Synthesis of Hybrid-Polypeptides m-PEO-b-poly(His-co-Gly) and m-PEO-b-poly(His-co-Ala) and Study of Their Structure and Aggregation. Influence of Hydrophobic Copolypeptides on the Properties of Poly(L-histidine)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Nim-Trityl Protected N-Carboxyanhydride of L-Histidine (Trt-His-NCA)
2.3. Synthesis of N-Carboxy Anhydride of Glycine (Gly-NCA)
2.4. Synthesis of N-Carboxy Anhydride of L-Alanine (Ala-NCA)
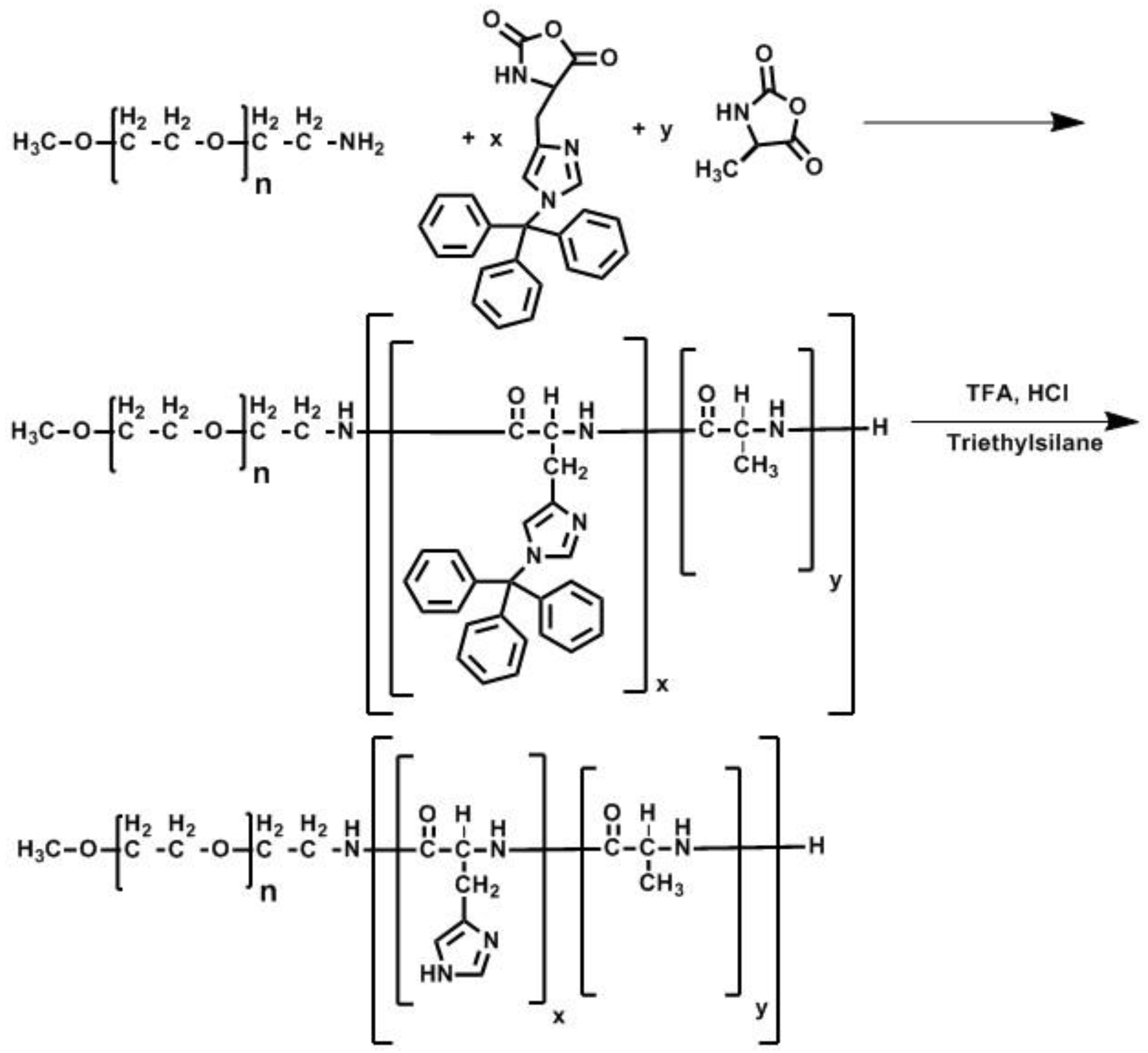
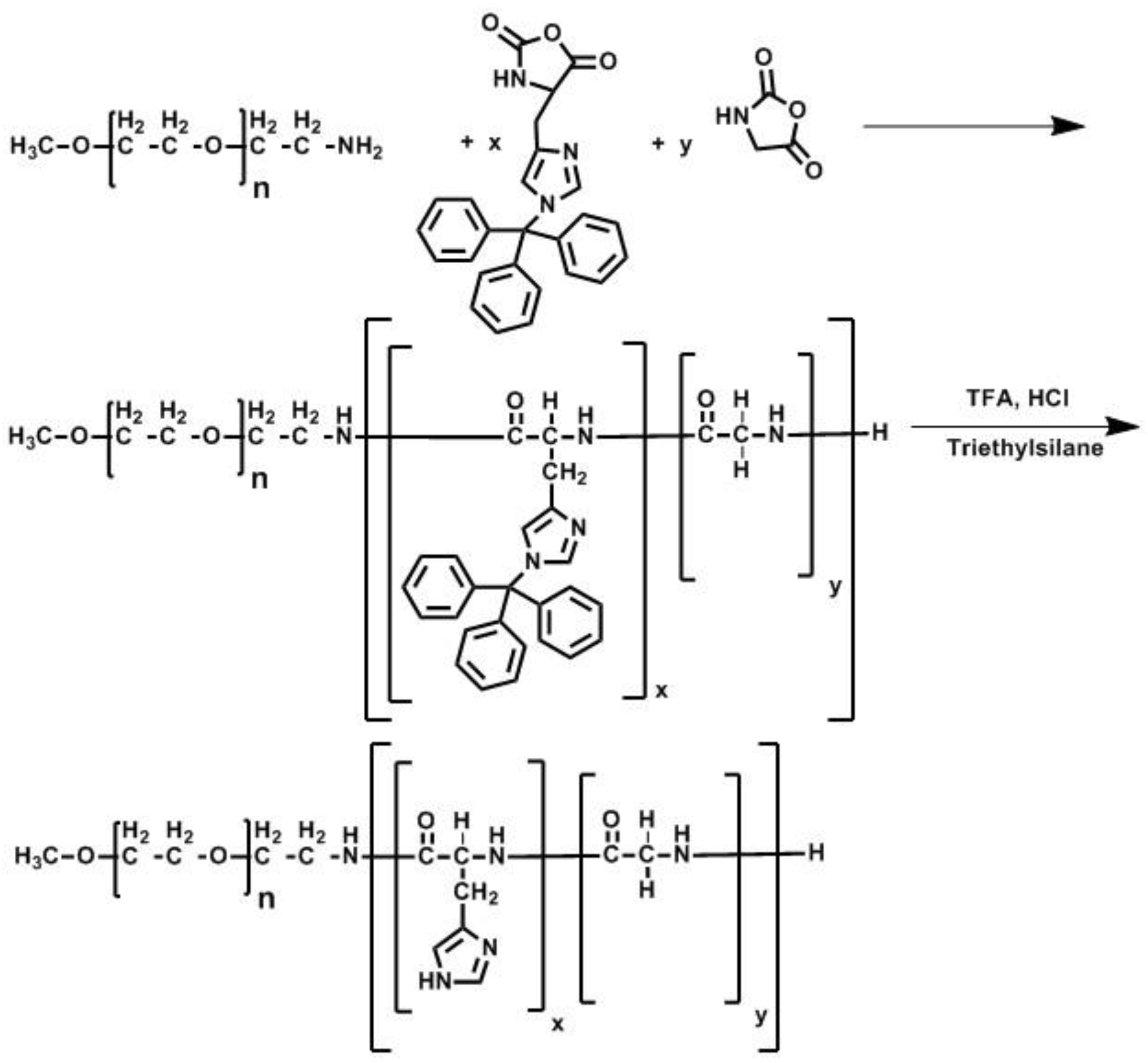
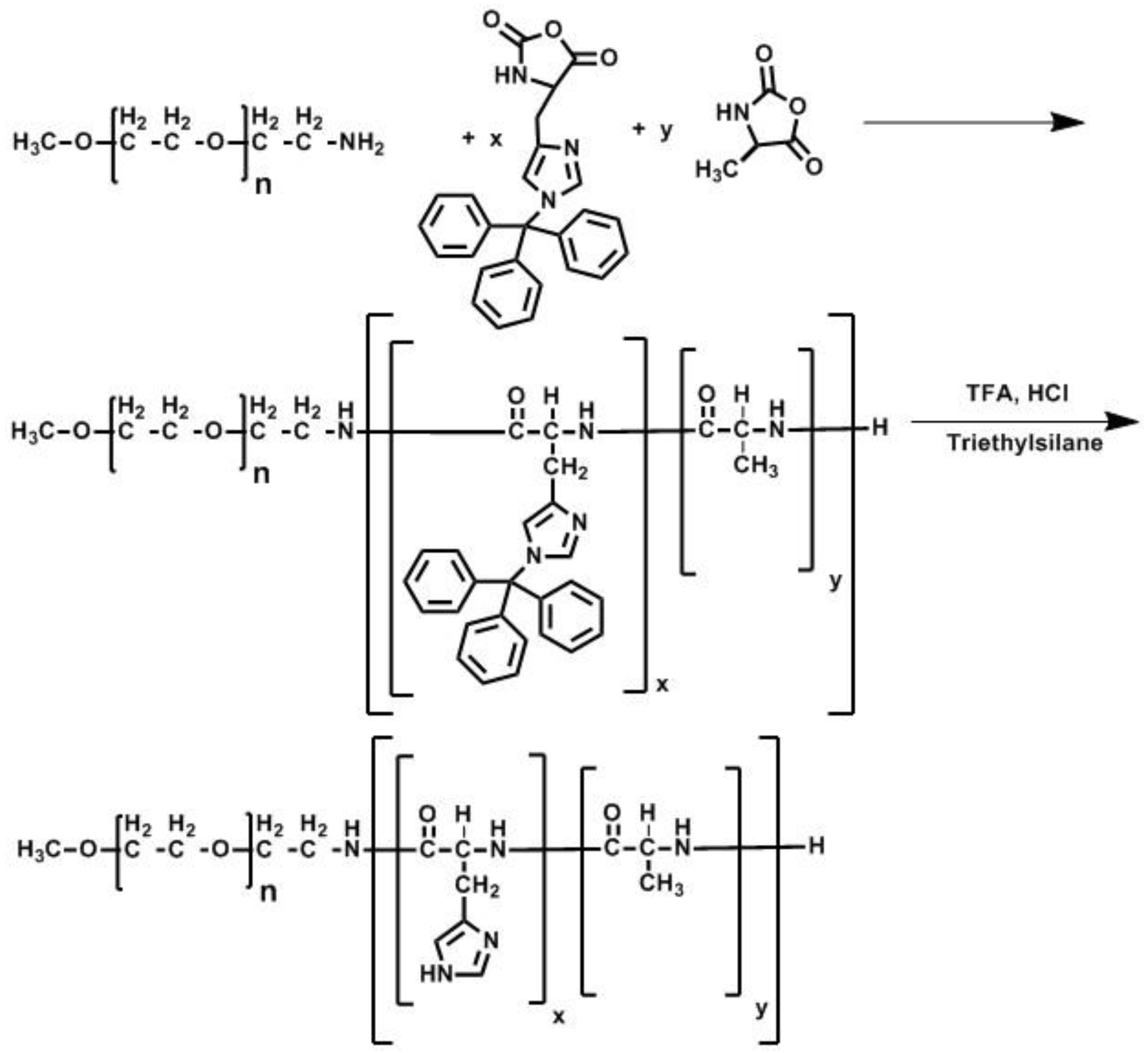
2.5. Polymer Synthesis
2.6. Characterization
3. Results and Discussion
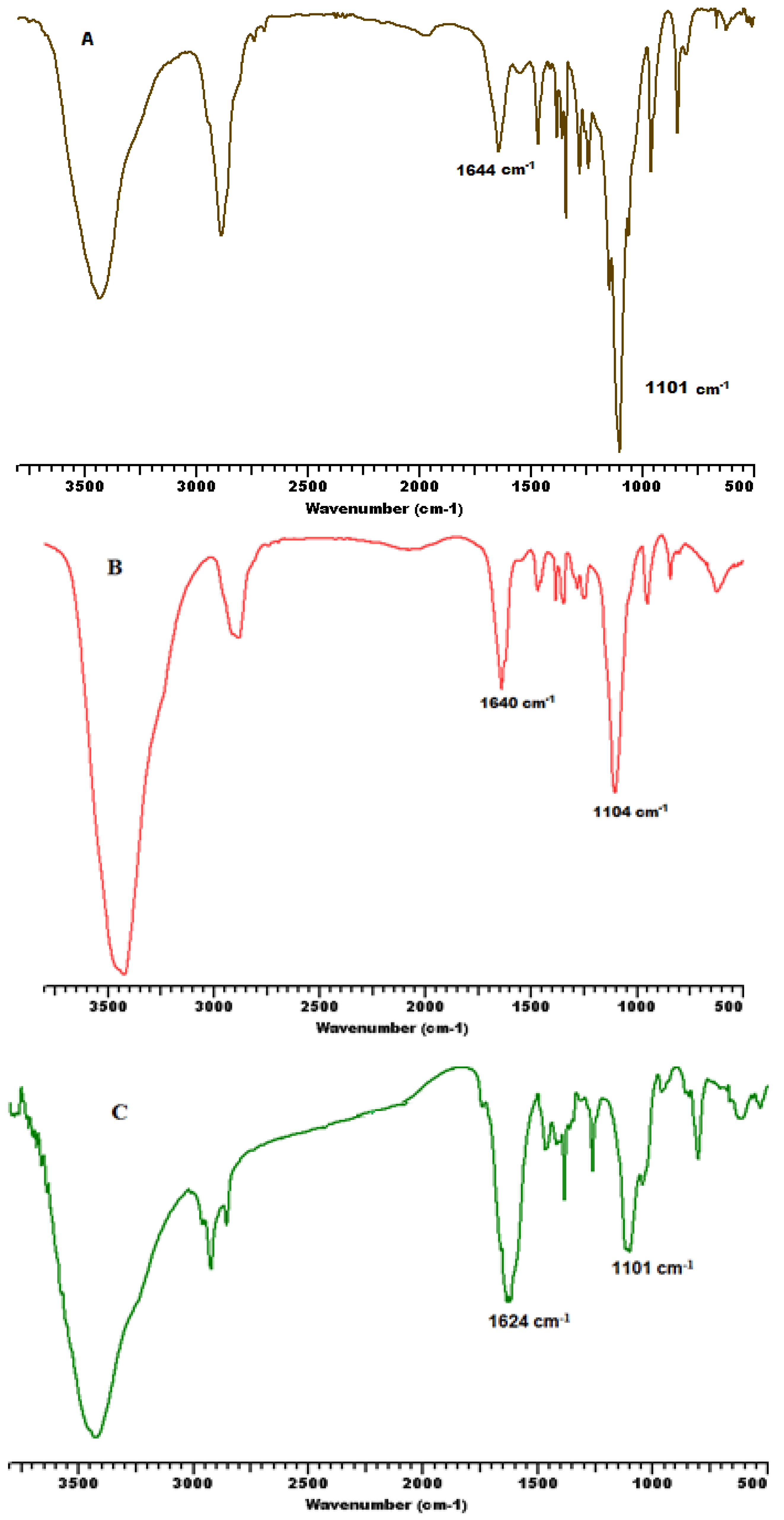
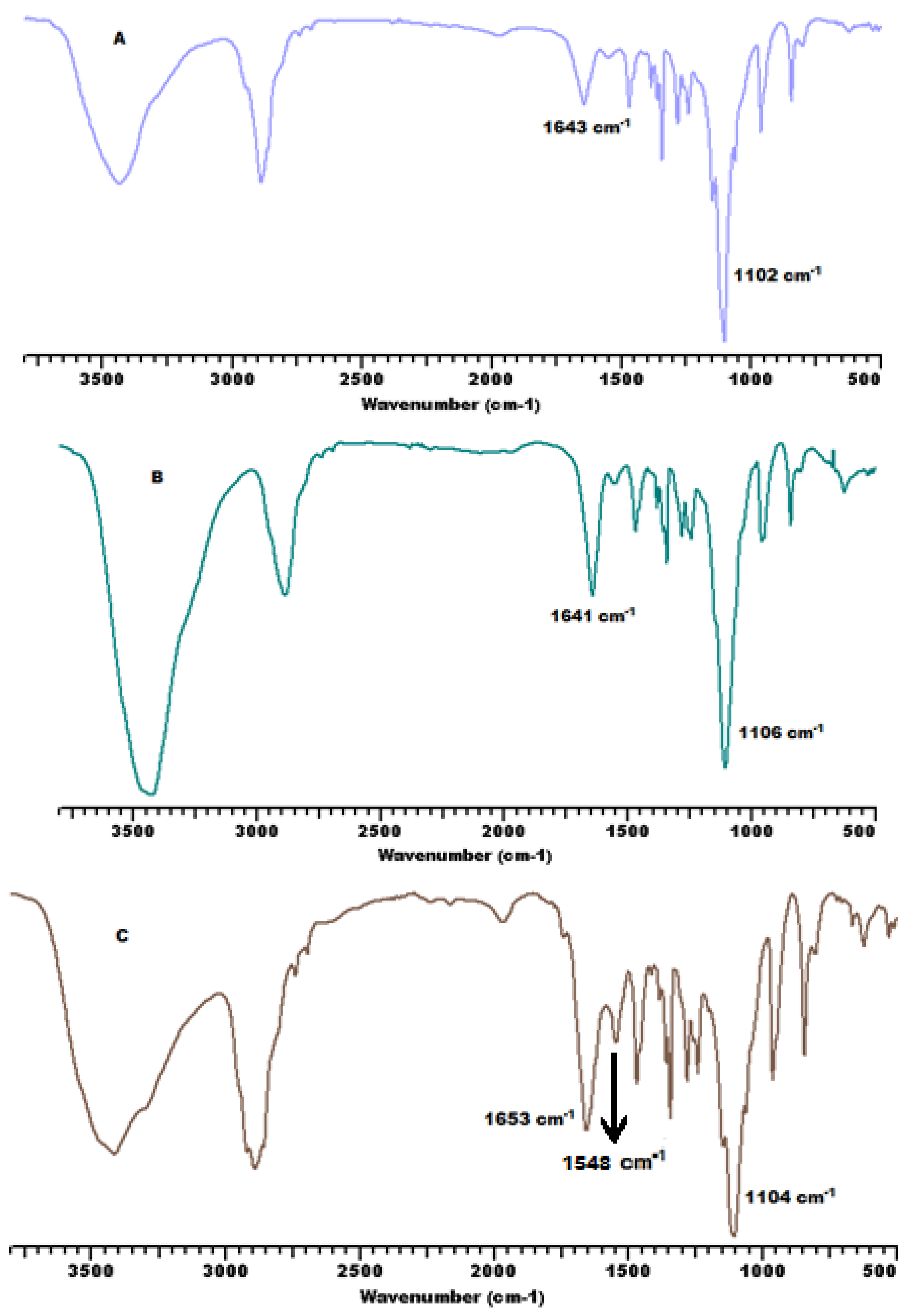
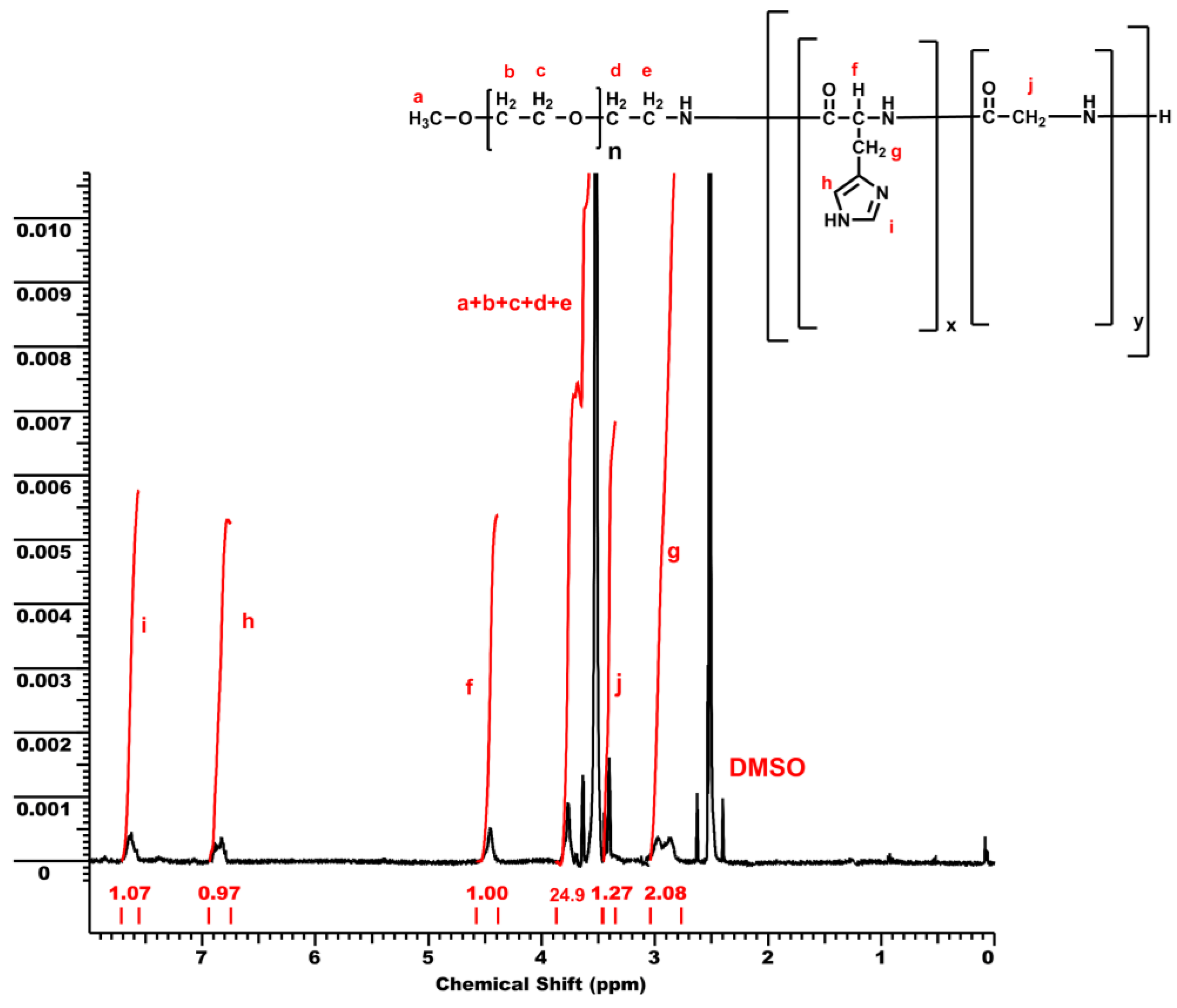
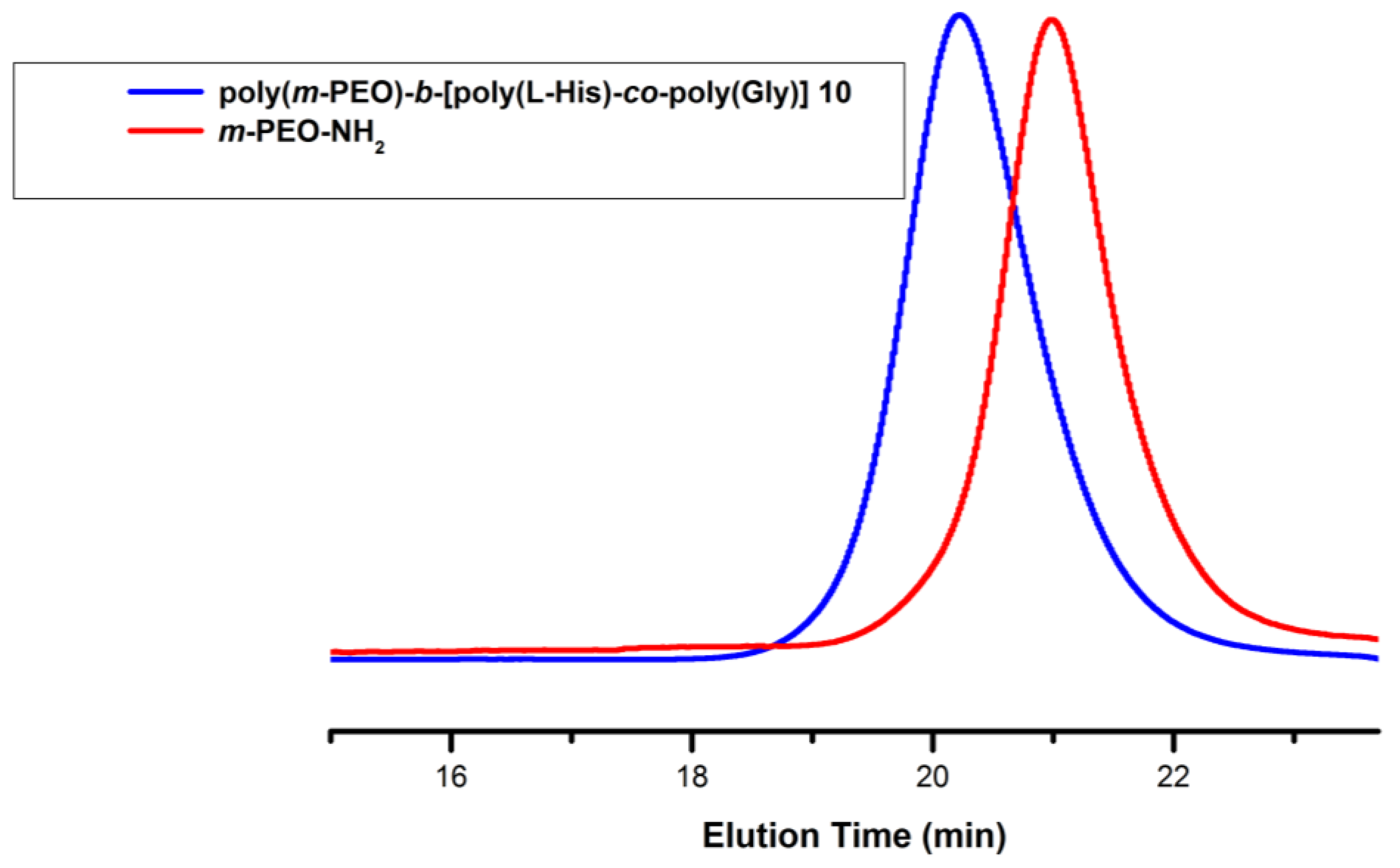
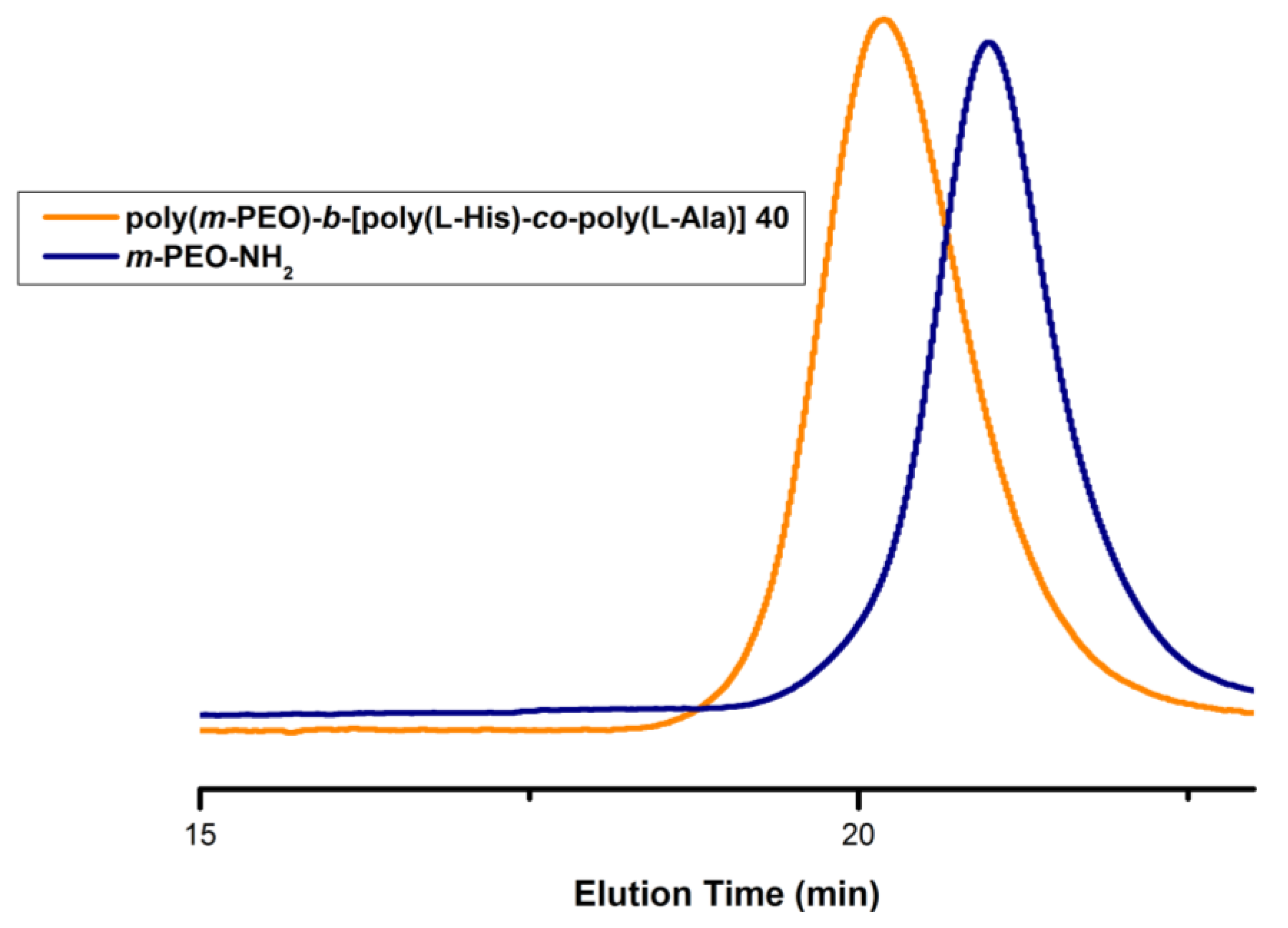
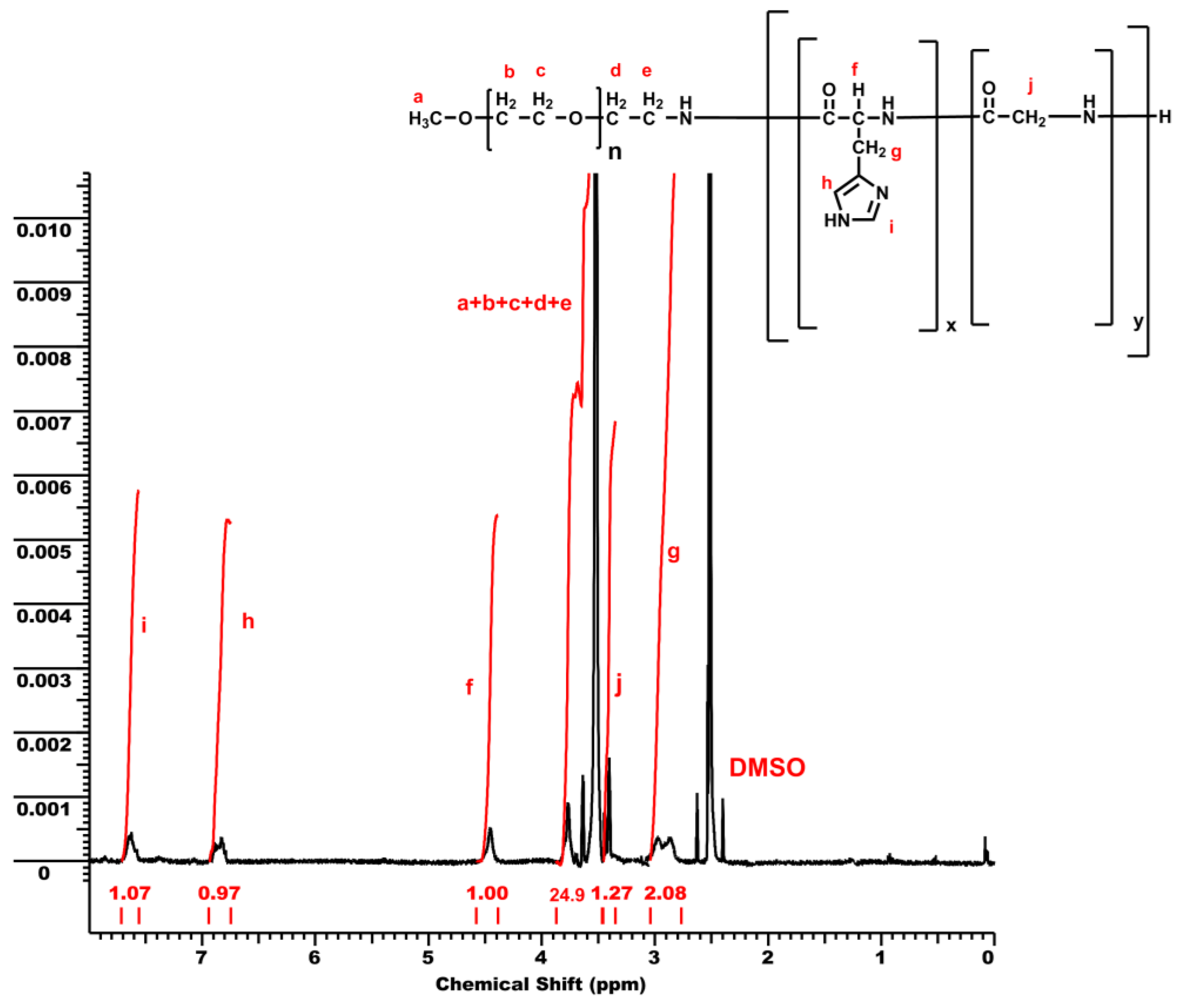
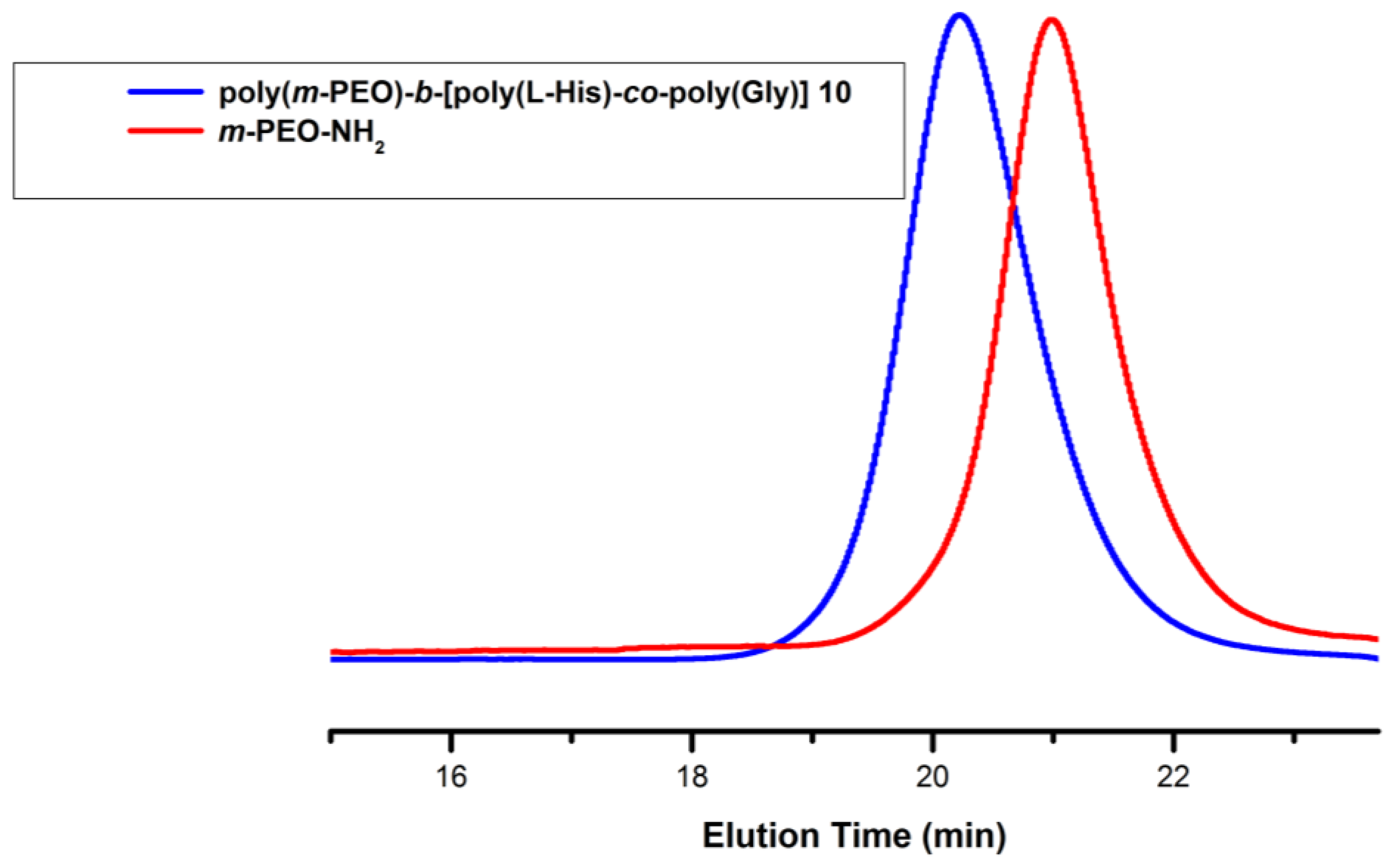
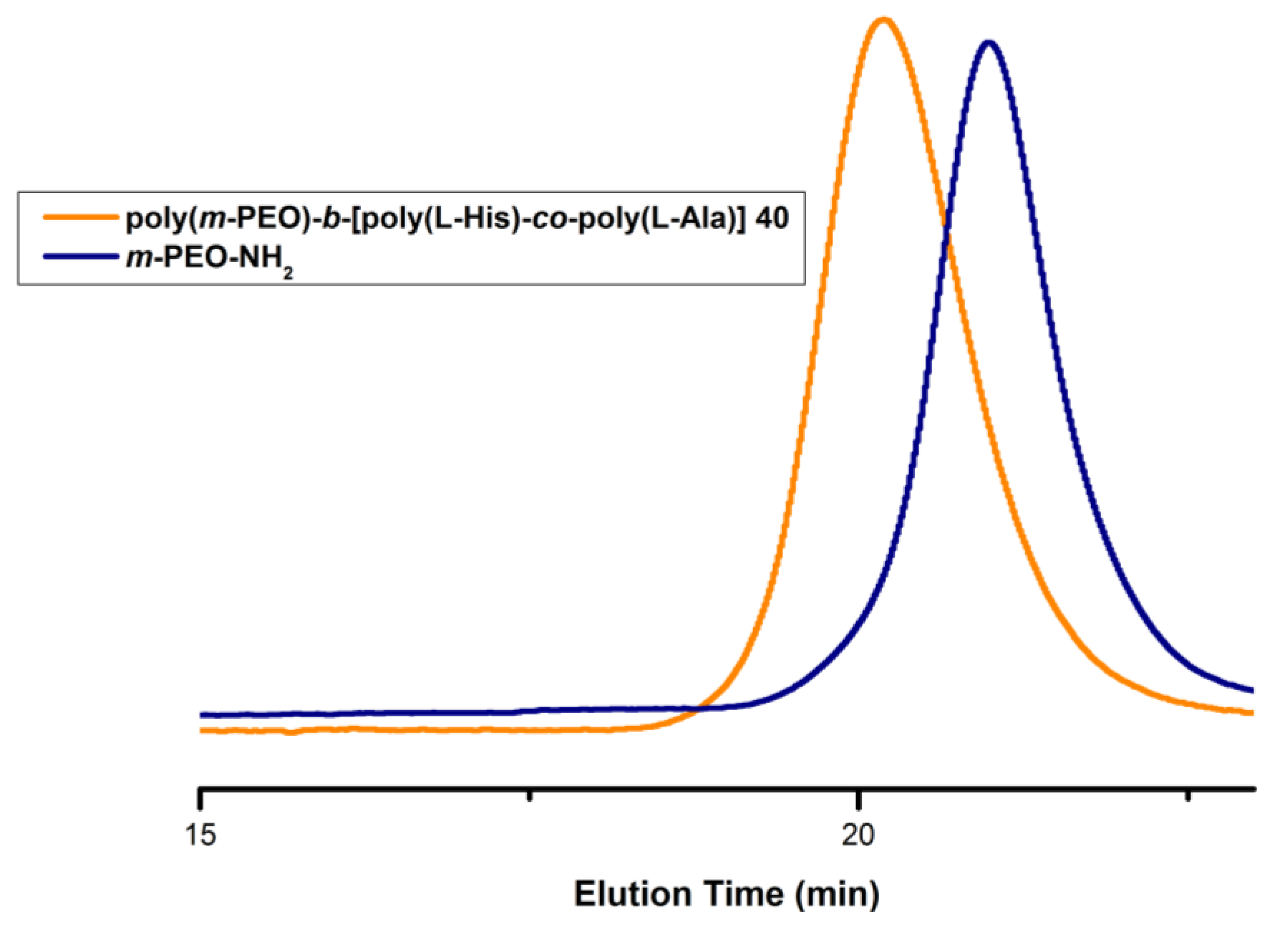
3.1. Synthesis
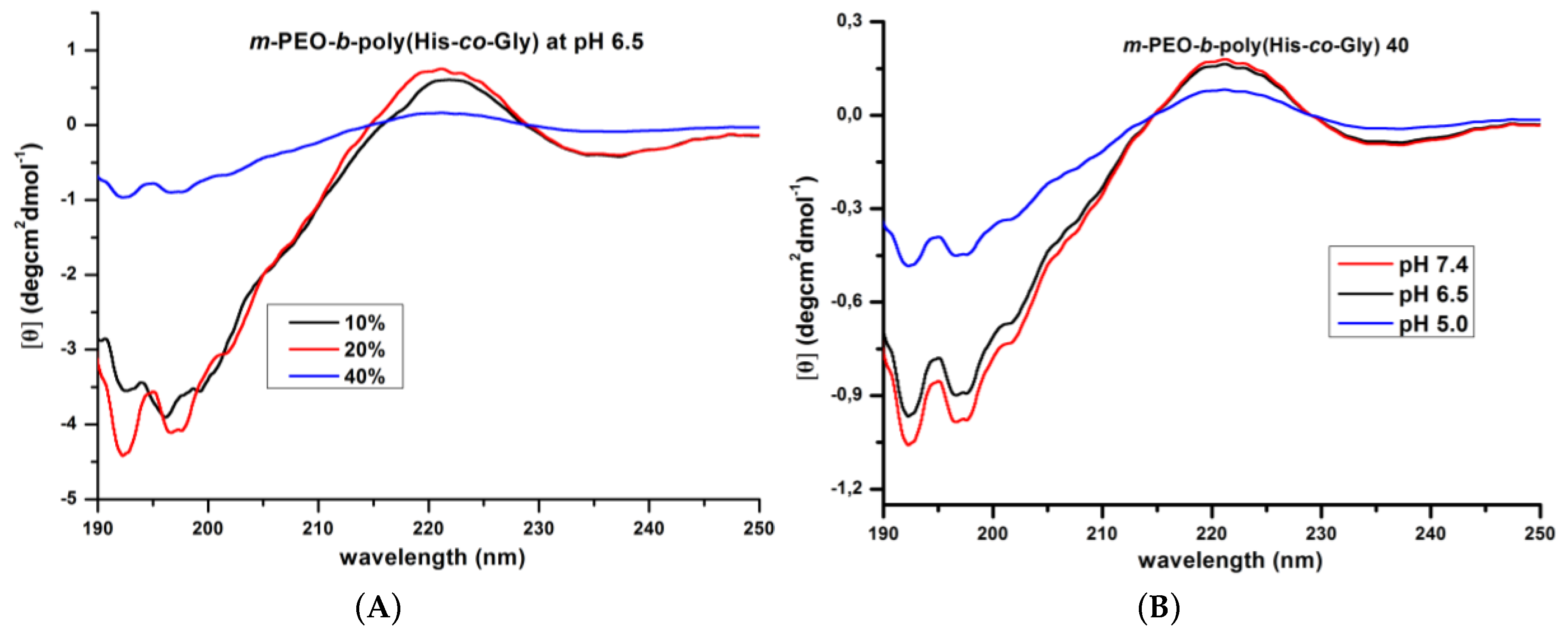
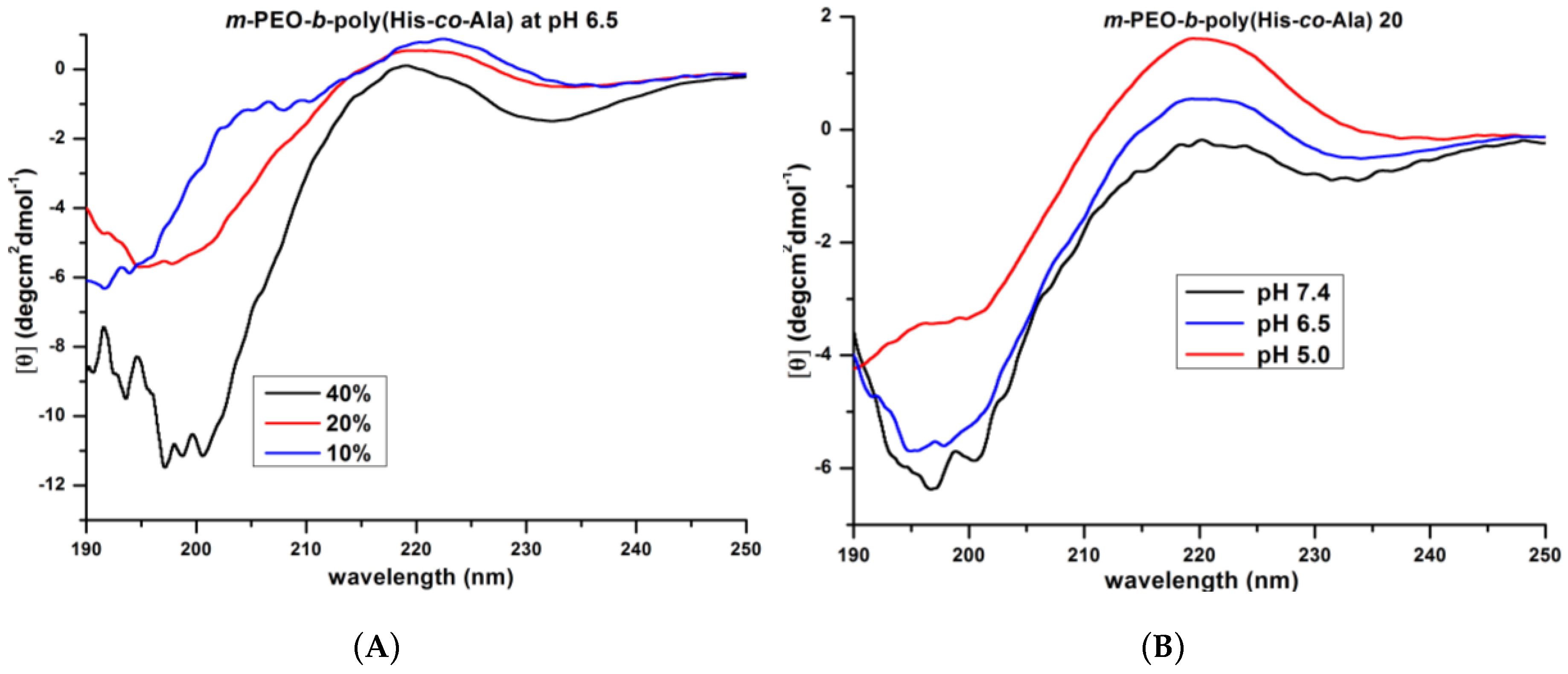
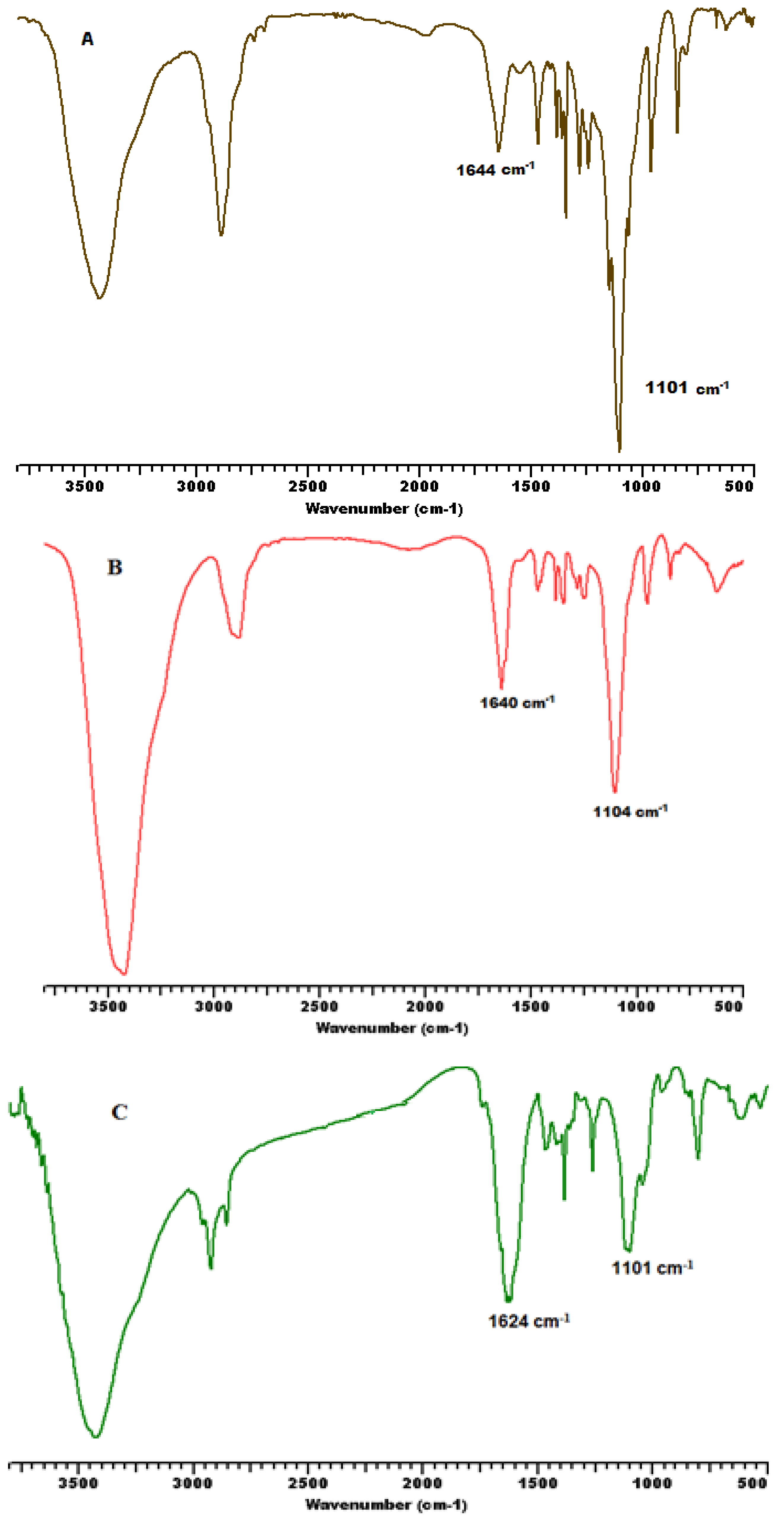
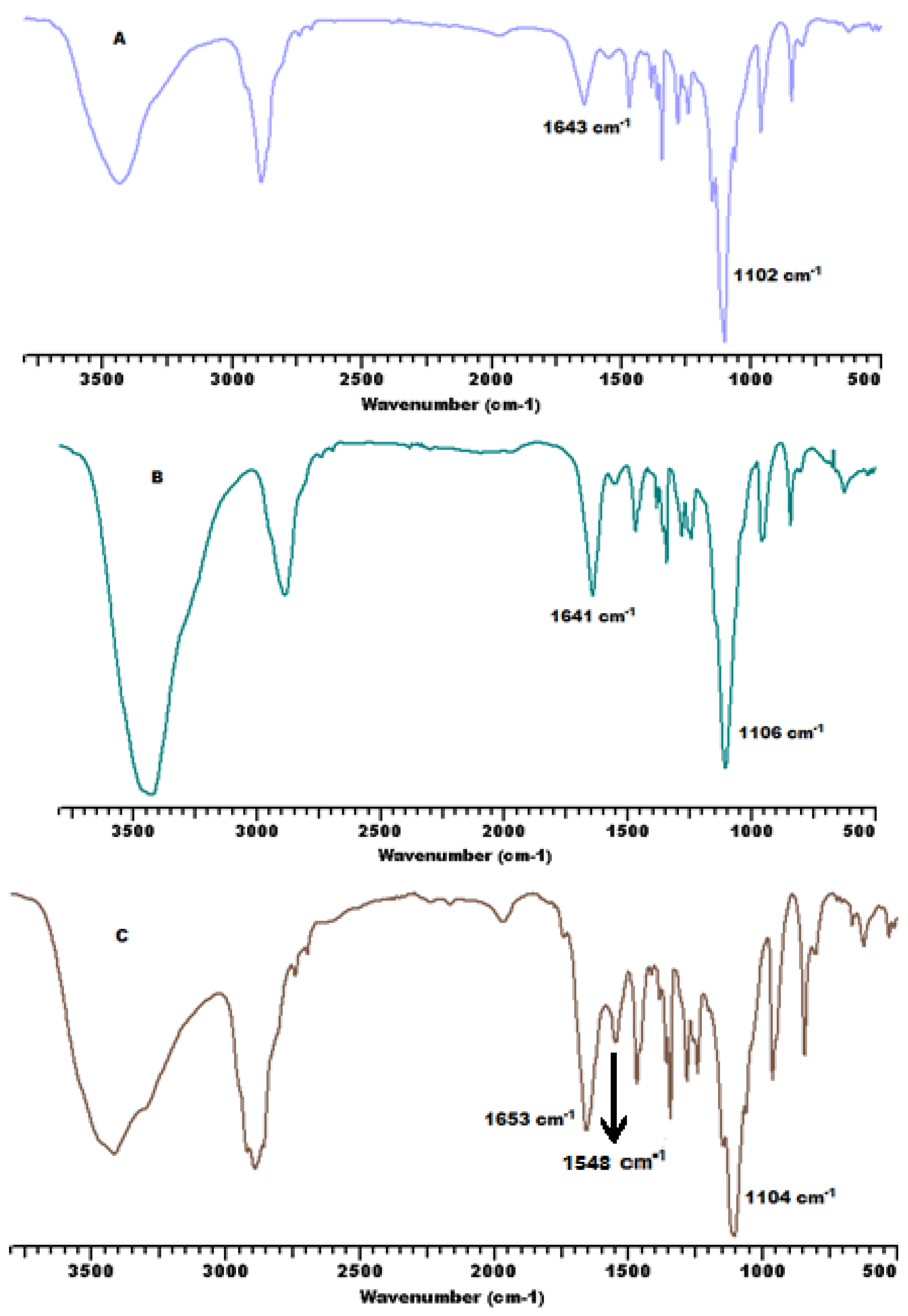
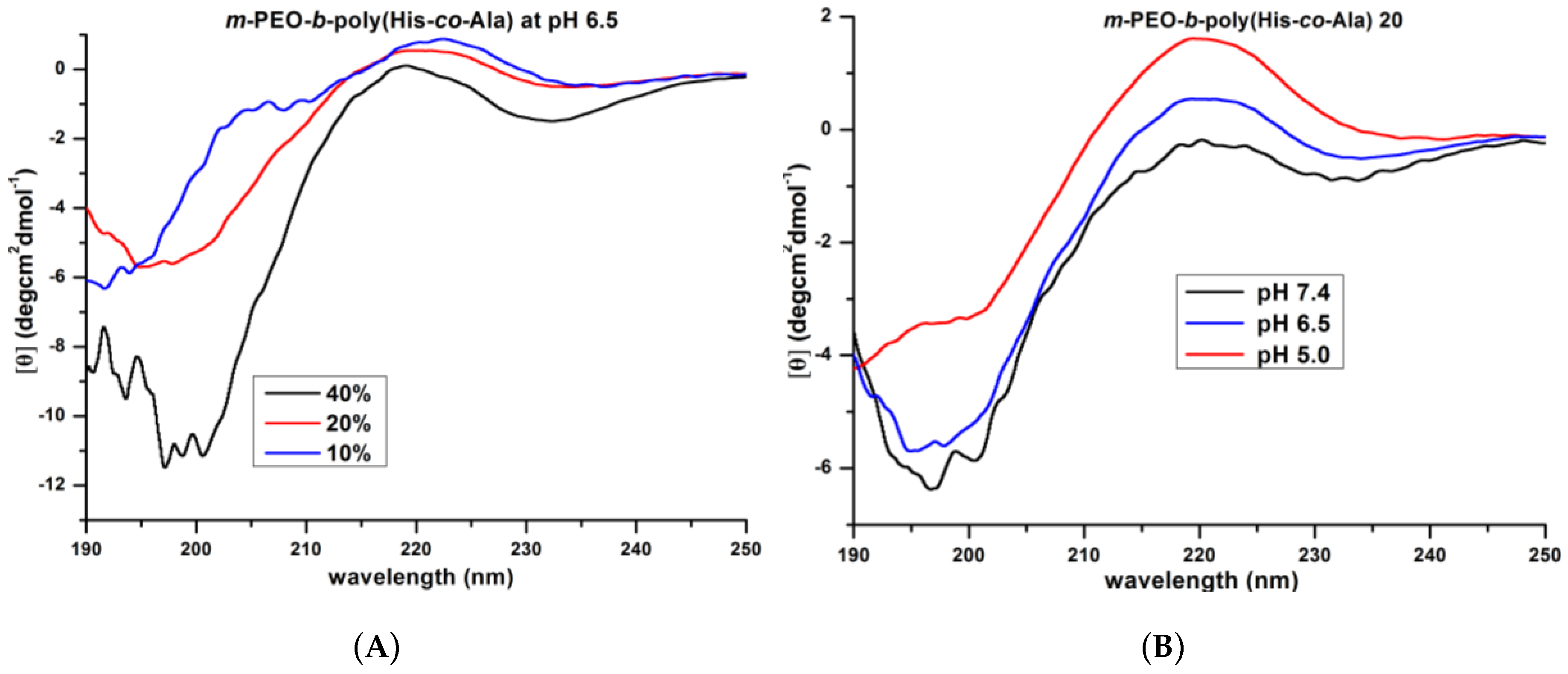
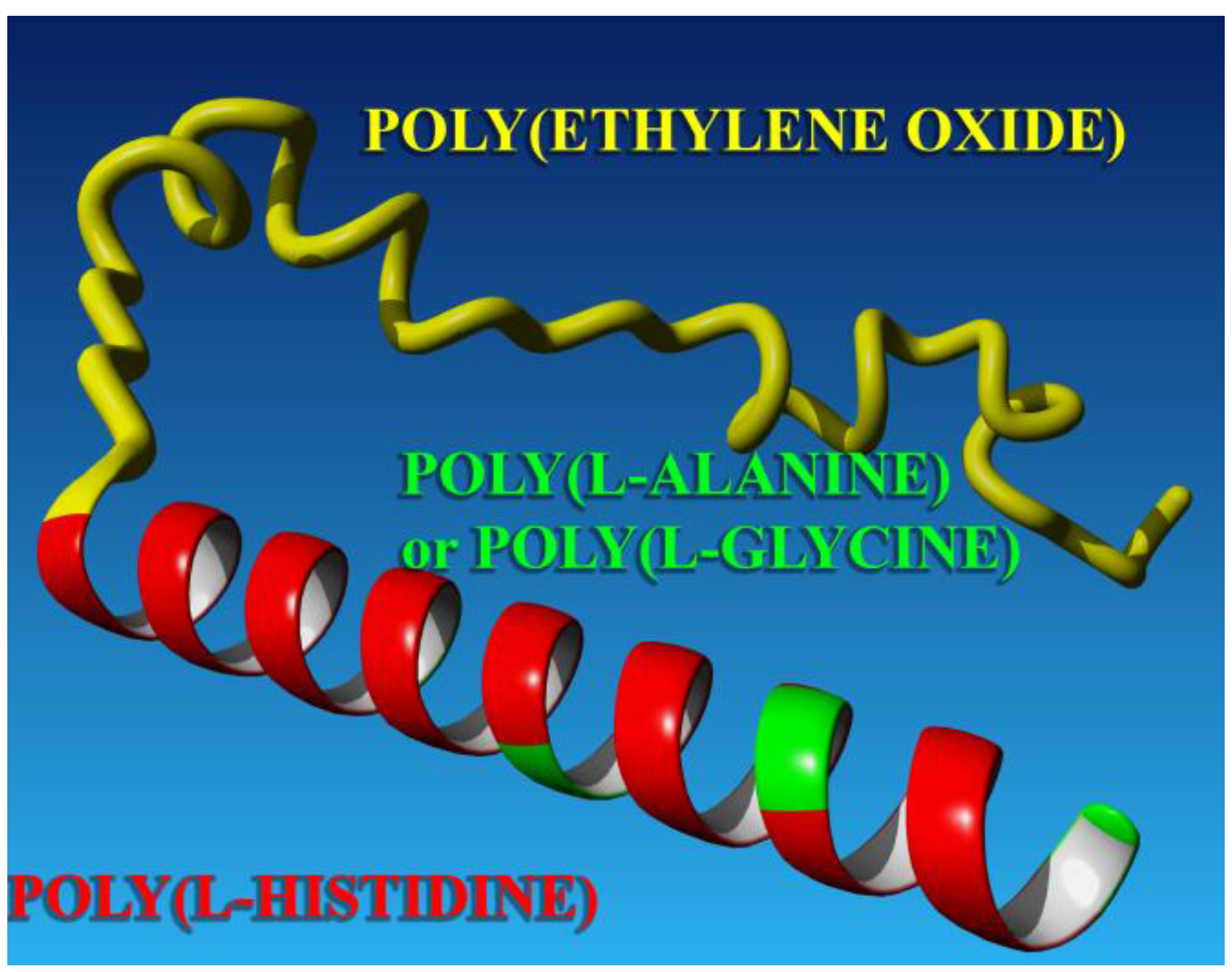
3.2. Secondary Structure
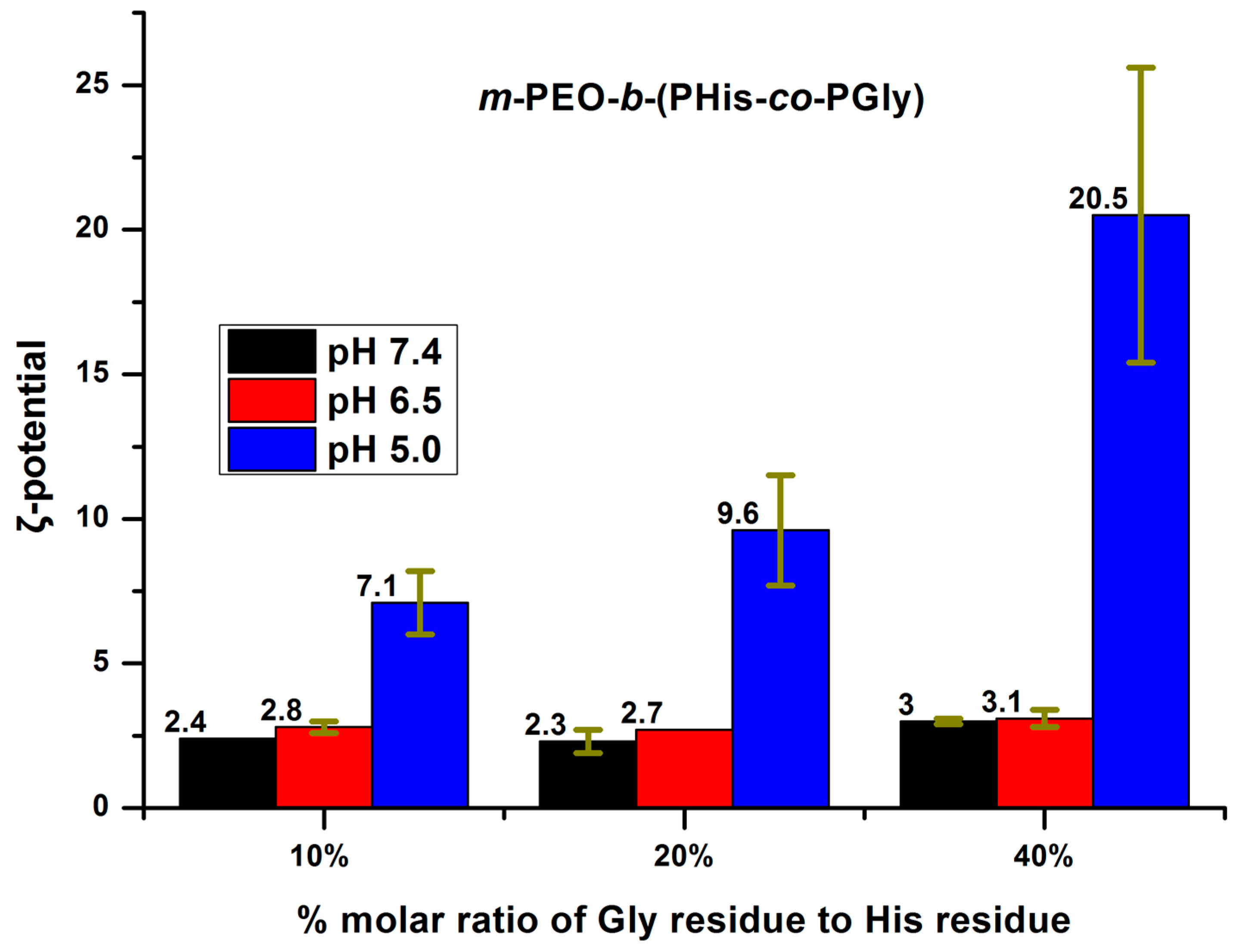
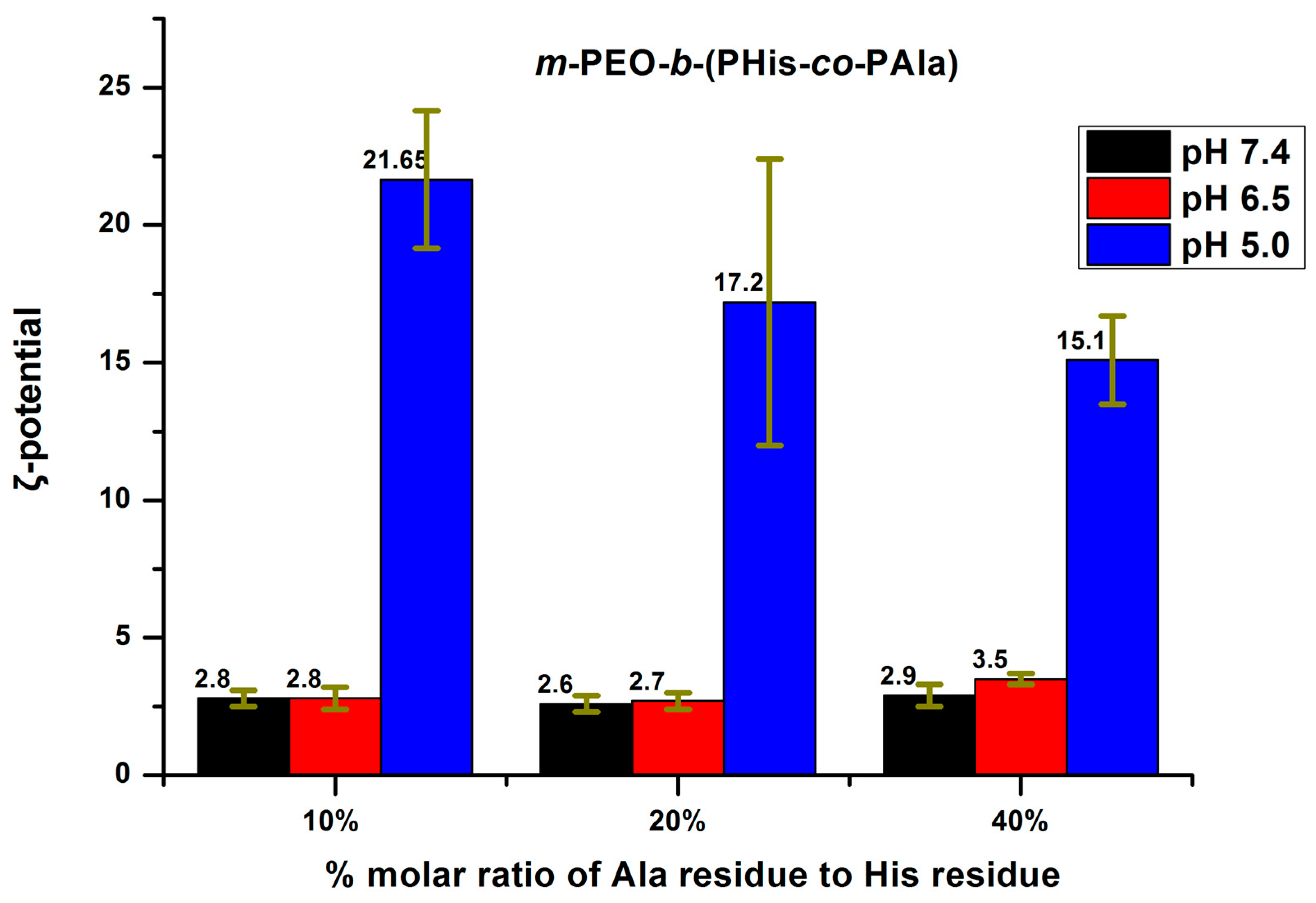
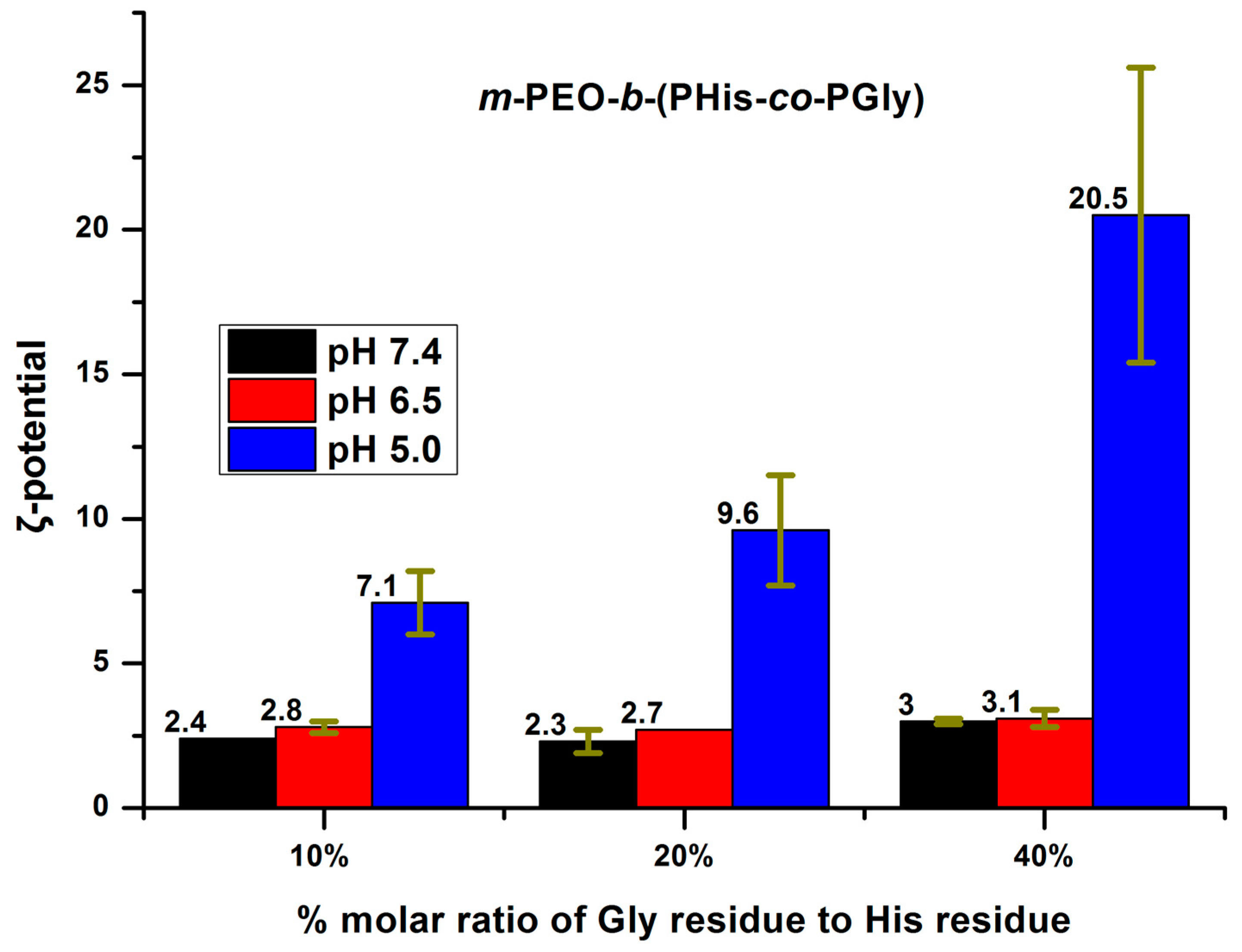
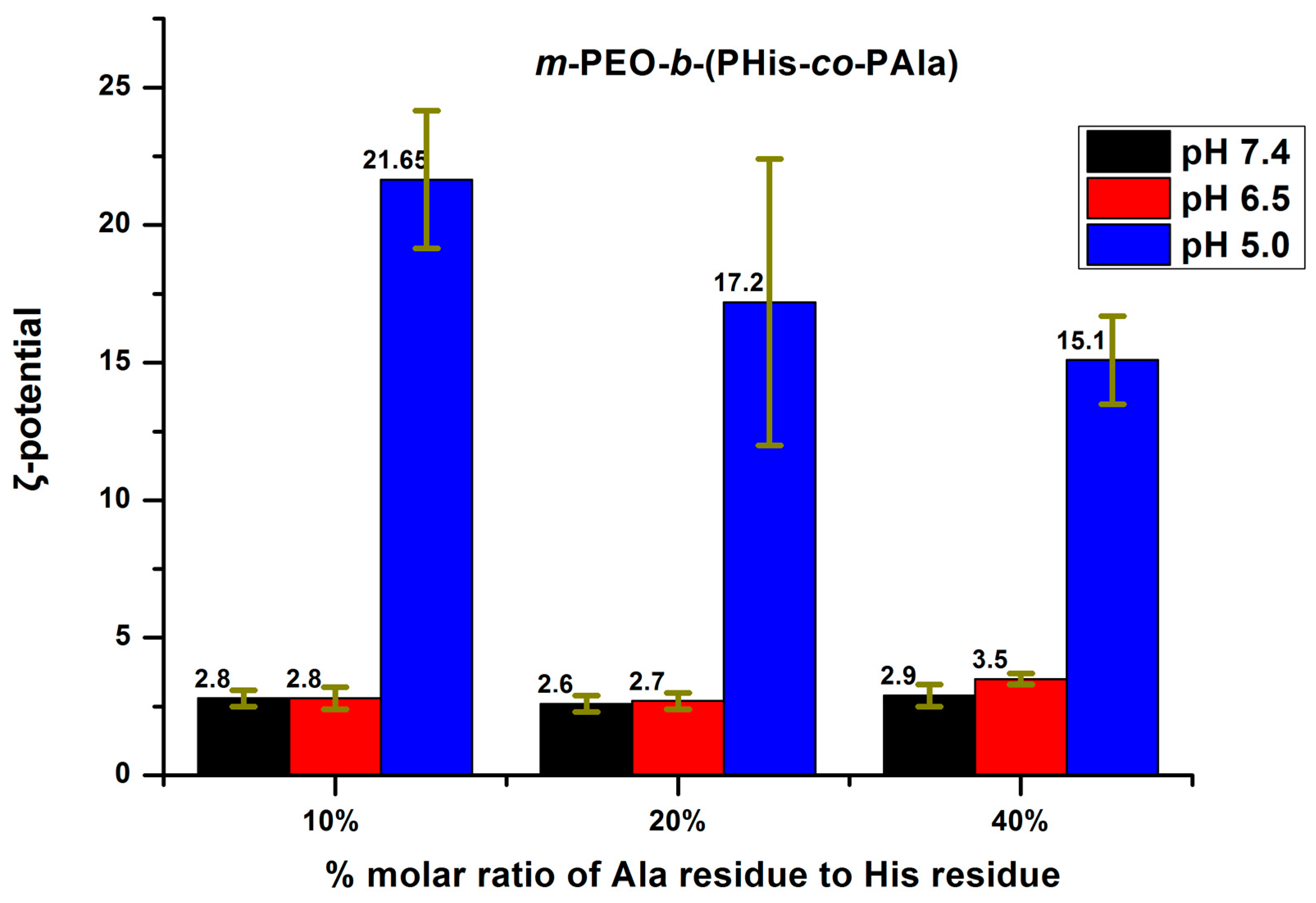
3.3. Aggregation Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, J.; Heise, A. Stimuli responsive synthetic polypeptides derived from n-carboxyanhydride (NCA) polymerisation. Chem. Soc. Rev. 2013, 42, 7373–7390. [Google Scholar] [CrossRef] [PubMed]
- He, C.L.; Zhuang, X.L.; Tang, Z.H.; Tian, H.Y.; Chen, X.S. Stimuli-sensitive synthetic polypeptide-based materials for drug and gene delivery. Adv. Healthc. Mater. 2012, 1, 48–78. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.L.; Lavasanifar, A.; Kwon, G.S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Checot, F.; Lecommandoux, S.; Gnanou, Y.; Klok, H.A. Water-soluble stimuli-responsive vesicles from peptide-based diblock copolymers. Angew. Chem. Int. Ed. 2002, 41, 1339–1343. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.; Shah, A.; Siddiq, M.; Kraatz, H.B. Polymeric micelles as drug delivery vehicles. RSC Adv. 2014, 4, 17028–17038. [Google Scholar] [CrossRef]
- Lee, J.S.; Feijen, J. Polymersomes for drug delivery: Design, formation and characterization. J. Control. Release 2012, 161, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef] [PubMed]
- Van Thienen, T.G.; Lucas, B.; Demeester, J.; De Smedt, S.C. On the synthesis and characterization of biodegradable dextran nanogels with tunable degradation properties. J. Control. Release 2006, 116, E12–E13. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Fu, X.H.; Fu, W.X.; Li, Z.B. Biodegradable stimuli-responsive polypeptide materials prepared by ring opening polymerization. Chem. Soc. Rev. 2015, 44, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorf, H.R. Polypeptides and 100 years of chemistry of alpha-amino acid N-carboxyanhydrides. Angew. Chem. Int. Ed. Engl. 2006, 45, 5752–5784. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Wu, J.; Cheng, R.; Meng, F.; Klok, H.-A.; Zhong, Z. Functional polypeptide and hybrid materials: Precision synthesis via α-amino acid n-carboxyanhydride polymerization and emerging biomedical applications. Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar] [CrossRef]
- Hehir, S.; Cameron, N.R. Recent advances in drug delivery systems based on polypeptides prepared fromn-carboxyanhydrides. Polym. Int. 2014, 63, 943–954. [Google Scholar] [CrossRef]
- Van Vlerken, L.E.; Vyas, T.K.; Amiji, M.M. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm. Res. 2007, 24, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Domb, A.; Quellec, P.; Blunk, T.; Muller, R.H.; Verbavatz, J.M.; Langer, R. The controlled intravenous delivery of drugs using peg-coated sterically stabilized nanospheres. Adv. Drug Deliv. Rev. 2012, 64, 316–326. [Google Scholar] [CrossRef]
- Veronese, F.M.; Pasut, G. Pegylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Hamidi, M.; Azadi, A.; Rafiei, P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006, 13, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Gref, R.; Minamitake, Y.; Peracchia, M.T.; Trubetskoy, V.; Torchilin, V.; Langer, R. Biodegradable long-circulating polymeric nanospheres. Science 1994, 263, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Patchornik, A.; Berger, A.; Katchalski, E. Poly-l-histidine. J. Am. Chem. Soc. 1957, 79, 5227–5230. [Google Scholar] [CrossRef]
- Jin, Y.D.; Gao, X.H. Spectrally tunable leakage-free gold nanocontainers. J. Am. Chem. Soc. 2009, 131, 17774–17776. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.D. Multifunctional compact hybrid au nanoshells: A new generation of nanoplasmonic probes for biosensing, imaging, and controlled release. Acc. Chem. Res. 2014, 47, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.H.; Lin, Y.F.; Lin, J.J.; Yu, C.S. Prediction of metal ion-binding sites in proteins using the fragment transformation method. PLoS ONE 2012, 7, e39252. [Google Scholar] [CrossRef] [PubMed]
- Zelikin, A.N.; Becker, A.L.; Johnston, A.P.R.; Wark, K.L.; Turatti, F.; Caruso, F. A general approach for DNA encapsulation in degradable polymer microcapsules. ACS Nano 2007, 1, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Mavrogiorgis, D.; Bilalis, P.; Karatzas, A.; Skoulas, D.; Fotinogiannopoulou, G.; Iatrou, H. Controlled polymerization of histidine and synthesis of well-defined stimuli responsive polymers. Elucidation of the structure–aggregation relationship of this highly multifunctional material. Polym. Chem. 2014, 5, 6256–6278. [Google Scholar] [CrossRef]
- Kim, G.M.; Bae, Y.H.; Jo, W.H. Ph-induced micelle formation of poly(histidine-co-phenylalanine)-block-poly(ethylene glycol) in aqueous media. Macromol. Biosci. 2005, 5, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Gaspard, J.; Silas, J.A.; Shantz, D.F.; Jan, J.-S. Supramolecular assembly of lysine-b-glycine block copolypeptides at different solution conditions. Supramol. Chem. 2010, 22, 178–185. [Google Scholar] [CrossRef]
- Papadopoulos, P.; Floudas, G.; Schnell, I.; Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Nanodomain-induced chain folding in poly(gamma-benzyl-l-glutamate)-b-polyglycine diblock copolymers. Biomacromolecules 2005, 6, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Tang, Z.; Zhang, D.; Song, W.; Duan, T.; Gu, J.; Chen, X. Poly(ornithine-co-arginine-co-glycine-co-aspartic acid): Preparation via nca polymerization and its potential as a polymeric tumor-penetrating agent. Macromol. Biosci. 2015, 15, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Joo, M.K.; Sohn, Y.S.; Jeong, B. Secondary structure effect of polypeptide on reverse thermal gelation and degradation of l/dl-poly(alanine)-poloxamer-l/dl-poly(alanine) copolymers. Macromolecules 2008, 41, 8204–8209. [Google Scholar] [CrossRef]
- Choi, Y.Y.; Joo, M.K.; Sohn, Y.S.; Jeong, B. Significance of secondary structure in nanostructure formation and thermosensitivity of polypeptide block copolymers. Soft Matter 2008, 4, 2383–2387. [Google Scholar] [CrossRef]
- Kim, E.H.; Joo, M.K.; Bahk, K.H.; Park, M.H.; Chi, B.; Lee, Y.M.; Jeong, B. Reverse thermal gelation of PAF-PLX-PAF block copolymer aqueous solution. Biomacromolecules 2009, 10, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Choi, B.G.; Moon, H.J.; Cho, S.H.; Jeong, B. Block sequence affects thermosensitivity and nano-assembly: PEG-l-PA-dl-PA and PEG-dl-PA-l-PA block copolymers. Soft Matter 2011, 7, 6515–6521. [Google Scholar] [CrossRef]
- Moon, H.J.; Choi, B.G.; Park, M.H.; Joo, M.K.; Jeong, B. Enzymatically degradable thermogelling poly(alanine-co-leucine)-poloxamer-poly(alanine-co-leucine). Biomacromolecules 2011, 12, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.Y.; Yeon, B.; Moon, H.J.; Jeong, B. Peg-l-paf and peg-d-paf: Comparative study on thermogellation and biodegradation. Macromolecules 2012, 45, 2007–2013. [Google Scholar] [CrossRef]
- Joo, M.K.; Ko, D.Y.; Jeong, S.J.; Park, M.H.; Shinde, U.P.; Jeong, B. Incorporation of d-alanine into poly(ethylene glycol) and l-poly(alanine-co-phenylalanine) block copolymers affects their nanoassemblies and enzymatic degradation. Soft Matter 2013, 9, 8014–8022. [Google Scholar] [CrossRef]
- Thornton, P.D.; Billah, S.M.R.; Cameron, N.R. Enzyme-degradable self-assembled hydrogels from polyalanine-modified poly(ethylene glycol) star polymers. Macromol. Rapid Commun. 2013, 34, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Sieber, P.; Riniker, B. Protection of histidine in peptide-synthesis—A reassessment of the trityl group. Tetrahedron Lett. 1987, 28, 6031–6034. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Hamley, I.W.; Kirkham, S.; Dehsorkhi, A.; Castelletto, V.; Adamcik, J.; Mezzenga, R.; Ruokolainen, J.; Mazzuca, C.; Gatto, E.; Venanzi, M.; et al. Self-assembly of a model peptide incorporating a hexa-histidine sequence attached to an oligo-alanine sequence, and binding to gold nta/nickel nanoparticles. Biomacromolecules 2014, 15, 3412–3420. [Google Scholar] [CrossRef] [PubMed]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. Polymer Handbook, 4th ed.; Wiley: New York, NY, USA, 1999. [Google Scholar]
- Gitsas, A.; Floudas, G.; Mondeshki, M.; Spiess, H.W.; Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Control of peptide secondary structure and dynamics in poly(gamma-benzyl-L-glutamate)-b-polyalanine peptides. Macromolecules 2008, 41, 8072–8080. [Google Scholar] [CrossRef]
- Johnson, R.P.; Jeong, Y.I.; John, J.V.; Chung, C.W.; Kang, D.H.; Selvaraj, M.; Suh, H.; Kim, I. Dual stimuli-responsive poly(n-isopropylacrylamide)-b-poly(L-histidine) chimeric materials for the controlled delivery of doxorubicin into liver carcinoma. Biomacromolecules 2013, 14, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Checot, F.; Brulet, A.; Oberdisse, J.; Gnanou, Y.; Mondain-Monval, O.; Lecommandoux, S. Structure of polypeptide-based diblock copolymers in solution: Stimuli-responsive vesicles and micelles. Langmuir 2005, 21, 4308–4315. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Mn 1st Block × 10−3 a | Mn 2nd Block × 10−3 b | Total Mn × 10−3 c | I d | Composition % (w/w) e |
|---|---|---|---|---|---|
| m-PEO-b-poly(His-co-Gly) 10 | 9.95 | 6.20 | 16.1 | 1.13 | PEO/His/Gly: 62/36/2 |
| m-PEO-b-poly(His-co-Gly) 20 | 9.95 | 5.75 | 15.6 | 1.11 | PEO/His/Gly: 62.1/34/3.9 |
| m-PEO-b-poly(His-co-Gly) 40 | 9.95 | 6.10 | 15.9 | 1.15 | PEO/His/Gly: 62/30/8 |
| m-PEO-b-poly(His-co-Ala) 10 | 9.95 | 6.30 | 16.1 | 1.10 | PEO/His/Ala: 62.8/35/2.2 |
| m-PEO-b-poly(His-co-Ala) 20 | 9.95 | 6.0 | 16.1 | 1.09 | PEO/His/Ala: 62.6/33/4.4 |
| m-PEO-b-poly (His-co-Ala) 40 | 9.95 | 6.30 | 16.2 | 1.14 | PEO/His/Ala: 62.7/27.5/9.8 |
| Sample | pH = 7.4 (Filtration) | pH = 6.5 | pH = 5.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RH (nm) | Poly | KCounts | RH (nm) | Poly | KCounts | RH (nm) | Poly | KCounts | |
| m-PEO-b-poly(His-co-Gly) 10 | 92.6 | 0.34 | 812 | 112.4 | 0.252 | 624 | 161.2 | 0.40 | 521 |
| m-PEO-b-poly(His-co-Gly) 20 | 113.1 | 0.24 | 873 | 128.1 | 0.21 | 723 | 223 | 0.44 | 432 |
| m-PEO-b-poly(His-co-Gly) 40 | 120.6 | 0.293 | 908 | 144.0 | 0.452 | 827 | 673.9 | 0.505 | 567 |
| m-PEO-b-poly(His-co-Ala) 10 | 96.5 | 0.309 | 582 | 132 | 0.462 | 430 | 143.3 | 0.394 | 307 |
| m-PEO-b-poly(His-co-Ala) 20 | 118.2 | 0.237 | 1013 | 163.7 | 0.416 | 908 | 165.3 | 0.36 | 497 |
| m-PEO-b-poly(His-co-Ala) 40 | 111.5 | 0.23 | 761 | 121 | 0.309 | 714 | (1) 175.5 (2) 1005 | 0.467 | 647 |
| Sample | pH = 7.4 (Filtration) | pH = 6.5 | pH = 5.0 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RG a | RH(0) b | RG/RH(0) | RG a | RH(0) b | RG/RH(0) | RG a | RH(0) b | RG/RH(0) | |
| m-PEO-b-poly(His-co-Gly) 10 | 126.1 | 180.7 | 0.69 | 142.6 | 288.4 | 0.49 | 197.1 | 898.6 | 0.22 |
| m-PEO-b-poly(His-co-Gly) 20 | 137.3 | 222.2 | 0.62 | 194.1 | 886.7 | 0.22 | 197.8 | 479.1 | 0.42 |
| m-PEO-b-poly(His-co-Gly) 40 | 152.6 | 191.1 | 0.95 | 184.5 | 347.7 | 0.53 | 221.7 | 743.7 | 0.30 |
| m-PEO-b-poly(His-co-Ala) 10 | 119.9 | 181.9 | 0.65 | 186.2 | 883.1 | 0.21 | 199 | 400.4 | 0.49 |
| m-PEO-b-poly(His-co-Ala) 20 | 123.1 | 153.9 | 0.80 | 188.6 | 860.6 | 0.21 | 194.4 | 483.6 | 0.40 |
| m-PEO-b-poly(His-co-Ala) 40 | 114.3 | 154.4 | 0.74 | 158.5 | 326.2 | 0.48 | 211.7 | 825.8 | 0.25 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoulas, D.; Stavroulaki, D.; Santorinaios, K.; Iatrou, H. Synthesis of Hybrid-Polypeptides m-PEO-b-poly(His-co-Gly) and m-PEO-b-poly(His-co-Ala) and Study of Their Structure and Aggregation. Influence of Hydrophobic Copolypeptides on the Properties of Poly(L-histidine). Polymers 2017, 9, 564. https://doi.org/10.3390/polym9110564
Skoulas D, Stavroulaki D, Santorinaios K, Iatrou H. Synthesis of Hybrid-Polypeptides m-PEO-b-poly(His-co-Gly) and m-PEO-b-poly(His-co-Ala) and Study of Their Structure and Aggregation. Influence of Hydrophobic Copolypeptides on the Properties of Poly(L-histidine). Polymers. 2017; 9(11):564. https://doi.org/10.3390/polym9110564
Chicago/Turabian StyleSkoulas, Dimitrios, Dimitra Stavroulaki, Konstantinos Santorinaios, and Hermis Iatrou. 2017. "Synthesis of Hybrid-Polypeptides m-PEO-b-poly(His-co-Gly) and m-PEO-b-poly(His-co-Ala) and Study of Their Structure and Aggregation. Influence of Hydrophobic Copolypeptides on the Properties of Poly(L-histidine)" Polymers 9, no. 11: 564. https://doi.org/10.3390/polym9110564