
A Differentiation-Related Gene Prognostic Index Contributes to Prognosis and Immunotherapy Evaluation in Patients with Hepatocellular Carcinoma


Abstract
:1. Introduction
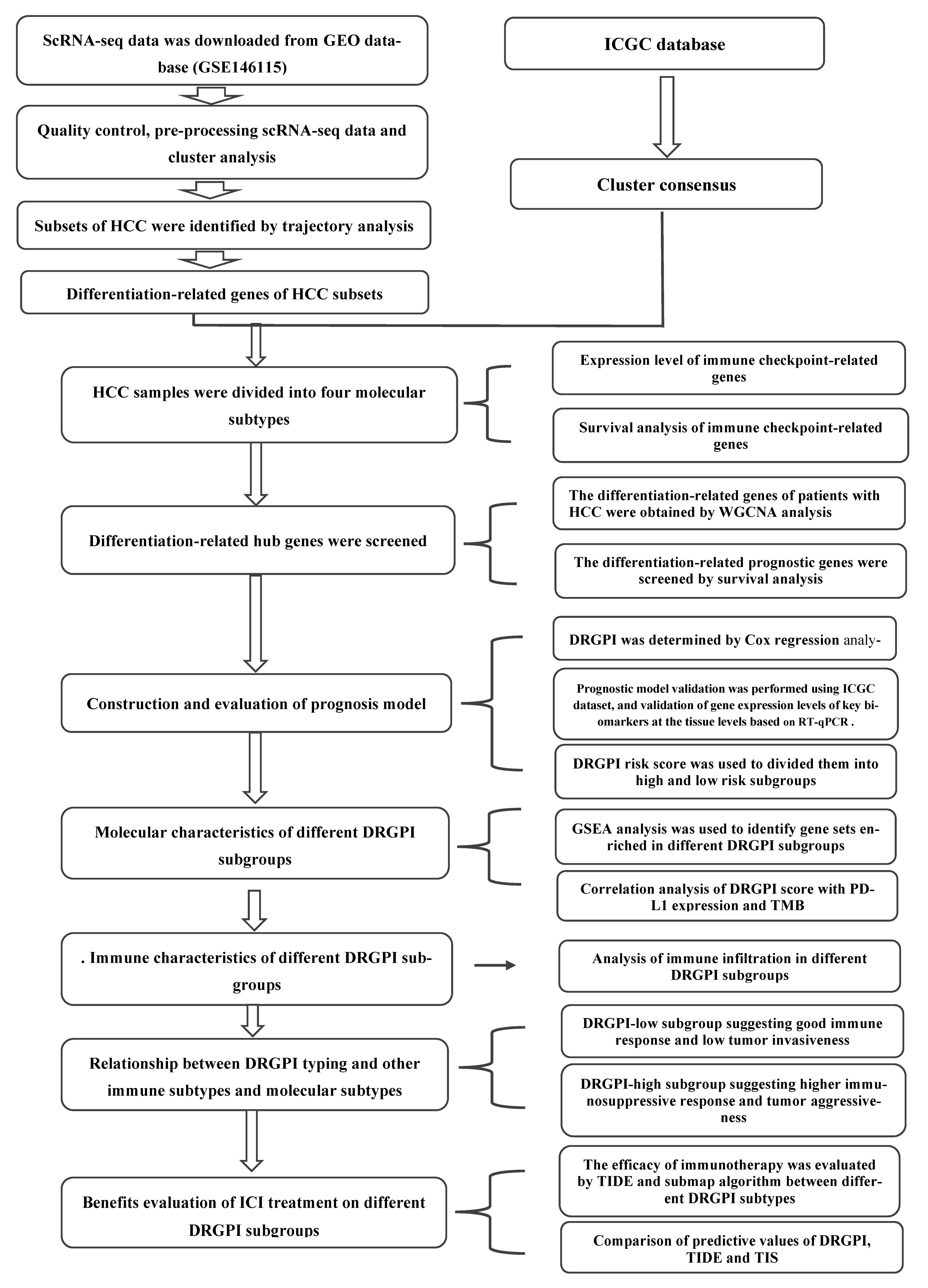
2. Materials and Methods
2.1. HCC Tissue Specimen Collection
2.2. ScRNA-Seq and Bulk RNA-Seq Data Acquisition and Preprocessing
2.3. Principal Component Analysis and Cell Annotation
2.4. HCC Cells Trajectory Analysis and Differentiation-Related Gene Identification
2.5. Differentiation-Related Gene Classification Based on Patients with HCC in ICGC Cohorts
2.6. CIBERSORT Estimation and the Expression of Immune Checkpoint Gene (ICGs) in Molecular Subtypes
2.7. Construction and Validation of a DRGPI and Nomogram
2.8. mRNA Extraction and RT-qPCR Analysis
2.9. Gene Set Enrichment Analysis (GSEA), Correlation Analysis, and Somatic Mutation Analysis of the DRGPI Subgroups
2.10. Comprehensive Analysis of Molecular and Immune Characteristics and ICI Therapy of Different DRGPI Subgroups
2.11. Statistical Analysis
3. Results
3.1. Annotation of 14 Cell Clusters Revealed High Cell Heterogeneity in HCC Preprocessing scRNA-Seq Data
3.2. Cell Trajectory Analysis Identified Seven HCC Subsets
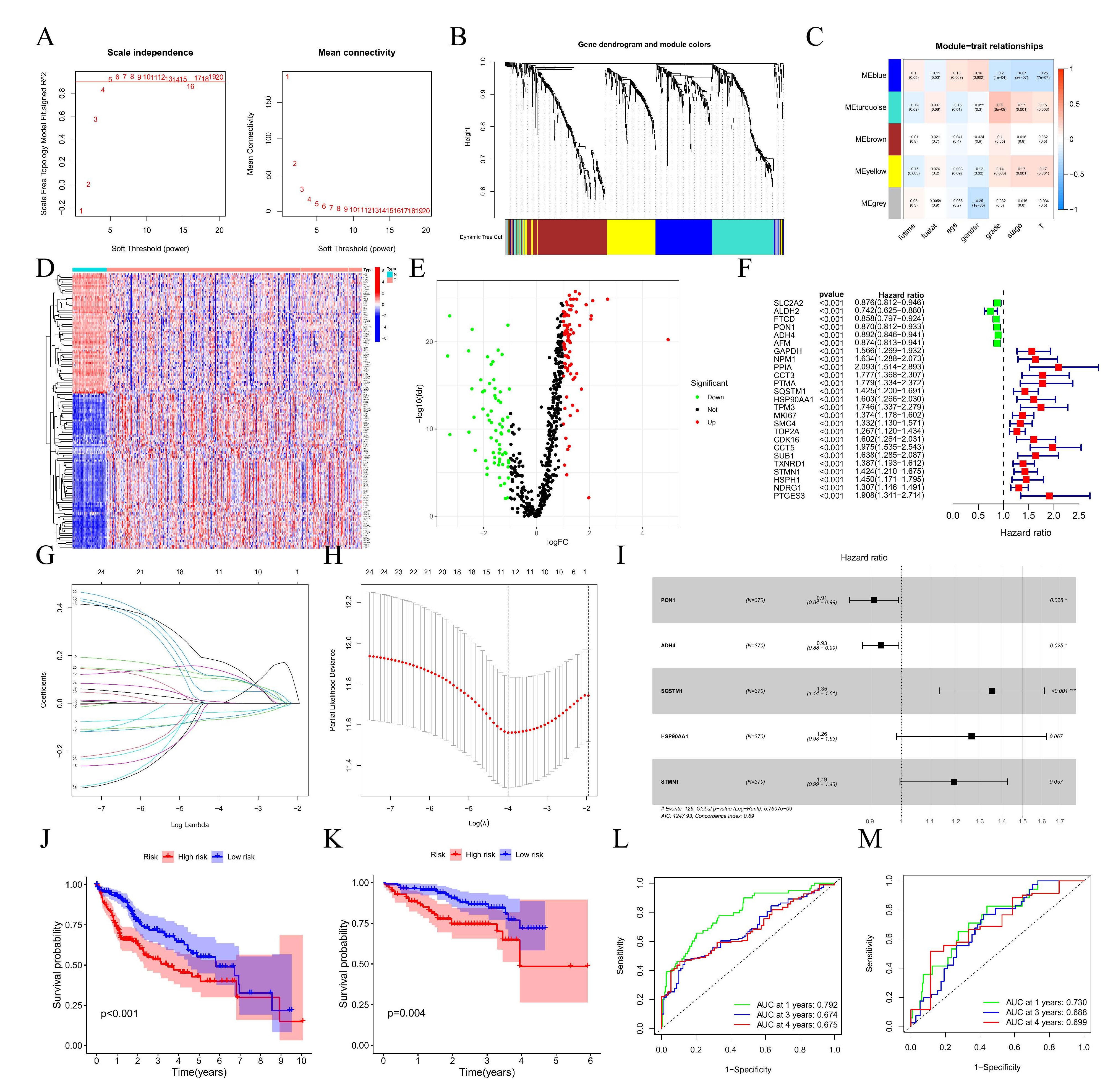
3.3. Four Molecular Subtypes in ICGC Database Based on HCC Differentiation-Related Gene (HDRG)
3.4. Analysis of Immune Cell Infiltration and ICGs Expression Levels of Four Molecular Subtypes
3.5. Construction, Evaluation, and Validation of a DRGPI-Based Prognostic Risk Model
3.6. Validation of the Expression Levels of Five Prognostic Genes
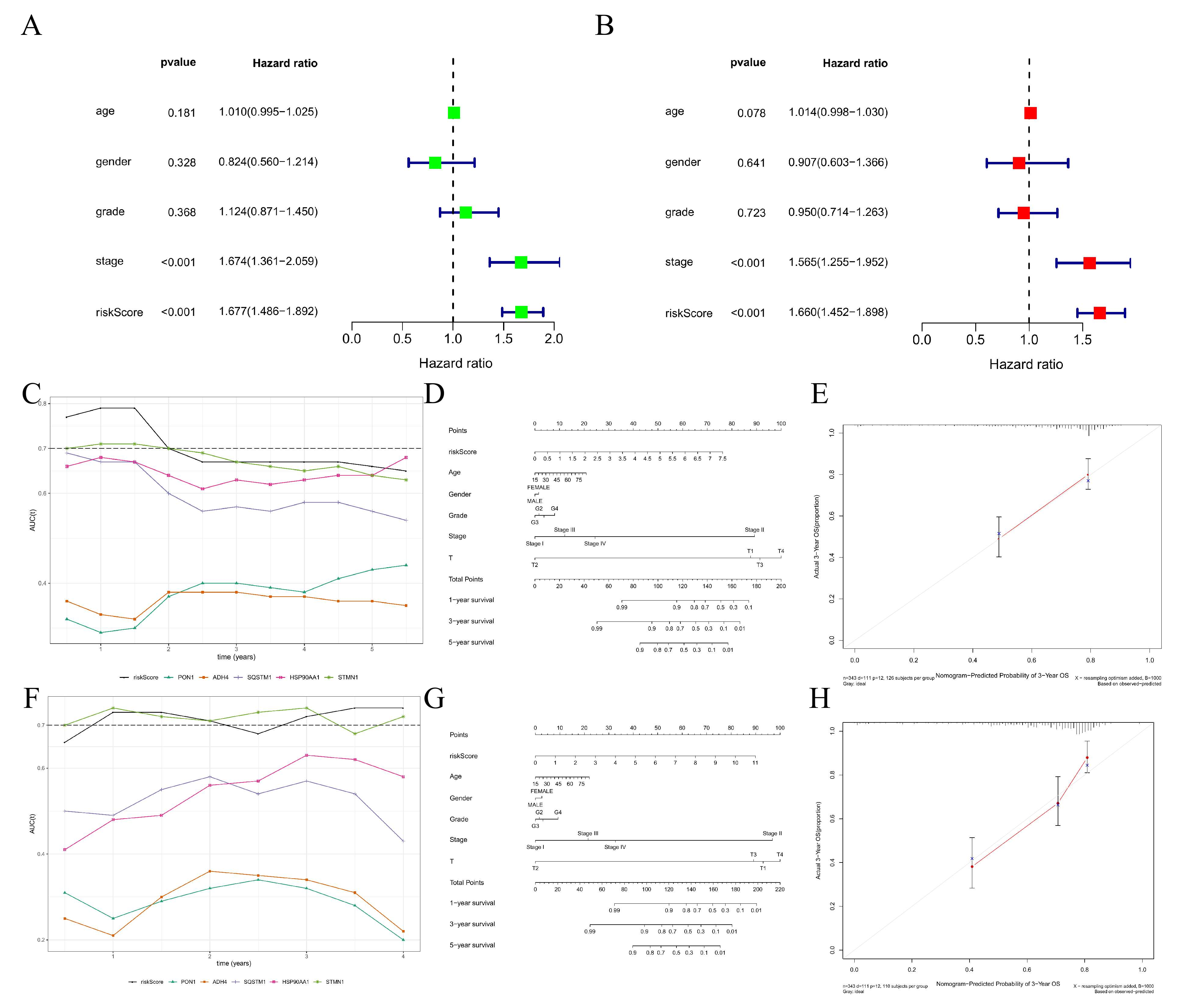
3.7. Construction and Efficiency Evaluation of a DRGPI-Based Nomogram
3.8. Molecular Characteristics of Different DRGPI Subgroups
3.9. Immune Characteristics of Different DRGPI Subgroups and Their Relationship with Other Immune Subtypes
3.10. The Benefit of ICI Therapy in Different DRGPI-Based Risk Subgroups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhou, D.; Luan, J.; Huang, C.; Li, J. Tumor-Associated Macrophages in Hepatocellular Carcinoma: Friend or Foe? Gut Liver 2021, 15, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Suh, J.H.; Choi, S.Y.; Kang, H.J.; Lee, H.H.; Ye, B.J.; Lee, G.R.; Jung, S.W.; Kim, C.J.; Lee-Kwon, W.; et al. Tonicity-responsive enhancer-binding protein promotes hepatocellular carcinogenesis, recurrence and metastasis. Gut 2019, 68, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ochi, H.; Hiraoka, A.; Hirooka, M.; Koizumi, Y.; Amano, M.; Azemoto, N.; Watanabe, T.; Yoshida, O.; Tokumoto, Y.; Mashiba, T.; et al. Direct-acting antivirals improve survival and recurrence rates after treatment of hepatocellular carcinoma within the Milan criteria. J. Gastroenterol. 2021, 56, 90–100. [Google Scholar] [CrossRef]
- Chiu, D.K.; Tse, A.P.; Xu, I.M.; Di Cui, J.; Lai, R.K.; Li, L.L.; Koh, H.Y.; Tsang, F.H.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Dauki, A.M.; Blachly, J.S.; Kautto, E.A.; Ezzat, S.; Abdel-Rahman, M.H.; Coss, C.C. Transcriptionally Active Androgen Receptor Splice Variants Promote Hepatocellular Carcinoma Progression. Cancer Res. 2020, 80, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Devalaraja, S.; To, T.K.J.; Folkert, I.W.; Natesan, R.; Alam, M.Z.; Li, M.; Tada, Y.; Budagyan, K.; Dang, M.T.; Zhai, L.; et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell 2020, 180, 1098–1114.e1016. [Google Scholar] [CrossRef]
- Machairas, N.; Tsilimigras, D.I.; Pawlik, T.M. Current Landscape of Immune Checkpoint Inhibitor Therapy for Hepatocellular Carcinoma. Cancers 2022, 14, 2018. [Google Scholar] [CrossRef]
- Lawal, G.; Xiao, Y.; Rahnemai-Azar, A.A.; Tsilimigras, D.I.; Kuang, M.; Bakopoulos, A.; Pawlik, T.M. The Immunology of Hepatocellular Carcinoma. Vaccines 2021, 9, 1184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, X.; Pan, C.; Zheng, X.; Hu, B.; Xie, R.; Hu, J.; Shang, X.; Yang, H. Single-cell RNA sequencing to track novel perspectives in HSC heterogeneity. Stem. Cell Res. Ther. 2022, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Krishna, C.; DiNatale, R.G.; Kuo, F.; Srivastava, R.M.; Vuong, L.; Chowell, D.; Gupta, S.; Vanderbilt, C.; Purohit, T.A.; Liu, M.; et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell 2021, 39, 662–677.e666. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Drobni, Z.D.; Zafar, A.; Quinaglia, T.; Hartmann, S.; Gilman, H.K.; Raghu, V.K.; Gongora, C.; Sise, M.E.; Alvi, R.M.; et al. Pericardial disease in patients treated with immune checkpoint inhibitors. J. Immunother. Cancer 2021, 9, e002771. [Google Scholar] [CrossRef]
- Xu, L.; Yu, W.; Xiao, H.; Lin, K. BIRC5 is a prognostic biomarker associated with tumor immune cell infiltration. Sci. Rep. 2021, 11, 390. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, C.; Liu, Z. Exploration the Significance of a Novel Immune-Related Gene Signature in Prognosis and Immune Microenvironment of Breast Cancer. Front. Oncol. 2020, 10, 1211. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.; Mukherjee, S.; Ebert, B.; Gillette, M.; Paulovich, A.; Pomeroy, S.; Golub, T.; Lander, E.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.; Kwon, A.Y.; Jeong, J.Y.; Kim, S.; Kang, H.; Park, J.; Kim, J.H.; Han, O.J.; Lim, S.M.; An, H.J. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci. Rep. 2020, 10, 643. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Yang, J.; Xu, D.; Gao, X.M.; Zhang, Z.; Hsu, J.L.; Li, C.W.; Lim, S.O.; Sheng, Y.Y.; Zhang, Y.; et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut 2019, 68, 1653–1666. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.M.; Long, G.V.; Blank, C.U. Learning from clinical trials of neoadjuvant checkpoint blockade. Nat. Med. 2020, 26, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Lisi, L.; Lacal, P.M.; Martire, M.; Navarra, P.; Graziani, G. Clinical experience with CTLA-4 blockade for cancer immunotherapy: From the monospecific monoclonal antibody ipilimumab to probodies and bispecific molecules targeting the tumor microenvironment. Pharmacol. Res. 2022, 175, 105997. [Google Scholar] [CrossRef] [PubMed]
- Oura, K.; Morishita, A.; Tani, J.; Masaki, T. Tumor Immune Microenvironment and Immunosuppressive Therapy in Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2021, 22, 5801. [Google Scholar] [CrossRef]
- Lu, C.; Rong, D.; Zhang, B.; Zheng, W.; Wang, X.; Chen, Z.; Tang, W. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol. Cancer 2019, 18, 130. [Google Scholar] [CrossRef] [Green Version]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Ren, L.; Li, J.; Wang, C.; Lou, Z.; Gao, S.; Zhao, L.; Wang, S.; Chaulagain, A.; Zhang, M.; Li, X.; et al. Single cell RNA sequencing for breast cancer: Present and future. Cell Death Discov. 2021, 7, 104. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Marsillach, J.; Camps, J.; Ferré, N.; Beltran, R.; Rull, A.; Mackness, B.; Mackness, M.; Joven, J. Paraoxonase-1 is related to inflammation, fibrosis and PPAR delta in experimental liver disease. BMC Gastroenterol. 2009, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Sorokin, A.V.; Kotani, K.; Elnabawi, Y.A.; Dey, A.K.; Sajja, A.P.; Yamada, S.; Ueda, M.; Harrington, C.L.; Baumer, Y.; Rodante, J.A.; et al. Association Between Oxidation-Modified Lipoproteins and Coronary Plaque in Psoriasis. Circ. Res. 2018, 123, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, F.; Oliveri, F.; Goud, H.K.; Mehkari, Z.; Mohammed, L.; Javed, M.; Althwanay, A.; Rutkofsky, I.H. Pleiotropic Effects of Statins in the Light of Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Cureus 2020, 12, e10446. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X.; Xia, Z.S.; Zhong, Y.Q. Gene therapy in pancreatic cancer. World J. Gastroenterol. 2014, 20, 13343–13368. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.R.; Zhang, M.Y.; Rao, H.L.; Pu, H.Y.; Zhang, H.Z.; Wang, H.Y. Identification of ADH4 as a novel and potential prognostic marker in hepatocellular carcinoma. Med. Oncol. 2012, 29, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.J.; Zhang, X.; Rodriguez-Velez, A.; Evans, T.D.; Razani, B. p62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid. Redox Signal. 2019, 31, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Xu, X.; Huang, D.; Cui, D.; Liu, L.; Liu, J.; He, Z.; Liu, J.; Zheng, S.; Luo, Y. Plasma Heat Shock Protein 90alpha as a Biomarker for the Diagnosis of Liver Cancer: An Official, Large-scale, and Multicenter Clinical Trial. EBioMedicine 2017, 24, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Shi, W.; Feng, L.; Dong, S.; Ning, Z.; Hua, Y.; Liu, L.; Chen, Z.; Meng, Z. FBXL6 governs c-MYC to promote hepatocellular carcinoma through ubiquitination and stabilization of HSP90AA1. Cell Commun. Signal. 2020, 18, 100. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; You, X.M.; Li, L.Q. Expression of HSP90AA1/HSPA8 in hepatocellular carcinoma patients with depression. OncoTargets Ther. 2018, 11, 3013–3023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhang, Z.; Huang, Y.; Wei, W.; Ning, S.; Li, J.; Liang, X.; Liu, K.; Zhang, L. Plasma HSP90AA1 Predicts the Risk of Breast Cancer Onset and Distant Metastasis. Front. Cell Dev. Biol. 2021, 9, 639596. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Li, H.; Wang, L.; Sun, W. An Immunohistochemical Study of Stathmin 1 Expression in Osteosarcoma Shows an Association with Metastases and Poor Patient Prognosis. Med. Sci. Monit. 2018, 24, 6070–6078. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Yokobori, T.; Altan, B.; Ide, M.; Mochiki, E.; Yanai, M.; Kimura, A.; Kogure, N.; Yanoma, T.; Suzuki, M.; et al. High STMN1 level is associated with chemo-resistance and poor prognosis in gastric cancer patients. Br. J. Cancer 2017, 116, 1177–1185. [Google Scholar] [CrossRef]
- Li, H.; Xu, Y.; Zhao, D. MicroRNA-193b regulates human ovarian cancer cell growth via targeting STMN1. Exp. Ther. Med. 2020, 20, 3310–3315. [Google Scholar] [CrossRef]
- Zhang, R.; Gao, X.; Zuo, J.; Hu, B.; Yang, J.; Zhao, J.; Chen, J. STMN1 upregulation mediates hepatocellular carcinoma and hepatic stellate cell crosstalk to aggravate cancer by triggering the MET pathway. Cancer Sci. 2020, 111, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Maimela, N.R.; Liu, S.; Zhang, Y. Fates of CD8+ T cells in Tumor Microenvironment. Comput. Struct. Biotechnol. J. 2019, 17, 1–13. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, A.; Feng, J.; Xu, Z.; Wang, P.; Ruan, P.; Luo, R.; Weng, Y.; Peng, M. Prognostic Impact of Memory CD8(+) T Cells on Immunotherapy in Human Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 698076. [Google Scholar] [CrossRef]
- Samaniego, R.; Gutiérrez-Seijo, A.; Sánchez-Mateos, P. Macrophages as prognostic biomarkers in human melanoma. Oncoscience 2018, 5, 179–180. [Google Scholar] [CrossRef]
- Diwanji, N.; Bergmann, A. Basement membrane damage by ROS- and JNK-mediated Mmp2 activation drives macrophage recruitment to overgrown tissue. Nat. Commun. 2020, 11, 3631. [Google Scholar] [CrossRef]
- Arvanitakis, K.; Koletsa, T.; Mitroulis, I.; Germanidis, G. Tumor-Associated Macrophages in Hepatocellular Carcinoma Pathogenesis, Prognosis and Therapy. Cancers 2022, 14, 226. [Google Scholar] [CrossRef]
- Li, X.; Wang, R.; Zhang, Y.; Han, S.; Gan, Y.; Liang, Q.; Ma, X.; Rong, P.; Wang, W.; Li, W. Molecular imaging of tumor-associated macrophages in cancer immunotherapy. Ther. Adv. Med. Oncol. 2022, 14, 17588359221076194. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Z.; Chang, Y.; Wang, K.; Kang, X.; Huang, R.; Zhang, Y.; Chen, J.; Zeng, F.; Wu, F.; et al. EFEMP2 indicates assembly of M0 macrophage and more malignant phenotypes of glioma. Aging 2020, 12, 8397–8412. [Google Scholar] [CrossRef] [PubMed]
- Farha, M.; Jairath, N.K.; Lawrence, T.S.; El Naqa, I. Characterization of the Tumor Immune Microenvironment Identifies M0 Macrophage-Enriched Cluster as a Poor Prognostic Factor in Hepatocellular Carcinoma. JCO Clin. Cancer Inform. 2020, 4, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Kuhn, C.; Ditsch, N.; Kolben, T.; Czogalla, B.; Beyer, S.; Trillsch, F.; Schmoeckel, E.; Mayr, D.; Mahner, S.; et al. Breast adipose tissue macrophages (BATMs) have a stronger correlation with breast cancer survival than breast tumor stroma macrophages (BTSMs). Breast Cancer Res. 2021, 23, 45. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- McGrail, D.J.; Garnett, J.; Yin, J.; Dai, H.; Shih, D.J.H.; Lam, T.N.A.; Li, Y.; Sun, C.; Li, Y.; Schmandt, R.; et al. Proteome Instability Is a Therapeutic Vulnerability in Mismatch Repair-Deficient Cancer. Cancer Cell 2020, 37, 371–386.e312. [Google Scholar] [CrossRef]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Coefficient | HR | 95%CL | p-Value | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| PON1 | −0.103 | 0.90 | 0.83 | 0.98 | 0.014 |
| ADH4 | −0.067 | 0.94 | 0.88 | 0.99 | 0.033 |
| SQSTM1 | 0.307 | 1.36 | 1.14 | 1.62 | 0.001 |
| HSP90AA1 | 0.252 | 1.29 | 1.00 | 1.65 | 0.048 |
| STMN1 | 0.180 | 1.20 | 1.00 | 1.43 | 0.049 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Liu, T.; Liu, Z.; Xiao, C.; Du, J.; Zuo, S.; Li, H.; Gu, H. A Differentiation-Related Gene Prognostic Index Contributes to Prognosis and Immunotherapy Evaluation in Patients with Hepatocellular Carcinoma. Cells 2022, 11, 2302. https://doi.org/10.3390/cells11152302
Xiao J, Liu T, Liu Z, Xiao C, Du J, Zuo S, Li H, Gu H. A Differentiation-Related Gene Prognostic Index Contributes to Prognosis and Immunotherapy Evaluation in Patients with Hepatocellular Carcinoma. Cells. 2022; 11(15):2302. https://doi.org/10.3390/cells11152302
Chicago/Turabian StyleXiao, Jingjing, Tao Liu, Zhenhua Liu, Chuan Xiao, Jun Du, Shi Zuo, Haiyang Li, and Huajian Gu. 2022. "A Differentiation-Related Gene Prognostic Index Contributes to Prognosis and Immunotherapy Evaluation in Patients with Hepatocellular Carcinoma" Cells 11, no. 15: 2302. https://doi.org/10.3390/cells11152302