
Interpenetration of Natural Polymer Aerogels by Supercritical Drying




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Preparation of Alginate/Gelatin Aerogels
2.1.1. Experimental Procedure to Produce Alginate Aerogel
2.1.2. Experimental Procedure to Produce Gelatin Aerogel
2.1.3. Experimental Procedure to Produce Alginate/Gelatin Aerogel
2.2. Supercritical Gel Drying
2.3. Analytical Methods
3. Results and Discussion
3.1. Alginate/Gelatin Aerogel Morphology
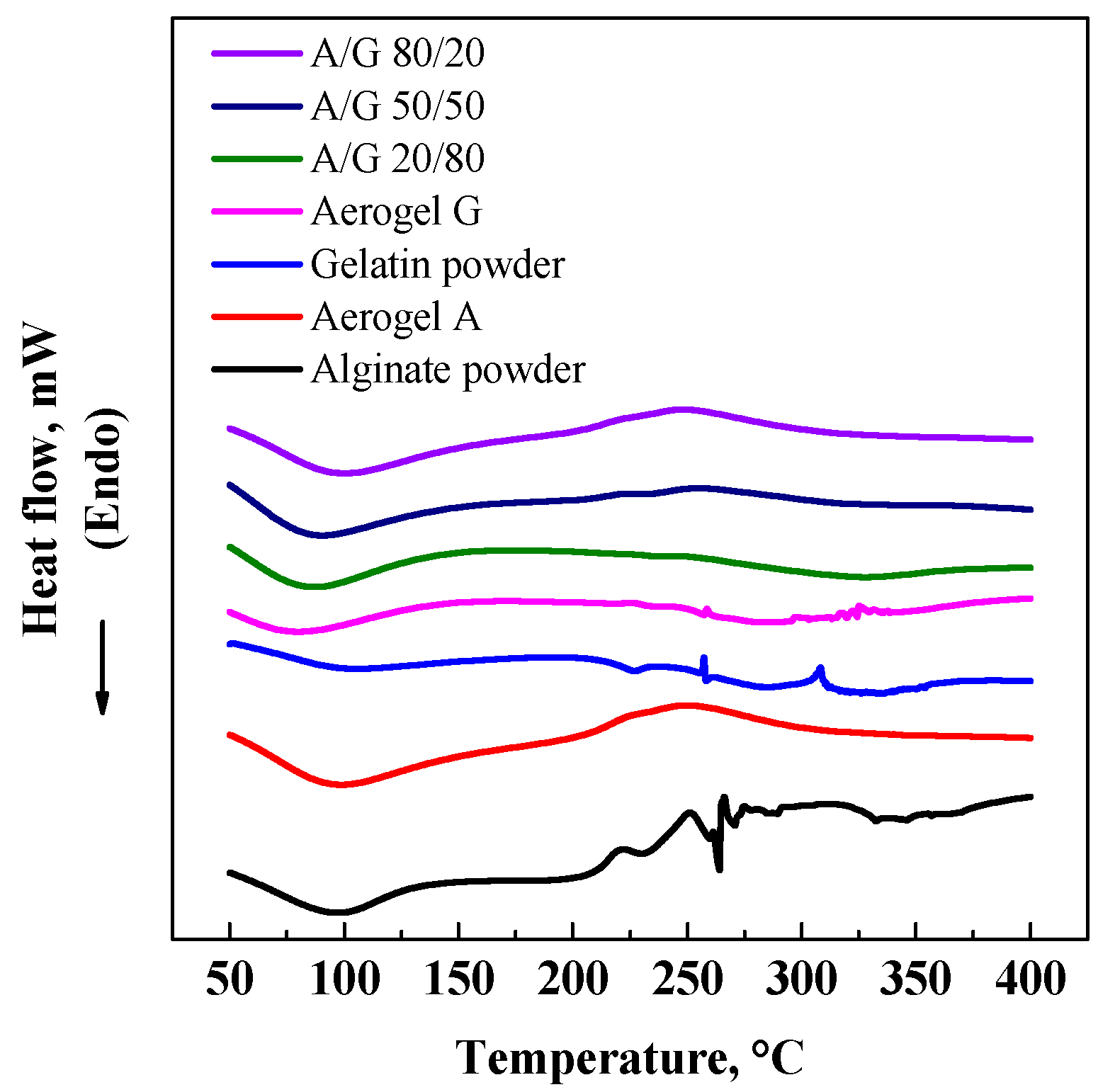
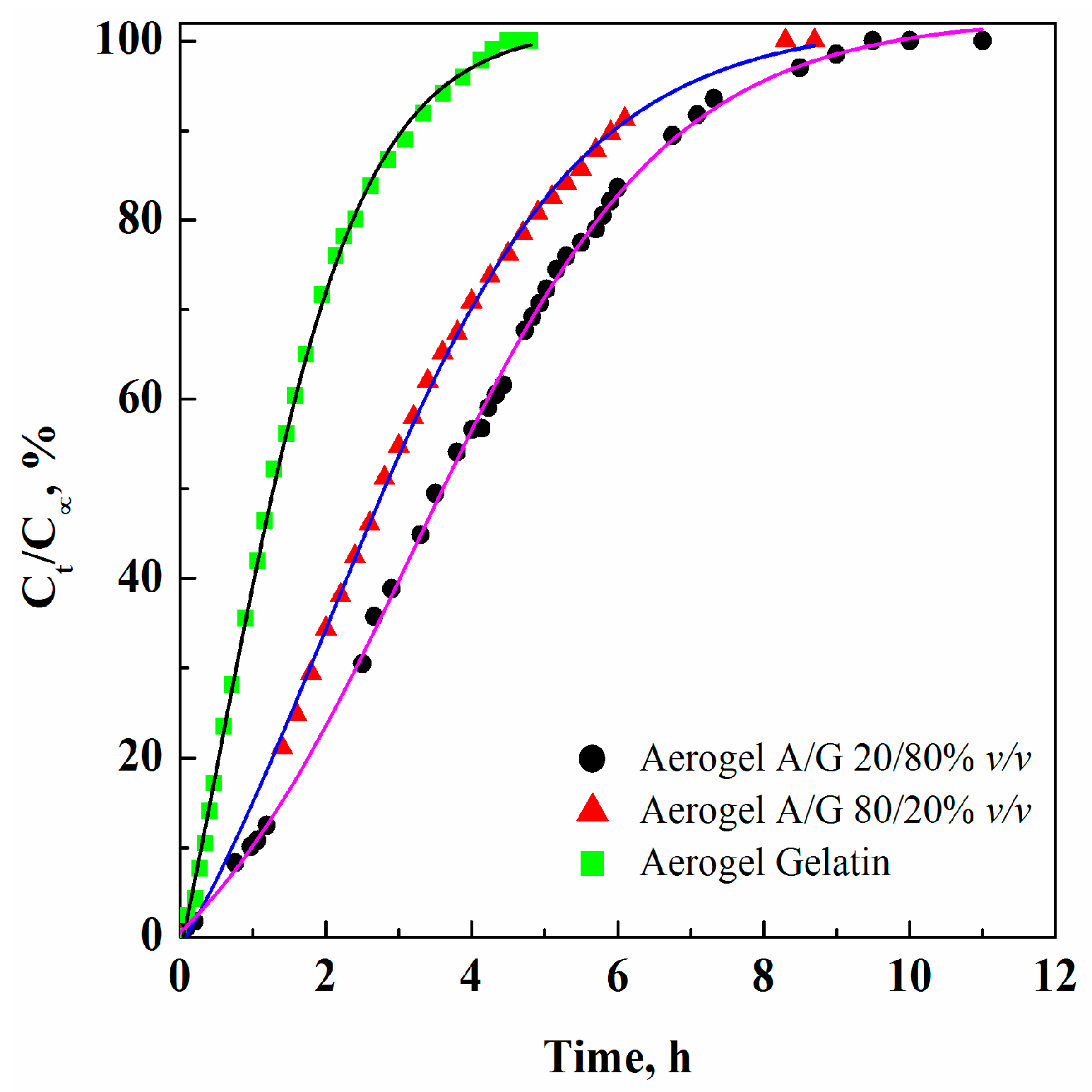
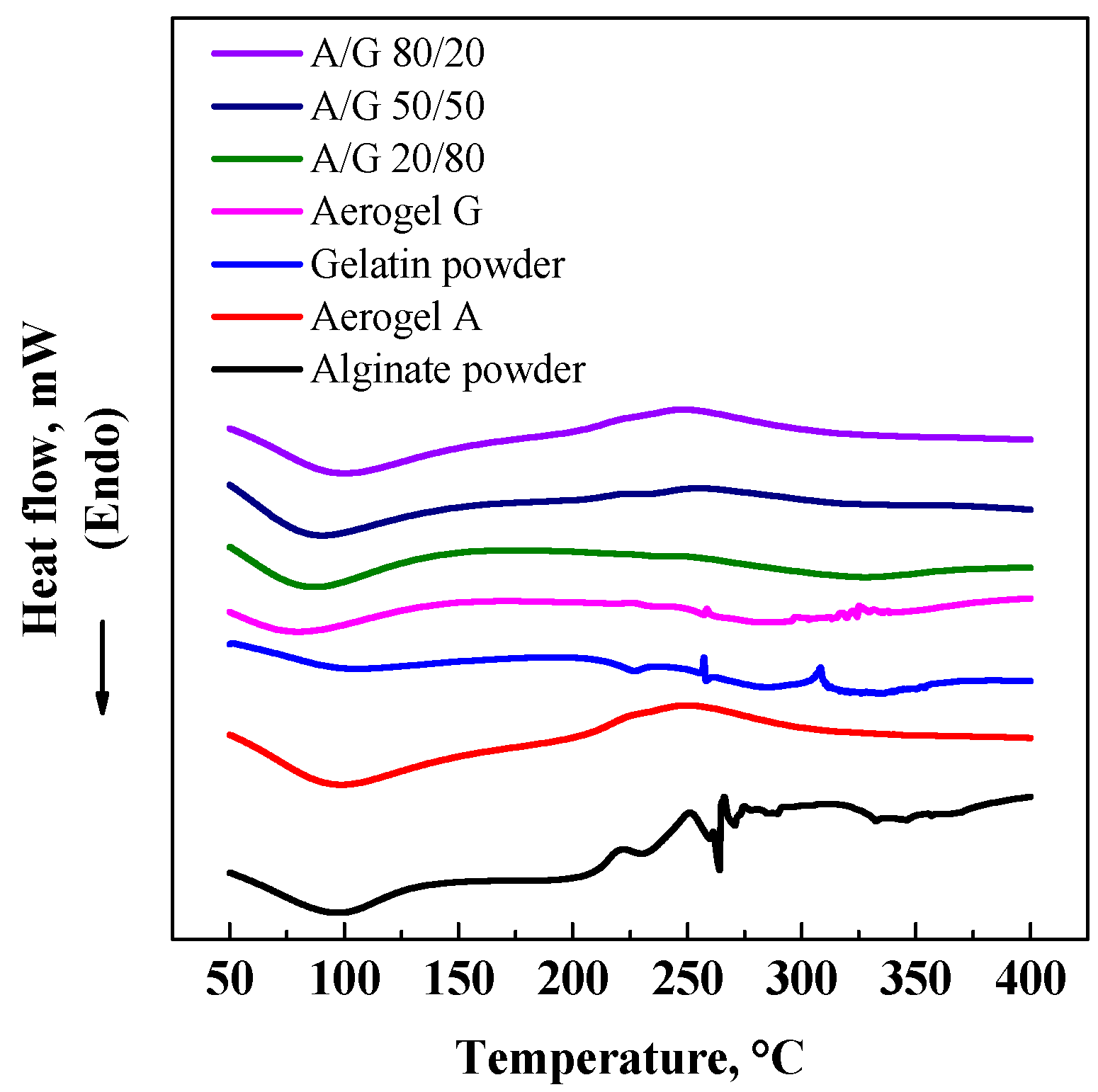
3.2. GTA Elimination from Alginate/Gelatin Aerogels
3.3. DSC and EDX Analyses
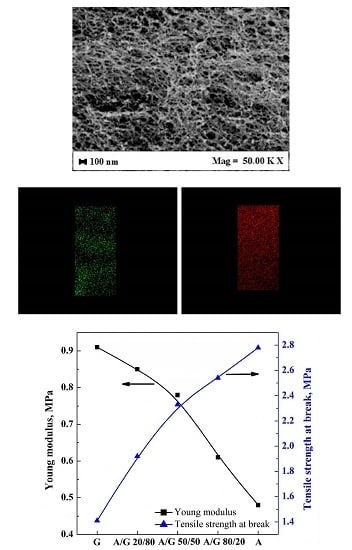
3.4. Mechanical Tests
4. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Autissier, A.; le Visage, C.; Pouzet, C.; Chaubet, F.; Letourneur, D. Fabrication of porous polysaccharide-based scaffolds using a combined freeze-drying/cross-linking process. Acta Biomater. 2010, 6, 3640–3648. [Google Scholar] [CrossRef] [PubMed]
- Jawad, H.; Lyon, A.R.; Harding, S.E.; Ali, N.N.; Boccaccini, A.R. Myocardial tissue engineering. Br. Med. Bull. 2008, 87, 31–47. [Google Scholar] [CrossRef] [PubMed]
- Inzana, J.A.; Olvera, D.; Fuller, S.M.; Kelly, J.P.; Graeve, O.A.; Schwarz, E.M.; Kates, S.L.; Awad, H.A. 3D printing of composite calcium phosphate and collagen scaffolds for bone regeneration. Biomaterials 2014, 35, 4026–4034. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Li, X.; Wen, P.; Zhang, Y.; Long, Y.; Wang, X.; Guo, Y.; Xing, F.; Gao, J. Preparation of aligned porous gelatin scaffolds by unidirectional freeze-drying method. Acta Biomater. 2010, 6, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Cao, Y. Tissue-engineering technology for tissue repair and regeneration. Compr Biotech. 2011, 5, 353–375. [Google Scholar]
- Nieponice, A.; Maul, T.M.; Cumer, J.M.; Soletti, L.; Vorp, D.A. Mechanical stimulation induces morphological and phenotypic changes in bone marrow-derived progenitor cells within a three-dimensional fibrin matrix. J. Biomed. Mater. Res. A 2007, 81, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Osathanon, T.; Linnes, M.L.; Rajachar, R.M.; Ratner, B.D.; Somerman, M.J.; Giachelli, C.M. Microporous nanofibrous fibrin-based scaffolds for bone tissue engineering. Biomaterials 2008, 29, 4091–4099. [Google Scholar] [CrossRef] [PubMed]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Poly. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Hussain, A.; Collins, G.; Yip, D.; Cho, C.H. Functional 3-D cardiac co-culture model using bioactive chitosan nanofiber scaffolds. Biotech. Bioeng. 2013, 110, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Papajová, E.; Bujdoš, M.; Chorvát, D.; Stach, M.; Lacík, I. Method for preparation of planar alginate hydrogels by external gelling using an aerosol of gelling solution. Carbohydr. Poly. 2012, 90, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.K.; Ma, P.X. Maintaining dimensions and mechanical properties of ionically crosslinked alginate hydrogel scaffolds in vitro. J. Biomed. Mater. Res. A 2008, 84, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, S.; Dubruel, P.; Schacht, E. Biopolymer-based hydrogels as scaffolds for tissue engineering applications: A review. Biomacromolecules 2011, 12, 1387–1408. [Google Scholar] [CrossRef] [PubMed]
- Whu, S.W.; Hung, K.-C.; Hsieh, K.-H.; Chen, C.-H.; Tsai, C.-L.; Hsu, S.-h. In vitro and in vivo evaluation of chitosan–gelatin scaffolds for cartilage tissue engineering. Mater. Sci. Eng. C 2013, 33, 2855–2863. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Kim, Y.; Lee, H.; Kim, G. A new hybrid scaffold constructed of solid freeform-fabricated pcl struts and collagen struts for bone tissue regeneration: Fabrication, mechanical properties, and cellular activity. J. Mater. Chem. 2012, 22, 15901–15909. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Poly. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, D.R.; Crompton, K.E.; Horne, M.K.; Finkelstein, D.I.; Forsythe, J.S. Neural tissue engineering of the cns using hydrogels. J. Biomed. Mater. Res. B 2008, 87B, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Freeman, I.; Cohen, S. The influence of the sequential delivery of angiogenic factors from affinity-binding alginate scaffolds on vascularization. Biomaterials 2009, 30, 2122–2131. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.D.; Kiick, K.L. Polysaccharide-modified synthetic polymeric biomaterials. Pept. Sci. 2010, 94, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Ganesh, N.; Selvamurugan, N.; Nair, S.V.; Furuike, T.; Tamura, H.; Jayakumar, R. Preparation and characterization of chitosan–gelatin/nanohydroxyapatite composite scaffolds for tissue engineering applications. Carbohydr. Poly. 2010, 80, 687–694. [Google Scholar] [CrossRef]
- Bigi, A.; Cojazzi, G.; Panzavolta, S.; Rubini, K.; Roveri, N. Mechanical and thermal properties of gelatin films at different degrees of glutaraldehyde crosslinking. Biomaterials 2001, 22, 763–768. [Google Scholar] [CrossRef]
- Nwe, N.; Furuike, T.; Tamura, H. Selection of a biopolymer based on attachment, morphology and proliferation of fibroblast NIH/3T3 cells for the development of a biodegradable tissue regeneration template: Alginate, bacterial cellulose and gelatin. Pro. Biochem. 2010, 45, 457–466. [Google Scholar] [CrossRef]
- Baldino, L.; Concilio, S.; Cardea, S.; De Marco, I.; Reverchon, E. Complete glutaraldehyde elimination during chitosan hydrogel drying by SC-CO2 processing. J. Supercrit. Fluid 2015, 103, 70–76. [Google Scholar] [CrossRef]
- Cheng, Y.; Lu, L.; Zhang, W.; Shi, J.; Cao, Y. Reinforced low density alginate-based aerogels: Preparation, hydrophobic modification and characterization. Carbohydr. Poly. 2012, 88, 1093–1099. [Google Scholar] [CrossRef]
- Yamamoto, M.; James, D.; Li, H.; Butler, J.; Rafii, S.; Rabbany, S. Generation of stable co-cultures of vascular cells in a honeycomb alginate scaffold. Tissue Eng. A 2009, 16, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; He, C.; Cao, L.; Feng, W.; Wang, H.; Mo, X.; Wang, J. Fabrication of gelatin–hyaluronic acid hybrid scaffolds with tunable porous structures for soft tissue engineering. Int. J. Biol. Macromol. 2011, 48, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Shivashankar, M.; Mandal, B.K. A review on interpenetrating polymer network. Int. J. Phram. Phram. Sci 2012, 4, 1–7. [Google Scholar]
- Yue, Y.M.; Xu, K.; Liu, X.G.; Chen, Q.; Sheng, X.; Wang, P.X. Preparation and characterization of interpenetration polymer network films based on poly(vinyl alcohol) and poly(acrylic acid) for drug delivery. J. Appl. Poly. Sci. 2008, 108, 3836–3842. [Google Scholar] [CrossRef]
- Sperling, L.; Mishra, V. The current status of interpenetrating polymer networks. Poly. Adv. Technol. 1996, 7, 197–208. [Google Scholar] [CrossRef]
- Wen, C.; Lu, L.; Li, X. Mechanically robust gelatin–alginate IPN hydrogels by a combination of enzymatic and ionic crosslinking approaches. Macromol. Mater. Eng. 2014, 299, 504–513. [Google Scholar] [CrossRef]
- Han, J.; Zhou, Z.; Yin, R.; Yang, D.; Nie, J. Alginate–chitosan/hydroxyapatite polyelectrolyte complex porous scaffolds: Preparation and characterization. Int. J. Biol. Macromol. 2010, 46, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Ionita, M.; Pandele, M.A.; Iovu, H. Sodium alginate/graphene oxide composite films with enhanced thermal and mechanical properties. Carbohydr. Poly. 2013, 94, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, E.D.; Yin, X.; Nair, K.; Sun, W. Fabrication, characterization, and biocompatibility of single-walled carbon nanotube-reinforced alginate composite scaffolds manufactured using freeform fabrication technique. J. Biomed. Mater. Res. B 2008, 87B, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, J.; Krause, A.; Möller, L.; Kensah, G.; Möwes, M.; Diekmann, A.; Martin, U.; Kirschning, A.; Gruh, I.; Dräger, G. Fully defined in situ cross-linkable alginate and hyaluronic acid hydrogels for myocardial tissue engineering. Biomaterials 2013, 34, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chan-Park, M.B. Hydrogel based on interpenetrating polymer networks of dextran and gelatin for vascular tissue engineering. Biomaterials 2009, 30, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Dinda, A.K.; Mishra, N.C. Fabrication and characterization of PCL/gelatin composite nanofibrous scaffold for tissue engineering applications by electrospinning method. Mater. Sci. Eng. C 2013, 33, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Rosellini, E.; Cristallini, C.; Barbani, N.; Vozzi, G.; Giusti, P. Preparation and characterization of alginate/gelatin blend films for cardiac tissue engineering. J. Biomed. Mater. Res. A 2009, 91, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zhang, H. Controlled freezing and freeze drying: A versatile route for porous and micro-/nano-structured materials. J. Chem. Technol. Biotechnol. 2011, 86, 172–184. [Google Scholar] [CrossRef]
- Prosapio, V.; Reverchon, E.; De Marco, I. Production of lysozyme microparticles to be used in functional foods, using an expanded liquid antisolvent process. J. Supercrit. Fluid 2016, 107, 106–113. [Google Scholar] [CrossRef]
- Campardelli, R.; Santo, I.E.; Albuquerque, E.C.; de Melo, S.V.; Della Porta, G.; Reverchon, E. Efficient encapsulation of proteins in submicro liposomes using a supercritical fluid assisted continuous process. J. Supercrit. Fluid 2016, 107, 163–169. [Google Scholar] [CrossRef]
- Reverchon, E.; Adami, R.; Campardelli, R.; Della Porta, G.; De Marco, I.; Scognamiglio, M. Supercritical fluids based techniques to process pharmaceutical products difficult to micronize: Palmitoylethanolamide. J. Supercrit. Fluid 2015, 102, 24–31. [Google Scholar] [CrossRef]
- Campardelli, R.; Baldino, L.; Reverchon, E. Supercritical fluids applications in nanomedicine. J. Supercrit. Fluid 2015, 101, 193–214. [Google Scholar] [CrossRef]
- Meneses, M.A.; Caputo, G.; Scognamiglio, M.; Reverchon, E.; Adami, R. Antioxidant phenolic compounds recovery from Mangifera indica L. by-products by supercritical antisolvent extraction. J. Food Eng. 2015, 163, 45–53. [Google Scholar] [CrossRef]
- Baldino, L.; Cardea, S.; Reverchon, E. Supercritical assisted enzymatic membranes preparation, for active packaging applications. J. Membr. Sci. 2014, 453, 409–418. [Google Scholar] [CrossRef]
- Baldino, L.; Sarno, M.; Cardea, S.; Irusta, S.; Ciambelli, P.; Santamaria, J.; Reverchon, E. Formation of cellulose acetate-graphene oxide nanocomposites by supercritical CO2 assisted phase inversion. Ind. Eng. Chem. Res. 2015, 54, 8147–8156. [Google Scholar] [CrossRef]
- García-González, C.A.; Concheiro, A.; Alvarez-Lorenzo, C. Processing of materials for regenerative medicine using supercritical fluid technology. Bioconj. Chem. 2015, 26, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- García-González, C.A.; Alnaief, M.; Smirnova, I. Polysaccharide-based aerogels—Promising biodegradable carriers for drug delivery systems. Carbohydr. Poly. 2011, 86, 1425–1438. [Google Scholar] [CrossRef]
- Mikkonen, K.S.; Parikka, K.; Ghafar, A.; Tenkanen, M. Prospects of polysaccharide aerogels as modern advanced food materials. Trends Food Sci. Technol. 2013, 34, 124–136. [Google Scholar] [CrossRef]
- Ulker, Z.; Erkey, C. An emerging platform for drug delivery: Aerogel based systems. J. Controll. Release 2014, 177, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Reverchon, E.; Pisanti, P.; Cardea, S. Nanostructured PLLA-hydroxyapatite scaffolds produced by a supercritical assisted technique. Ind. Eng. Chem. Res. 2009, 48, 5310–5316. [Google Scholar] [CrossRef]
- Brunner, G. Gas Extraction: An Introduction to Fundamentals of Supercritical Fluids and the Application to Separation Processes; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Dhandayuthapani, B.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Polymeric scaffolds in tissue engineering application: A review. Int. J. Poly. Sci. 2011. [Google Scholar] [CrossRef]
- Reverchon, E.; Cardea, S. Supercritical fluids in 3-D tissue engineering. J. Supercrit. Fluid 2012, 69, 97–107. [Google Scholar] [CrossRef]
- Barnes, C.P.; Sell, S.A.; Boland, E.D.; Simpson, D.G.; Bowlin, G.L. Nanofiber technology: Designing the next generation of tissue engineering scaffolds. Adv. Drug deliv. Rev. 2007, 59, 1413–1433. [Google Scholar] [CrossRef] [PubMed]
- Speer, D.P.; Chvapil, M.; Eskelson, C.; Ulreich, J. Biological effects of residual glutaraldehyde in glutaraldehyde-tanned collagen biomaterials. J. Biomed. Mater. Res. 1980, 14, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.P. Mechanical properties of normal and diseased cerebrovascular system. J. Vasc. Interv. Neurol. 2009, 2, 155–162. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aerogel | Porosity (%) | CGTAmax@5 h (ppm) | CGTAmax@8 h (ppm) | Bulk density (g/cm3) | Skeletal density (g/cm3) | Specific surface area (m2/g) |
|---|---|---|---|---|---|---|
| G | 95.0 ± 3.2 | 4.2 | 1.4 | 0.016 | 0.315 | 227 |
| A/G 20/80% v/v | 92.1 ± 3.1 | 6.6 | 2.8 | 0.026 | 0.327 | 235 |
| A/G 50/50% v/v | 89.9 ± 2.8 | 9.5 | 5.1 | 0.034 | 0.335 | 248 |
| A/G 80/20% v/v | 88.3 ± 2.7 | 21.5 | 6.8 | 0.041 | 0.342 | 260 |
| A | 84.8 ± 1.9 | – | – | 0.055 | 0.366 | 271 |
| Aerogel | Young modulus (MPa) | Tensile strength at break (MPa) |
|---|---|---|
| G | 0.91 ± 0.11 | 1.41 ± 0.15 |
| A/G 20/80% v/v | 0.85 ± 0.08 | 1.92 ± 0.18 |
| A/G 50/50% v/v | 0.78 ± 0.06 | 2.33 ± 0.25 |
| A/G 80/20% v/v | 0.61 ± 0.05 | 2.54 ± 0.30 |
| A | 0.48 ± 0.03 | 2.78 ± 0.36 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldino, L.; Concilio, S.; Cardea, S.; Reverchon, E. Interpenetration of Natural Polymer Aerogels by Supercritical Drying. Polymers 2016, 8, 106. https://doi.org/10.3390/polym8040106
Baldino L, Concilio S, Cardea S, Reverchon E. Interpenetration of Natural Polymer Aerogels by Supercritical Drying. Polymers. 2016; 8(4):106. https://doi.org/10.3390/polym8040106
Chicago/Turabian StyleBaldino, Lucia, Simona Concilio, Stefano Cardea, and Ernesto Reverchon. 2016. "Interpenetration of Natural Polymer Aerogels by Supercritical Drying" Polymers 8, no. 4: 106. https://doi.org/10.3390/polym8040106