
Protective Effects of Necrostatin-1 in Acute Pancreatitis: Partial Involvement of Receptor Interacting Protein Kinase 1

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Acute Pancreatitis
2.3. Histological Analysis and Biochemical Measurements of Acute Pancreatitis
2.4. Cell Preparation and Solutions
2.5. Immunofluorescence
2.6. Western Blotting
2.7. Measurement of Necrosis and Apoptosis
2.8. Measurement of Cytosolic Calcium and Reactive Oxygen Species
2.9. Statistical Analysis
3. Results
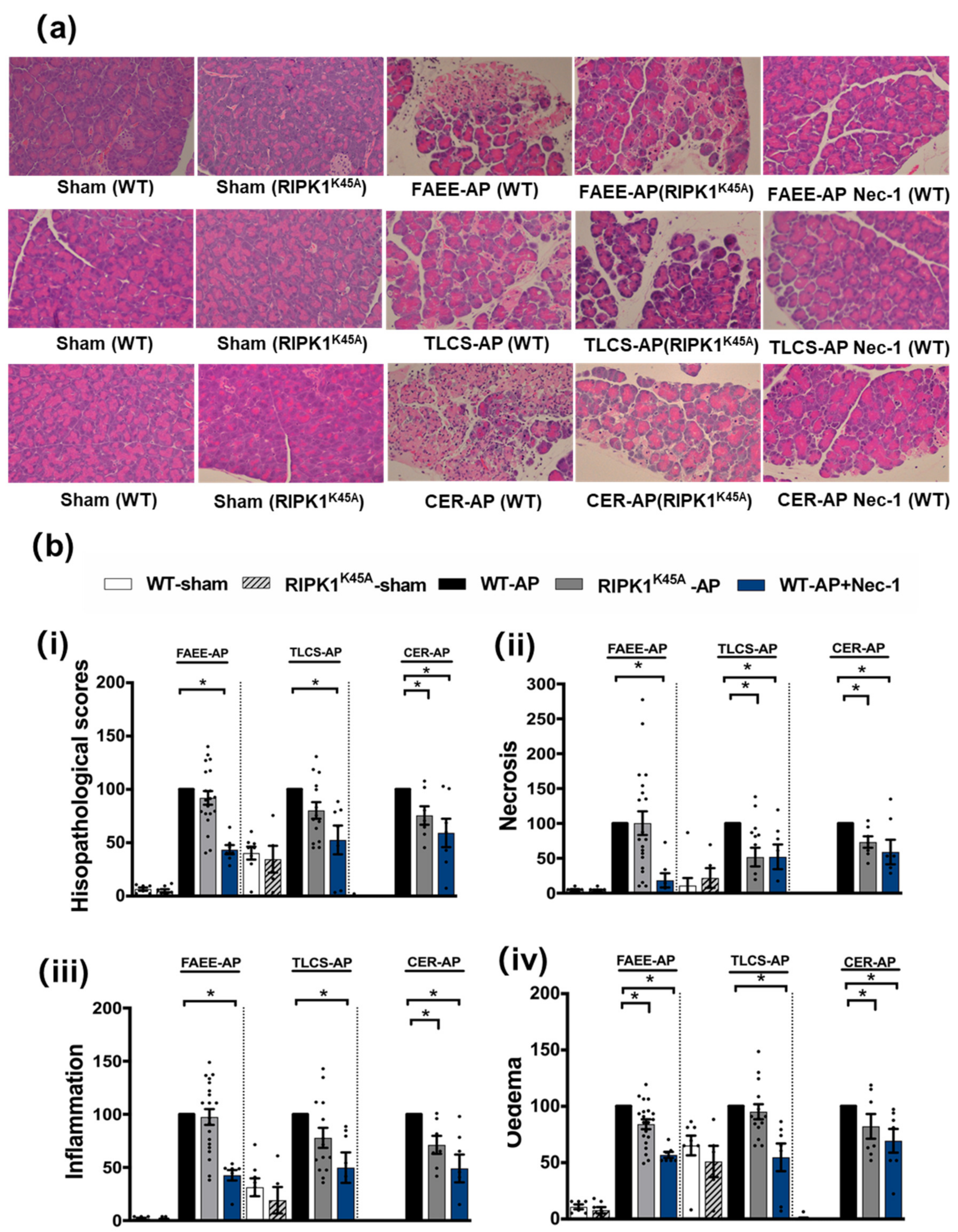
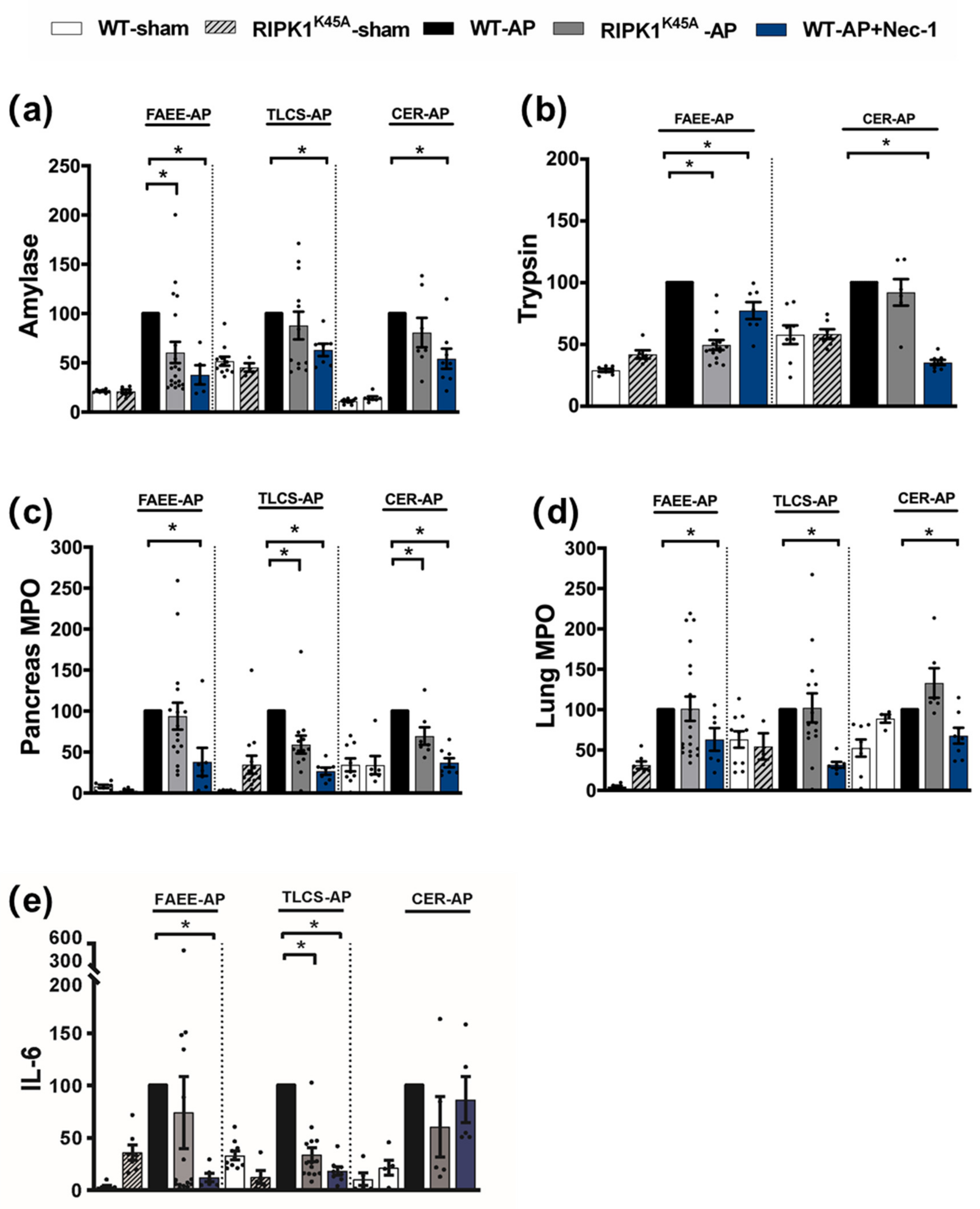
3.1. Effects of Genetic and Pharmacological RIPK1 Inhibition in FAEE-AP, TLCS-AP and CER-AP
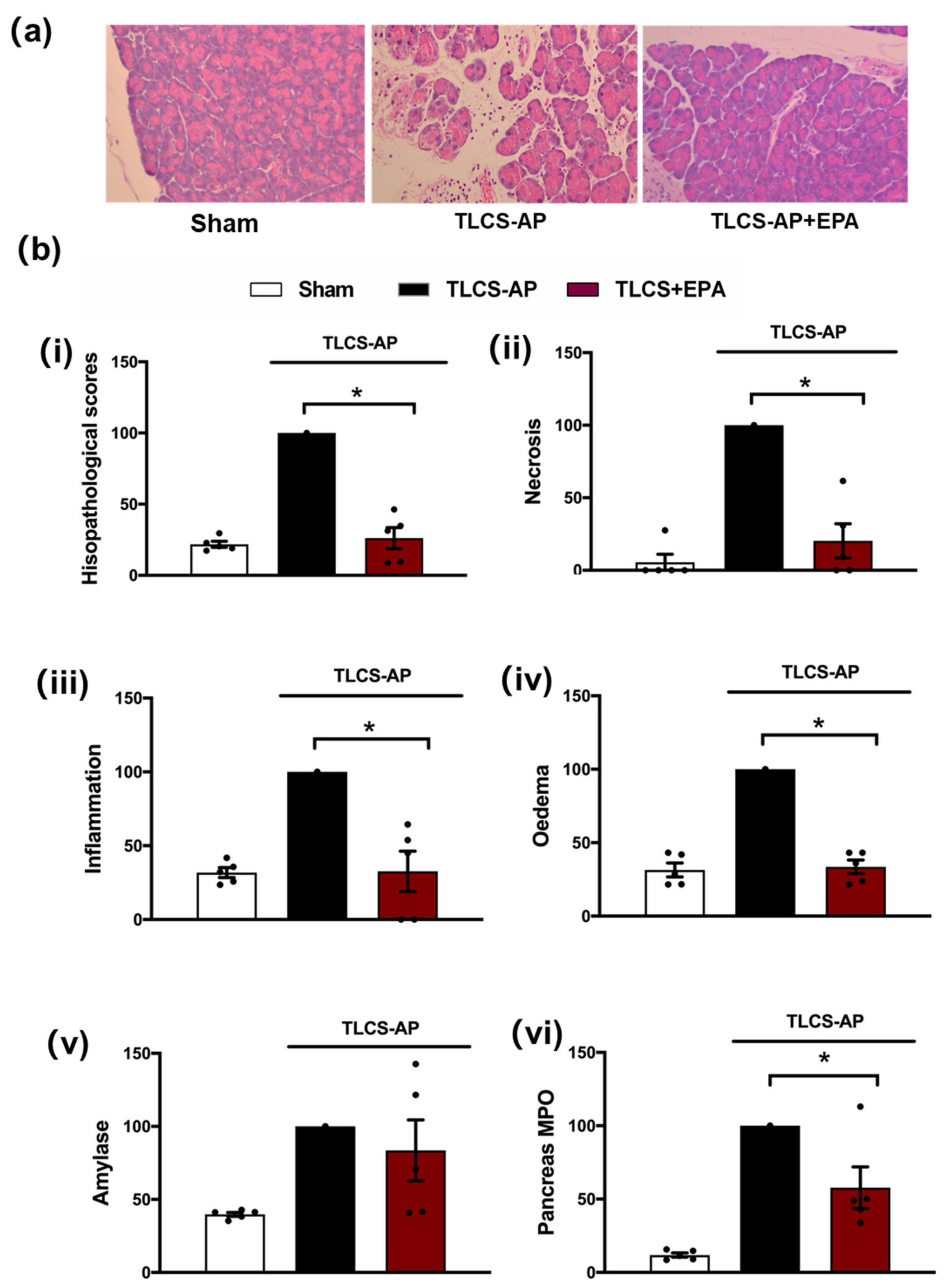
3.2. Protective Effects of IDO Inhibition in TLCS-AP
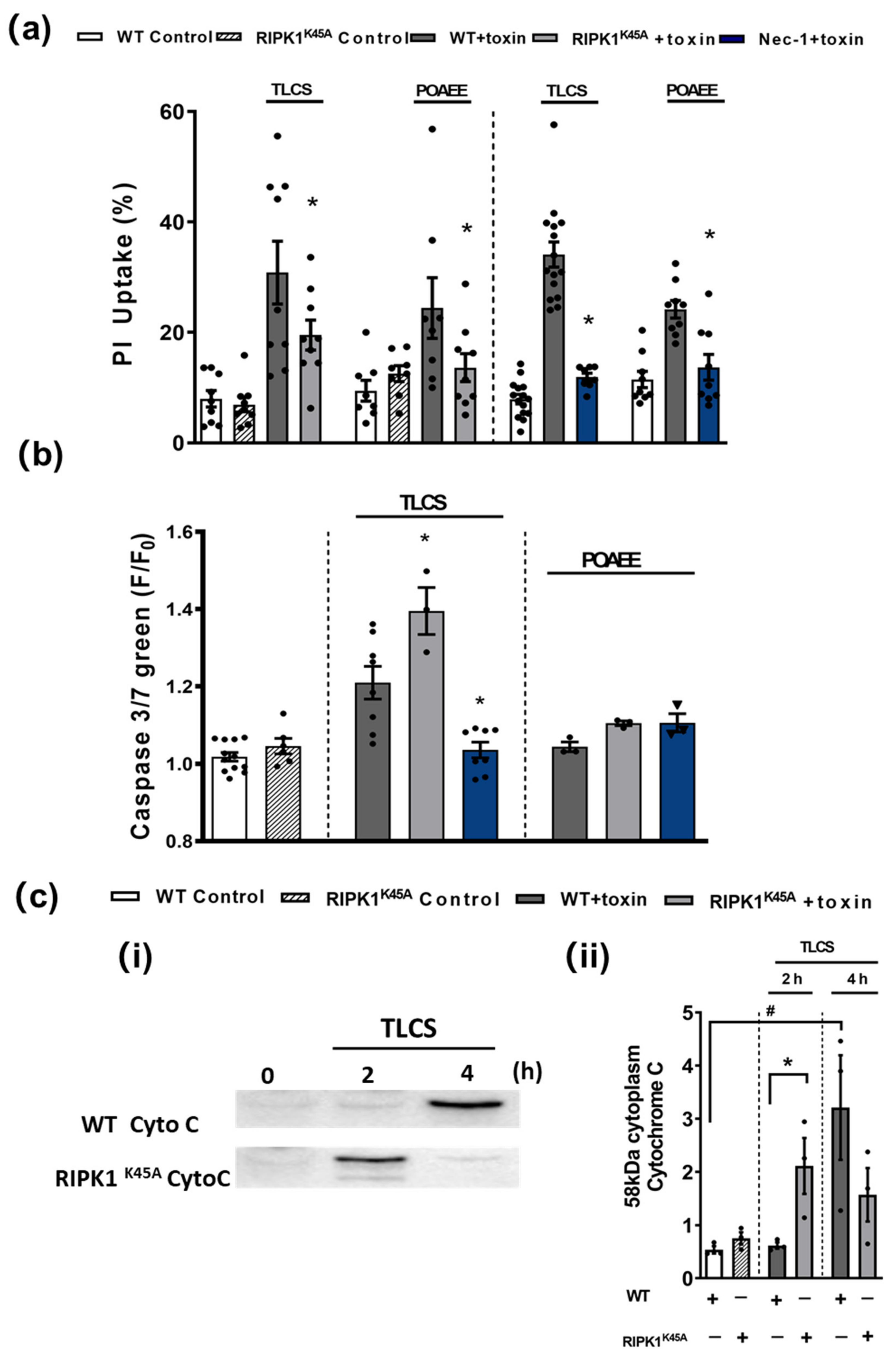
3.3. Comparative Effects of RIPK1K45A and Nec-1 on PAC Cell Death
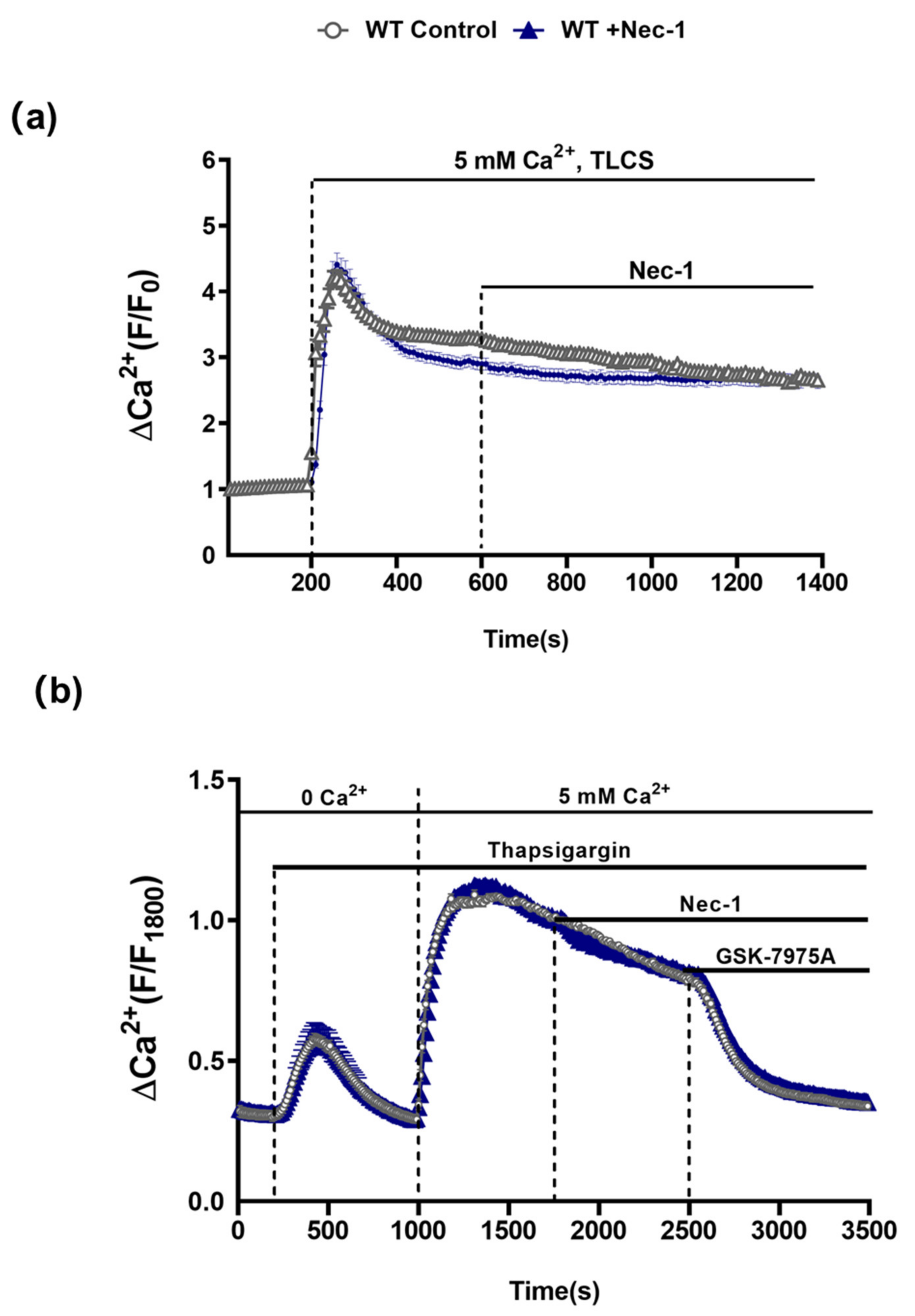
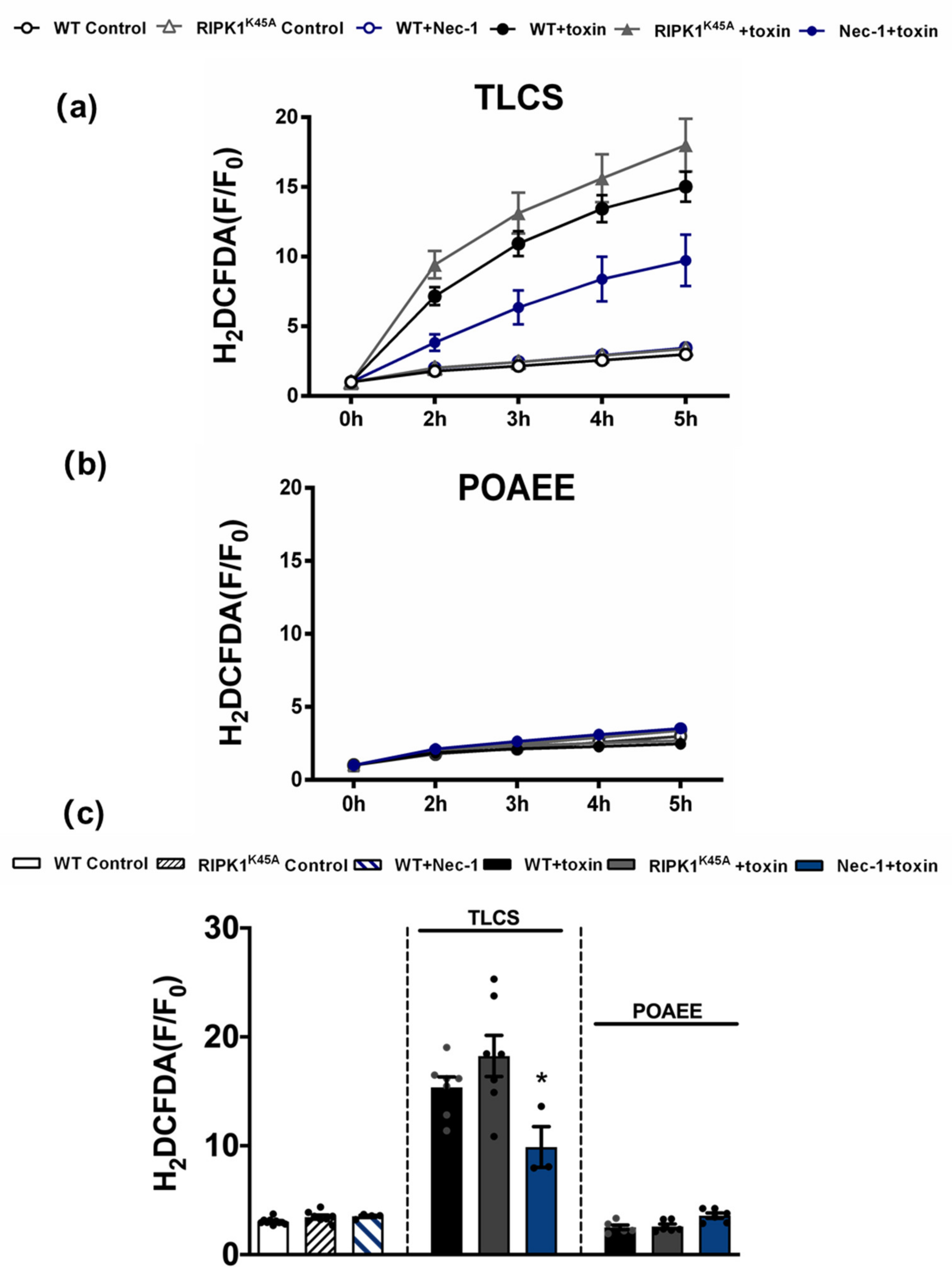
3.4. Comparative Effects of RIPK1K45A and Nec-1 on Intracellular Ca2+ and ROS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology 2019, 156, 254–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criddle, D.N. Reactive oxygen species, Ca(2+) stores and acute pancreatitis; a step closer to therapy? Cell Calcium. 2016, 60, 180–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, R.; Mareninova, O.A.; Odinokova, I.V.; Huang, W.; Murphy, J.; Chvanov, M.; Javed, M.A.; Wen, L.; Booth, D.M.; Cane, M.C.; et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: Inhibition prevents acute pancreatitis by protecting production of ATP. Gut 2016, 65, 1333–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Booth, D.M.; Cane, M.C.; Chvanov, M.; Javed, M.A.; Elliott, V.L.; Armstrong, J.A.; Dingsdale, H.; Cash, N.; Li, Y.; et al. Fatty acid ethyl ester synthase inhibition ameliorates ethanol-induced Ca2+-dependent mitochondrial dysfunction and acute pancreatitis. Gut 2014, 63, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Pallagi, P.; Madacsy, T.; Varga, A.; Maleth, J. Intracellular Ca(2+) Signalling in the Pathogenesis of Acute Pancreatitis: Recent Advances and Translational Perspectives. Int. J. Mol. Sci. 2020, 21, 4005. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef] [Green Version]
- Mifflin, L.; Ofengeim, D.; Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat. Rev. Drug Discov. 2020, 19, 553–571. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Y.; Jenkins, L.W.; Kochanek, P.M.; Clark, R.S. Bench-to-bedside review: Apoptosis/programmed cell death triggered by traumatic brain injury. Crit. Care 2005, 9, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Louhimo, J.; Steer, M.L.; Perides, G. Necroptosis Is an Important Severity Determinant and Potential Therapeutic Target in Experimental Severe Pancreatitis. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 519–535. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fan, C.; Zhang, Y.; Yu, X.; Wu, X.; Zhang, X.; Zhao, Q.; Zhang, H.; Xie, Q.; Li, M.; et al. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell Death Differ. 2017, 24, 1459–1469. [Google Scholar] [CrossRef]
- Wu, J.F.; Huang, Z.; Ren, J.M.; Zhang, Z.R.; He, P.; Li, Y.X.; Ma, J.H.; Chen, W.Z.; Zhang, Y.Y.; Zhou, X.J.; et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013, 23, 994–1006. [Google Scholar] [CrossRef] [Green Version]
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576. [Google Scholar] [CrossRef]
- Linkermann, A.; Brasen, J.H.; De Zen, F.; Weinlich, R.; Schwendener, R.A.; Green, D.R.; Kunzendorf, U.; Krautwald, S. Dichotomy between RIP1- and RIP3-mediated necroptosis in tumor necrosis factor-alpha-induced shock. Mol. Med. 2012, 18, 577–586. [Google Scholar] [CrossRef]
- Zou, C.; Xiong, Y.; Huang, L.Y.; Song, C.L.; Wu, X.A.; Li, L.L.; Yang, S.Y. Design, Synthesis, and Biological Evaluation of 1-Benzyl-1H-pyrazole Derivatives as Receptor Interacting Protein 1 Kinase Inhibitors. Chem. Biol. Drug Des. 2016, 87, 569–574. [Google Scholar] [CrossRef]
- Walker, A.L.; Ancellin, N.; Beaufils, B.; Bergeal, M.; Binnie, M.; Bouillot, A.; Clapham, D.; Denis, A.; Haslam, C.P.; Holmes, D.S.; et al. Development of a Series of Kynurenine 3-Monooxygenase Inhibitors Leading to a Clinical Candidate for the Treatment of Acute Pancreatitis. J. Med. Chem. 2017, 60, 3383–3404. [Google Scholar] [CrossRef] [Green Version]
- Mole, D.J.; Webster, S.P.; Uings, I.; Zheng, X.; Binnie, M.; Wilson, K.; Hutchinson, J.P.; Mirguet, O.; Walker, A.; Beaufils, B.; et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 2016, 22, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Daley-Bauer, L.P.; Thapa, R.J.; Mandal, P.; Berger, S.B.; Huang, C.; Sundararajan, A.; Guo, H.; Roback, L.; Speck, S.H.; et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. USA 2014, 111, 7753–7758. [Google Scholar] [CrossRef] [Green Version]
- Perides, G.; van Acker, G.J.; Laukkarinen, J.M.; Steer, M.L. Experimental acute biliary pancreatitis induced by retrograde infusion of bile acids into the mouse pancreatic duct. Nat. Protoc. 2010, 5, 335–341. [Google Scholar] [CrossRef]
- Koblish, H.K.; Hansbury, M.J.; Bowman, K.J.; Yang, G.; Neilan, C.L.; Haley, P.J.; Burn, T.C.; Waeltz, P.; Sparks, R.B.; Yue, E.W.; et al. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol. Cancer Ther. 2010, 9, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Voronina, S.; Javed, M.A.; Awais, M.; Szatmary, P.; Latawiec, D.; Chvanov, M.; Collier, D.; Huang, W.; Barrett, J.; et al. Inhibitors of ORAI1 prevent cytosolic calcium-associated injury of human pancreatic acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology 2015, 149, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Booth, D.M.; Murphy, J.A.; Mukherjee, R.; Awais, M.; Neoptolemos, J.P.; Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Criddle, D.N. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 2011, 140, 2116–2125. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef]
- Northington, F.J.; Chavez-Valdez, R.; Graham, E.M.; Razdan, S.; Gauda, E.B.; Martin, L.J. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J. Cereb. Blood Flow Metab. 2011, 31, 178–189. [Google Scholar] [CrossRef]
- Xu, X.; Chua, K.W.; Chua, C.C.; Liu, C.F.; Hamdy, R.C.; Chua, B.H. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 2010, 1355, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Linkermann, A.; Brasen, J.H.; Himmerkus, N.; Liu, S.; Huber, T.B.; Kunzendorf, U.; Krautwald, S. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012, 81, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.C.; Davidson, S.M.; Lim, S.Y.; Simpkin, J.C.; Hothersall, J.S.; Yellon, D.M. Necrostatin: A potentially novel cardioprotective agent? Cardiovasc Drugs Ther. 2007, 21, 227–233. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Y.; Bai, G.; Li, H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2011, 2, e115. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Ma, C.; Qiao, J.; Zhang, C. Necroptosis contributes to the NMDA-induced excitotoxicity in rat’s cultured cortical neurons. Neurosci. Lett. 2008, 447, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding cell death signals in inflammation and immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Vanden Berghe, T.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Epperly, M.; Watkins, S.C.; Greenberger, J.S.; Kagan, V.E.; Bayir, H. Necrostatin-1 rescues mice from lethal irradiation. Biochim. Biophys. Acta. 2016, 1862, 850–856. [Google Scholar] [CrossRef]
- Wu, J.; Mulatibieke, T.; Ni, J.; Han, X.; Li, B.; Zeng, Y.; Wan, R.; Wang, X.; Hu, G. Dichotomy between Receptor-Interacting Protein 1- and Receptor-Interacting Protein 3-Mediated Necroptosis in Experimental Pancreatitis. Am. J. Pathol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Teng, X.; Degterev, A.; Jagtap, P.; Xing, X.; Choi, S.; Denu, R.; Yuan, J.; Cuny, G.D. Structure-activity relationship study of novel necroptosis inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 5039–5044. [Google Scholar] [CrossRef]
- Li, Z.; Ma, B.; Lu, M.; Qiao, X.; Sun, B.; Zhang, W.; Xue, D. Construction of network for protein kinases that play a role in acute pancreatitis. Pancreas 2013, 42, 607–613. [Google Scholar] [CrossRef]
- Polykratis, A.; Hermance, N.; Zelic, M.; Roderick, J.; Kim, C.; Van, T.-M.; Lee, T.H.; Chan, F.K.M.; Pasparakis, M.; Kelliher, M.A. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 2014, 193, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Jouan-Lanhouet, S.; Riquet, F.; Duprez, L.; Vanden Berghe, T.; Takahashi, N.; Vandenabeele, P. Necroptosis, in vivo detection in experimental disease models. Semin. Cell Dev. Biol. 2014, 35, 2–13. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.W.; Kaiser, W.J.; Mocarski, E.S. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012, 11, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774. [Google Scholar] [CrossRef] [Green Version]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; et al. Menadione-induced Reactive Oxygen Species Generation via Redox Cycling Promotes Apoptosis of Murine Pancreatic Acinar Cells. J. Biol. Chem. 2006, 281, 40485–40492. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chen, X.D.; Yu, J.; Chi, J.L.; Long, F.W.; Yang, H.W.; Chen, K.L.; Lv, Z.Y.; Zhou, B.; Peng, Z.H.; et al. Deletion Of XIAP reduces the severity of acute pancreatitis via regulation of cell death and nuclear factor-kappaB activity. Cell Death Dis. 2017, 8, e2685. [Google Scholar] [CrossRef]
- Kaiser, A.M.; Saluja, A.K.; Sengupta, A.; Saluja, M.; Steer, M.L. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 1995, 269, C1295–C1304. [Google Scholar] [CrossRef]
- Mareninova, O.A.; Sung, K.F.; Hong, P.; Lugea, A.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Cell death in pancreatitis: Caspases protect from necrotizing pancreatitis. J. Biol. Chem. 2006, 281, 3370–3381. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.C.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.V.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J. Biol. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [Green Version]
- Voronina, S.; Longbottom, R.; Sutton, R.; Petersen, O.H.; Tepikin, A. Bile acids induce calcium signals in mouse pancreatic acinar cells: Implications for bile-induced pancreatic pathology. J. Physiol. 2002, 540, 49–55. [Google Scholar] [CrossRef]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in mice reduces store-operated Ca2+ influx and the severity of acute pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Takemoto, K.; Hatano, E.; Iwaisako, K.; Takeiri, M.; Noma, N.; Ohmae, S.; Toriguchi, K.; Tanabe, K.; Tanaka, H.; Seo, S.; et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014, 4, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Shindo, R.; Kakehashi, H.; Okumura, K.; Kumagai, Y.; Nakano, H. Critical contribution of oxidative stress to TNFalpha-induced necroptosis downstream of RIPK1 activation. Biochem. Biophys. Res. Commun. 2013, 436, 212–216. [Google Scholar] [CrossRef]
- Fanczal, J.; Pallagi, P.; Gorog, M.; Diszhazi, G.; Almassy, J.; Madacsy, T.; Varga, A.; Csernay-Biro, P.; Katona, X.; Toth, E.; et al. TRPM2-mediated extracellular Ca(2+) entry promotes acinar cell necrosis in biliary acute pancreatitis. J. Physiol. 2020, 598, 1253–1270. [Google Scholar] [CrossRef] [Green Version]
- Acovic, A.; Gazdic, M.; Jovicic, N.; Harrell, C.R.; Fellabaum, C.; Arsenijevic, N.; Volarevic, V. Role of indoleamine 2,3-dioxygenase in pathology of the gastrointestinal tract. Therap. Adv. Gastroenterol. 2018, 11, 1756284818815334. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.W.; Terentis, A.C.; King, N.J.; Thomas, S.R. Role of indoleamine 2,3-dioxygenase in health and disease. Clin. Sci. 2015, 129, 601–672. [Google Scholar] [CrossRef]
- Taher, Y.A.; Piavaux, B.J.; Gras, R.; van Esch, B.C.; Hofman, G.A.; Bloksma, N.; Henricks, P.A.; van Oosterhout, A.J. Indoleamine 2,3-dioxygenase-dependent tryptophan metabolites contribute to tolerance induction during allergen immunotherapy in a mouse model. J. Allergy Clin. Immunol. 2008, 121, 983–991. [Google Scholar] [CrossRef]
- Choi, B.K.; Asai, T.; Vinay, D.S.; Kim, Y.H.; Kwon, B.S. 4-1BB-mediated amelioration of experimental autoimmune uveoretinitis is caused by indoleamine 2,3-dioxygenase-dependent mechanisms. Cytokine 2006, 34, 233–242. [Google Scholar] [CrossRef]
- Fallarino, F.; Volpi, C.; Zelante, T.; Vacca, C.; Calvitti, M.; Fioretti, M.C.; Puccetti, P.; Romani, L.; Grohmann, U. IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J. Immunol. 2009, 183, 6303–6312. [Google Scholar] [CrossRef] [Green Version]
- El-Zaatari, M.; Bass, A.J.; Bowlby, R.; Zhang, M.; Syu, L.J.; Yang, Y.; Grasberger, H.; Shreiner, A.; Tan, B.; Bishu, S.; et al. Indoleamine 2,3-Dioxygenase 1, Increased in Human Gastric Pre-Neoplasia, Promotes Inflammation and Metaplasia in Mice and Is Associated With Type II Hypersensitivity/Autoimmunity. Gastroenterology 2018, 154, 140–153. [Google Scholar] [CrossRef]
- Xu, H.; Oriss, T.B.; Fei, M.; Henry, A.C.; Melgert, B.N.; Chen, L.; Mellor, A.L.; Munn, D.H.; Irvin, C.G.; Ray, P.; et al. Indoleamine 2,3-dioxygenase in lung dendritic cells promotes Th2 responses and allergic inflammation. Proc. Natl. Acad. Sci. USA 2008, 105, 6690–6695. [Google Scholar] [CrossRef] [Green Version]
- Scott, G.N.; DuHadaway, J.; Pigott, E.; Ridge, N.; Prendergast, G.C.; Muller, A.J.; Mandik-Nayak, L. The immunoregulatory enzyme IDO paradoxically drives B cell-mediated autoimmunity. J. Immunol. 2009, 182, 7509–7517. [Google Scholar] [CrossRef] [Green Version]
- Yue, E.W.; Sparks, R.; Polam, P.; Modi, D.; Douty, B.; Wayland, B.; Glass, B.; Takvorian, A.; Glenn, J.; Zhu, W.; et al. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of indoleamine 2,3-dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J. Am. Coll. Surg. 2008, 206, 849–854, discussion 854–846. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; Costantino, C.L.; Metz, R.; Muller, A.J.; Prendergast, G.C.; Yeo, C.J.; Brody, J.R. Genotyping and expression analysis of IDO2 in human pancreatic cancer: A novel, active target. J. Am. Coll. Surg. 2009, 208, 781–787, discussion 787–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skouras, C.; Zheng, X.; Binnie, M.; Homer, N.Z.; Murray, T.B.; Robertson, D.; Briody, L.; Paterson, F.; Spence, H.; Derr, L.; et al. Increased levels of 3-hydroxykynurenine parallel disease severity in human acute pancreatitis. Sci. Rep. 2016, 6, 33951. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Davidson, S.M.; Mocanu, M.M.; Yellon, D.M.; Smith, C.C. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007, 21, 467–469. [Google Scholar] [CrossRef] [Green Version]
- Shalbueva, N.; Mareninova, O.A.; Gerloff, A.; Yuan, J.; Waldron, R.T.; Pandol, S.J.; Gukovskaya, A.S. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2013, 144, 437–446. [Google Scholar] [CrossRef] [Green Version]
- Toth, E.; Maleth, J.; Zavogyan, N.; Fanczal, J.; Grassalkovich, A.; Erdos, R.; Pallagi, P.; Horvath, G.; Tretter, L.; Balint, E.R.; et al. Novel mitochondrial transition pore inhibitor N-methyl-4-isoleucine cyclosporin is a new therapeutic option in acute pancreatitis. J. Physiol. 2019, 597, 5879–5898. [Google Scholar] [CrossRef] [Green Version]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small Molecule Inhibitors of Cyclophilin D To Protect Mitochondrial Function as a Potential Treatment for Acute Pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Criddle, D.N. Keeping mitochondria happy-benefits of a pore choice in acute pancreatitis. J. Physiol. 2019, 597, 5741–5742. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ouyang, Y.; Wen, L.; Armstrong, J.A.; Chvanov, M.; Latawiec, D.; Cai, W.; Awais, M.; Mukherjee, R.; Huang, W.; Gough, P.J.; et al. Protective Effects of Necrostatin-1 in Acute Pancreatitis: Partial Involvement of Receptor Interacting Protein Kinase 1. Cells 2021, 10, 1035. https://doi.org/10.3390/cells10051035
Ouyang Y, Wen L, Armstrong JA, Chvanov M, Latawiec D, Cai W, Awais M, Mukherjee R, Huang W, Gough PJ, et al. Protective Effects of Necrostatin-1 in Acute Pancreatitis: Partial Involvement of Receptor Interacting Protein Kinase 1. Cells. 2021; 10(5):1035. https://doi.org/10.3390/cells10051035
Chicago/Turabian StyleOuyang, Yulin, Li Wen, Jane A. Armstrong, Michael Chvanov, Diane Latawiec, Wenhao Cai, Mohammad Awais, Rajarshi Mukherjee, Wei Huang, Peter J. Gough, and et al. 2021. "Protective Effects of Necrostatin-1 in Acute Pancreatitis: Partial Involvement of Receptor Interacting Protein Kinase 1" Cells 10, no. 5: 1035. https://doi.org/10.3390/cells10051035