1. Introduction
Sake is an alcoholic beverage that originated in Japan and has been made for centuries. It is made by parallel double fermentation of sake rice with a high starch content, mainly by the action of two types of microorganisms [
1]. The microorganisms inoculated are
Aspergillus oryzae and the yeast
Saccharomyces cerevisiae. Genome sequence analysis has revealed that sake yeast is a different clade (i.e., subspecies) than wine or ale brewer’s yeasts [
2]. Sake yeast also originated in Japan and is specifically tolerant to high alcohol concentration and low pH, with a high fermentation ability for making sake with excellent flavor characteristics. These characteristics indicate that sake yeast has been domesticated for a unique purpose in a geographically isolated situation.
According to recent statistics from Japan’s National Tax Agency, more than 1400 sake breweries exist in Japan and more than 10,000 sake brands are sold globally. The variety in sake brewing arises from the existence of many kinds of sake yeast isolates that have been bred by the National Research Institute of Brewing, prefectural brewing institutes, and private research laboratories in sake breweries [
3]. Some of the isolates developed by the National Research Institute of Brewing are known as kyokai yeast and are distributed by the Brewing Society of Japan. To begin development, excellent yeasts suitable for sake brewing were selected from the yeasts used in sake breweries. Then, “parallel” or “serial” breeding techniques were used to meet social demands and fulfill consumer taste expectations. An example of parallel breeding output is non-foam-forming yeast. The excellent sake yeasts that were previously used in sake breweries caused foam to accumulate in the upper layer of the fermentation tank. However, this foam production creates a need for large tanks and increases the labor requirements among sake brewery workers. In the breeding selection method developed by Ouchi and colleagues [
4], non-foam-forming yeasts were bred in parallel from several excellent yeast isolates: non-foam-forming kyokai strain numbers 601 (hereafter designated K601), K701, K901, K1001, and K1401 were bred from strains K6, K7, K9, K10, and K14, respectively [
5]. By contrast, serial breeding involves the creation of a single strain with several excellent traits through a successive breeding operation. The non-foam-forming strain K1601 originated from strains K7, K9, and K10, and produces excellent sake that is highly aromatic and slightly acidic. Strain K1601 was used to breed strain K1801, which produces considerable
ginjo aroma [
6]. Strain K1801 was then used to breed strain K1901, which produces less ethyl carbamate, a group 2A carcinogen [
7]. Because sake yeast is diploid, breeding efficiency is limited despite the use of appropriate breeding selection methods. Therefore, serial breeding requires long durations of time and considerable effort.
Comparison of the genome sequences of kyokai yeast has revealed many mutations that accumulated during the breeding process [
2]. The whole-genome sequence of strain K7 was compared with the sequence of the canonical non-foam-forming strain K701. This comparison identified heterozygous and homozygous nonsynonymous variants in 73 and 5 genes, respectively, although the two strains have a parent–child relationship [
8]. Strain K701 was isolated in the early 1970s, and both isolates have accumulated mutations in parallel over nearly 50 years. However, unexpected mutations were presumably introduced during the breeding of the canonical non-foam-forming strain. To maintain consistent brewing properties, other than the properties of interest, unexpected mutations should be minimized.
The relationship between yeast genotype and phenotype has been studied by multiple researchers, revealing several genes important for brewing characteristics. Shimoi, et al. found that the non-foam-forming strain K701 has a chromosomal translocation in the
AWA1 region that encodes the mannoprotein present in the cell wall [
9]. The hydrophobicity of the cell surface is altered by the loss of
AWA1 activity, such that carbon dioxide is no longer bound to the cell surface, leading to the non-foam-forming phenotype [
9]. Kitamoto, et al. revealed that the loss of the arginase gene (
CAR1) results in reduced urea production, thereby eliminating the potential carcinogen ethyl carbamate derived from urea that forms during the storage of sake [
7]. Ichikawa, et al. found that sake yeast strains producing the strong
ginjo aroma of ethyl caproic acid have the Gly1250Ser mutation in the
FAS2 gene [
10], which encodes the alpha subunit of fatty acid synthase. This dominant
FAS2 mutation was thought to decrease fatty acid synthesis, resulting in decreased carbon chain elongation and increased amounts of caproic acid and caproyl-CoA, precursors of ethyl caproate [
10]. Furthermore, sake yeast defective in the
MDE1 gene reportedly exhibits reduced production of dimethyl trisulfide (DMTS), a strong, unpleasant odor known as
hineka [
11]. Evidence-based genetic engineering based on genotype–phenotype relationships enables the construction of sake yeast strains with superior properties. However, homozygous alleles must be prepared when introducing recessive mutations and markers must be prepared for selection of the recombinant yeast.
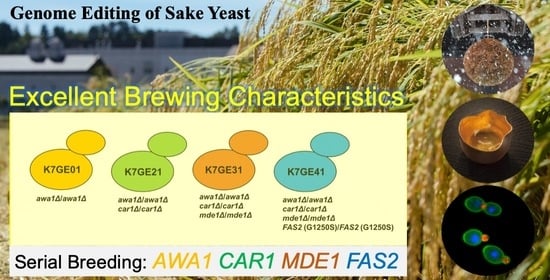
In this study, we attempted to construct sake yeast with excellent brewing characteristics by using genome editing technology. We introduced eight mutations into K7 to construct non-foam-forming sake yeast with three additional excellent brewing characteristics. To construct a non-foam-forming strain that makes sake without producing carcinogens or an unpleasant odor, instead producing a sweet ginjo aroma, we introduced the car1∆/car1∆, mde1∆/mde1∆, and FAS2 (G1250S)/FAS2 (G1250S) mutations into awa1∆/awa1∆, in that order. A small-scale fermentation test showed that the desired sake was achieved. Based on whole-genome sequencing and morphological analyses of the genome-edited strains, we propose that genome editing technology can be applied for the effective construction of sake yeast strains by serial breeding.
2. Materials and Methods
2.1. Strains and Media
The sake yeast strains used in this study were the foaming isolate K7 and genome-edited, non-foam-forming isolate K7GE01 [
8]. The genome-edited strains constructed in this study were K7GE21, K7GE31, and K7GE41. Yeast strains were cultivated at 30 °C in yeast extract peptone dextrose medium containing 1% (
w/v) Bacto yeast extract (BD Biosciences, Palo Alto, CA, USA), 2% (
w/v) Bacto peptone (BD Biosciences), and 2% glucose to transform yeast trains, extract yeast DNA, and prepare precultures for fermentation tests. Geneticin (Takara, Kyoto, Japan) was added at 350 μg/mL to yeast extract peptone dextrose agar plates containing 2% agar (Shouei, Tokyo, Japan) after autoclaving.
2.2. Application of Genome Editing Techniques to Sake Yeast Strains
We applied genome editing techniques to the
AWA1,
CAR1,
MDE1, and
FAS2 genes located on chromosomes XV, XVI, X, and XVI, respectively. pCAS-Pro-AWA1 plasmid co-expressing nuclease protein Cas9 and Ribozyme-sgRNA that guides the Cas9 to the target
AWA1 sequence were described in Ohnuki, et al. [
8]. We constructed the pCAS-Pro-CAR1 plasmid from pCAS Prolin-URA3 [
12] with the
CAR1 target sequence (
Supplementary Materials Table S1) using a restriction-free cloning method [
13]. The repair DNA used had a length of 150 bp (
CAR1 donor sense and
CAR1 donor antisense for K7GE21) and was generated by annealing equimolar amounts of single-stranded oligonucleotides as follows: the mixture was first denatured at 100 °C for 5 min and then allowed to cool to 25 °C with a ramp of 0.1 °C/s [
14]. We constructed pCAS-Pro-MDE1 and pCAS-Pro-FAS2 (G1250S) plasmids in a similar manner.
To measure the genome editing efficiency of each sake yeast strain, K7 was co-transformed with the pCAS-Pro-AWA1, pCAS-Pro-CAR1, pCAS-Pro-MDE1, and pCAS-Pro-FAS2 (G1250S) plasmids, as well as the corresponding repair DNAs, using an improved lithium acetate transformation method. To construct strain K7GE21, K7GE01 was co-transformed with the pCAS-Pro-CAR1 plasmid and the corresponding repair DNA. After selection of G418
r colonies, we amplified the genomes of the yeast transformants by colony polymerase chain reaction (PCR) (primers described in
Supplementary Materials Table S2) to confirm the presence of desired fragments. We amplified PCR fragments of 755 and 1757 bp with deleted and intact copies of the
CAR1 gene, respectively. When both deleted and intact copies were detected, we re-examined the heterozygosity after reisolating single colonies. After we confirmed the presence of the
car1∆/
car1∆ alleles, we allowed the pCAS-Pro-CAR1 plasmid to be spontaneously eliminated, producing the genome-edited strain, K7GE21. We constructed strains K7GE31 and K7GE41 in a similar manner by using pCAS-Pro-MDE1 and pCAS-Pro-FAS2 (G1250S) plasmids.
2.3. Whole-Genome Sequencing
We extracted DNA from strains K7, K7GE01, K7GE21, K7GE31, and K7GE41 to determine their whole-genome sequences. First, we isolated high-molecular-weight DNA (10 µg, ~24 kb fragments) with a Genomic-tip 100/G kit (QIAGEN, Germantown, MD, USA), in accordance with the manufacturer’s instructions. We estimated the purity of the DNA samples using a spectrophotometer (Nano Drop; Thermo Fisher Scientific, Waltham, MA, USA). The whole-genome sequencing was outsourced to GeneBay (Yokohama, Japan). The DNA samples were sent to Novogene (Singapore) to prepare a PCR-free, paired-end, sequencing library and for whole-genome sequence analysis (2 × 150 bp) using an Illumina NovaSeq 6000 sequencing platform (Illumina, San Diego, CA, USA) at ~100-fold nominal coverage. The adapter contamination was removed and the low-quality bases trimmed. We obtained the K7 reference genome (NRIB_SYGD, txid721032) from the Sake Yeast Genome Database (
https://nribf1.nrib.go.jp/SYGD/, ver. 1.0) and prepared it for use in sequencing data analysis. The software packages that we used for sequencing data analysis were: Sequence Alignment/Map Tools (ver. 1.11) [
15] to convert the “sam” format to “bam” format and to modify information concerning the paired reads; Burrows–Wheeler Aligner (ver. 0.7.17) [
16] for mapping reads to the K7 reference genome; Picard-tools (ver. 2.25.0;
https://broadinstitute.github.io/picard) to remove duplicate reads; Genome Analysis TK (ver. 4.1.8.0) [
17] to rearrange the bam format, extract mutation candidates, identify and filter variants relative to K7, and identify the mutations. Finally, variants were annotated manually with the aid of SnpEff software (ver. 4.3;
https://pcingola.github.io/SnpEff/) [
18].
2.4. Fluorescence Staining, Microscopy, and Image Processing
We cultivated cells of strains K7, K7GE01, K7GE21, K7GE31, and K7GE41 until the early log phase (<5 × 10
6) and fixed them with medium containing 3.7% (
w/v) formaldehyde (Wako, Osaka, Japan). We then triple-stained cells with fluorescein isothiocyanate-conjugated concanavalin A (Sigma, St. Louis, MO, USA) for the cell wall, rhodamine-phalloidin (Invitrogen, Carlsbad, CA, USA) for the actin cytoskeleton, and 4′,6-diamidino-2-phenylindole (Sigma) for nuclear DNA, as described previously [
19]. We acquired fluorescence microscopy images of the cells using a microscope (Axio Imager) equipped with a special lens (6100 EC Plan-Neofluar; Carl Zeiss, Oberkochen, Germany), a cooled-charge-coupled device camera (CoolSNAP HQ; Roper Scientific Photometrics, Tucson, AZ, USA), and appropriate software (AxioVision; Carl Zeiss). We analyzed the micrographs of the cells with image processing software designed for diploid cells (CalMorph, ver. 1.3) [
20]. We obtained the morphological data of 501 traits from the single-cell data. Descriptions of each trait have been presented previously [
19]. The CalMorph user manual is available at the
S. cerevisiae Morphological Database (
https://www.yeast.ib.k.u-tokyo.ac.jp/CalMorph/).
2.5. Principal Component Analysis (Pca) for Dimensional Reduction and Calculation of the Euclidean Distance in the Degenerated Morphological Space
Morphological data obtained in this study (strains K7, K7GE01, K7GE21, K7GE31, and K7GE41) were used for statistical analyses. There were eight samples for strain K7, and four each for strains K7GE01, K7GE21, K7GE31, and K7GE41, respectively. We applied PCA to the mean Z-values in each strain for all 501 traits that were calculated using a general linear model. From the PCA of the five sake yeasts, the cumulative contribution ratios of the first 11 principal components reached 90%.
The Euclidean distance [
21] was used to assess morphological differences between two strains; this distance is near zero if the cell morphology of the two strains is similar, but is otherwise larger. The Euclidean distance between each strain was calculated from the principal component scores of the first 11 principal components (cumulative contribution ratio 90%), as described previously [
22]. To calculate the principal component scores of each strain, the Z-values from each independent experiment in each strain were projected onto the 11 principal components. The mean values and standard deviations were calculated from the Euclidean distances of each replicate from the center of the parental strain in the orthogonal morphological space of 11 PCs.
2.6. Small-Scale Fermentation Test
A sake mash was prepared by mixing 72.8 g of pregelatinized rice (corresponding to 100 g of white rice), 19.2 g of dried koji (rice with Aspergillus oryzae mold, corresponding to 20 g of white rice), 136 μL of 90% lactic acid, and 170 mL of water containing 1 × 109 precultured yeast cells. The mash (three replicates) was incubated at 15 °C for 20 days without shaking. The fermentation was monitored daily by quantifying the amount of evolved CO2, measuring the weight loss of the sake mash. After the sake fermentation, the mash was collected in 50-mL centrifuge tubes and centrifuged at 15 °C, 5000 rpm for 15 min. The supernatant was filtered with microfiber cloth to yield the sake product and stored at −80 °C.
2.7. Component Analysis of Sake Made in the Fermentation Test
The sake meter value (SMV) was calculated after measuring the density of the sake relative to water. Density was measured with a density/specific gravity meter (DA-650; Kyoto Denshi, Kyoto, Japan) equipped with an autosampler (CHD-502; Kyoto Electronics Manufacturing, Kyoto, Japan). A thawed sample of 10 mL or more was added to a 20-mL vial for measurement. SMV was calculated as SMV = (1/specific gravity − 1) × 1443. A more negative SMV indicates a higher specific gravity and sugar content.
The ethanol concentration was measured with a gas chromatograph (6890 N; Agilent Technologies, Santa Clara, CA, USA) equipped with an auto injector (7683B; Agilent). As an internal standard, 1% isopropanol (960 μL) was mixed with the sample (40 μL).
Aroma components, such as ethyl acetate, 1-propanol, isobutanol, isoamyl alcohol, isoamyl acetate, and ethyl caproate, were measured by a gas chromatograph (6890 N; Agilent) equipped with a head space sampler (HSS 7697A; Agilent). Internal standards (100 μL) were mixed with the samples (900 μL).
Acidity and amino acid contents were measured with an automatic titrator (COM-1700; Hiranuma, Ibaraki, Japan). Measurements were performed with 10 mL of the samples.
Organic acids, including malic acid, succinic acid, lactic acid, citric acid, acetic acid, and phosphoric acid, were measured by a liquid chromatograph (CBM40 equipped with an SCR-102H column; Shimadzu, Kyoto, Japan). A thawed 1 mL sample was injected for measurement.
The precursor of dimethyl trisulfide (1,2-dihydroxy-5-(methylsulfinyl) -pentan-3-one; DMTS-P1) was analyzed by LC-MS (LCMS8040, Shimadzu) using (ethyl-d3)-DMTS-P1 as an internal standard [
23,
24].
2.8. PCA of Sake Components
We performed PCA using 16 or 19 parameters on the mean values yielded by component analysis of 12 samples (four strains × three samples) of sake produced in the small-scale fermentation test. The mean value of each strain, and the value of each sample, were plotted with two components. Among the correlation coefficients obtained by mapping the values of the 12 samples, we considered those with p < 0.05 after Bonferroni correction to be statistically significant.
2.9. Structural Changes Predicted Based on the N532K Change in Nrd1
The secondary and tertiary protein structures of Nrd1 of the S288C reference genome (Saccharomyces Genome Database; SGD) were predicted by Iterative Threading Assembly Refinement (I-TASSER) [
25]. The first ranked PDB files, defined by the C- and TM-scores [
26], were visualized by UCFS Chimera [
27].
4. Discussion
We applied genome editing technology to sake yeast isolates in a previous study [
8] with the aim of performing parallel breeding by deletion of
AWA1 in strains K6, K7, K9, and K10. In the present study, we attempted to introduce
car1∆/
car1∆,
mde1∆/
mde1∆, and
FAS2 (G1250S)/
FAS2 (G1250S) mutations into a K7-derived diploid non-foam-forming sake yeast strain (
awa1∆/
awa1∆) by means of genome editing technology. We finally created a serial breeding strain (K7GE41) that contained eight mutations. Analyses of the components of the sake made with the genome-edited strains revealed that urea, the precursor of ethyl carbamate, was dramatically reduced as expected for
car1∆/
car1∆; DMTS-P1, the precursor of
hineka (DMTS), was hardly detected as expected for
mde1∆/
mde1∆; and a large amount of ethyl caproate was produced as expected for
FAS2 (G1250S)/
FAS2 (G1250S). Although serial breeding is often considered to be laborious and time-consuming, this study is the first successful attempt to generate a sake yeast strain with these four excellent brewing characteristics. Our findings demonstrate that genome editing technology is extremely effective for the breeding of sake yeast strains.
4.1. Variation of Genome Editing Efficiency during Sake Yeast Breeding
After sake yeast breeding, we found that the genome editing efficiencies at the
AWA1,
CAR1,
MDE1, and
FAS2 loci varied from 16% to 96%. Previous studies have shown that the efficiency and specificity of genome editing are affected by the host strain, CRISPR-Cas9/sgRNA system to express ribozyme, and sgRNA design [
8]. In particular, unlike laboratory strains such as S288C, the choice of tRNA promoter used for the expression of the ribozyme was important in the polyploid industrial S. cerevisiae strain ATCC4124 for improving tolerance and productivity [
12]. We performed all genome editing of sake yeast using the same pCas9-Pro-based plasmid harboring the tRNAPro promoter used in ATCC4124 [
12]. Because a maximum genome editing efficiency of 96% was achieved, we consider there to be sufficient compatibility between the tRNAPro and sake yeast. For the design of sgRNA, we consistently followed the method described by Ohnuki, et al. [
8] but the use of an alternative protospacer adjacent motif sequence may change the genome editing efficiency. Furthermore, genes present in the heterochromatin and nucleosome regions tend to be less efficient in genome editing [
33,
34]. The
AWA1 gene, which exhibited the lowest genome editing efficiency, is in the heterochromatin region near the telomere region on chromosome XV. Furthermore, regarding
AWA1, dynamic genomic changes were observed in almost all transformants, although many colonies did not have the correct gene deletion, which implied that the cleavages were rarely repaired in the desired manner. This may be due to the fact that the
AWA1 gene deletion was made after the deletion of a large (6 kb) genomic fragment. Another notable feature during the genome editing of sake yeast strains was the appearance of a high frequency of homozygous mutations, such that we observed no heterogeneous mutations in more than 400 transformants. This could be caused by strong tRNAPro promoter activity for ribozyme expression, or by the loss of homozygosity frequently observed in the sake yeast genome [
2].
4.2. Changes in the Components of Sake due to FAS2 (G1250S) and car1∆
Sake made by yeast isolates with the
FAS2 (G1250S)/
FAS2 (G1250S) mutation was previously shown to contain a large amount of ethyl caproate. However, it was unclear whether those yeast isolates harbor any extra mutations. By comparing the genome-edited strains K7GE41 and K7GE31, we have elucidated the characteristics of sake affected by
FAS2 (G1250S)/
FAS2 (G1250S). The sake made with K7GE41 had a high concentration of pyruvic acid and a low SMV (indicating glucose retention). In our small fermentation test, K7GE41 exhibited a slow fermentation rate, which may lead to delayed glucose consumption and the accumulation of intermediate metabolites such as pyruvic acid. Sake made with K7GE41 contained elevated and reduced amounts of ethyl caproate and ethyl acetate, respectively. Because these two aromatic compounds are synthesized by the same ethyl esterase, encoded by
EEB1, competitive inhibition may have contributed to these changes. Another aroma component, isoamyl acetate, was also reduced in sake made by K7GE41. A previous study [
35] also showed an inverse relationship between isoamyl acetate and ethyl caproate, so negative feedback regulation might occur between the isoamyl acetate and ethyl caproate biosyntheses. Finally, the sake made by K7GE41 contained more amino acids.
FAS2 encodes a catalytic subunit of fatty acid synthase, so the introduction of the
FAS2 (G1250S) mutation presumably affects the membrane, thereby increasing the proportion of autolyzed yeast in mash. Indeed, the proportion of viable K7GE41 in the mash was lower than the corresponding proportions of other strains (data not shown). Therefore, autolysis of K7GE41 may cause intracellular amino acid release into the mash.
Deletion of the CAR1 gene encoding arginase in strains K7GE21, K7GE31, and K7GE41 unexpectedly caused a reduction of isoamyl alcohol, an aroma component synthesized from leucine via keto acid. Although the detailed mechanism of metabolic regulation is unknown, the amount of leucine-derived isoamyl alcohol may have decreased due to the effect of reduced arginine metabolism. LOH was introduced into K7GE31, but there were no obvious holistic morphological changes or component changes of sake during this step. This implies that the phenotypic changes due to this LOH were negligible. Overall, our study revealed unexpected links between the modified genes and brewing properties. By applying genome editing technology to sake yeast strains, our findings have clarified several characteristics of sake affected by FAS2 (G1250S) and car1∆.
4.3. Effectiveness of Genome Editing in Serial Breeding
We generated strains K7GE21, K7GE31, and K7GE41 by serial breeding in this study, based on the previously constructed strain K7GE01. A novel genome editing technology enables simultaneous manipulation of multiple genes [
12], but this technique requires storage of intermediate sake yeast strains produced by serial breeding. The intermediate strains can be utilized for brewing to make a variety of sake. We first constructed strain K7GE01 because societal demands require improved productivity. We then created strain K7GE21 because safety is widely regarded as important. Some people like the
ginjo aroma but others do not, so we made strain K7GE31 without
hineka, and finally constructed strain K7GE41. These varieties generated by genome editing can be chosen according to the desired properties of sake. It is possible to breed sake yeast conventionally with similar, excellent brewing characteristics, but breeding in this manner is laborious and time-consuming. In addition, many off-target mutations may accumulate, and there is a risk that the resulting yeast will exhibit unexpected brewing characteristics.
4.4. Breeding of Sake Yeast in the Future
We employed evidence-based breeding using genome editing technology in this study, using our accumulated knowledge of sake brewing. K7GE41 is an optimal sake yeast strain with ideal brewing characteristics, and the sake yeast strains generated during this breeding process are also useful for making a wide variety of sake. This technology will enable the creation of a wider variety of sake yeast by changing the yeast strains initially used. Strains K6, K9, and K10 are from the same lineage of sake yeasts as strain K7, but their brewing characteristics are distinct. Therefore, it may be useful to generate genome-edited yeast strains from each sake yeast. In addition to the yeast strains distributed by the Brewing Society of Japan, the same genome editing technology could be applied to the strains owned by each prefecture and sake brewery. Furthermore, genome editing technology can be applied to design or anticipate changes in metabolic pathways during sake yeast. A long-term goal of sake yeast breeding is to create a strain that exhibits enhanced ethanol resistance. It would also be useful to create yeast that produces high levels of both ethyl caproate and isoamyl acetate. The regulations regarding genome-edited food and beverages have been amended in many countries including Japan. If a foreign gene is not inserted, it is now possible to sell genome-edited food and beverages without a safety review in Japan by simply making a voluntary notification. Therefore, the breeding of sake yeast using genome editing may be actively pursued in the future, and both evidence- and prediction-based breeding of sake yeast are expected to be actively explored.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






