 9015–9030. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.J.; Emans, P.J.; Surtel, D.A.M.; van der Kraan, P.M.; van Rhijn, L.W.; Welting, T.J.M. BAPX-1/NKX-3.2 Acts as a Chondrocyte Hypertrophy Molecular Switch in Osteoarthritis. Arthritis Rheumatol. 2015, 67, 2944–2956. [Google Scholar] [CrossRef] [PubMed]
- Hirao, M.; Tamai, N.; Tsumaki, N.; Yoshikawa, H.; Myoui, A. Oxygen Tension Regulates Chondrocyte Differentiation and Function during Endochondral Ossification. J. Biol. Chem. 2006, 281, 31079–31092. [Google Scholar] [CrossRef] [PubMed]
- Manferdini, C.; Maumus, M.; Gabusi, E.; Piacentini, A.; Filardo, G.; Peyrafitte, J.-A.; Jorgensen, C.; Bourin, P.; Fleury-Cappellesso, S.; Facchini, A.; et al. Adipose-Derived Mesenchymal Stem Cells Exert Antiinflammatory Effects on Chondrocytes and Synoviocytes From Osteoarthritis Patients Through Prostaglandin E2. Arthritis Care Res. 2013, 65, 1271–1281. [Google Scholar] [CrossRef]
- Kuroda, K.; Kabata, T.; Hayashi, K.; Maeda, T.; Kajino, Y.; Iwai, S.; Fujita, K.; Hasegawa, K.; Inoue, D.; Sugimoto, N.; et al. The paracrine effect of adipose-derived stem cells inhibits osteoarthritis progression. BMC Musculoskelet. Disord. 2015, 16, 236. [Google Scholar] [CrossRef]
- Murdoch, A.D.; Grady, L.M.; Ablett, M.P.; Katopodi, T.; Meadows, R.S.; Hardingham, T.E. Chondrogenic Differentiation of Human Bone Marrow Stem Cells in Transwell Cultures: Generation of Scaffold-Free Cartilage. Stem Cells 2007, 25, 2786–2796. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- De Pieri, A.; Rochev, Y.; Zeugolis, D.I. Scaffold-free cell-based tissue engineering therapies: Advances, shortfalls and forecast. NPJ Regen. Med. 2021, 6, 1–15. [Google Scholar] [CrossRef]
- Singh, Y.P.; Moses, J.C.; Bhardwaj, N.; Mandal, B.B. Overcoming the Dependence on Animal Models for Osteoarthritis Therapeutics–The Promises and Prospects of In Vitro Models. Adv. Heal. Mater. 2021, 10, 2100961. [Google Scholar] [CrossRef] [PubMed]
- Samvelyan, H.J.; Hughes, D.; Stevens, C.; Staines, K.A. Models of Osteoarthritis: Relevance and New Insights. Calcif. Tissue Res. 2020, 109, 243–256. [Google Scholar] [CrossRef]
- Meng, X.; Leslie, P.; Zhang, Y.; Dong, J. Stem cells in a three-dimensional scaffold environment. SpringerPlus 2014, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.; Midha, S.; Sharma, A.; Ghosh, S. Silk-Based Bioinks for 3D Bioprinting. Adv. Healthc. Mater. 2018, 7, e1701204. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, J.; Magli, S.; Rabbachin, L.; Sampaolesi, S.; Nicotra, F.; Russo, L. 3D Extracellular Matrix Mimics: Fundamental Concepts and Role of Materials Chemistry to Influence Stem Cell Fate. Biomacromolecules 2020, 21, 1968–1994. [Google Scholar] [CrossRef] [PubMed]
- Murab, S.; Chameettachal, S.; Bhattacharjee, M.; Das, S.; Kaplan, D.L.; Ghosh, S. Matrix-Embedded Cytokines to Simulate Osteoarthritis-Like Cartilage Microenvironments. Tissue Eng. Part A 2013, 19, 1733–1753. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Tsukamoto, I.; Inoue, S.; Hashimoto, K.; Akagi, M. Cyclic compressive loading activates angiotensin II type 1 receptor in articular chondrocytes and stimulates hypertrophic differentiation through a G-protein-dependent pathway. FEBS Open Bio 2018, 8, 962–973. [Google Scholar] [CrossRef]
- Dai, M.; Sui, B.; Xue, Y.; Liu, X.; Sun, J. Cartilage repair in degenerative osteoarthritis mediated by squid type II collagen via immunomodulating activation of M2 macrophages, inhibiting apoptosis and hypertrophy of chondrocytes. Biomaterials 2018, 180, 91–103. [Google Scholar] [CrossRef]
- Chameettachal, S.; Midha, S.; Ghosh, S. Regulation of Chondrogenesis and Hypertrophy in Silk Fibroin-Gelatin-Based 3D Bioprinted Constructs. ACS Biomater. Sci. Eng. 2016, 2, 1450–1463. [Google Scholar] [CrossRef]
- Murphy, S.V.; Atala, A. 3D bioprinting of tissues and organs. Nat. Biotechnol. 2014, 32, 773–785. [Google Scholar] [CrossRef]
- Sanchez-Adams, J.; Leddy, H.A.; McNulty, A.L.; O’Conor, C.J.; Guilak, F. The Mechanobiology of Articular Cartilage: Bearing the Burden of Osteoarthritis. Curr. Rheumatol. Rep. 2014, 16, 1–9. [Google Scholar] [CrossRef]
- Grodzinsky, A.J.; Levenston, M.E.; Jin, M.; Frank, E.H. Cartilage Tissue Remodeling in Response to Mechanical Forces. Annu. Rev. Biomed. Eng. 2000, 2, 691–713. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Yu, Y.E.; Zhang, X.; Watts, T.; Zhou, B.; Wang, J.; Wang, T.; Zhao, W.; Chiu, K.-Y.; et al. Subchondral Trabecular Rod Loss and Plate Thickening in the Development of Osteoarthritis. J. Bone Miner. Res. 2017, 33, 316–327. [Google Scholar] [CrossRef]
- Boyd, S.K.; Müller, R.; Zernicke, R.F. Mechanical and Architectural Bone Adaptation in Early Stage Experimental Osteoarthritis. J. Bone Miner. Res. 2002, 17, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Candrian, C.; Vonwil, D.; Barbero, A.; Bonacina, E.; Miot, S.; Farhadi, J.; Wirz, D.; Dickinson, S.; Hollander, A.; Jakob, M.; et al. Engineered cartilage generated by nasal chondrocytes is responsive to physical forces resembling joint loading. Arthritis Care Res. 2007, 58, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yao, S.; Alini, M.; Grad, S. Different response of articular chondrocyte subpopulations to surface motion. Osteoarthr. Cartil. 2007, 15, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Mauck, R.; Wang, C.-B.; Oswald, E.; Ateshian, G.; Hung, C. The role of cell seeding density and nutrient supply for articular cartilage tissue engineering with deformational loading. Osteoarthr. Cartil. 2003, 11, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-Y.; Tanaka, N.; Ohkuma, S.; Iwabuchi, Y.; Tanne, Y.; Kamiya, T.; Kunimatsu, R.; Huang, Y.-C.; Yoshioka, M.; Mitsuyoshi, T.; et al. Applying an excessive mechanical stress alters the effect of subchondral osteoblasts on chondrocytes in a co-culture system. Eur. J. Oral Sci. 2010, 118, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, C.; Simon, M. In Vitro Human Joint Models Combining Advanced 3D Cell Culture and Cutting-Edge 3D Bioprinting Technologies. Cells 2021, 10, 596. [Google Scholar] [CrossRef]
- Lee, D.; Erickson, A.; You, T.; Dudley, A.T.; Ryu, S. Pneumatic microfluidic cell compression device for high-throughput study of chondrocyte mechanobiology. Lab Chip 2018, 18, 2077–2086. [Google Scholar] [CrossRef]
- Rosser, J.; Bachmann, B.; Jordan, C.; Ribitsch, I.; Haltmayer, E.; Gueltekin, S.; Junttila, S.; Galik, B.; Gyenesei, A.; Haddadi, B.; et al. Microfluidic nutrient gradient–based three-dimensional chondrocyte culture-on-a-chip as an in vitro equine arthritis model. Mater. Today Bio 2019, 4, 100023. [Google Scholar] [CrossRef] [PubMed]
- Occhetta, P.; Mainardi, A.; Votta, E.; Vallmajo-Martin, Q.; Ehrbar, M.; Martin, I.; Barbero, A.; Rasponi, M. Hyperphysiological compression of articular cartilage induces an osteoarthritic phenotype in a cartilage-on-a-chip model. Nat. Biomed. Eng. 2019, 3, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Paggi, C.A.; Venzac, B.; Karperien, M.; Leijten, J.C.; Le Gac, S. Monolithic microfluidic platform for exerting gradients of compression on cell-laden hydrogels, and application to a model of the articular cartilage. Sens. Actuators B Chem. 2020, 315, 127917. [Google Scholar] [CrossRef]
- Mansoorifar, A.; Gordon, R.; Bergan, R.C.; Bertassoni, L.E. Bone-on-a-Chip: Microfluidic Technologies and Microphysiologic Models of Bone Tissue. Adv. Funct. Mater. 2020, 31, 2006796. [Google Scholar] [CrossRef] [PubMed]
- Pirosa, A.; Gottardi, R.; Alexander, P.G.; Puppi, D.; Chiellini, F.; Tuan, R.S. An in vitro chondro-osteo-vascular triphasic model of the osteochondral complex. Biomaterials 2021, 272, 120773. [Google Scholar] [CrossRef]
- Lin, Z.; Li, Z.; Li, E.N.; Li, X.; Del Duke, C.J.; Shen, H.; Hao, T.; O’Donnell, B.; Bunnell, B.A.; Goodman, S.B.; et al. Osteochondral Tissue Chip Derived From iPSCs: Modeling OA Pathologies and Testing Drugs. Front. Bioeng. Biotechnol. 2019, 7, 411. [Google Scholar] [CrossRef]
- Lin, H.; Lozito, T.P.; Alexander, P.G.; Gottardi, R.; Tuan, R.S. Stem Cell-Based Microphysiological Osteochondral System to Model Tissue Response to Interleukin-1β. Mol. Pharm. 2014, 11, 2203–2212. [Google Scholar] [CrossRef]
- Whelan, I.T.; Moeendarbary, E.; A Hoey, D.; Kelly, D.J. Biofabrication of vasculature in microphysiological models of bone. Biofabrication 2021, 13, 032004. [Google Scholar] [CrossRef]
- Li, T.; Liu, B.; Chen, K.; Lou, Y.; Jiang, Y.; Zhang, D. Small molecule compounds promote the proliferation of chondrocytes and chondrogenic differentiation of stem cells in cartilage tissue engineering. Biomed. Pharmacother. 2020, 131, 110652. [Google Scholar] [CrossRef]
- Chen, S.; Borowiak, M.; Fox, J.L.; Maehr, R.; Osafune, K.; Davidow, L.; Lam, K.; Peng, L.F.; Schreiber, S.L.; Rubin, L.L.; et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat. Chem. Biol. 2009, 5, 258–265. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, W.; Laurent, T.; Ding, S. Small molecules, big roles–the chemical manipulation of stem cell fate and somatic cell reprogramming. J. Cell Sci. 2012, 125, 5609–5620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Siclari, V.A.; Lan, S.; Zhu, J.; Koyama, E.; Dupuis, H.L.; Enomoto-Iwamoto, M.; Beier, F.; Qin, L. The Critical Role of the Epidermal Growth Factor Receptor in Endochondral Ossification. J. Bone Miner. Res. 2011, 26, 2622–2633. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, N.; Han, C.-Y.E.; Cam, L.; Lee, J.I.; Pretorius, J.; Fisher, S.; Rosenfeld, R.; Scully, S.; Nishinakamura, R.; Duryea, D.; et al. A novel chordin-like BMP inhibitor, CHL2, expressed preferentially in chondrocytes of developing cartilage and osteoarthritic joint cartilage. Development 2004, 131, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Hong, C.C.; Sachidanandan, C.; Babitt, J.L.; Deng, D.Y.; Hoyng, S.A.; Lin, H.Y.; Bloch, K.D.; Peterson, R.T. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 2008, 4, 33–41. [Google Scholar] [CrossRef]
- Caron, M.M.; Ripmeester, E.G.; Akker, G.V.D.; Wijnands, N.K.P.; Steijns, J.; Surtel, D.A.; Cremers, A.; Emans, P.J.; van Rhijn, L.W.; Welting, T.J. Discovery of bone morphogenetic protein 7-derived peptide sequences that attenuate the human osteoarthritic chondrocyte phenotype. Mol. Ther.-Methods Clin. Dev. 2021, 21, 247–261. [Google Scholar] [CrossRef]
- Yang, Y.R.; Yang, X.F.; Duan, H.C.; Qiao, J.Q. Cyclooxygenase-2 Inhibitor Rofecoxib Prevents Chondrocytes against Hypertrophy via Wnt/β-Catenin Pathway. J. Biol. Regul. Homeost. Agents 2020, 34, 785–794. [Google Scholar]
- Lories, R.J.; Monteagudo, S. Review Article: Is Wnt Signaling an Attractive Target for the Treatment of Osteoarthritis? Rheumatol. Ther. 2020, 7, 259–270. [Google Scholar] [CrossRef]
- Deshmukh, V.; O’Green, A.; Bossard, C.; Seo, T.; Lamangan, L.; Ibanez, M.; Ghias, A.; Lai, C.; Do, L.; Cho, S.; et al. Modulation of the Wnt pathway through inhibition of CLK2 and DYRK1A by lorecivivint as a novel, potentially disease-modifying approach for knee osteoarthritis treatment. Osteoarthr. Cartil. 2019, 27, 1347–1360. [Google Scholar] [CrossRef]
- Nagao, M.; Hamilton, J.L.; Kc, R.; Berendsen, A.D.; Duan, X.; Cheong, C.W.; Li, X.; Im, H.-J.; Olsen, B.R. Vascular Endothelial Growth Factor in Cartilage Development and Osteoarthritis. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Marsano, A.; da Cunha, C.M.M.; Ghanaati, S.; Gueven, S.; Centola, M.; Tsaryk, R.; Barbeck, M.; Stuedle, C.; Barbero, A.; Helmrich, U.; et al. Spontaneous In Vivo Chondrogenesis of Bone Marrow-Derived Mesenchymal Progenitor Cells by Blocking Vascular Endothelial Growth Factor Signaling. Stem Cells Transl. Med. 2016, 5, 1730–1738. [Google Scholar] [CrossRef]
- Da Cunha, C.M.M.; Perugini, V.; Bernegger, P.; Centola, M.; Barbero, A.; Guildford, A.L.; Santin, M.; Banfi, A.; Martin, I.; Marsano, A. Vascular Endothelial Growth Factor Sequestration Enhances In Vivo Cartilage Formation. Int. J. Mol. Sci. 2017, 18, 2478. [Google Scholar] [CrossRef]
- Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling Pathways in Cartilage Repair. Int. J. Mol. Sci. 2014, 15, 8667–8698. [Google Scholar] [CrossRef]
- Markway, B.D.; Cho, H.; Johnstone, B. Hypoxia promotes redifferentiation and suppresses markers of hypertrophy and degeneration in both healthy and osteoarthritic chondrocytes. Arthritis Res. Ther. 2013, 15, R92. [Google Scholar] [CrossRef]
- Szojka, A.R.A.; Li, D.X.; Sopcak, M.E.J.; Ma, Z.; Kunze, M.; Mulet-Sierra, A.; Adeeb, S.M.; Westover, L.; Jomha, N.M.; Adesida, A.B. Mechano-Hypoxia Conditioning of Engineered Human Meniscus. Front. Bioeng. Biotechnol. 2021, 9, 739438. [Google Scholar] [CrossRef]
- Rodríguez, L.A.G.; Hernández-Díaz, S. The risk of upper gastrointestinal complications associated with nonsteroidal anti-inflammatory drugs, glucocorticoids, acetaminophen, and combinations of these agents. Arthritis Res. Ther. 2000, 3, 98–101. [Google Scholar] [CrossRef]
- Solomon, S.D.; Wittes, J.; Finn, P.V.; Fowler, R.; Viner, J.; Bertagnolli, M.M.; Arber, N.; Levin, B.; Meinert, C.L.; Martin, B.; et al. Cardiovascular Risk of Celecoxib in 6 Randomized Placebo-Controlled Trials. Circulation 2008, 117, 2104–2113. [Google Scholar] [CrossRef]
- Evans, C.H.; Kraus, V.B.; Setton, L.A. Progress in intra-articular therapy. Nat. Rev. Rheumatol. 2013, 10, 11–22. [Google Scholar] [CrossRef]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Bryant, S.J. The role of chondroitin sulfate in regulating hypertrophy during MSC chondrogenesis in a cartilage mimetic hydrogel under dynamic loading. Biomaterials 2018, 190–191, 51–62. [Google Scholar] [CrossRef]
- Bian, L.; Hou, C.; Tous, E.; Rai, R.; Mauck, R.L.; Burdick, J.A. The influence of hyaluronic acid hydrogel crosslinking density and macromolecular diffusivity on human MSC chondrogenesis and hypertrophy. Biomaterials 2013, 34, 413–421. [Google Scholar] [CrossRef]
- Bian, L.; Zhai, D.Y.; Tous, E.; Rai, R.; Mauck, R.L.; Burdick, J.A. Enhanced MSC chondrogenesis following delivery of TGF-β3 from alginate microspheres within hyaluronic acid hydrogels in vitro and in vivo. Biomaterials 2011, 32, 6425–6434. [Google Scholar] [CrossRef]
- Bello, A.B.; Kim, Y.; Park, S.; Muttigi, M.S.; Kim, J.; Park, H.; Lee, S. Matrilin3/TGFβ3 gelatin microparticles promote chondrogenesis, prevent hypertrophy, and induce paracrine release in MSC spheroid for disc regeneration. NPJ Regen. Med. 2021, 6, 1–13. [Google Scholar] [CrossRef]
- Ferguson, C.M. Smad2 and 3 Mediate Transforming Growth Factor-1-Induced Inhibition of Chondrocyte Maturation. Endocrinology 2000, 141, 4728–4735. [Google Scholar] [CrossRef]
- Jin, Y.; Koh, R.H.; Kim, S.-H.; Kim, K.M.; Park, G.K.; Hwang, N.S. Injectable anti-inflammatory hyaluronic acid hydrogel for osteoarthritic cartilage repair. Mater. Sci. Eng. C 2020, 115, 111096. [Google Scholar] [CrossRef]
- Van Buul, G.M.; Koevoet, W.L.; Kops, N.; Bos, P.K.; Verhaar, J.; Weinans, H.; Bernsen, M.; Van Osch, G.J. Platelet-Rich Plasma Releasate Inhibits Inflammatory Processes in Osteoarthritic Chondrocytes. Am. J. Sports Med. 2011, 39, 2362–2370. [Google Scholar] [CrossRef]
- Bendinelli, P.; Matteucci, E.; Dogliotti, G.; Corsi, M.M.; Banfi, G.; Maroni, P.; Desiderio, M.A. Molecular basis of anti-inflammatory action of platelet-rich plasma on human chondrocytes: Mechanisms of NF-κB inhibition via HGF. J. Cell. Physiol. 2010, 225, 757–766. [Google Scholar] [CrossRef]
- Lee, H.-R.; Shon, O.-J.; Park, S.-I.; Kim, H.-J.; Kim, S.; Ahn, M.-W.; Do, S.H. Platelet-Rich Plasma Increases the Levels of Catabolic Molecules and Cellular Dedifferentiation in the Meniscus of a Rabbit Model. Int. J. Mol. Sci. 2016, 17, 120. [Google Scholar] [CrossRef]
- Hou, M.; Zhang, Y.; Zhou, X.; Liu, T.; Yang, H.; Chen, X.; He, F.; Zhu, X. Kartogenin prevents cartilage degradation and alleviates osteoarthritis progression in mice via the miR-146a/NRF2 axis. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Valiani, A.; Izadi, M.A.; Bahramian, H.; Esfandiari, E.; Hashemibeni, B. Comparison between the effect of kartogenin and TGFβ3 on chondrogenesis of human adipose- derived stem cells in fibrin scaffold. Bratisl. Med. J. 2018, 118, 591–597. [Google Scholar] [CrossRef]
- Blanco, M.N.F.; Bastiaansen-Jenniskens, Y.; Narcisi, R.; van Osch, G. Effect of inflammation on hypertrophy to human articular chondrocytes. Osteoarthr. Cartil. 2020, 28, S113–S114. [Google Scholar] [CrossRef]
- Cecil, D.L.; Johnson, K.; Rediske, J.; Lotz, M.; Schmidt, A.M.; Terkeltaub, R. Inflammation-Induced Chondrocyte Hypertrophy Is Driven by Receptor for Advanced Glycation End Products. J. Immunol. 2005, 175, 8296–8302. [Google Scholar] [CrossRef]
- Jahangir, S.; Eglin, D.; Pötter, N.; Ravari, M.K.; Stoddart, M.J.; Samadikuchaksaraei, A.; Alini, M.; Eslaminejad, M.B.; Safa, M. Inhibition of hypertrophy and improving chondrocyte differentiation by MMP-13 inhibitor small molecule encapsulated in alginate-chondroitin sulfate-platelet lysate hydrogel. Stem Cell Res. Ther. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Ando, A.; Suda, H.; Hagiwara, Y.; Onoda, Y.; Chimoto, E.; Saijo, Y.; Itoi, E. Reversibility of Immobilization-Induced Articular Cartilage Degeneration after Remobilization in Rat Knee Joints. Tohoku J. Exp. Med. 2011, 224, 77–85. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Type of Analyses | Marker | Technique |
|---|---|---|
| Composition of the extracellular matrix | Glycosaminoglycans (GAG) | Histology (Safranin-O staining) Biochemistry (DMMB GAG quantification) [30] |
| Calcium (Ca) deposit | Histology (Alizarin red staining) Biochemistry (Ca quantification) [31] | |
| COLX COL10A1 | Immunostaining Western blot In-situ hybridisation [32,33] | |
| MMP-1, -2, -13 MMP1, MMP2, MMP13 | Immunostaining Western blot qRT-PCR [34,35] | |
| MMP-derived fragment of type II collagen | Histology [34,35] | |
| Biochemical components | RAMAN spectroscopy [36,37] | |
| Properties of the tissue and or ECM | Collagen fiber breakdown | Scanning electron microscopy [38] Histology Transmission electron microscopy |
| Matrix stiffness | Atomic Force Microscopy [39] | |
| Assessments of (sub)cellular properties | Hypertrophic markers (ALP, IHH) | qRT-PCR [40] |
| (Frizzled-related Protein (FRZB), Gremlin (GREM1), Dickkopf-I, (DKK1) (decreased expression in OA) | qRT-PCR [41] | |
| RUNX2 (total) RUNX2 (nuclear) | qRT-PCR Western blot [42] | |
| Phosphorylated Smad1, Smad5, and Smad9 | Immunostaining Western blot [43] | |
| Bone morphogenetic protein type I receptors, Activin A receptor like type 2,3,6 (ALK2,3,6) | Immunostaining Western blot [35,44] | |
| Transglutaminase 2 (TG2) | Immunostaining Western blot [45] | |
| Analyses of the degradome and the extracellular vesicles | Ca released | Biochemical quantification [46] |
| ALP activity | Biochemical quantification [46] | |
| MMPs activity | Biochemical quantification Zymography [47] | |
| MMP-derived fragment of type II collagen and aggrecan | Immunoassay [48] |
| Signaling Pathway | Role in OA Progression | Pathway Specific Inhibitor | Mechanism of Action of Inhibitor/Activator |
|---|---|---|---|
| IHH/parathyroid hormone-related protein (PTHrP) signaling | IHH signaling activates OA hypertrophy aided by RUNX2. PTHrP selectively inhibits hypertrophy by acting in a negative feedback loop with IHH [51,52]. | HDAC4 (Inhibitor) | Downregulates RUNX2 expression and thus regulates the IHH signaling pathway [53]. |
| Ipriflavone (Inhibitor) | Blocks IHH pathway [54]. | ||
| WNT signaling | Binding of Frizzled receptor and low-density lipoprotein receptor-related protein (LRP) 5/6 to WNT ligand enhances nuclear translocation of Beta-Catenin (β-catenin) and causes the expression of RUNX2, further initiating hypertrophy [29]. Non-canonical Wnts (e.g., Wnt5A) play a dual role. Wnt5A activates hypertrophy during the initial stages of chondrogenic differentiation, while in later stages inhibits RUNX2 expression [55,56]. | DKK1 (Inhibitor) | Interacts with low-density lipoprotein receptor proteins (LRP-5 and LRP-6), and inhibits the formation of the WNT-Fz-LRP complex [41]. |
| FRZB (Inhibitor) | Develops a non-functional complex with Frizzled receptors inhibiting WNT/β-catenin signaling [41]. | ||
| EPZ005687 (Inhibitor) | Inhibits enhancer of zeste homolog 2 (EZH2), a histone methyltransferase that is involved in the induction of hypertrophic OA, by blocking WNT/β-catenin signaling [57]. | ||
| PKF118-130 (Inhibitor) | Inhibits WNT signaling by inhibiting nuclear translocation of β-catenin, thus enhancing the chondrogenic marker expression, while reducing the expression of hypertrophic markers [58]. | ||
| A stapled peptide derived from the Bcl9 homology domain-2) (SAH-Bcl9), Stapled β -catenin binding domain of Axin (StAx-35R) (Inhibitor) | These small molecule inhibitors inhibit canonical WNT signaling, thereby inhibiting hypertrophic chondrocyte shift, increasing the gene expression of SOX9 and ACAN, and decreasing the expression of COL10A1 [59]. | ||
| Transforming growth factor-β (TGF-β) signaling | High TGF-β1 levels have been observed in OA patients leading to osteophyte development and chondrocyte hypertrophy [60,61]. | SB505124 (Inhibitor) | Blocks TGF-β type I receptor, thus inhibiting TGF-β activity and reducing the degeneration of OA articular cartilage [62]. |
| BMP signaling | Increased phosphorylation of intracellular SMAD proteins (SMAD1/5/8) leads to enhanced nuclear translocation of SMAD4, inducing hypertrophy. Increased BMP-2 protein expression has been detected in human OA cartilage [8,63]. | LDN193189 (Inhibitor) | Blocks BMP signaling by selective inhibition of ALK2/3 and suppresses hypertrophic OA traits, thereby reducing the expression of COLX and MMP-13 [35]. |
| Matrilin-3 (Inhibitor) | Inhibits binding of BMP-2 with its receptor by interacting with BMP-2 ligand, thus inhibiting downstream BMP signaling and decreasing the hypertrophic marker COLX [64]. | ||
| Noggin (Inhibitor) | Blocks BMP-2 activity by inhibiting the binding of BMP-2 with its receptors, reducing cartilage degradation in OA [65]. | ||
| SMAD7 (Inhibitor) | Inhibits Smad pathways in chondrocytes in vivo. Smad7 deficiency leads to a reduction in the hypertrophic zone [66]. | ||
| Calcium signaling | A rise in extracellular calcium and increased activity of calcium-sensing receptors have been linked to COLX up-regulation during OA. The binding of calcium to calmodulin activates Calcium/calmodulin-dependent protein kinase, inducing hypertrophy [67,68,69]. | - | - |
| Integrin signaling | Overexpression of integrin pathway modulators RhoA/Rock suppresses ALP and mineralisation in chondrocytes [70,71,72]. | Function-blocking anti-integrin β1 antibody (Inhibitor) | Inhibits COLX expression and hypertrophy [73]. |
| Notch signaling | Enhanced mRNA expression of Notch ligand Jagged 1 and its receptor Notch 1 in human OAACs has been identified [51,74]. | N- [N-(3,5-diflurophenylacetate)-L-alanyl]-(S)-phenylglycine t-butyl ester (DAPT). (Inhibitor) | Intra-articular injection of DAPT in mouse knee reduced hypertrophic OA progression [51,74]. |
| MAPK pathway (p38, c-Jun N-terminal (JNK) kinase, and extra-cellular-regulated kinases (ERK)) | MAP kinases act as key mediators that regulate the expression of MMPs during OA. Activation of p38 represses COLX expression and OA progression. Phosphorylation of ERK1/2 increases in OA with an increased hypertrophic phenotype [75,76,77]. | U0126 (Inhibitor) Sprouty RTK signaling antagonist 4 (Inhibitor MAPK) | Inhibits the MEK-ERK pathway leading to reduced pERK levels and diminished expression of RUNX2, COL10A1, ADAMTS5, and MMP-13 [77]. Inhibits MAPK pathway leading to inhibiton of chondrocyte hypertrophy [78]. |
| AMP-activated protein kinase (AMPK)/PI3K/AKT signaling pathway | Reduced AMPK and PI3K-AKT expression have been observed in OA articular cartilage [79,80,81]. | Asiatic acid (Inhibitor PI3K/AKT) (Activator) | Inhibits the phosphorylation status of PI3K/AKT and activates the phosphorylation of AMK, contributing to the reduction in hypertrophy [82]. |
| FGF signaling | Enhanced FGF23 and FGF1 in OA chondrocytes [29]. | G141 (Inhibitor) | Reduces the expression of hypertrophic markers MMP-13 and COL10A1 and reduces cartilage degradation [83]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chawla, S.; Mainardi, A.; Majumder, N.; Dönges, L.; Kumar, B.; Occhetta, P.; Martin, I.; Egloff, C.; Ghosh, S.; Bandyopadhyay, A.; et al. Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies. Cells 2022, 11, 4034. https://doi.org/10.3390/cells11244034
Chawla S, Mainardi A, Majumder N, Dönges L, Kumar B, Occhetta P, Martin I, Egloff C, Ghosh S, Bandyopadhyay A, et al. Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies. Cells. 2022; 11(24):4034. https://doi.org/10.3390/cells11244034
Chicago/Turabian StyleChawla, Shikha, Andrea Mainardi, Nilotpal Majumder, Laura Dönges, Bhupendra Kumar, Paola Occhetta, Ivan Martin, Christian Egloff, Sourabh Ghosh, Amitabha Bandyopadhyay, and et al. 2022. "Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies" Cells 11, no. 24: 4034. https://doi.org/10.3390/cells11244034
APA StyleChawla, S., Mainardi, A., Majumder, N., Dönges, L., Kumar, B., Occhetta, P., Martin, I., Egloff, C., Ghosh, S., Bandyopadhyay, A., & Barbero, A. (2022). Chondrocyte Hypertrophy in Osteoarthritis: Mechanistic Studies and Models for the Identification of New Therapeutic Strategies. Cells, 11(24), 4034. https://doi.org/10.3390/cells11244034