
Calreticulin and the Heart


Department of Biochemistry, University of Alberta, Edmonton, AB T6G 1H7, Canada
*
Authors to whom correspondence should be addressed.
Cells 2022, 11(11), 1722; https://doi.org/10.3390/cells11111722
Submission received: 4 April 2022
/
Revised: 13 May 2022
/
Accepted: 17 May 2022
/
Published: 24 May 2022
(This article belongs to the Special Issue Cardiac Growth Control and Heart Cell Death)
Abstract
:Calreticulin is an endoplasmic Ca2+ binding protein and molecular chaperone. As a cardiac embryonic gene, calreticulin is essential for heart development. The protein supports Ca2+-dependent signaling events that are critical to cardiomyocyte differentiation and cardiogenesis. The increased expression of calreticulin and endoplasmic reticulum/sarcoplasmic reticulum Ca2+ capacity produces cardiomyocytes with enhanced efficiency, and detrimental mechanical stretching of cardiac fibroblasts, leading to cardiac pathology. Deletion of the calreticulin gene in adult cardiomyocytes results in left ventricle dilation, an impaired electrocardiogram, and heart failure. These observations indicate that a well-adjusted endoplasmic reticulum and calreticulin-dependent Ca2+ pool in cardiomyocytes are critical for the maintenance of proper cardiac function.
1. Introduction
Cardiovascular disease is one of the major health burdens in developed countries. Key causes of heart failure are ischemic heart disease and myocardial infarction, which damage the heart muscle, thereby compromising heart function. Ca2+ is essential for normal heart function, and Ca2+ dysregulation is one of the hallmarks of a failing heart. Cycling of Ca2+ in cardiomyocytes drives muscle excitation–contraction (E-C) coupling. The sarcoplasmic reticulum (SR), a functionally specialized form of the endoplasmic reticulum (ER) and a component of the cellular reticular network, is the source of Ca2+ for muscle contraction and relaxation [1]. This membrane possesses a complex collection of Ca2+ regulatory proteins that control and regulate the E-C coupling of the cardiac muscle. Calsequestrin, the ryanodine receptor/Ca2+ channel (RyR), junctin, junctate, sarcalumenin, and histidine-rich protein are examples of proteins that are unique to the SR and play an important role in SR Ca2+ handling. Ca2+ release from the SR via the RyR initiates muscle contraction [2]. The muscle relaxes when Ca2+ is decreased in the cytoplasm by the action of SR-associated Ca2+-ATPase (SERCA), a plasma membrane Na+/Ca2+ exchanger, and plasma membrane Ca2+-ATPase [3]. As important components of the cellular reticular network, cardiomyocytes also contain functional ER, which supports important cellular functions, such as lipid biosynthesis, protein synthesis, folding, and post-translational modification. Whether these typical ER-associated functions are shared between highly organized and functionally specialized SR and the perinuclear network of ER-like membranes remains to be established [4]. The potential contribution of the ER and ER homeostasis to cardiac pathophysiology also remains to be explored. Here, we focus on calreticulin, a major Ca2+ binding protein of the ER, and its impact on cardiac physiology/pathology, and we provide evidence that ER functions are essential components of cardiomyocyte biology.
2. Calreticulin and Heart Development
Calreticulin is an ER-resident Ca2+-binding chaperone present in a number of diverse species [5]. The protein binds Ca2+ in the ER lumen with high capacity and participates in the folding of newly synthesized proteins and glycoproteins. Calreticulin, together with calnexin (an integral ER membrane chaperone similar to calreticulin) and PDIA3 (also known as ERp57), constitute a so-called “calreticulin/calnexin cycle”, which is responsible for the folding and quality control of newly synthesized glycoproteins [6,7,8,9,10]. Calreticulin is highly expressed in embryonic hearts, but despite this, the expression of calreticulin is sharply downregulated in adult cardiomyocytes, which rely on Ca2+ to carry out mechanical functions in the heart. Calreticulin protein contains functionally specialized domains including an N-terminal globular domain and an extended arm proline-rich domain (P-domain), which are responsible for the chaperone function of the protein. In addition, the C-terminal highly acid C-domain is responsible for Ca2+ binding and buffering [5].
Cardiac development is a well-controlled molecular and morphogenetic event, and even small perturbations in this process can have devastating consequences in the form of congenital heart disease [11]. In mice, calreticulin deficiency is embryonic lethal at embryonic day 14.5 due to impaired development of the ventricular wall and septum [12,13]. In vitro and in vivo biochemical and cell biological studies have indicated that Ca2+ handling by calreticulin is responsible for the embryonic lethality in calreticulin-deficient mice [12,14]. This finding is underscored by generating a rescue mouse model system with a constitutively active expression of calcineurin in a calreticulin-deficient mouse, which allows the development of viable embryos with live birth, indicating a critical role for calreticulin in supporting Ca2+/calcineurin-dependent transcriptional events during cardiac development [14]. Intriguingly, calreticulin-deficient mice that have been rescued with an expression of calcineurin have died postnatally, with marked changes in their energy metabolism being observed in the absence of calreticulin [14,15].
The ultrastructure of the myofibrils is disorganized in the developing heart in the absence of calreticulin, and the transcriptional function of MEF2C shows impaired nuclear localization, further supporting the involvement of a critical Ca2+ and calreticulin-dependent checkpoint in cardiac myofibrillogenesis [13]. A role for calreticulin in cardiogenesis is further supported by studies with calreticulin-deficient embryonic stem (ES) cells [13]. In the absence of calreticulin, there are impaired Ca2+-dependent transcriptional activities and impaired myofibrillogenesis due to decreased activity of the muscle-specific transcription factor MEF2C [13]. Inhibition of Wnt signaling is necessary to maintain ES cells in a pluripotent state [16]. In calreticulin-deficient ES cells, Wnt signaling is disrupted, indicating the importance of calreticulin and Ca2+ signaling during early cardiac development [17]. Calreticulin-deficient ES cells remain either pluripotent and/or in an undefined state so they are unable to properly differentiate into cardiomyocytes. Furthermore, calreticulin-deficient ES cells express a specific set of miRNAs [18].
miRNA plays a significant role in cardiovascular differentiation, function, and disease and has been the target of intense scrutiny [19,20,21]. Ingenuity Pathway Analysis of miRNA that was identified in calreticulin-deficient ES cells indicates that the top canonical pathways affected in the absence of calreticulin are Wnt signaling, TGFβ signaling, and cardiac hypertrophy markers (Table 1). One of the main families of miRNA affected in the absence of calreticulin is the miR-302 family [17,22]. The miR-302 family is a polycistronic group nestled on chromosome 3 that can induce and maintain ES cell pluripotency [22]. Recent studies demonstrate that in a human mast cell line (HMC-1 cells), miR-302e decreased in abundance after an increase in cytoplasmic Ca2+ concentration, leading to inflammation and upregulation of a RelA protein, which is part of the NF-κB family [23]. In calreticulin-deficient ES cells, the miR-302 family is increased, potentially playing an anti-inflammatory role by influencing the NF-κB inflammatory pathway. Is the Ca2+ binding function of calreticulin involved, or is its role in ER-dependent stress responses, including unfolded protein response (UPR), responsible? Likely, both are involved. Many miRNAs are targets of the nuclease activity of the ER stress sensor IRE1α, driving apoptotic events that are due to ER stress [24]. In fibroblasts that are deficient in the plasma membrane Ca2+ channel ORAI1, which is responsible for Ca2+ entry from the extracellular space due to store-operated Ca2+ entry, there is an increase in several miRNAs with target degradation that is dependent on intracellular Ca2+ levels [25].
Evidence of the importance of ER Ca2+ and the Ca2+ binding function of calreticulin in cardiac development comes from studies of GRP94, another ER Ca2+ binding chaperone and resident protein. GRP94 and calreticulin have many similar features. Firstly, GRP94 has a highly acidic C-terminal domain that binds approximately 20 moles of Ca2+ per mole of protein [26]. Second, GRP94 deficiency in mice is embryonic lethal due to impaired cardiac development [27]. Lastly, GRP94-deficient ES cells are unable to differentiate efficiently into cardiomyocytes [27]. This provides additional evidence that the Ca2+ binding function of calreticulin and proper ER Ca2+ homeostasis are essential for cardiac development.
Recently, somatic mutations of the calreticulin gene were discovered in patients with essential thrombocythemia and primary myelofibrosis [28]. The most common mutations were a 52-bp deletion (del52) and a 5-bp insertion (ins5), both of which led to a frameshift and a modified Ca2+ binding C-domain [28]. Calreticulin mutations result in the loss of the amino acid sequence (KDEL) that is responsible for ER retrieval and changes from negatively charged Ca2+ binding residues to a large cluster of positively charged amino acids in the C-domain [28]. Importantly, a homozygous mouse model for knock-in of the del52 mutant (no acidic residues in the Ca2+ binding C-domain) is embryonic lethal in a way similar to that seen for silencing of the calreticulin gene [12,29]. These findings fully support the conclusion that the loss of Ca2+ binding to calreticulin that affects cellular Ca2+ signaling is sufficient to induce embryonic lethality in mice [12,14]. This further strengthens our notion that ER-associated Ca2+-dependent events are critical during cardiogenesis and play different roles in fully differentiated cardiomyocytes (Figure 1).
3. Calreticulin in the Adult Heart
Calreticulin is highly expressed in the developing heart, but it is only a minor component in an adult heart [30]. Interestingly, an increased abundance of calreticulin in adult hearts is associated with failing and hypertrophied human hearts [31,32]. In mice, forced overexpression of calreticulin in cardiomyocytes increases cardiomyocyte ER/SR Ca2+ capacity and mechanical work potential but also activates the IRE1α branch of the UPR and eventually leads to cardiomyopathy [33,34]. Calreticulin overexpression also causes a reduction in the abundance of gap junction protein in the heart, indicating a defect in cell–cell communication [33]. There is an impaired expression of Ca2+ signaling proteins such as triadin and junctin, as well as the gap junction proteins connexin 43 and 45. Ca2+-handling proteins such as calsequestrin, SERCA, and the RyR, are downregulated in calreticulin-overexpressing hearts, while calmodulin, calcineurin, and MEF2C are increased [33]. Additionally, impaired gap junctions, aberrant Ca2+ signaling, and arrhythmia have been observed in a non-inducible calreticulin overexpression mouse model system [35]. While increasing calreticulin abundance in adult cardiomyocytes improves ER/SR Ca2+ capacity and delays store-operated Ca2+ entry, it also stimulates the UPR, which promotes an increase in cardiac TGFβ abundance that, in turn, induces increased collagen deposition and severe cardiac fibrosis [33,34]. This is due to the mechanical stretching of cardiac fibroblasts because of enhanced cardiomyocyte efficiency and the activation of the IRE1α branch of the UPR pathway [36]. Interestingly, inhibition of IRE1α activation with tauroursodeoxycholic acid (TUDCA), a proteostasis promoter [37], prevents the development of cardiac fibrosis in hearts that are overexpressing calreticulin [34,38].
Considering that calreticulin was initially identified as a component of the fetal gene program in the heart, it is not surprising that up-regulation of the calreticulin gene induces cardiac remodeling. Activation of the fetal gene program is an adaptive state that supports intrinsic cell survival pathways in the heart [39,40]. The fetal gene program is normally active during embryonic development and is necessary for the embryo to survive under hypoxic conditions. As the heart grows in utero, it is subjected to increases in hemodynamic load, a low oxygen environment, and a changing metabolic landscape. The decrease in oxygen tension specifically turns on a transcription factor, HIF1α, which is stabilized under low oxygen conditions. HIF1α targets a specific subgroup of promoters for a variety of proteins, including proteins involved in proliferation, metabolism, and angiogenesis [41]. One of these downstream targets is calreticulin [42]. Increased abundance of calreticulin likely triggers changes in the metabolic capacity that lead to a disruption in reactive oxygen species and fluctuations in Ca2+ uptake and efflux from the mitochondria, as well as disruptions in cellular Ca2+ homeostasis in general. Increased abundance of calreticulin also provides an enhanced Ca2+ binding/buffering capacity in the lumen of the ER, thereby supporting homeostatic recovery. Alternatively, hypoxia-induced changes in redox potential inside the lumen of the ER will affect protein translation, folding, assembly, and posttranslational modifications, triggering activation of the UPR, an ER stress-coping response, and increased expression of the calreticulin chaperone to support protein quality control events. Calreticulin expression is turned on during ER stress by Ca2+ signaling pathways, such as activation of the G-coupled receptors or disruption of Ca2+ stores. This Ca2+ store depletion-dependent induction of expression is reliant on new protein synthesis, implying transcriptional activation is necessary. This is likely via an ER stress element, CCAAT-N9-CCACG, that is recognized by the ER stress transcription factor ATF6 [43,44]. During ER stress, ATF6 also turns on a regulator of calcineurin called RCAN1, thereby suppressing calcineurin-dependent pathways [45]. It appears that either hypoxia or ER stress up-regulates the expression of calreticulin and fine-tunes downstream Ca2+-dependent transcriptional responses.
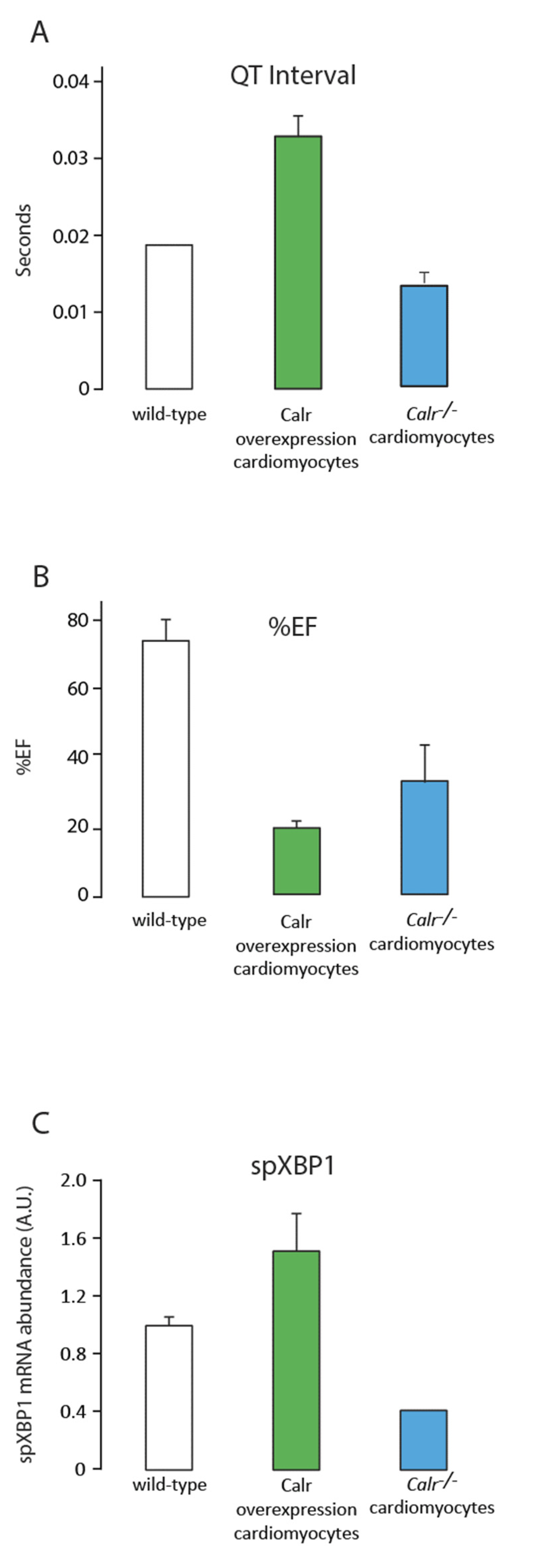
The Cre:LoxP tamoxifen-inducible system combined with the myosin heavy-chain-promoter-driven expression of Cre recombinase [33,46,47] has been used to silence the calreticulin gene in adult cardiomyocytes. ECHO analysis of adult hearts from mice with calreticulin gene knockout showed severe left ventricle dilation (Figure 1 and Figure 2; Table 2). Interestingly, both the calreticulin-deficient mouse model and the calreticulin-overexpressing mouse model exhibited significantly reduced Ejection Fraction and Fractional Shortening (Figure 2 and Figure 3; Table 3). An electrocardiography recording (ECG analysis) revealed a reduction in the QT interval in adult hearts with a silenced calreticulin gene (Figure 1, Figure 2 and Figure 3; Table 3), while the QT interval was increased in the calreticulin-overexpressing hearts [33,48,49]. Short QT syndrome is associated with sudden death and atrial fibrillation [50]. This suggests that the calreticulin Ca2+ binding capacity is affecting the depolarization and repolarization of ventricle cardiomyocytes. The ECG analysis supported the idea that up-regulation of calreticulin (increased ER Ca2+ capacity) or deletion of calreticulin (reduced ER Ca2+ capacity) in the adult heart impairs systolic and diastolic functions. Calreticulin deficiency in adult cardiomyocytes has also resulted in a 50% reduction in spXBP1 mRNA compared to a 30% increase in spXBP1 when calreticulin is overexpressed (Figure 3, [34]), indicating a connection between the expression of calreticulin and activation of IRE1α, an ER stress sensor.
4. Conclusions and Future Challenges
- ER Ca2+ capacity/homeostasis is essential for cardiac development and leads to heart disease when dysregulated.
- Calreticulin supports Ca2+-dependent signaling events that are critical to cardiomyocyte differentiation and cardiogenesis.
- Calreticulin is a major Ca2+ binding protein in the ER/SR of the developing heart and is downregulated after birth when calsequestrin, a muscle-specific Ca2+ binding/storage protein, is upregulated to supply Ca2+ that supports E-C coupling.
- The increased expression of calreticulin and an increased ER/SR Ca2+ capacity produce cardiomyocytes with enhanced efficiency, triggering detrimental mechanical stretching of cardiac fibroblasts and leading to cardiac pathology.
- The calreticulin-dependent Ca2+ pool in adult cardiomyocytes must be controlled as any increase or decrease in calreticulin results in heart pathology.
- Resolving the specific functions of ER versus SR in muscle cells remains a challenge.
- The role of calreticulin mutants needs to be explored for a better understanding of their role in cardiac pathophysiology.
- Do other ER-associated chaperones play a role in cardiomyocyte Ca2+ homeostasis?
- Further understanding of the role of ER/SR lumenal Ca2+ homeostasis will allow for the development of more targeted approaches to combat heart disease.
Author Contributions
J.G. and M.M. analyzed data and wrote the manuscript; W.-A.W. and A.R. carried out mouse experimentation, designed experiments, and analyzed data. All authors have read and agreed to the published version of the manuscript.
Funding
Research in our laboratory is supported by a generous donation from the Kenneth and Sheelagh McCourt family, the University Hospital Foundation; the Canadian Institutes of Health Research grants PS-153325; Natural Sciences and Engineering Research Council of Canada grant RGPIN-2019-04908; and SynAD. W.-A.W. was supported by a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship and an Alberta Cancer Foundation Scholarship.
Institutional Review Board Statement
All methods were carried out in accordance with relevant guidelines and regulations and approved by Biosafety Officers in the Department of Environment, Health and Safety at the University of Alberta. All animal experiments were carried out according to the University of Alberta Animal Policy and Welfare Committee and the Canadian Council on Animal Care Guidelines. The approval for the use of mice in research was granted by the Animal Care and Use Committee for Health Sciences, a University of Alberta ethics review committee (Permit AUP297).
Informed Consent Statement
Not Applicable.
Data Availability Statement
Data Available on Request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fabiato, A.; Fabiato, F. Calcium and cardiac excitation-contraction coupling. Annu. Rev. Physiol. 1979, 41, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Benitah, J.P.; Perrier, R.; Mercadier, J.J.; Pereira, L.; Gomez, A.M. RyR2 and Calcium Release in Heart Failure. Front. Physiol. 2021, 12, 734210. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Despa, S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J. Pharmacol. Sci. 2006, 100, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalak, M.; Opas, M. Endoplasmic and sarcoplasmic reticulum in the heart. Trends Cell. Biol. 2009, 19, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef]
- High, S.; Lecomte, F.J.; Russell, S.J.; Abell, B.M.; Oliver, J.D. Glycoprotein folding in the endoplasmic reticulum: A tale of three chaperones? FEBS Lett. 2000, 476, 38–41. [Google Scholar] [CrossRef]
- Zapun, A.; Jakob, C.A.; Thomas, D.Y.; Bergeron, J.J. Protein folding in a specialized compartment: The endoplasmic reticulum. Struct. Fold. Des. 1999, 7, R173–R182. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, J.J.M.; Brenner, M.B.; Thomas, D.Y.; Williams, D.B. Calnexin: A membrane-bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 1994, 19, 124–128. [Google Scholar] [CrossRef]
- Saito, Y.; Ihara, Y.; Leach, M.R.; Cohen-Doyle, M.F.; Williams, D.B. Calreticulin functions in vitro as a molecular chaperone for both glycosylated and non-glycosylated proteins. EMBO J. 1999, 18, 6718–6729. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Frickel, E.M. Calnexin, calreticulin, and ERp57: Teammates in glycoprotein folding. Cell Biochem. Biophys. 2003, 39, 223–247. [Google Scholar] [CrossRef]
- Morton, S.U.; Quiat, D.; Seidman, J.G.; Seidman, C.E. Genomic frontiers in congenital heart disease. Nat. Rev. Cardiol. 2022, 19, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Mesaeli, N.; Nakamura, K.; Zvaritch, E.; Dickie, P.; Dziak, E.; Krause, K.H.; Opas, M.; MacLennan, D.H.; Michalak, M. Calreticulin is essential for cardiac development. J. Cell Biol. 1999, 144, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Puceat, M.; Perez-Terzic, C.; Mery, A.; Nakamura, K.; Michalak, M.; Krause, K.-H.; Jaconi, M.E. Calreticulin reveals a critical Ca2+ checkpoint in cardiac myofibrillogenesis. J. Cell Biol. 2002, 158, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Nakamura, K.; Lynch, J.; Opas, M.; Olson, E.N.; Agellona, L.B.; Michalak, M. Cardiac specific expression of calcineurin reverses embryonic lethality in calreticulin-deficient mouse. J. Biol. Chem. 2002, 277, 50776–50779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, J.M.; Chilibeck, K.; Qui, Y.; Michalak, M. Assembling pieces of the cardiac puzzle; calreticulin and calcium-dependent pathways in cardiac development, health, and disease. Trends Cardiovasc. Med. 2006, 16, 65–69. [Google Scholar] [CrossRef]
- Sokol, S.Y. Maintaining embryonic stem cell pluripotency with Wnt signaling. Development 2011, 138, 4341–4350. [Google Scholar] [CrossRef] [Green Version]
- Groenendyk, J.; Michalak, M. Disrupted WNT signaling in mouse embryonic stem cells in the absence of calreticulin. Stem Cell Rev. 2014, 10, 191–206. [Google Scholar] [CrossRef]
- Groenendyk, J.; Michalak, M. A genome-wide siRNA screen identifies novel phospho-enzymes affecting Wnt/beta-catenin signaling in mouse embryonic stem cells. Stem Cell Rev. 2011, 7, 910–926. [Google Scholar] [CrossRef]
- Groenendyk, J.; Fan, X.; Peng, Z.; Kurgan, L.; Michalak, M. Endoplasmic reticulum and the microRNA environment in the cardiovascular system. Can. J. Physiol. Pharmacol. 2019, 97, 515–527. [Google Scholar] [CrossRef]
- Olson, E.N. MicroRNAs as therapeutic targets and biomarkers of cardiovascular disease. Sci. Transl. Med. 2014, 6, 239ps233. [Google Scholar] [CrossRef] [Green Version]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimi, K.; Fuchtbauer, A.C.; Fathi, F.; Mowla, S.J.; Fuchtbauer, E.M. Expression of the miR-302/367 microRNA cluster is regulated by a conserved long non-coding host-gene. Sci. Rep. 2021, 11, 11115. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Jiang, L.; Hu, Q.; Li, Y. MiR-302e attenuates allergic inflammation in vitro model by targeting RelA. Biosci. Rep. 2018, 38, BSR20180025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassler, J.; Cao, S.S.; Kaufman, R.J. IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev. Cell 2012, 23, 921–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenendyk, J.; Fan, X.; Peng, Z.; Ilnytskyy, Y.; Kurgan, L.; Michalak, M. Genome-wide analysis of thapsigargin-induced microRNAs and their targets in NIH3T3 cells. Genom. Data 2014, 2, 325–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macer, D.R.; Koch, G.L. Identification of a set of calcium-binding proteins in reticuloplasm, the luminal content of the endoplasmic reticulum. J. Cell Sci. 1988, 91, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Wanderling, S.; Simen, B.B.; Ostrovsky, O.; Ahmed, N.T.; Vogen, S.M.; Gidalevitz, T.; Argon, Y. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 2007, 18, 3764–3775. [Google Scholar] [CrossRef]
- Prins, D.; Gonzalez Arias, C.; Klampfl, T.; Grinfeld, J.; Green, A.R. Mutant Calreticulin in the Myeloproliferative Neoplasms. Hemasphere 2020, 4, e333. [Google Scholar] [CrossRef]
- Balligand, T.; Achouri, Y.; Pecquet, C.; Gaudray, G.; Colau, D.; Hug, E.; Rahmani, Y.; Stroobant, V.; Plo, I.; Vainchenker, W.; et al. Knock-in of murine Calr del52 induces essential thrombocythemia with slow-rising dominance in mice and reveals key role of Calr exon 9 in cardiac development. Leukemia 2020, 34, 510–521. [Google Scholar] [CrossRef]
- Martinho-Dias, D.; Leite-Moreira, A.; Castro-Chaves, P. Calreticulin in the Heart: From Embryological Development to Cardiac Pathology. Curr. Mol. Med. 2016, 16, 12–22. [Google Scholar] [CrossRef]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Dai, W.; Zhang, L.; Juhaeri, J.; Wang, Y.; Caubel, P. Risk of Cardiovascular Events, Stroke, Congestive Heart Failure, Interstitial Lung Disease, and Acute Liver Injury: Dronedarone versus Amiodarone and Other Antiarrhythmics. J. Atr. Fibrillation 2013, 6, 890. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Oka, T.; Hunter, B.; Robinson, A.; Papp, S.; Nakamura, K.; Srisakuldee, W.; Nickel, B.E.; Light, P.E.; Dyck, J.R.; et al. Calreticulin induces dilated cardiomyopathy. PLoS ONE 2013, 8, e56387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenendyk, J.; Lee, D.; Jung, J.; Dyck, J.R.; Lopaschuk, G.D.; Agellon, L.B.; Michalak, M. Inhibition of the Unfolded Protein Response Mechanism Prevents Cardiac Fibrosis. PLoS ONE 2016, 11, e0159682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Robertson, M.; Liu, G.; Dickie, P.; Nakamura, K.; Guo, J.Q.; Duff, H.J.; Opas, M.; Kavanagh, K.; Michalak, M. Complete heart block and sudden death in mice overexpressing calreticulin. J. Clin. Invest. 2001, 107, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Groenendyk, J.; Wang, Q.; Wagg, C.; Lee, D.; Robinson, A.; Barr, A.; Light, P.E.; Lopaschuk, G.D.; Agellon, L.B.; Michalak, M. Selective enhancement of cardiomyocyte efficiency results in a pernicious heart condition. PLoS ONE 2020, 15, e0236457. [Google Scholar] [CrossRef]
- Vega, H.; Agellon, L.B.; Michalak, M. The rise of proteostasis promoters. IUBMB Life 2016, 68, 943–954. [Google Scholar] [CrossRef]
- Rani, S.; Sreenivasaiah, P.K.; Kim, J.O.; Lee, M.Y.; Kang, W.S.; Kim, Y.S.; Ahn, Y.; Park, W.J.; Cho, C.; Kim, D.H. Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress. PLoS ONE 2017, 12, e0176071. [Google Scholar] [CrossRef] [Green Version]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the fetal gene program: A suggested metabolic link to gene expression in the heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Guimaraes-Camboa, N.; Stowe, J.; Aneas, I.; Sakabe, N.; Cattaneo, P.; Henderson, L.; Kilberg, M.S.; Johnson, R.S.; Chen, J.; McCulloch, A.D.; et al. HIF1alpha Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Dev. Cell 2015, 33, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Xie, P.; Hao, N.; Zhang, M.; Liu, Y.; Liu, P.; Semenza, G.L.; He, J.; Zhang, H. HIF-1-regulated expression of calreticulin promotes breast tumorigenesis and progression through Wnt/beta-catenin pathway activation. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waser, M.; Mesaeli, N.; Spencer, C.; Michalak, M. Regulation of calreticulin gene expression by calcium. J. Cell Biol. 1997, 138, 547–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belmont, P.J.; Tadimalla, A.; Chen, W.J.; Martindale, J.J.; Thuerauf, D.J.; Marcinko, M.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (RCAN1), a novel ATF6-inducible gene. J. Biol. Chem. 2008, 283, 14012–14021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuhiro, K.; Satouh, Y.; Nozawa, K.; Isotani, A.; Fujihara, Y.; Hirashima, Y.; Matsumura, H.; Takumi, K.; Miyano, T.; Okabe, M.; et al. Calreticulin is required for development of the cumulus oocyte complex and female fertility. Sci. Rep. 2015, 5, 14254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heine, H.L.; Leong, H.S.; Rossi, F.M.; McManus, B.M.; Podor, T.J. Strategies of conditional gene expression in myocardium: An overview. Methods Mol. Med. 2005, 112, 109–154. [Google Scholar] [CrossRef]
- Lankipalli, R.S.; Zhu, T.; Guo, D.; Yan, G.X. Mechanisms underlying arrhythmogenesis in long QT syndrome. J. Electrocardiol. 2005, 38, 69–73. [Google Scholar] [CrossRef]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Schimpf, R.; Borggrefe, M.; Wolpert, C. Clinical and molecular genetics of the short QT syndrome. Curr. Opin. Cardiol. 2008, 23, 192–198. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of a relationship between the expression of calreticulin and cardiac development. Calreticulin is abundant in the developing heart, and the expression of the calreticulin gene declines during cardiogenesis. In the adult heart, calreticulin is only a minor Ca2+ binding protein and calsequestrin is a major SR Ca2+ binding and storage protein. Calreticulin deficiency is embryonic lethal in mice. In the adult heart, either an increased abundance of calreticulin or calreticulin deficiency leads to cardiac pathology and heart failure.
Figure 1.
Schematic representation of a relationship between the expression of calreticulin and cardiac development. Calreticulin is abundant in the developing heart, and the expression of the calreticulin gene declines during cardiogenesis. In the adult heart, calreticulin is only a minor Ca2+ binding protein and calsequestrin is a major SR Ca2+ binding and storage protein. Calreticulin deficiency is embryonic lethal in mice. In the adult heart, either an increased abundance of calreticulin or calreticulin deficiency leads to cardiac pathology and heart failure.
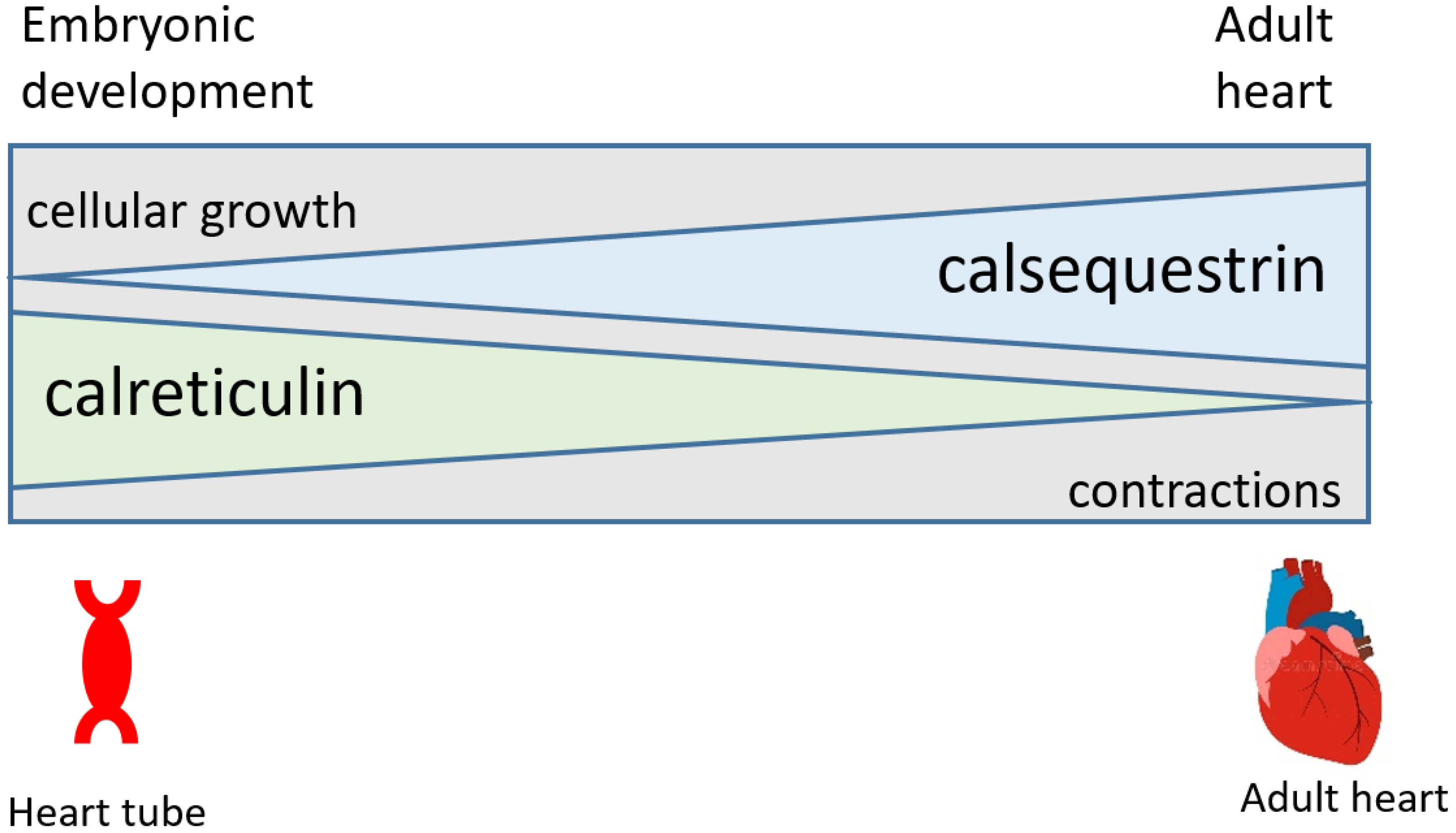
Figure 2.
ECHO and ECG analyses of hearts with a silenced calreticulin gene in adult cardiomyocytes. Mice with a calreticulin gene containing two loxP sites flanking exons 4–7 [46] were cross-bred with αMHC (myosin heavy chain)-Cre mice (C57BL/6). To delete exons 4–7 and silence the calreticulin gene in cardiomyocytes, mice were fed tamoxifen [33]. (A). Representative M-mode echocardiography (ECHO) images of wild-type and calreticulin knockout (Calr−/−) hearts from mice fed tamoxifen for 2 weeks (n = 3). (B). Electrocardiogram (ECG) traces of electrical activity in wild-type and calreticulin knockout (Calr−/−) hearts after 2 weeks of tamoxifen treatment. Representative electrocardiography recording images of hearts from wild-type and Calr−/− mice fed tamoxifen for 2 weeks (n = 3).
Figure 2.
ECHO and ECG analyses of hearts with a silenced calreticulin gene in adult cardiomyocytes. Mice with a calreticulin gene containing two loxP sites flanking exons 4–7 [46] were cross-bred with αMHC (myosin heavy chain)-Cre mice (C57BL/6). To delete exons 4–7 and silence the calreticulin gene in cardiomyocytes, mice were fed tamoxifen [33]. (A). Representative M-mode echocardiography (ECHO) images of wild-type and calreticulin knockout (Calr−/−) hearts from mice fed tamoxifen for 2 weeks (n = 3). (B). Electrocardiogram (ECG) traces of electrical activity in wild-type and calreticulin knockout (Calr−/−) hearts after 2 weeks of tamoxifen treatment. Representative electrocardiography recording images of hearts from wild-type and Calr−/− mice fed tamoxifen for 2 weeks (n = 3).
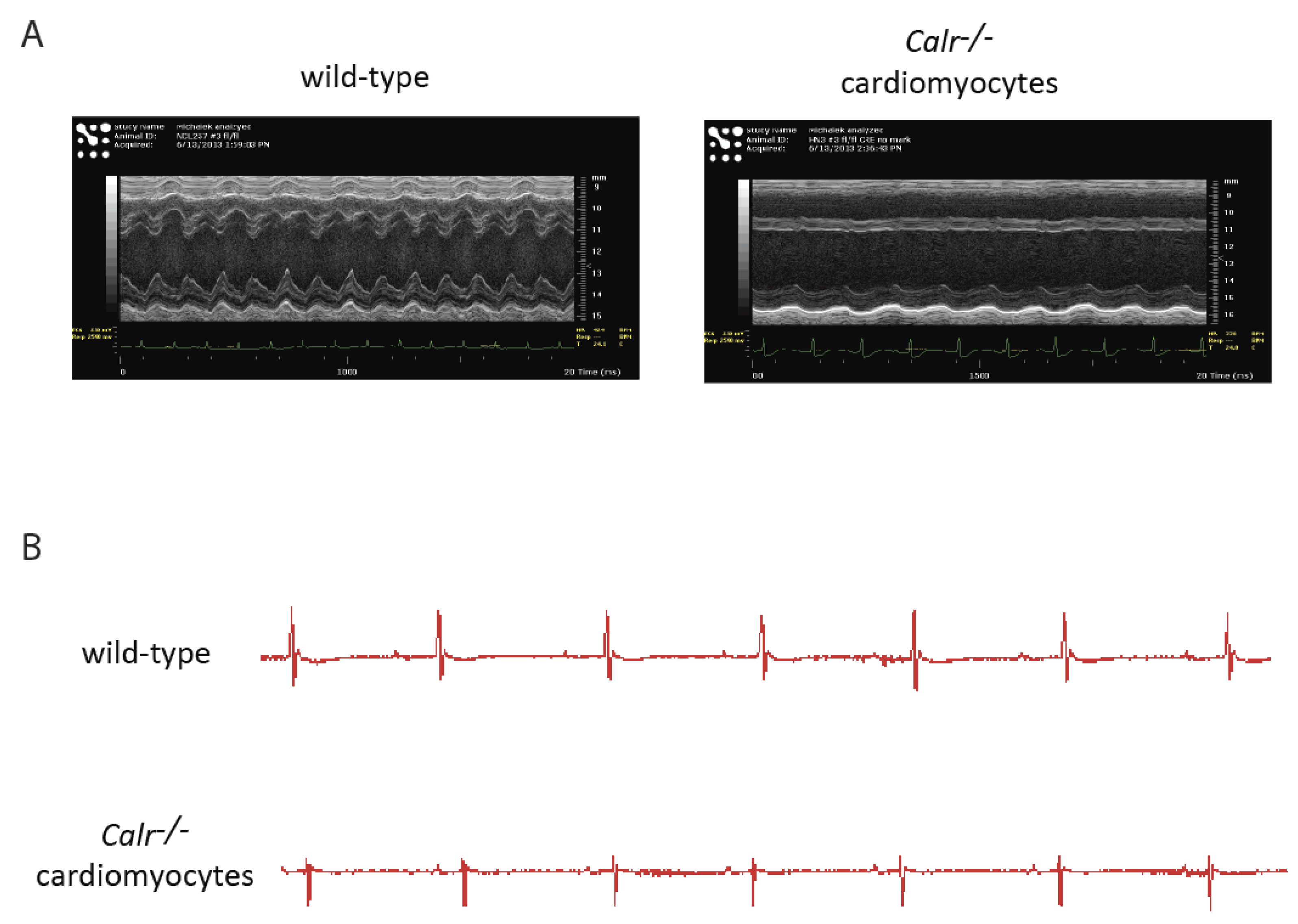
Figure 3.
Comparison of wild-type, calreticulin-overexpressing, and Calr−/− mouse model systems. Data from three mouse models after 2 weeks of tamoxifen treatment to induce the conditional knockout of calreticulin in cardiomyocytes (Calr−/−) or 3 weeks of tamoxifen treatment for conditional overexpression of calreticulin in cardiomyocytes. (A). QT interval from the ECG data (Figure 2B); (B). percent Ejection Fraction (%EF) from the echocardiogram analysis (Figure 2A); (C). abundance of spliced XBP1 (spXBP1) mRNA, a measure of IRE1α activation and an ER stress sensor (n = 3).
Figure 3.
Comparison of wild-type, calreticulin-overexpressing, and Calr−/− mouse model systems. Data from three mouse models after 2 weeks of tamoxifen treatment to induce the conditional knockout of calreticulin in cardiomyocytes (Calr−/−) or 3 weeks of tamoxifen treatment for conditional overexpression of calreticulin in cardiomyocytes. (A). QT interval from the ECG data (Figure 2B); (B). percent Ejection Fraction (%EF) from the echocardiogram analysis (Figure 2A); (C). abundance of spliced XBP1 (spXBP1) mRNA, a measure of IRE1α activation and an ER stress sensor (n = 3).

{kind=link}
{kind=link}
{kind=link}
Table 1.
Ingenuity Pathway Analysis.
| Molecular and Cellular Functions | Number of Molecules |
| Cellular Growth and Proliferation | 883 |
| Cellular Development | 763 |
| Cellular Movement | 584 |
| Top Canonical Pathways | Ratio |
| Wnt Signaling | 40/63 |
| TGF-β Signaling | 52/86 |
| Cardiac Hypertrophy Signaling | 106/259 |
Ingenuity Pathway Analysis of 31 differentially expressed miRNAs in calreticulin-deficient ES cells, targeting 6942 genes. miRNA expression was analyzed in calreticulin-deficient ES cells and Ingenuity Pathway Analysis was carried out on the miRNAs that were differentially expressed.
Table 2.
Echocardiogram Analysis.
| Wild-Type | Calr−/− | Calr OE | |
|---|---|---|---|
| Body weight (g) | 20.725 ± 0.245 | 16.285 ± 0.595 | 20.71 ± 0.581 |
| % EF | 75.485 ± 7.765 | 22.775 ± 11.875 | 15.40556 ± 4.430 |
| % FS | 44.035 ± 7.125 | 10.5 ± 5.710 | 10.62471 ± 2.200 |
| LV Mass (g) | 73.875 ± 0.615 | 66.22 ± 8.650 | 85.10569 ± 4.049 |
Calreticulin-deficient hearts (Calr−/−); hearts with an increased abundance of calreticulin (Calr OE); ejection fraction (EF); fractional shortening (FS); left ventricle (LV).
Table 3.
Electrocardiogram Analysis.
| Wild-Type | Calr−/− | Calr OE | ||
|---|---|---|---|---|
| RR Interval | (s) | 0.152 ± 0.013 | 0.159 ± 0.034 | 0.132 ± 0.011 |
| Heart Rate | (BPM) | 403.173 ± 32.553 | 388.470 ± 70.599 | 486.023 ± 50.192 |
| PR Interval | (s) | 0.036 ± 0.004 | 0.038 ± 0.003 | 0.031 ± 0.005 |
| P Duration | (s) | 0.017 ± 0.004 | 0.019 ± 0.001 | 0.013 ± 0.003 |
| QRS Interval | (s) | 0.009 ± 0.001 | 0.009 ± 0.002 | 0.013 ± 0.001 |
| QT Interval | (s) | 0.020 ± 0.002 | 0.017 ± 0.002 | 0.031 ± 0.004 |
| JT Interval | (s) | 0.010 ± 0.004 | 0.008 ± 0.001 | 0.016 ± 0.005 |
| P Amplitude | (mV) | 0.068 ± 0.033 | 0.085 ± 0.010 | 0.060 ± 0.032 |
| ST Height | (mV) | 0.063 ± 0.035 | 0.046 ± 0.025 | −0.170 ± 0.147 |
Calreticulin-deficient hearts (Calr−/−); hearts with an increased abundance of calreticulin (Calr OE); RR Interval (the time elapsed between two successive R waves); Beat per minute (BPM); PR Interval (the time between atrial depolarization and ventricular depolarization); P Duration (the period that covers the earliest deflection to the latest deflection); QRS Interval (ventricular contraction); QT Interval (the beginning of ventricular depolarization to the end of ventricular repolarization); JT Interval (the period of time that covers the end of the J wave to the end of the T wave); P Amplitude (the height of the initial deflection P wave); ST Height (the height between the bottom of the S dip to the top of the T wave).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Groenendyk, J.; Wang, W.-A.; Robinson, A.; Michalak, M. Calreticulin and the Heart. Cells 2022, 11, 1722. https://doi.org/10.3390/cells11111722
AMA Style
Groenendyk J, Wang W-A, Robinson A, Michalak M. Calreticulin and the Heart. Cells. 2022; 11(11):1722. https://doi.org/10.3390/cells11111722
Chicago/Turabian StyleGroenendyk, Jody, Wen-An Wang, Alison Robinson, and Marek Michalak. 2022. "Calreticulin and the Heart" Cells 11, no. 11: 1722. https://doi.org/10.3390/cells11111722
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.