
Core/Shell Conjugated Polymer/Quantum Dot Composite Nanofibers through Orthogonal Non-Covalent Interactions

Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials and General Methods
2.2. OPV(Organic Photovoltaics) Fabrication and Testing
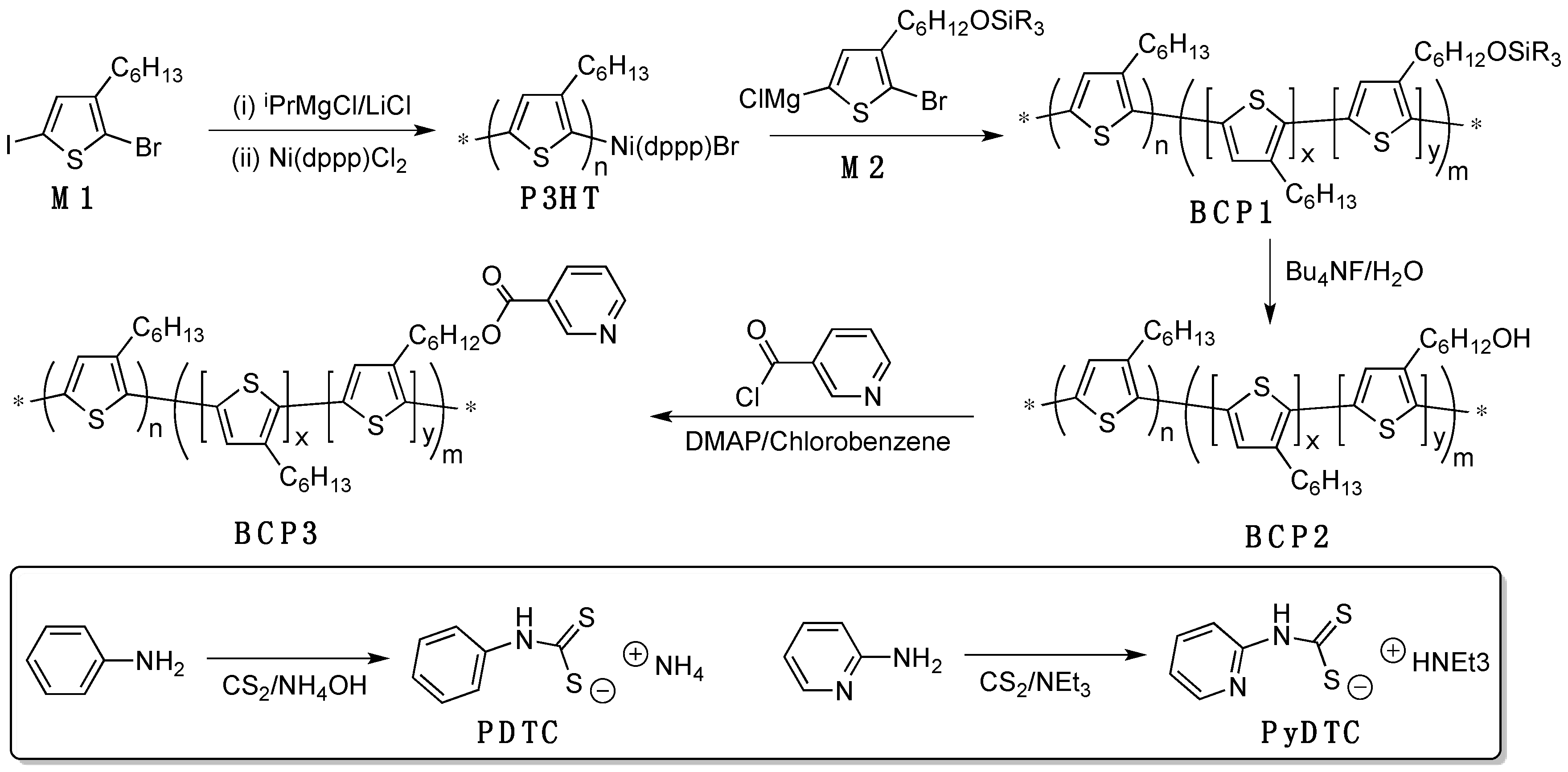
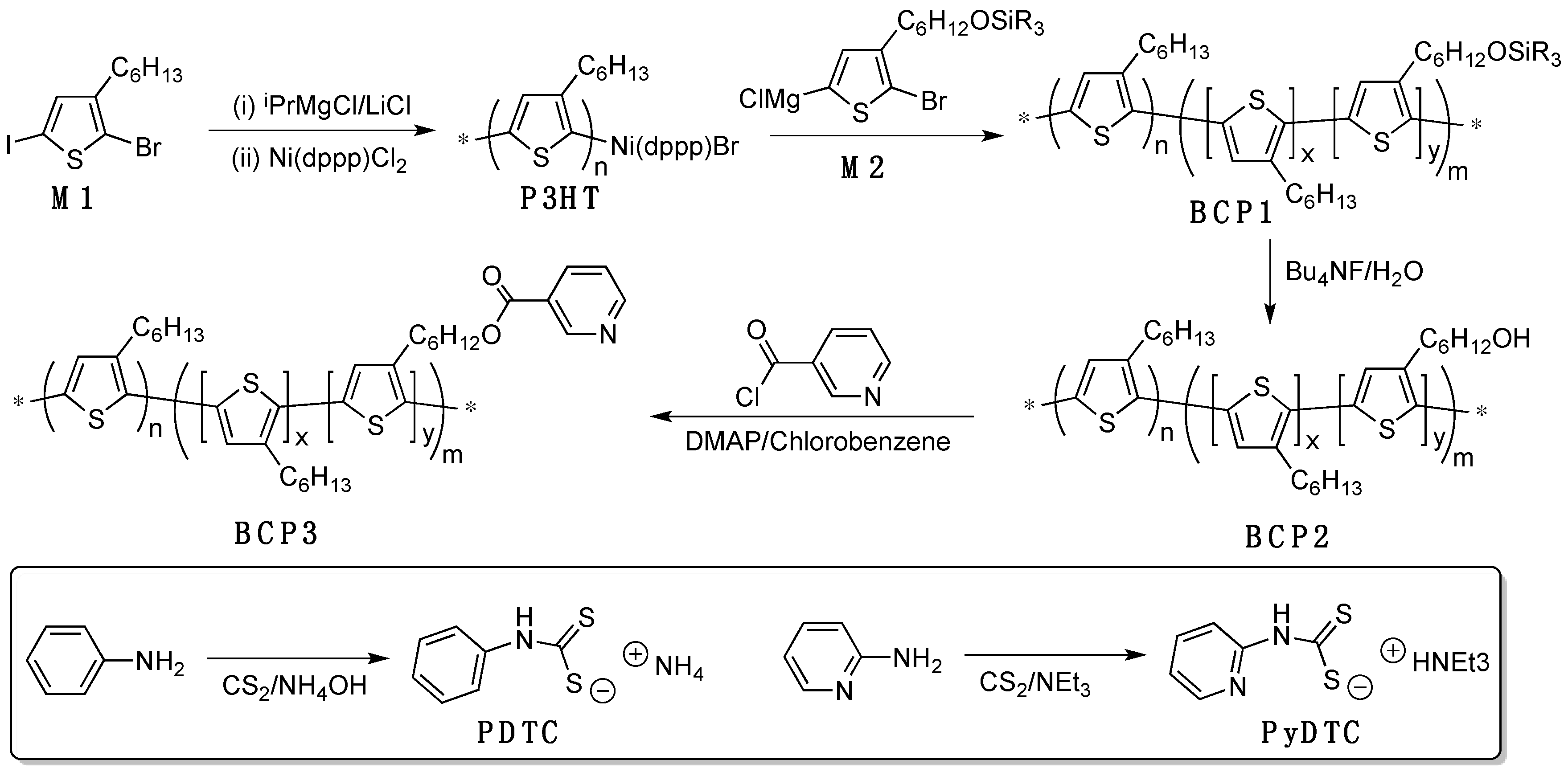
2.3. Detailed Synthetic Procedures
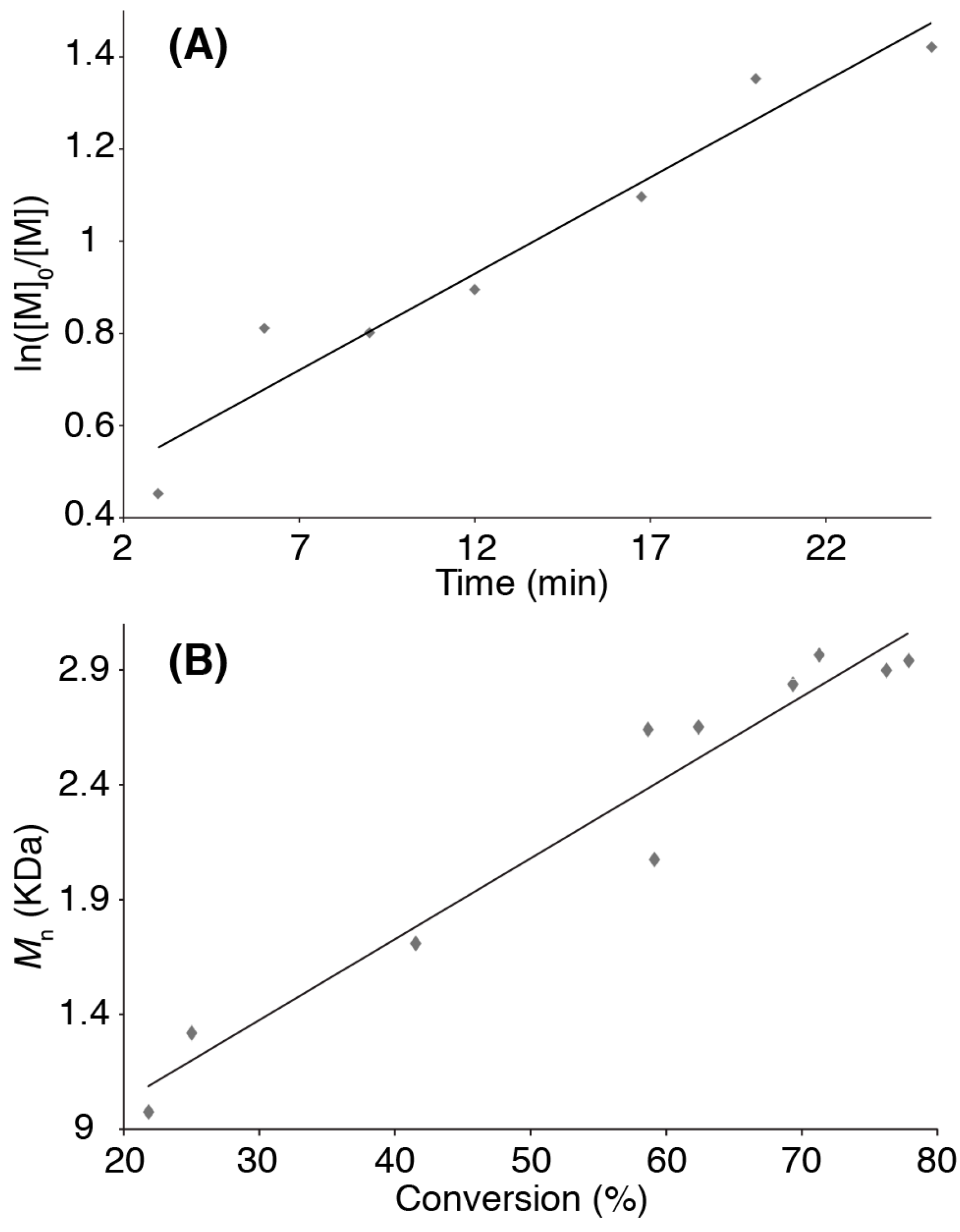
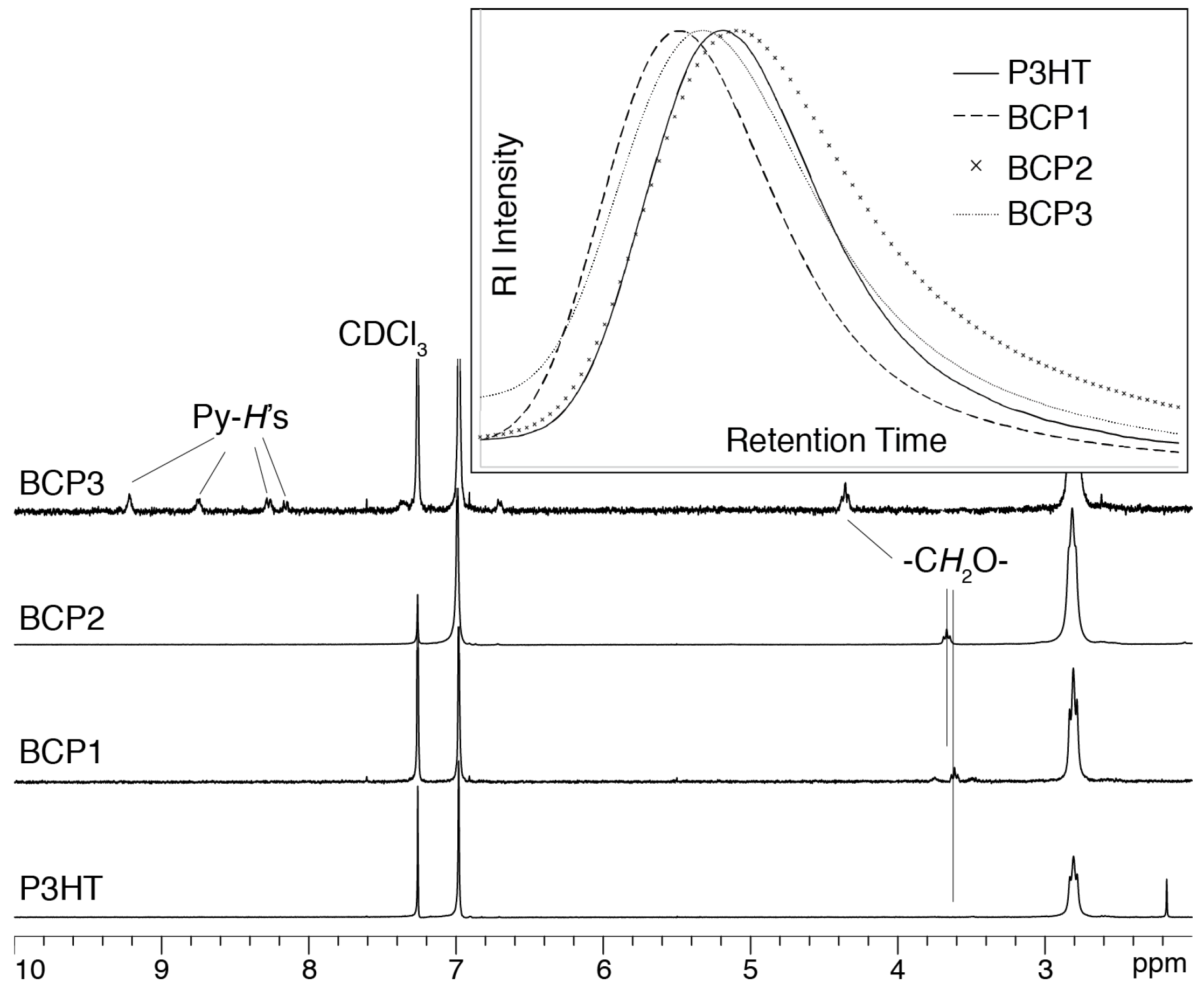
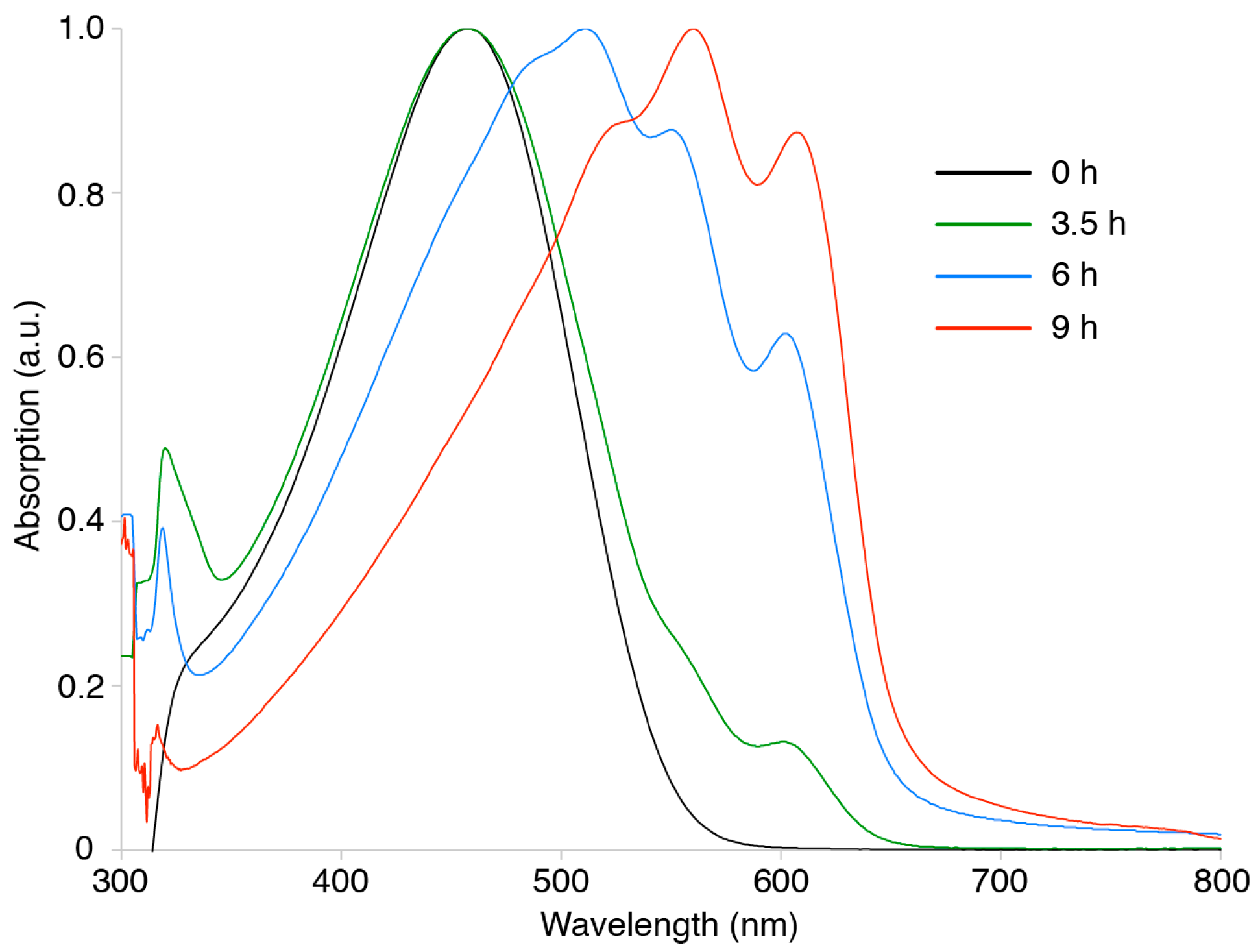
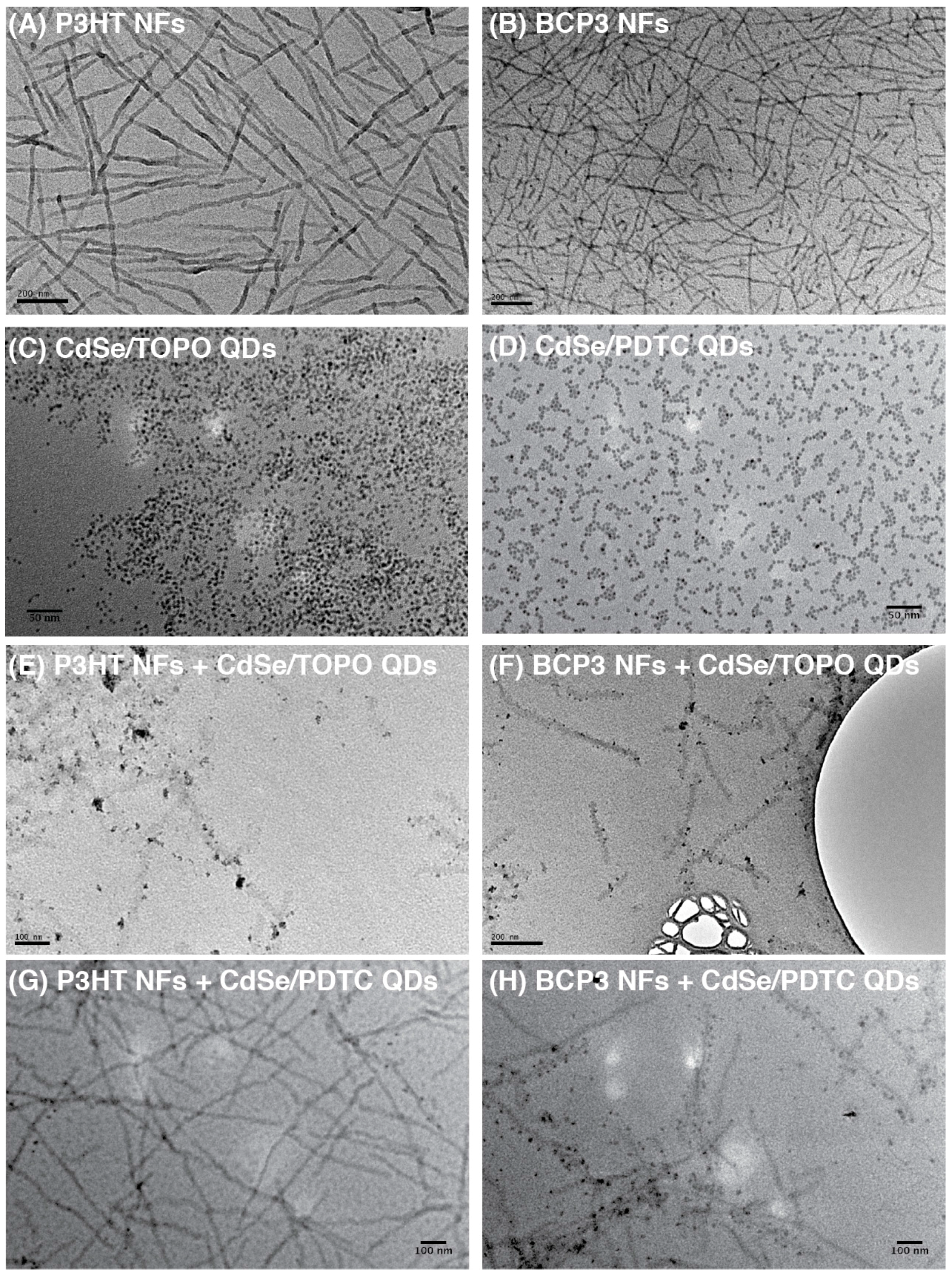
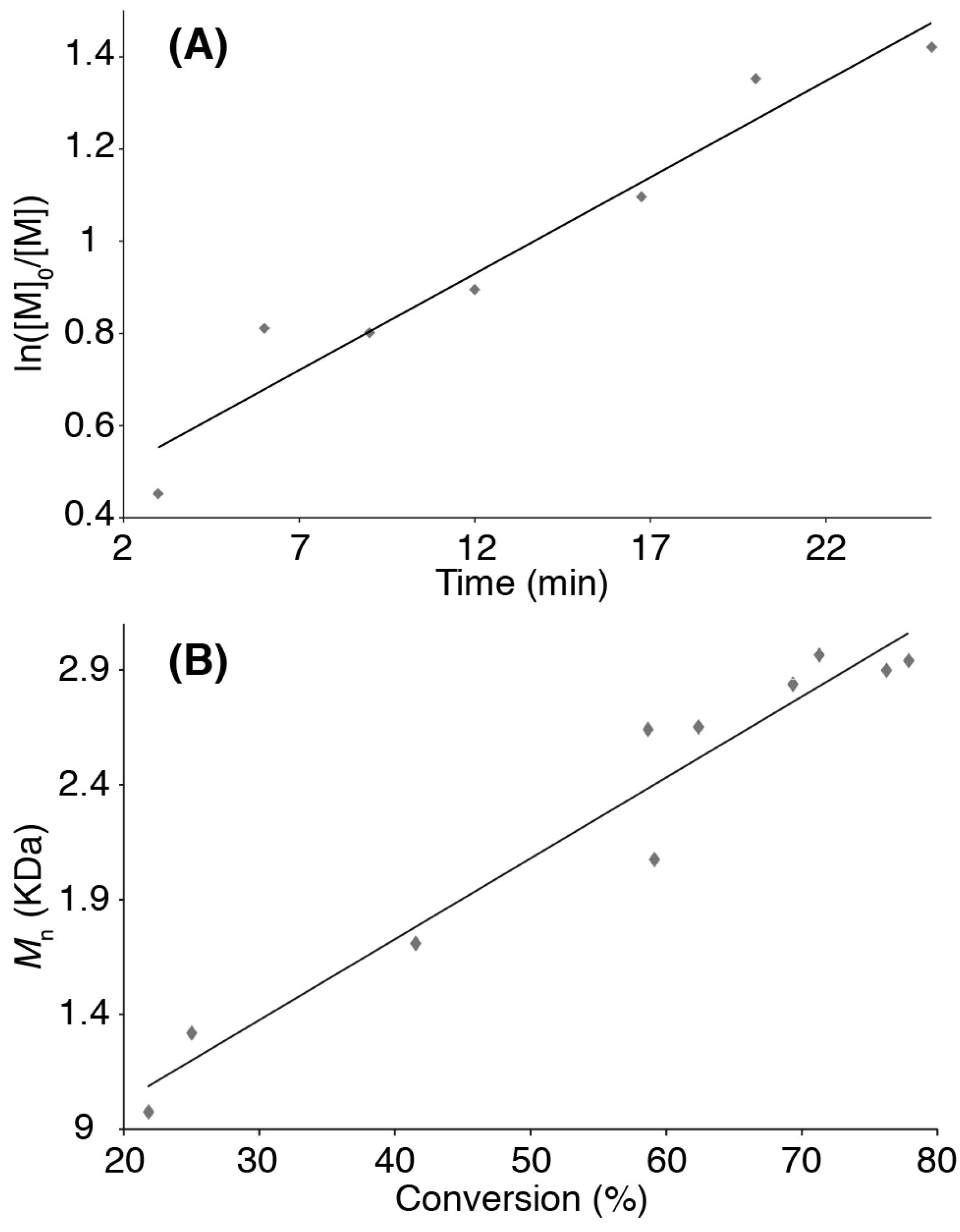
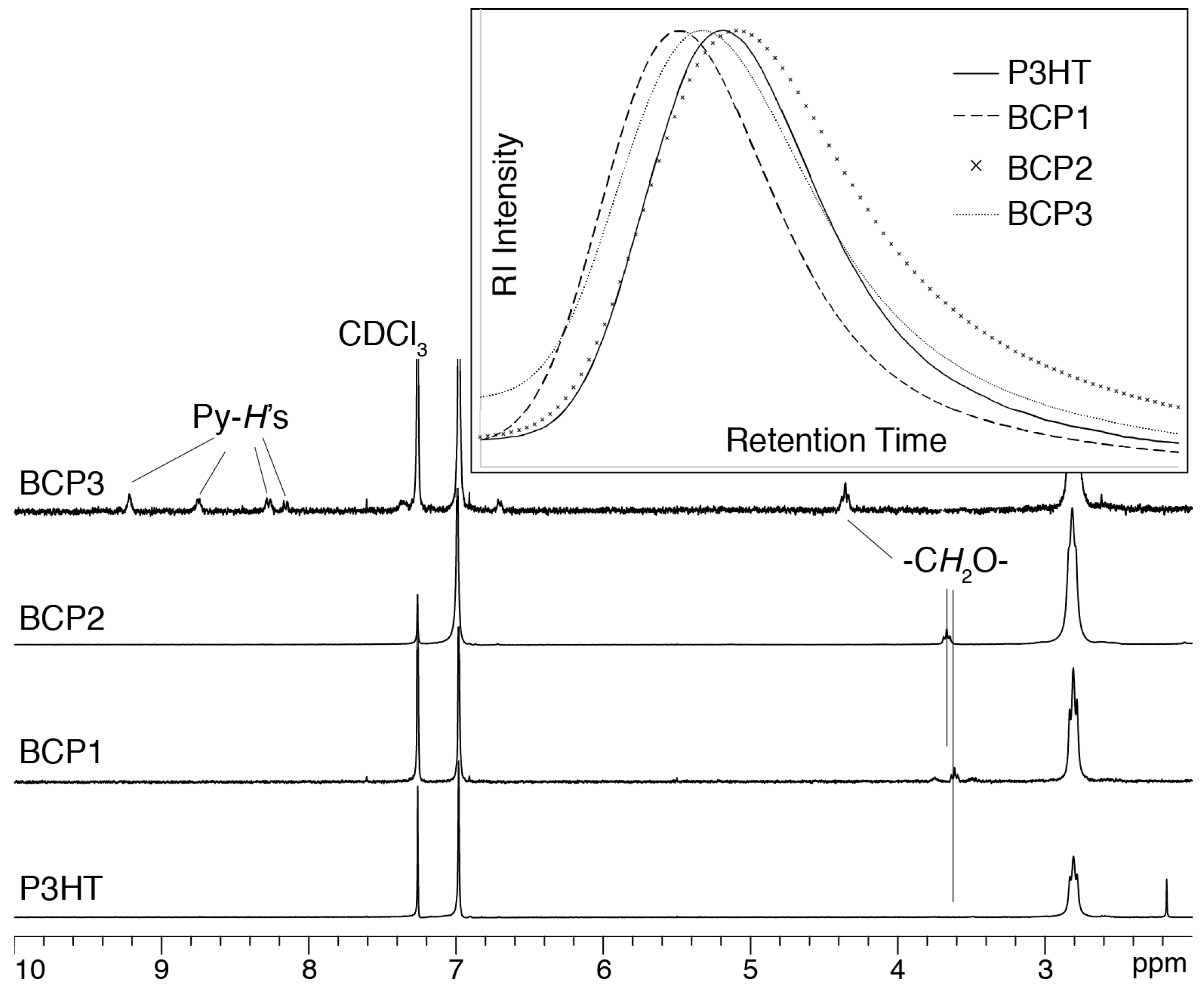
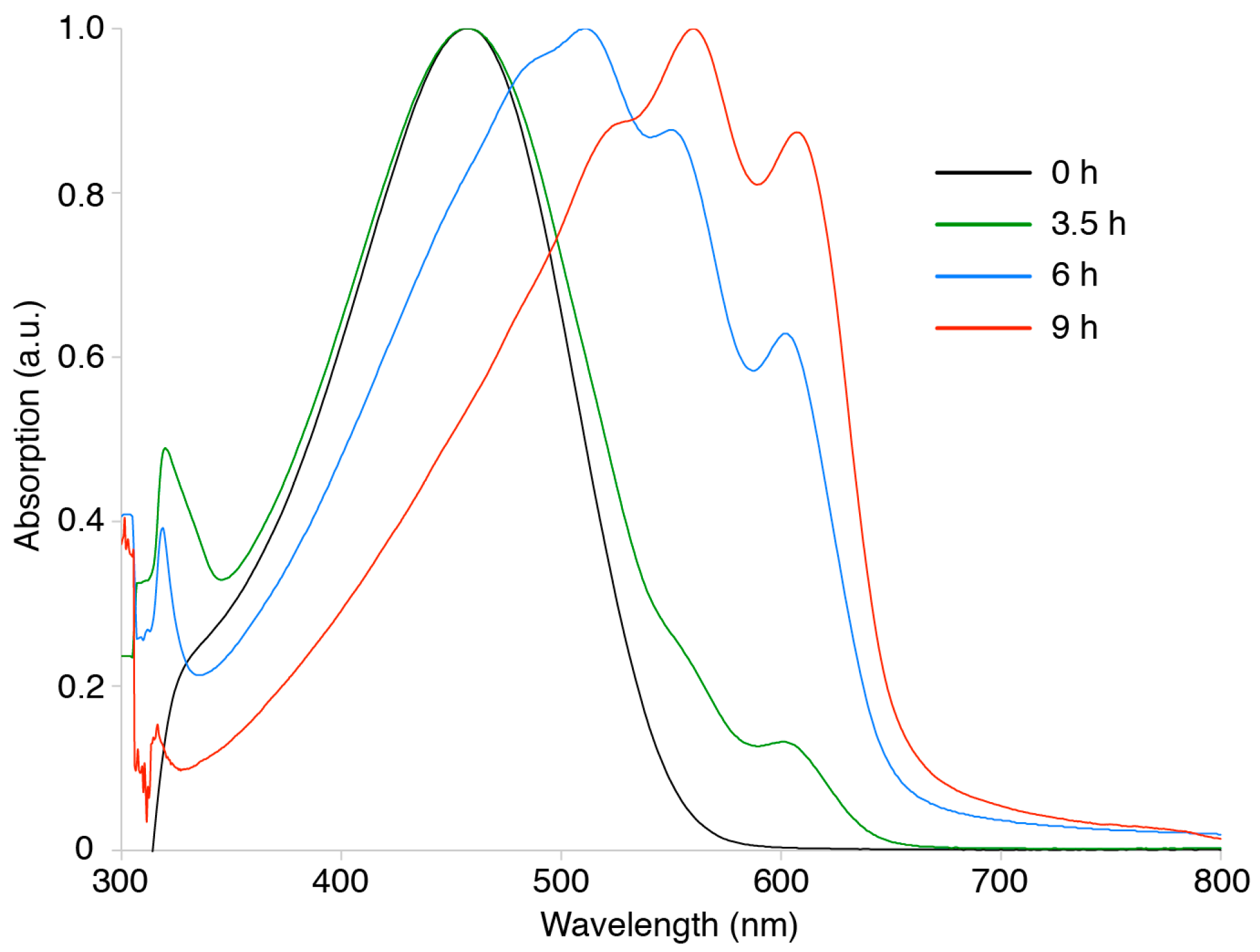
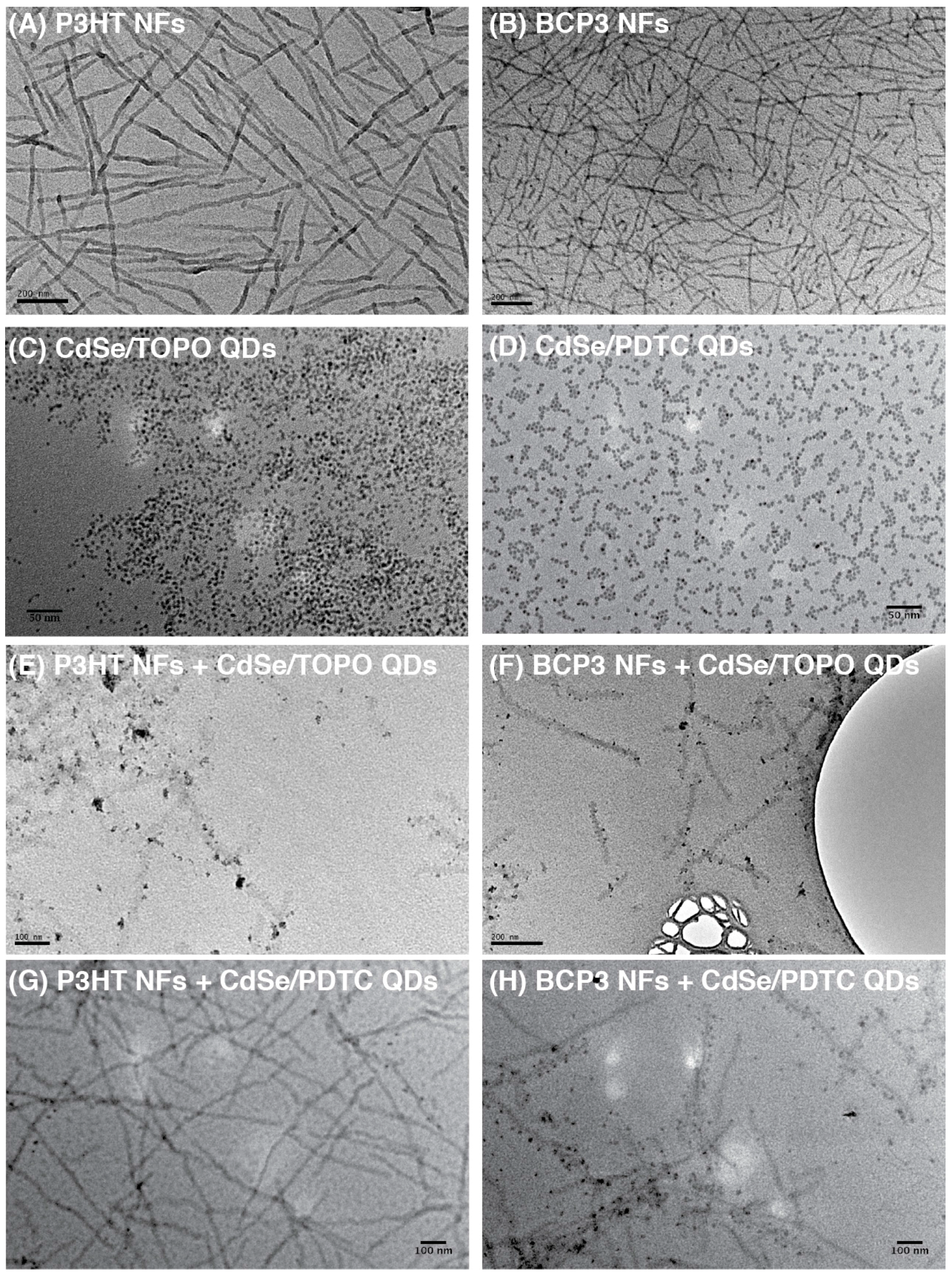
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Skotheim, T.A.; Reynolds, J.R. Handbook of Conducting Polymers, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Nielsen, T.D.; Cruickshank, C.; Forged, S.; Thorsen, J.; Krebs, F.C. Business, Market and Intellectual Property Analysis of Polymer Solar Cells. Sol. Energy Mater. Sol. Cells 2010, 94, 1553–1571. [Google Scholar] [CrossRef]
- Jørgensen, M.; Carlé, J.E.; Søndergaard, R.R.; Lauritzen, M.; Dagnæs-Hansen, N.A.; Byskov, S.L.; Andersen, T.R.; Larsen-Olsen, T.T.; Böttiger, A.P.L.; Andreasen, B.; et al. The State of Organic Solar Cells—A Meta Analysis. Sol. Energy Mater. Sol. Cells 2013, 119, 84–93. [Google Scholar] [CrossRef]
- You, J.; Dou, L.; Yoshimura, K.; Kato, T.; Ohya, K.; Moriarty, T.; Emery, K.; Chen, C.-C.; Gao, J.; Li, G.; et al. A Polymer Tandem Solar Cell with 10.6% Power Conversion Efficiency. Nat. Commun. 2013, 4, 1446. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Chen, C.-C.; Hong, Z.; Yoshimura, K.; Ohya, K.; Xu, R.; Ye, S.; Gao, J.; Li, G.; Yang, Y. 10.2% Power Conversion Effi ciency Polymer Tandem Solar Cells Consisting of Two Identical Sub-Cells. Adv. Mater. 2013, 25, 3973–3978. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Peng, R.; Ai, L.; Zhang, X.; Ge, Z. Efficient Polymer Solar Cells Employing a Non-Conjugated Small-Molecule Electrolyte. Nat. Photonics 2015, 9, 520–524. [Google Scholar] [CrossRef]
- Liu, Y.; Page, Z.A.; Russell, T.P.; Emrick, T. Finely Tuned Polymer Interlayers Enhance Solar Cell Efficiency. Angew. Chem. Int. Ed. 2015, 54, 11485–11489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Fang, J.; Lu, K.; Wang, Z.; Ma, W.; Wei, Z. Conjugated Polymer-Small Molecule Alloy Leads to High Efficient Ternary Organic Solar Cells. J. Am. Chem. Soc. 2015, 137, 8176–8183. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Jiang, K.; Yang, G.; Liu, J.; Li, Z.; Lin, H.; Liu, Y.; Zhao, J.; Zhang, J.; Huang, F.; et al. Terthiophene-Based D–A Polymer with an Asymmetric Arrangement of Alkyl Chains That Enables Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2015, 137, 14149–14157. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-D.; Cui, C.; Li, Y.-Q.; Zhou, L.; Ou, Q.-D.; Li, C.; Li, Y.; Tang, J.-X. Single-Junction Polymer Solar Cells Exceeding 10% Power Conversion Efficiency. Adv. Mater. 2015, 27, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-M.; Hong, Z.; Li, G.; Yang, Y. Recent Progress in Polymer Solar Cells: Manipulation of Polymer:Fullerene Morphology and the Formation of Efficient Inverted Polymer Solar Cells. Adv. Mater. 2009, 21, 1434–1449. [Google Scholar] [CrossRef]
- Brabec, C.J.; Heeney, M.; McCulloch, I.; Nelson, J. Influence of Blend Microstructures on Bulk Heterojunction Organic Photovoltaics Performance. Chem. Soc. Rev. 2011, 40, 1185–1199. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Kramer, E.J.; Heeger, A.J.; Bazan, G.C. Bulk Heterojunction Solar Cells: Morphology and Performance Relationships. Chem. Rev. 2014, 114, 7006–7043. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.; Huettner, S.; Thelakkat, M. Donor-Acceptor Block Copolymers for Photovoltaic Applications. J. Mater. Chem. 2010, 20, 10788–10797. [Google Scholar] [CrossRef]
- Miyanishi, S.; Zhang, Y.; Tajima, K.; Hashimoto, K. Fullerene Attached All-Semiconducting Diblock Copolymers for Stable Single-Component Polymer Solar Cells. Chem. Commun. 2010, 46, 6723–6725. [Google Scholar] [CrossRef] [PubMed]
- Topham, P.D.; Parnell, A.J.; Hiorns, R.C. Block Copolymer Strategies for Solar Cell Technology. J. Polym. Sci. B Polym. Phys. 2011, 49, 1131–1156. [Google Scholar] [CrossRef]
- Chan, S.-H.; Lai, C.-S.; Chen, H.-L.; Ting, C.; Chen, C.-P. Highly Efficient P3HT: C60 Solar Cell Free of Annealing Process. Macromolecules 2011, 44, 8886–8891. [Google Scholar] [CrossRef]
- Botiz, I.; Schaller, R.D.; Verduzco, R.; Darling, S.B. Optoelectronic Properties and Charge Transfer in Donor/Acceptor All-Conjugated Diblock Copolymers. J. Phys. Chem. C 2011, 115, 9260–9266. [Google Scholar] [CrossRef]
- Verduzco, R.; Botiz, I.; Pickel, D.L.; Michael Kilbey, S., II; Hong, K.; Dimasi, E.; Darling, S.B. Polythiophene-block-polyfluorene and Polythiophene-blockpoly(fluorene-co-benzothiadiazole): Insights into the Self-Assembly of All-Conjugated Block Copolymers. Macromolecules 2011, 44, 530–539. [Google Scholar] [CrossRef]
- Ramos, A.M.; Rispens, M.T.; van Duren, J.K.J.; Hummelen, J.C.; Janssen, R.A.J. Photoinduced Electron Transfer and Photovoltaic Devices of a Conjugated Polymer with Pendant Fullerenes. J. Am. Chem. Soc. 2001, 123, 6714–6715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Svensson, M.; Andersson, M.R.; Maggini, M.; Bucella, S.; Menna, E.; Inganäs, O. Soluble Polythiophenes with Pendant Fullerene Groups as Double Cable Materials for Photodiodes. Adv. Mater. 2001, 13, 1871–1874. [Google Scholar] [CrossRef]
- Tan, Z.; Hou, J.; He, Y.; Zhou, E.; Yang, C.; Li, Y. Synthesis and Photovoltaic Properties of a Donor-Acceptor Double-Cable Polythiophene with High Content of C60 Pendant. Macromolecules 2007, 40, 1868–1873. [Google Scholar] [CrossRef]
- Chan, S.-H.; Chen, C.-P.; Chao, T.-C.; Ting, C.; Lin, C.-S.; Ko, B.-T. Synthesis, Characterization, and Photovoltaic Properties of Novel Semiconducting Polymers with Thiophene-Phenylene-Thiophene (TPT) as Coplanar Units. Macromolecules 2008, 41, 5519–5526. [Google Scholar] [CrossRef]
- Li, M.; Xu, P.; Yang, J.; Yang, S. Donor-π-Acceptor Double-Cable Polythiophenes Bearing Fullerene Pendant with Tunable Donor/Acceptor Ratio: A Facile Postpolymerization. J. Mater. Chem. 2010, 20, 3953–3960. [Google Scholar] [CrossRef]
- Yao, K.; Chen, L.; Li, F.; Wang, P.; Chen, Y. Cooperative Assembly Donor/Acceptor System Induced by Intermolecular Hydrogen Bonds Leading to Oriented Nanomorphology for Optimized Photovoltaic Performance. J. Phys. Chem. C 2012, 116, 714–721. [Google Scholar] [CrossRef]
- Lin, Y.; Lim, J.A.; Wei, Q.; Mannsfeld, S.C.B.; Briseno, A.L.; Watkins, J.J. Cooperative Assembly of Hydrogen-Bonded Diblock Copolythiophene/Fullerene Blends for Photovoltaic Devices with Well-Defined Morphologies and Enhanced Stability. Chem. Mater. 2012, 24, 622–632. [Google Scholar] [CrossRef]
- Li, F.; Yang, J.; Qin, Y. Synthesis and Characterization of Polythiophene Block Copolymer and Fullerene Derivative Capable of “Three-Point” Complementary Hydrogen Bonding Interactions and Their Application in Bulk-Heterojunction Solar Cells. J. Polym. Sci. A Polym. Chem. 2013, 51, 3339–3350. [Google Scholar] [CrossRef]
- Li, F.; Yager, K.G.; Dawson, N.M.; Yang, J.; Malloy, K.J.; Qin, Y. Complementary Hydrogen Bonding and Block Copolymer Self-Assembly in Cooperation toward Stable Solar Cells with Tunable Morphologies. Macromolecules 2013, 46, 9021–9031. [Google Scholar] [CrossRef]
- Lai, Y.-C.; Ohshimizu, K.; Takahashi, A.; Hsu, J.-C.; Higashihara, T.; Ueda, M.; Chen, W.-C. Synthesis of All-Conjugated Poly(3-hexylthiophene)-block-poly(3-(4′-(3″,7″-dimethyloctyloxy)-3ʹ-pyridinyl)thiophene) and Its Blend for Photovoltaic Applications. J. Polym. Sci. A Polym. Chem. 2011, 49, 2577–2587. [Google Scholar] [CrossRef]
- Li, F.; Yager, K.G.; Dawson, N.M.; Jiang, Y.-B.; Malloy, K.J.; Qin, Y. Stable and Controllable Polymer/Fullerene Composite Nanofibers through Cooperative Noncovalent Interactions for Organic Photovoltaics. Chem. Mater. 2014, 26, 3747–3756. [Google Scholar] [CrossRef]
- Li, F.; Yager, K.G.; Dawson, N.M.; Jiang, Y.-B.; Malloy, K.J.; Qin, Y. Nano-Structuring Polymer/Fullerene Composites through the Interplay of Conjugated Polymer Crystallization, Block Copolymer Self-Assembly and Complementary Hydrogen Bonding Interactions. Polym. Chem. 2015, 6, 721–731. [Google Scholar] [CrossRef]
- Li, F.; Dawson, N.M.; Jiang, Y.-B.; Malloy, K.J.; Qin, Y. Conjugated Polymer/Fullerene Nanostructures through Cooperative Non-Covalent Interactions for Organic Solar Cells. Polymer 2015, 76, 220–229. [Google Scholar] [CrossRef]
- Nozik, A.J. Quantum Dot Solar Cells. Physica E 2002, 14, 115–120. [Google Scholar] [CrossRef]
- Milliron, D.J.; Gur, I.; Alivisatos, A.P. Hybrid Organic-Nanocrystal Solar Cells. MRS Bull. 2005, 30, 41–44. [Google Scholar] [CrossRef]
- Rupasov, V.I.; Klimov, V.I. Carrier Multiplication in Semiconductor Nanocrystals via Intraband Optical Transitions Involving Virtual Biexciton States. Phys. Rev. B 2007, 76, 125321. [Google Scholar] [CrossRef]
- Kamat, P.V. Quantum Dot Solar Cells. Semiconductor Nanocrystals as Light Harvesters. J. Phys. Chem. C 2008, 112, 18737–18753. [Google Scholar] [CrossRef]
- Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P.V. Quantum Dot Solar Cells. Harvesting Light Energy with CdSe Nanocrystals Molecularly Linked to Mesoscopic TiO2 Films. J. Am. Chem. Soc. 2006, 128, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kamat, P.V.; Kuno, M. A CdSe Nanowire/Quantum Dot Hybrid Architecture for Improving Solar Cell Performance. Adv. Funct. Mater. 2010, 20, 1464–1472. [Google Scholar] [CrossRef]
- Dayal, S.; Kopodakis, N.; Olson, D.C.; Ginley, D.S.; Rumbles, G. Photovoltaic Devices with a Low Band Gap Polymer and CdSe Nanostructures Exceeding 3% Efficiency. Nano Lett. 2010, 10, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.H.; Kamat, P.V. CdSe Quantum Dot/Fullerene Hybrid Nanocomposite for Solar Energy Conversion: Electron Transfer and Photoelectrochemistry. ACS Nano 2011, 5, 9421–9427. [Google Scholar] [CrossRef] [PubMed]
- Klem, E.J.D.; Gregory, C.W.; Cunninghan, G.B.; Hall, S.; Temple, D.S.; Lewis, J.S. Planar PdS Quantum Dot/C60 Heterojunction Photovoltaic Devices with 5.2% Power Conversion Efficiency. Appl. Phys. Lett. 2012, 100, 173109. [Google Scholar] [CrossRef]
- Zang, H.; Routh, P.K.; Alam, R.; Maye, M.M.; Cotlet, M. Core Size Dependent Hole Transfer from a Photoexcited CdSe/ZnS Quantum Dot to a Conductive Polymer. Chem. Commun. 2014, 50, 5958–5960. [Google Scholar] [CrossRef] [PubMed]
- El-Ballouli, A.O.; Alarousu, E.; Bernardi, M.; Aly, S.M.; Lagrow, A.P.; Bakr, O.M.; Mohammed, O.F. Quantum Confinement-Tunable Ultrafast Charge Transfer at the PbS Quantum Dot and Phenyl-C61-butyric Acid Methyl Ester Interface. J. Am. Chem. Soc. 2014, 136, 6952–6959. [Google Scholar] [CrossRef] [PubMed]
- Shallcross, R.C.; Chawla, G.S.; Marikkar, F.S.; Tolbert, S.; Pyun, J.; Armstrong, N.R. Efficient CdSe Nanocrystal Diffraction Gratings Prepared by Microcontact Molding. ACS Nano 2009, 3, 3629–3637. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.W.; Qu, L.; Guo, W.; Peng, X. Experimental Determination of the Extinction Coefficient of CdTe, CdSe, and CdS Nanocrystals. Chem. Mater. 2003, 15, 2854–2860. [Google Scholar] [CrossRef]
- Sheina, E.E.; Liu, J.; Iovu, M.C.; Laird, D.W.; McCullough, R.D. Chain Growth Mechanism for Regioregular Nickel-Initiated Cross-Coupling Polymerizations. Macromolecules 2004, 37, 3526–3528. [Google Scholar] [CrossRef]
- Iovu, M.C.; Sheina, E.E.; Gil, R.R.; McCullough, R.D. Experimental Evidence for the Quasi-“Living” Nature of the Grignard Metathesis Method for the Synthesis of Regioregular Poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Yokozawa, T.; Yokoyama, A. Chain-Growth Condensation Polymerization for the Synthesis of Well-Defined Condensation Polymers and π-Conjugated Polymers. Chem. Rev. 2009, 109, 5595–5619. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Peng, X. Control of Photoluminescence Porperties of CdSe Nanocrystals in Growth. J. Am. Chem. Soc. 2002, 124, 2049–2055. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.T.; Weiss, E.A. Relaxation of Exciton Confinement in CdSe Quantum Dots by Modification with a Conjugated Dithiocarbamate Ligand. ACS Nano 2010, 4, 3195–3200. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.D.; Homan, S.B.; Kodaimati, M.; He, C.; Nepomnyashchii, A.B.; Swenson, N.K.; Lian, S.; Calzada, R.; Weiss, E.A. Electronic Processes within Quantum Dot-Molecule Complexes. Chem. Rev. 2016, 116, 12865–12919. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blends b | PCE (%) c | JSC (mA/cm2) d | VOC (V) e | FF (%) f |
|---|---|---|---|---|
| P3HT BHJ | 0.17 ± 0.03 (0.19) | 1.89 ± 0.156 (2.00) | 0.27 ± 0.00 (0.27) | 33 ± 3 (31) |
| P3HT NF | 0.53 ± 0.21 (0.79) | 3.95 ± 1.54 (5.99) | 0.51 ± 0.02 (0.54) | 27 ± 2 (28) |
| BCP3 BHJ | 0.78 ± 0.08 (0.87) | 4.23 ± 0.618 (4.92) | 0.49 ± 0.01 (0.50) | 38 ± 1 (40) |
| BCP3 NF | 0.42 ± 0.06 (0.46) | 3.26 ± 1.15 (4.35) | 0.34 ± 0.04 (0.39) | 41 ± 14 (58) |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, B.W.; Meng, L.; Fetrow, C.; Qin, Y. Core/Shell Conjugated Polymer/Quantum Dot Composite Nanofibers through Orthogonal Non-Covalent Interactions. Polymers 2016, 8, 408. https://doi.org/10.3390/polym8120408
Watson BW, Meng L, Fetrow C, Qin Y. Core/Shell Conjugated Polymer/Quantum Dot Composite Nanofibers through Orthogonal Non-Covalent Interactions. Polymers. 2016; 8(12):408. https://doi.org/10.3390/polym8120408
Chicago/Turabian StyleWatson, Brad W., Lingyao Meng, Chris Fetrow, and Yang Qin. 2016. "Core/Shell Conjugated Polymer/Quantum Dot Composite Nanofibers through Orthogonal Non-Covalent Interactions" Polymers 8, no. 12: 408. https://doi.org/10.3390/polym8120408
APA StyleWatson, B. W., Meng, L., Fetrow, C., & Qin, Y. (2016). Core/Shell Conjugated Polymer/Quantum Dot Composite Nanofibers through Orthogonal Non-Covalent Interactions. Polymers, 8(12), 408. https://doi.org/10.3390/polym8120408