
Prophylactic Treatment of Undernourished Mice with Cotrimoxazole Induces a Different Profile of Dysbiosis with Functional Metabolic Alterations

 , and
, and
Abstract
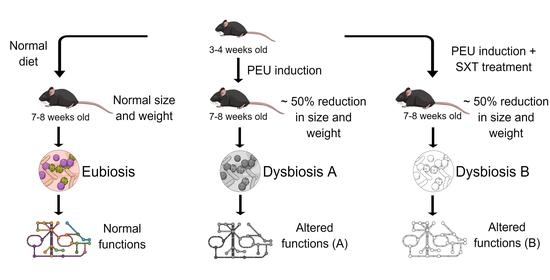
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal and Study Design
2.2. Fecal Sampling and DNA Extraction
2.3. 16S rRNA Gene Sequence and Analysis
2.4. Statistical Analysis of 16S rRNA Sequencing Data
3. Results
3.1. Changes in Body Weight and Daily Weight Gain in C57BL/6 Mice during Undernourishment Induction
3.2. Effects of Undernourishment on the Microbiota Composition and Function
3.3. Effects of Cotrimoxazole (SXT) on the Gut Microbiome
3.4. The Use of Cotrimoxazole during Undernourishment in Infant Mice Does Not Revert Undernutrition Effects on Microbiota but Results in a Distinct Profile of Dysbiosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayashi, C.; Krasevec, J.; Kumapley, R. Levels and Trends in Child Malnutrition: Key Findings of the 2021 Edition of the Joint Child Malnutrition Estimates; United Nations Children’s Fund, World Health Organization, World Bank Group, International Bank for Reconstruction and Development, Eds.; World Health Organization: Geneva, Switzerland, 2021; ISBN (UNICEF) 978-92-806-5219-2. [Google Scholar]
- Giallourou, N.; Fardus-Reid, F.; Panic, G.; Veselkov, K.; McCormick, B.J.J.; Olortegui, M.P.; Ahmed, T.; Mduma, E.; Yori, P.P.; Mahfuz, M.; et al. Metabolic maturation in the first 2 years of life in resource-constrained settings and its association with postnatal growths. Sci. Adv. 2020, 6, eaay5969. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.K.; Zambruni, M.; Melby, C.L.; Melby, P.C. Impact of Childhood Malnutrition on Host Defense and Infection. Clin. Microbiol. Rev. 2017, 30, 919–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.; Huq, S.; Yatsunenko, T.; Haque, R.; Mahfuz, M.; Alam, M.A.; Benezra, A.; DeStefano, J.; Meier, M.F.; Muegge, B.D.; et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature 2014, 510, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.J.; Pulendran, B. The potential of the microbiota to influence vaccine responses. J. Leukoc. Biol. 2017, 103, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell. Infect. Microbiol. 2021, 11, 625913. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wu, L.; Huntington, N.D.; Zhang, X. Crosstalk Between Gut Microbiota and Innate Immunity and Its Implication in Autoimmune Diseases. Front. Immunol. 2020, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Tooley, K.L. Effects of the human gut microbiota on cognitive performance, brain structure and function: A narrative review. Nutrients 2020, 12, 3009. [Google Scholar] [CrossRef]
- Jeong, S. Factors influencing development of the infant microbiota: From prenatal period to early infancy. Clin. Exp. Pediatr. 2021, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Planer, J.D.; Liu, J.; Rao, S.; Yatsunenko, T.; Trehan, I.; Manary, M.J.; Liu, T.-C.; Stappenbeck, T.S.; Maleta, K.M.; et al. Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci. Transl. Med. 2015, 7, 276ra24. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.D.J.; Berkley, J.A. Severe acute malnutrition and infection. Paediatr. Int. Child Health 2014, 34, S1–S29. [Google Scholar] [CrossRef]
- World Health Organization. Management of Severe Malnutrition: A Manual for Physicians and Other Senior Health Workers; World Health Organization: Geneva, Switzerland, 1999. [Google Scholar]
- Williams, P.C.M.; Berkley, J.A. Sever Acute Malnutrition Update: Current WHO Guidelines and the WHO Essential Medicine List for Children; World Health Organization: Geneva, Switzerland, 2016; pp. 1–40. [Google Scholar]
- A Berkley, J.; Ngari, M.; Thitiri, J.; Mwalekwa, L.; Timbwa, M.; Hamid, F.; Ali, R.; Shangala, J.; Mturi, N.; Jones, K.; et al. Daily co-trimoxazole prophylaxis to prevent mortality in children with complicated severe acute malnutrition: A multicentre, double-blind, randomised placebo-controlled trial. Lancet Glob. Health 2016, 4, e464–e473. [Google Scholar] [CrossRef] [Green Version]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, J.; Guarner, F.; Fernandez, L.B.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.D.; Thitiri, J.; Ngari, M.; Berkley, J.A. Childhood malnutrition: Toward an understanding of infections, inflammation, and antimicrobials. Food Nutr. Bull. 2014, 35, S64–S70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, A.; Woodward, B. Thymic Epithelial Cells of Severely Undernourished Mice: Accumulation of Cholesteryl Esters and Absence of Cytoplasmic Vacuoles. Exp. Biol. Med. 1985, 178, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. The R Project for Statistical Computing. R Foundation for Statistical Computing Web-Site. Available online: www.R-project.org (accessed on 10 December 2021).
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 10 December 2021).
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Lin, H.; Das Peddada, S. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef]
- Dinh, D.M.; Ramadass, B.; Kattula, D.; Sarkar, R.; Braunstein, P.; Tai, A.; Wanke, C.A.; Hassoun, S.; Kane, A.V.; Naumova, E.N.; et al. Longitudinal Analysis of the Intestinal Microbiota in Persistently Stunted Young Children in South India. PLoS ONE 2016, 11, e0155405. [Google Scholar] [CrossRef]
- Million, M.; Alou, M.T.; Khelaifia, S.; Bachar, D.; Lagier, J.-C.; Dione, N.; Brah, S.; Hugon, P.; Lombard, V.; Armougom, F.; et al. Increased Gut Redox and Depletion of Anaerobic and Methanogenic Prokaryotes in Severe Acute Malnutrition. Sci. Rep. 2016, 6, 26051. [Google Scholar] [CrossRef]
- Smith, M.I.; Yatsunenko, T.; Manary, M.J.; Trehan, I.; Mkakosya, R.; Cheng, J.; Kau, A.L.; Rich, S.S.; Concannon, P.; Mychaleckyj, J.C.; et al. Gut Microbiomes of Malawian Twin Pairs Discordant for Kwashiorkor. Science 2013, 339, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Monira, S.; Nakamura, S.; Gotoh, K.; Izutsu, K.; Watanabe, H.; Alam, N.H.; Endtz, H.P.; Cravioto, A.; Ali, S.I.; Nakaya, T.; et al. Gut Microbiota of Healthy and Malnourished Children in Bangladesh. Front. Microbiol. 2011, 2, 228. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, T.; Gupta, S.S.; Bhattacharya, T.; Yadav, D.; Barik, A.; Chowdhury, A.; Das, B.; Mande, S.S.; Nair, G.B. Gut Microbiomes of Indian Children of Varying Nutritional Status. PLoS ONE 2014, 9, e95547. [Google Scholar] [CrossRef]
- Chu, Y.; Sun, S.; Huang, Y.; Gao, Q.; Xie, X.; Wang, P.; Li, J.; Liang, L.; He, X.; Jiang, Y.; et al. Metagenomic analysis revealed the potential role of gut microbiome in gout. npj Biofilms Microbiomes 2021, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Z.; Hu, B.; Huang, W.; Yuan, C.; Zou, L. Response of gut microbiota to metabolite changes induced by endurance exercise. Front. Microbiol. 2018, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Douglas, G.M.; Hayes, M.G.; MacDonald, J.; Desai, D.K.; Allward, N.; Jones, C.M.A.; Wright, R.J.; Dhanani, A.S.; Comeau, A.M.; et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat. Commun. 2022, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- A Ciliberto, M.; Sandige, H.; Ndekha, M.J.; Ashorn, P.; Briend, A.; Ciliberto, H.M.; Manary, M.J. Comparison of home-based therapy with ready-to-use therapeutic food with standard therapy in the treatment of malnourished Malawian children: A controlled, clinical effectiveness trial. Am. J. Clin. Nutr. 2005, 81, 864–870. [Google Scholar] [CrossRef] [Green Version]
- Bahwere, P.; Levy, J.; Hennart, P.; Donnen, P.; Lomoyo, W.; Dramaix-Wilmet, M.; Hemelof, W.; Butzler, J.-P.; De Mol, P. Community-acquired bacteremia among hospitalized children in rural central Africa. Int. J. Infect. Dis. 2001, 5, 180–188. [Google Scholar] [CrossRef]
- Berkley, J.A.; Lowe, B.S.; Mwangi, I.; Williams, T.; Bauni, E.; Mwarumba, S.; Ngetsa, C.; Slack, M.P.; Njenga, S.; Hart, C.A.; et al. Bacteremia among Children Admitted to a Rural Hospital in Kenya. N. Engl. J. Med. 2005, 352, 39–47. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.W.; Moodley-Govender, E.; Berla, B.; Kelkar, T.; Wang, B.; Sun, X.; Daniels, B.; Coutsoudis, A.; Trehan, I.; Dantas, G. Cotrimoxazole Prophylaxis Increases Resistance Gene Prevalence and α-Diversity but Decreases β-Diversity in the Gut Microbiome of Human Immunodeficiency Virus–Exposed, Uninfected Infants. Clin. Infect. Dis. 2020, 71, 2858–2868. [Google Scholar] [CrossRef] [Green Version]
- Bourke, C.D.; Gough, E.K.; Pimundu, G.; Shonhai, A.; Berejena, C.; Terry, L.; Baumard, L.; Choudhry, N.; Karmali, Y.; Bwakura-Dangarembizi, M.; et al. Cotrimoxazole reduces systemic inflammation in HIV infection by altering the gut microbiome and immune activation. Sci. Transl. Med. 2019, 11, eaav0537. [Google Scholar] [CrossRef]
- Willmann, M.; Vehreschild, M.J.G.T.; Biehl, L.M.; Vogel, W.; Dörfel, D.; Hamprecht, A.; Seifert, H.; Autenrieth, I.B.; Peter, S. Distinct impact of antibiotics on the gut microbiome and resistome: A longitudinal multicenter cohort study. BMC Biol. 2019, 17, 76. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Duncan, S.H.; McCrae, S.I.; Millar, J.; Jackson, M.S.; Flint, H.J. Restricted Distribution of the Butyrate Kinase Pathway among Butyrate-Producing Bacteria from the Human Colon. J. Bacteriol. 2004, 186, 2099–2106. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-Y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrzosek, L.; Miquel, S.; Noordine, M.-L.; Bouet, S.; Chevalier-Curt, M.J.; Robert, V.; Philippe, C.; Bridonneau, C.; Cherbuy, C.; Robbe-Masselot, C.; et al. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 2013, 11, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.Y.; Zheng, Y.; Huang, Z.; Wang, X.M.; Yang, H. Lactococcus nasutitermitis sp. nov. isolated from a termite gut. Int. J. Syst. Evol. Microbiol. 2016, 66, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ji, H.; Wang, S.; Liu, H.; Zhang, W.; Zhang, D.; Wang, Y. Probiotic Lactobacillus plantarum promotes intestinal barrier function by strengthening the epithelium and modulating gut microbiota. Front. Microbiol. 2018, 9, 1953. [Google Scholar] [CrossRef] [Green Version]
- Schwiertz, A.; Hold, G.; Duncan, S.H.; Gruhl, B.; Collins, M.D.; Lawson, P.A.; Flint, H.J.; Blaut, M. Anaerostipes caccae gen. nov., sp. nov., a New Saccharolytic, Acetate-utilising, Butyrate-producing Bacterium from Human Faeces. Syst. Appl. Microbiol. 2002, 25, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Delcour, A.H. Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2009, 1794, 808–816. [Google Scholar] [CrossRef] [Green Version]
- Vatanen, T.; Kostic, A.D.; D’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.-M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Metges, C.C. Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 2000, 130, 1857S–1864S. [Google Scholar] [CrossRef]
- Tomé, D.; Bos, C. Lysine requirement through the human life cycle. J. Nutr. 2007, 137, 1642S–1645S. [Google Scholar] [CrossRef] [Green Version]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ji, B.; Babaei, P.; Das, P.; Lappa, D.; Ramakrishnan, G.; Fox, T.E.; Haque, R.; Petri, W.A.; Bäckhed, F.; et al. Gut microbiota dysbiosis is associated with malnutrition and reduced plasma amino acid levels: Lessons from genome-scale metabolic modeling. Metab. Eng. 2018, 49, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eslabão, L.B.; Gubert, G.F.; Beltrame, L.C.; Mello, I.M.A.; Bruna-Romero, O.; Zárate-Bladés, C.R. Prophylactic Treatment of Undernourished Mice with Cotrimoxazole Induces a Different Profile of Dysbiosis with Functional Metabolic Alterations. Cells 2022, 11, 2278. https://doi.org/10.3390/cells11152278
Eslabão LB, Gubert GF, Beltrame LC, Mello IMA, Bruna-Romero O, Zárate-Bladés CR. Prophylactic Treatment of Undernourished Mice with Cotrimoxazole Induces a Different Profile of Dysbiosis with Functional Metabolic Alterations. Cells. 2022; 11(15):2278. https://doi.org/10.3390/cells11152278
Chicago/Turabian StyleEslabão, Lívia Budziarek, Gabriela Farias Gubert, Lucas Cafferati Beltrame, Isis M. A. Mello, Oscar Bruna-Romero, and Carlos R. Zárate-Bladés. 2022. "Prophylactic Treatment of Undernourished Mice with Cotrimoxazole Induces a Different Profile of Dysbiosis with Functional Metabolic Alterations" Cells 11, no. 15: 2278. https://doi.org/10.3390/cells11152278