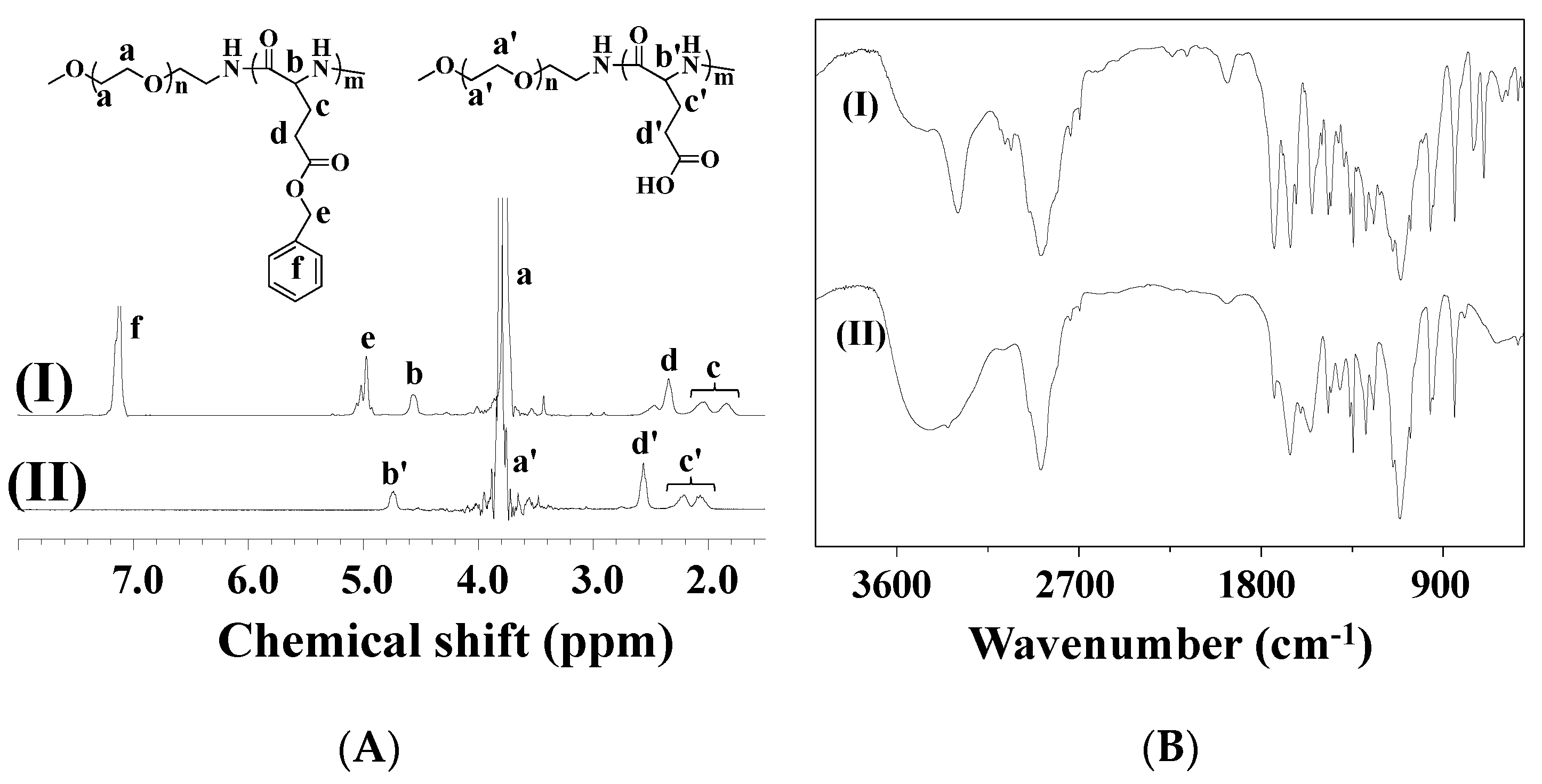
3.2. pH-Sensitive EPI Release in Vitro and Optimized Biocompatibility of mPEG-b-PGA/EPI
In order to confirm the acid-sensitivity of mPEG-
b-PGA/EPI, the
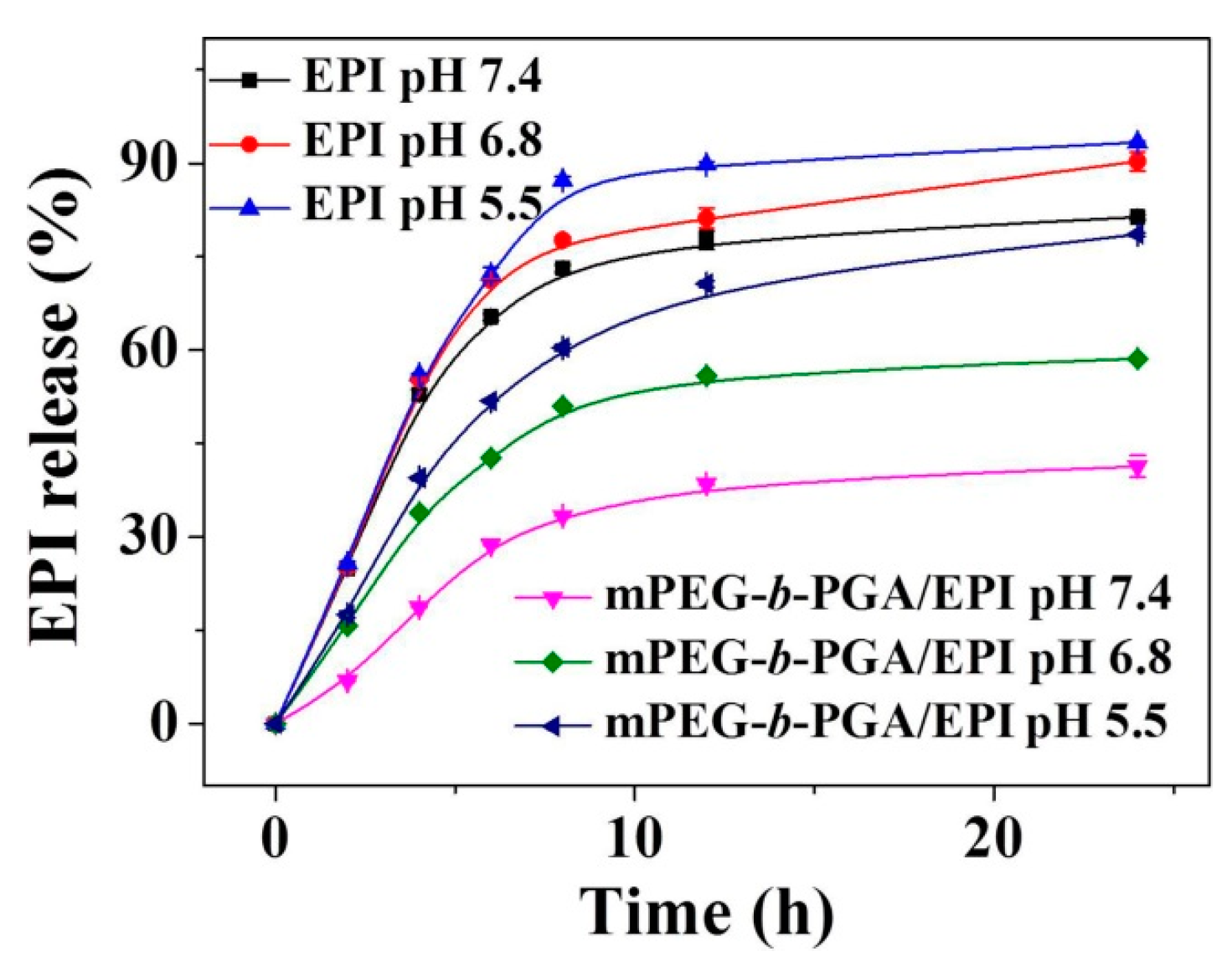
in vitro release experiments of EPI were carried out in PBS at pH 5.5, 6.8, and 7.4, 37 °C. As shown in
Figure 3, the free DOX release from dialysis bag was more than 70% in PBS at all the test pHs within 8 h, indicating the feasibility of release proposal. The cumulative release of EPI in different conditions from mPEG-
b-PGA/EPI gradually increased in a time-dependent manner, and was obviously slower than that of free EPI. Furthermore, the complex micelle release EPI more rapidly, and the cumulative release was obviously higher as the decrease of pH. In physiological pH environment (
i.e., pH 7.4), only 41.3% of loaded EPI released after 24 h incubation, while the cumulative release reached 58.6 and 78.5%, respectively, for the same duration at pH 6.8 and 5.5, which mimicked the intratumoral and intracellular acidic microenvironments. These results might attribute to the disruption of electrostatic interaction between EPI and mPEG-
b-PGA because of the deionization of PGA moiety (p
Ka = 4.5) and ionization of EPI (p
Ka = 7.7) as the decrease of pH [
16,
27]. In addition, the increased hydrophilicity of EPI in acid condition, which was much lower than the p
Ka of EPI, also resulted in the acidity-accelerated drug release [
16]. Thus, mPEG-
b-PGA/EPI with pH-sensitive property could realize the controlled drug release and might perform better pharmacokinetic characteristics
in vivo compared with free EPI. In addition, the limited premature EPI release and relative stability of mPEG-
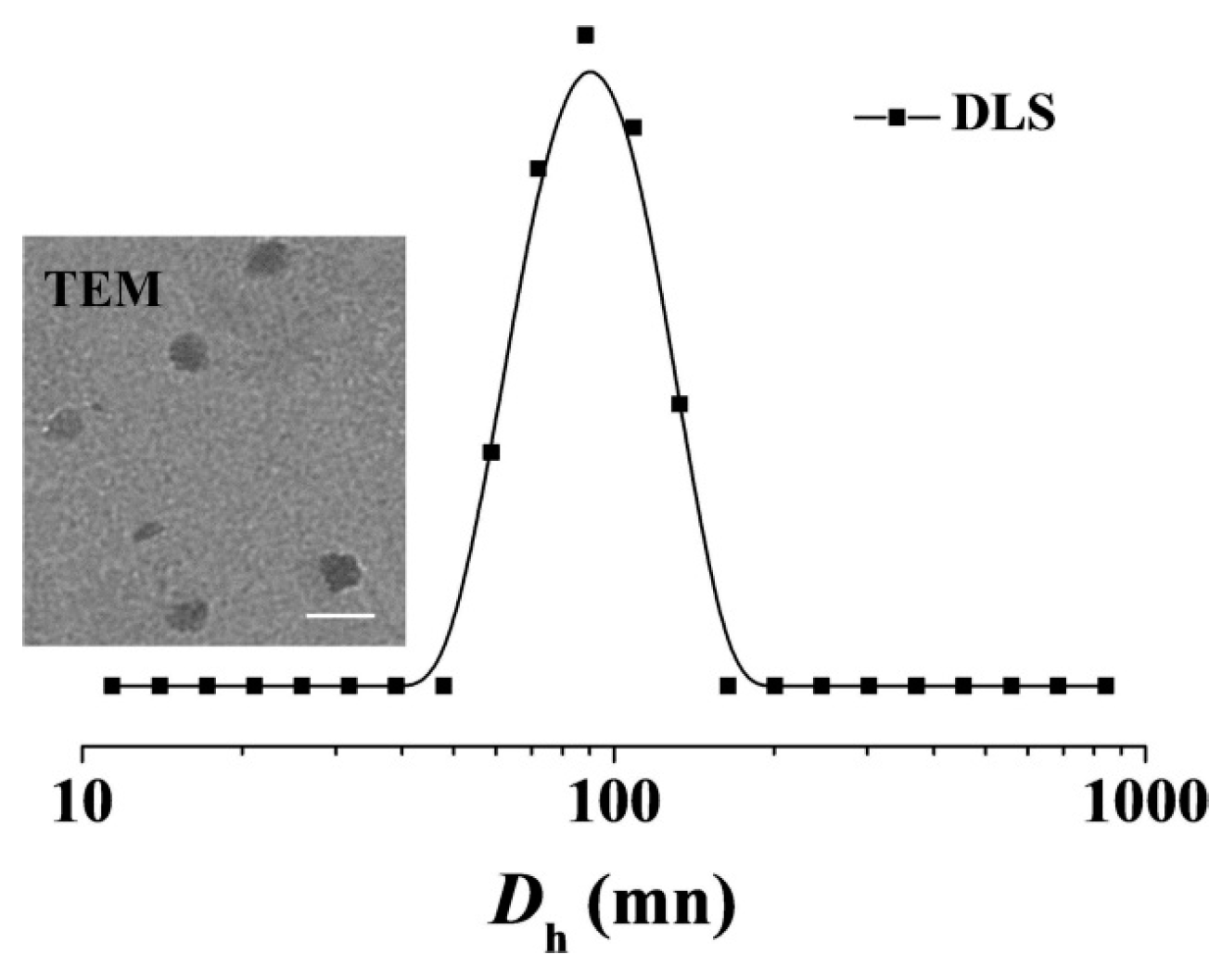
b-PGA/EPI in PBS and BSA/PBS of pH 7.4 at 25 °C were confirmed by monitoring the
Dh through DLS. As shown in
Supplementary Figure S1, the laden micelle exhibited nearly constant diameters during the incubation in both PBS and BSA/PBS solutions for 72 h, which indicated that the encapsulated drug less released prematurely from mPEG-
b-PGA/EPI at pH 7.4.
Figure 3.
Time- and pH-dependent EPI release profiles of free EPI and mPEG-b-PGA/EPI in PBS of pH 7.4, 6.8, and 5.5 at 37 °C. Data were presented as mean ± SD (n = 3).
Figure 3.
Time- and pH-dependent EPI release profiles of free EPI and mPEG-b-PGA/EPI in PBS of pH 7.4, 6.8, and 5.5 at 37 °C. Data were presented as mean ± SD (n = 3).
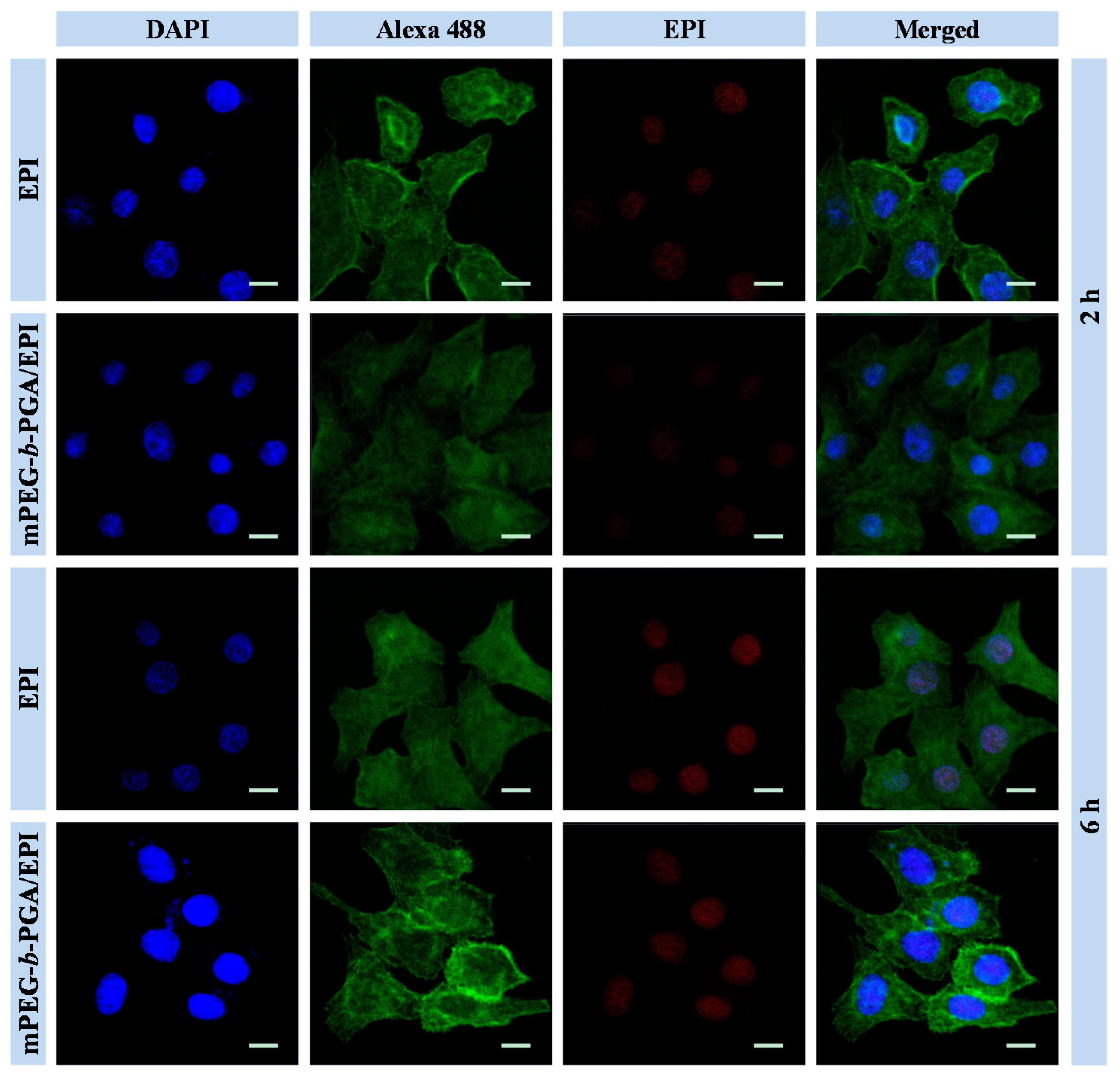
Both CLSM and FCM were employed to detect the cellular uptake and intracellular EPI release from mPEG-
b-PGA/EPI. HepG2 cells were cultivated with free EPI or mPEG-
b-PGA/EPI at an EPI equivalent dose of 10.0 μg·mL
−1 for 2 or 6 h. As shown in CLSM results in
Figure 4, the red EPI fluorescence of the two forms of EPI mainly concentrated in the nuclei of HepG2 cells, coinciding with the location of DAPI (blue). After 2 h culture, the red fluorescence intensity of HepG2 cells in the free EPI group is obviously higher than that in the mPEG-
b-PGA/EPI group, indicating free EPI could be uptake faster. There may be two reasons causing these results. For the first, the polymeric nanoparticles are mainly uptaken by cells via endocytosis pathway, while the small molecule agents via diffusion approach, which was energy-independent and easily coming up [
28]. For the other one, the EPI in the hydrophobic core of nanoparticles exhibits weaker fluorescence intensity due to the self-quenching effect of fluorescent molecules compared with free EPI at the same concentration [
25,
29]. When the culture period was extended to 6 h, the red fluorescence intensity in both of the two groups increased somewhat. However, the increase amplitude in the mPEG-
b-PGA/EPI group, whose red fluorescence intensity was nearly close to that of free EPI group, was more obviously. This might attribute to the increased endocytosis of loading micelle and the rapid EPI release in endosomes where the pH is much lower than that in normal physiological microenvironment, while free EPI effused quickly after uptake [
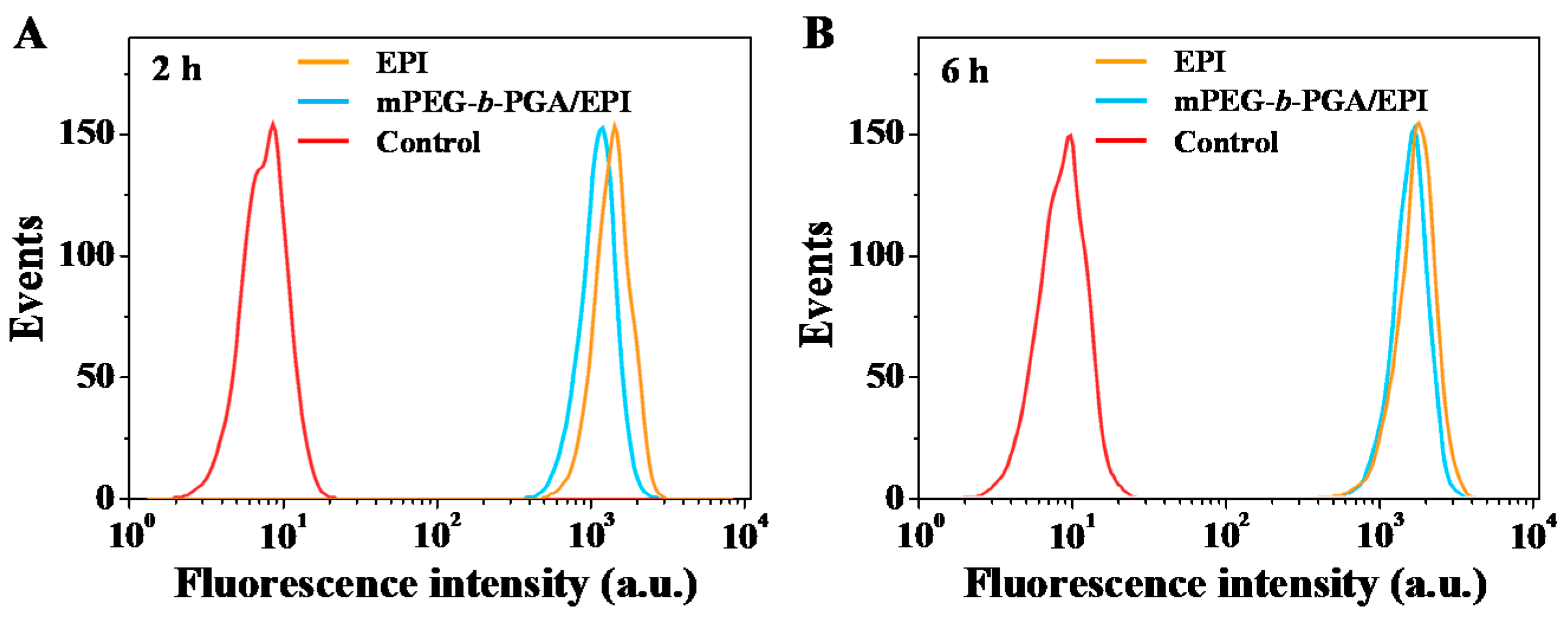
30]. The date of FCM further confirmed the results of CLSM (
Figure 5). The signal intensity of EPI in HepG2 cells dealt with mPEG-
b-PGA/EPI was lower than that with free EPI at both of the test durations (
i.e., 2 and 6 h). Additionally, at the 6 h point-in-time, the signal intensities of the two drug formulation groups were much closed.
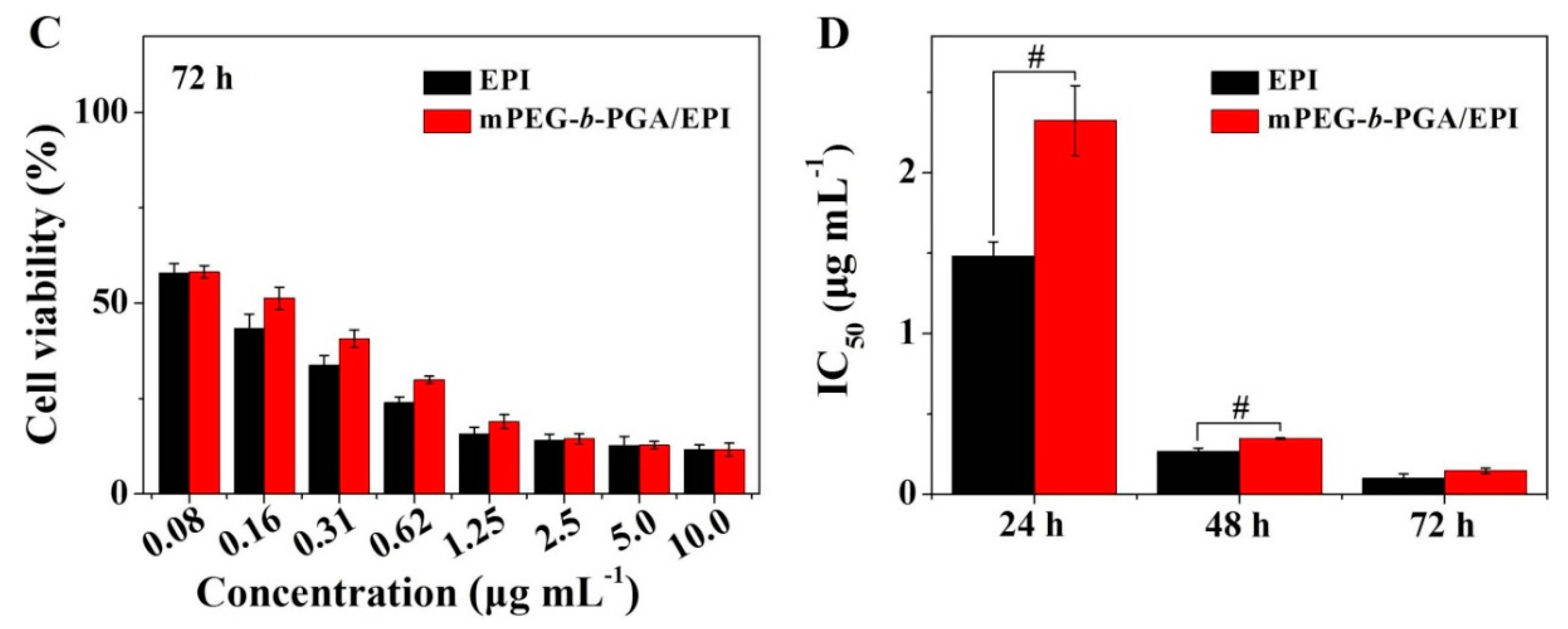
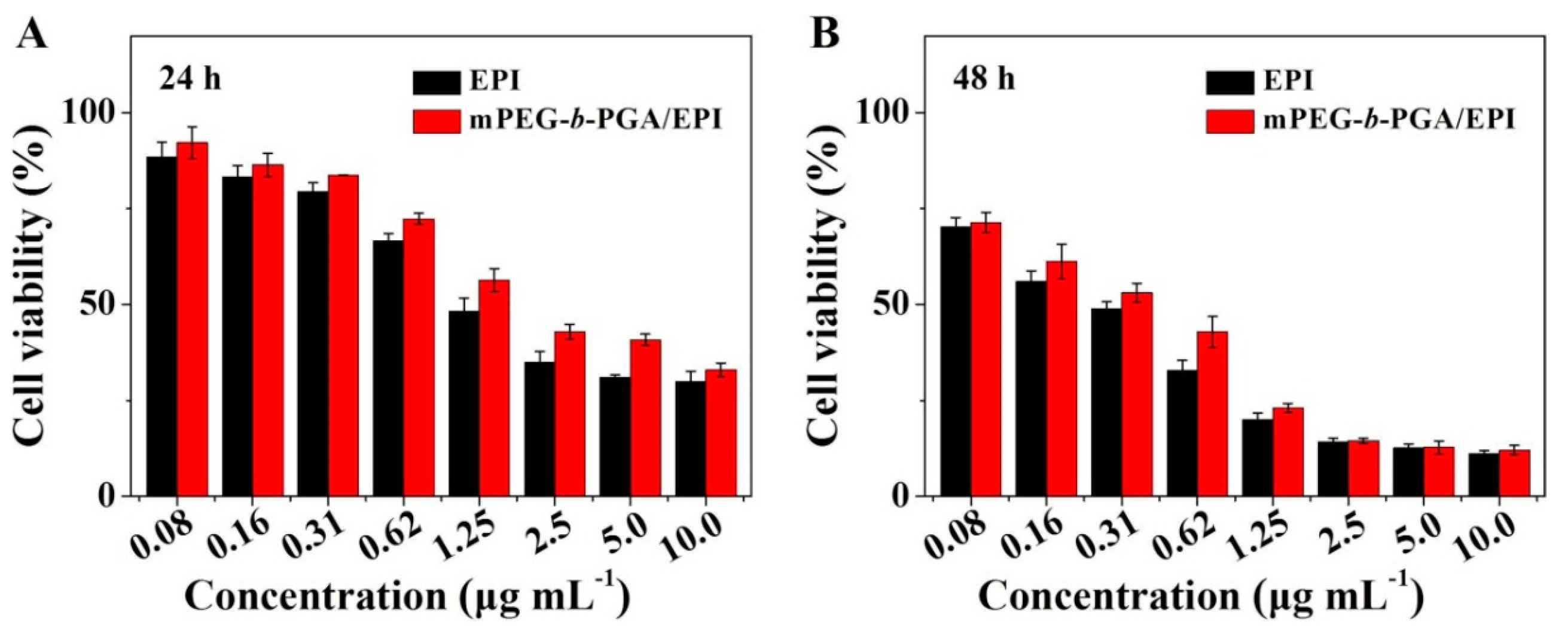
The MTT colorimetric assay was universally used to detect the cytotoxicity of materials and drugs
in vitro. In this study, MTT assay was also carried out toward HepG2 cells. As shown in
Supplementary Figure S2, mPEG-
b-PGA possessed nice cytocompatibility. Even dealt with the maximum concentration of 100.0 μg·mL
−1 for 72 h, the cells could keep their viability for more than 90%. It indicated that the polypeptide-based block copolymer was a kind of safe materials for nanocarrier construction as reported previously [
14,
30]. On drug-related MTT assay, both free EPI and mPEG-
b-PGA/EPI exhibited typically time- and agent concentration-dependent cytotoxicity effects (
Figure 6). It was worth noted that mPEG-
b-PGA/EPI revealed a weaker cell viability inhibition effect compared with free EPI at equivalent drug concentration and test time (
i.e., 24, 48, or 72 h), which may be led by the slow agent release rate and cellular uptake as shown in the EPI release and cellular uptake experiments. However, this gap diminished gradually with the extension of time, especially in the groups cultured with high concentrations of EPI (≥2.5 μg·mL
−1). The half-maximal inhibitory concentration (IC
50) values of different drug formulations also confirmed the above declarations. The IC
50 values of mPEG-
b-PGA/EPI at 24 and 48 h were 2.3 and 0.3 μg·mL
−1, respectively, which were significantly higher than those of free EPI at 1.5 and 0.2 μg·mL
−1 (
p < 0.01), respectively, whereas the difference of IC
50 values between mPEG-
b-PGA/EPI and free EPI was not significant at 72 h, which might be caused by the sustained drug release from the micelle and increased intracellular drug accumulation with time.
Figure 4.
Cellular uptake microimages of HepG2 cells after incubation with free EPI or mPEG-b-PGA/EPI for 2 or 6 h. Scale bar: 25.0 μm.
Figure 4.
Cellular uptake microimages of HepG2 cells after incubation with free EPI or mPEG-b-PGA/EPI for 2 or 6 h. Scale bar: 25.0 μm.
Figure 5.
FCM profiles of HepG2 cells incubated with free EPI, mPEG-b-PGA/EPI, or PBS (as control) for 2 (A) or 6 h (B).
Figure 5.
FCM profiles of HepG2 cells incubated with free EPI, mPEG-b-PGA/EPI, or PBS (as control) for 2 (A) or 6 h (B).
Figure 6.
In vitro cytotoxicities of free EPI and mPEG-b-PGA/EPI toward HepG2 cells after incubation for 24 (A); 48 (B); or 72 h (C); and the corresponding IC50 values (D). Data were presented as a mean ± SD (n = 3; # p < 0.01).
Figure 6.
In vitro cytotoxicities of free EPI and mPEG-b-PGA/EPI toward HepG2 cells after incubation for 24 (A); 48 (B); or 72 h (C); and the corresponding IC50 values (D). Data were presented as a mean ± SD (n = 3; # p < 0.01).
The excellent blood compatibility of drug-loaded nanoparticles is an important premise for their clinical applications, because most of them will be administrated through intravenous injection [
31]. Several studies have verified that the polypeptide-based block copolymer can improve the blood compatibility of free drugs [
32,
33]. In this study, the hemolysis assay was performed through spectroscopy based on the previous report [
32]. As shown in
Supplementary Figure S3C, there was no hemolysis in the mPEG-
b-PGA group of various dosages, and the hemolysis ratio was nearly 0% even at the highest polymer concentration of 5.0 mg·mL
−1. What is more, mPEG-
b-PGA/EPI could relieve the hemolysis induced by high dosages of free EPI (> 0.03 mg·mL
−1) and exhibited excellent blood compatibility (
Supplementary Figure S3A,B). The ameliorative effect might be attributable to the protection of hydrophilic shell and the negatively charged surface [
16].
3.3. Upregulated Tolerability of mPEG-b-PGA/EPI in Vivo
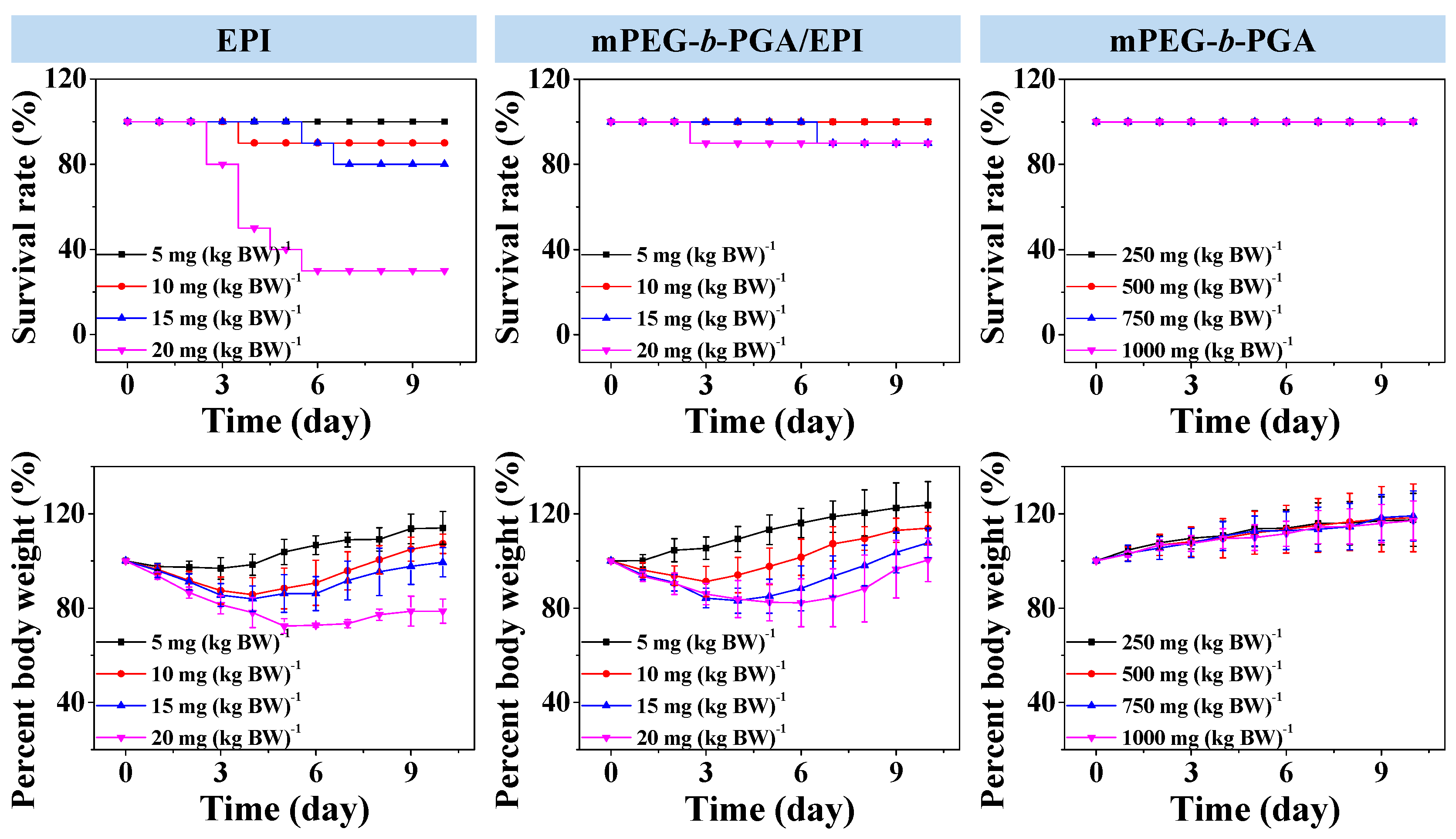
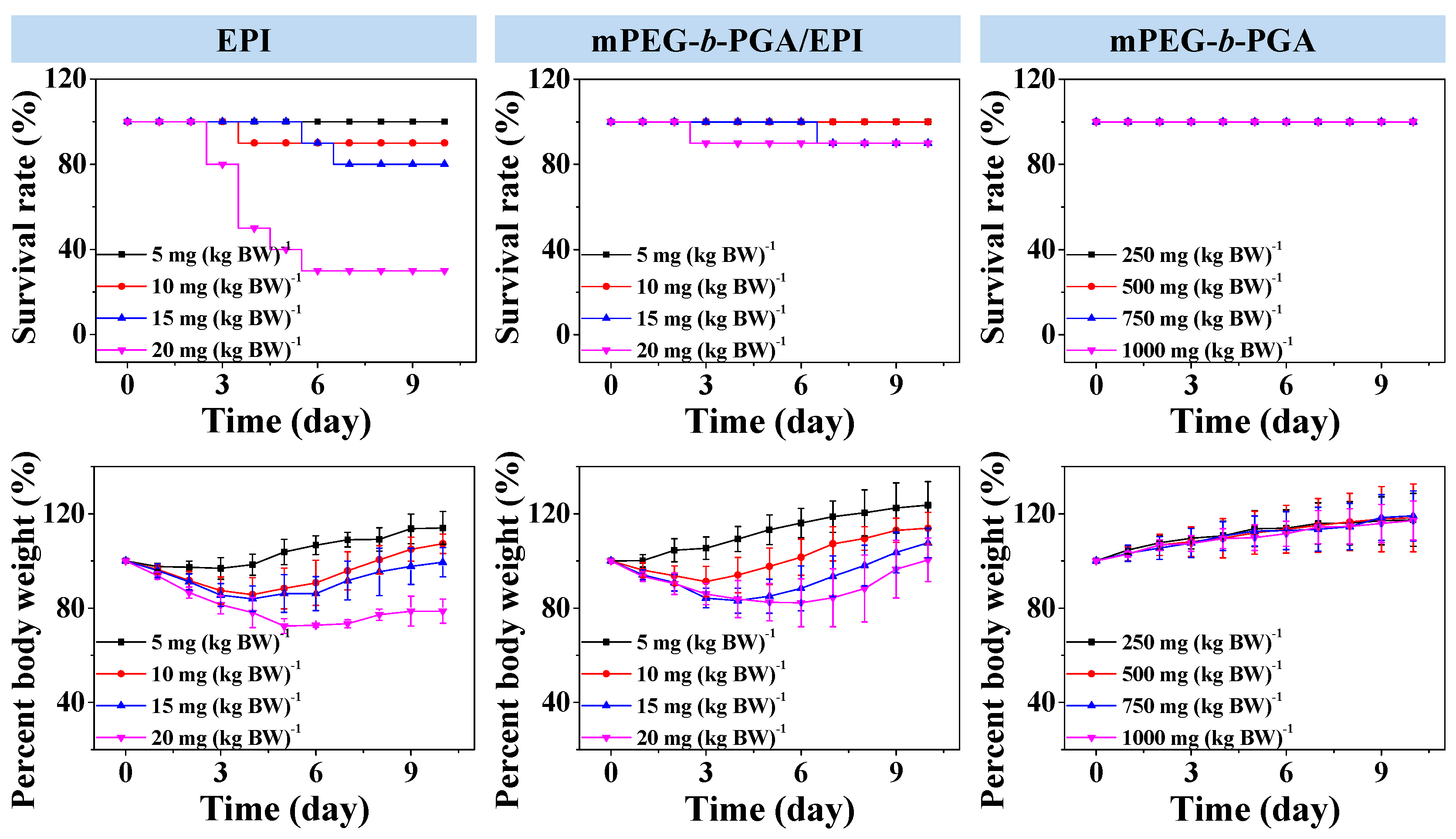
MTD, which reflects the toxicity and tolerability, is one of the most important indices for the in vivo applications of various therapeutic agents. Usually, a higher MTD predicts better efficacy. In this study, the MTDs were estimated by a single intravenous injection of mPEG-b-PGA at doses of 250.0, 500.0, 750.0, and 1000.0 mg per kg body weight (mg·(kg·BW)−1), and free EPI and mPEG-b-PGA/EPI at 5.0, 10.0, 15.0, and 20.0 mg EPI equivalent per kg body weight to Kunming mouse. The survival rates and body weights of mice were monitored for 10 days after intravenous injection.
The survival rate and body weight change situations in MTD tests were showed in
Figure 7. In terms of survival rate, no death appeared in various doses of mPEG-
b-PGA group. The survival rates of free EPI group at doses of 5.0, 10.0, 15.0, and 20.0 mg·(kg·BW)
−1 were 100%, 90%, 80%, and 30%, respectively, at the end of all the tests. In contrast, there were no dead animals in the mPEG-
b-PGA/EPI group at both low and medium doses (
i.e., 5.0 and 10.0 mg·(kg·BW)
−1), while the survival rates of high dose groups (
i.e., 15.0 and 20.0 mg·(kg·BW)
−1) were both high, up to 90% after the same observation period. In terms of body weight, a stable increase could be seen in the mPEG-
b-PGA group, while there were various decrease levels of body weights in the drug-treated groups except low dose of mPEG-
b-PGA/EPI group (
i.e., 5.0 mg·(kg·BW)
−1). However, the decrease extent of mPEG-
b-PGA/EPI group was smaller, and the recovery was more rapid compared with that of free EPI group. At the end of all the tests, the percentage body weights to the original values of mPEG-
b-PGA/EPI group at dose of 5.0, 10.0, 15.0, and 20.0 mg·(kg·BW)
−1 were 123.6% ± 10.0%, 113.9% ± 6.8%, 107.7% ± 6.3%, and 100.5% ± 9.3%, respectively, which were significantly higher than those of free EPI group at the equivalent dose (
i.e., 114.0% ± 7.0%, 107.2% ± 4.1%, 99.4% ± 6.1%, and 78.7% ± 5.1%, respectively). The MTD was estimated according to the threshold at which all animals survived and the loss of body weight was below 15% [
24]. The MTD of free EPI was determined to be between 5.0 to 10.0 mg·(kg·BW)
−1, which was lower than that in previous studies due to different animal species in tests [
34]. However, mPEG-
b-PGA/EPI was able to upregulate the MTD of EPI to between 10.0 and 15.0 mg·(kg·BW)
−1, which was nearly two times higher than that of free drug. Therefore, mPEG-
b-PGA possessed excellent biocompatibility, and mPEG-
b-PGA/EPI was a better formulation than free EPI for clinical use. In addition, the diets of mice were quantified during the MTD test. As shown in
Supplementary Figure S4, the variation trend of food and water in all groups were consistent with the change of body weight, which further confirmed the biocompatibility of mPEG-
b-PGA and the advantage of mPEG-
b-PGA/EPI.
Figure 7.
Survival rates and percent body weights of Kunming mice after intravenous injection of free EPI or mPEG-b-PGA/EPI at EPI equivalent dose of 5.0, 10.0, 15.0, or 20.0 mg·(kg·BW)−1, or mPEG-b-PGA at dose of 250.0, 500.0, 750.0, or 1000.0 mg·(kg·BW)−1 (n = 10).
Figure 7.
Survival rates and percent body weights of Kunming mice after intravenous injection of free EPI or mPEG-b-PGA/EPI at EPI equivalent dose of 5.0, 10.0, 15.0, or 20.0 mg·(kg·BW)−1, or mPEG-b-PGA at dose of 250.0, 500.0, 750.0, or 1000.0 mg·(kg·BW)−1 (n = 10).
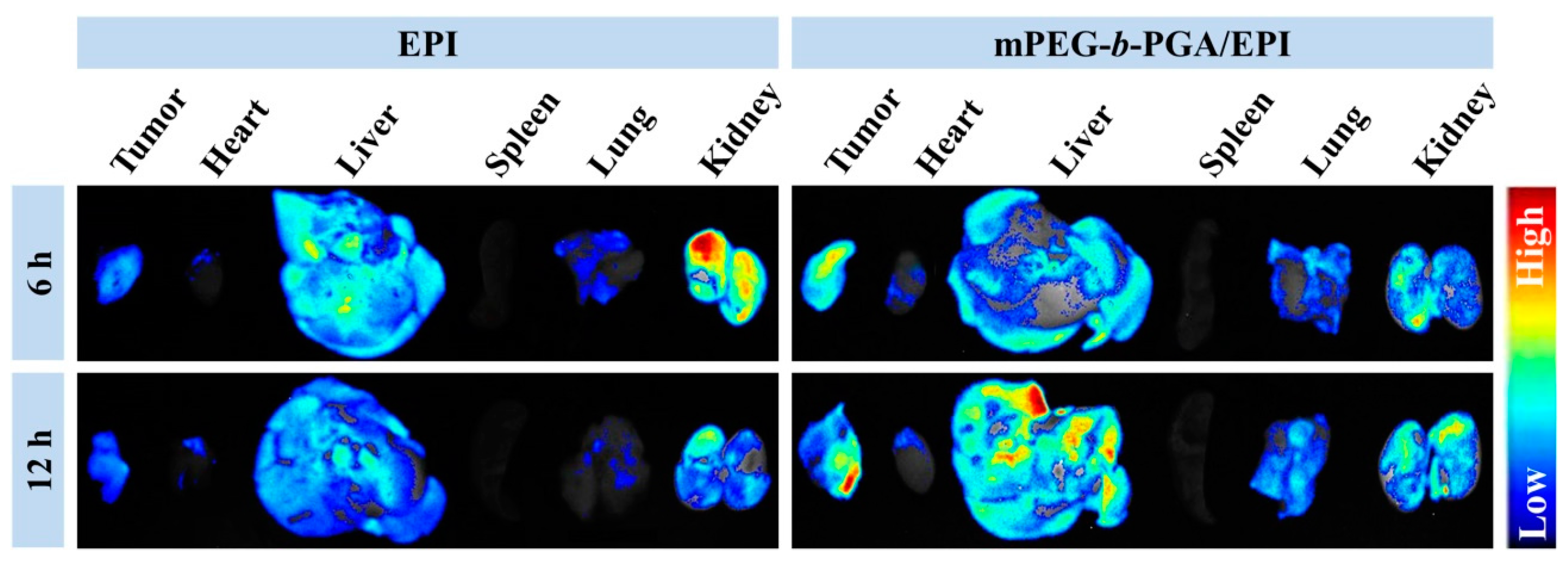
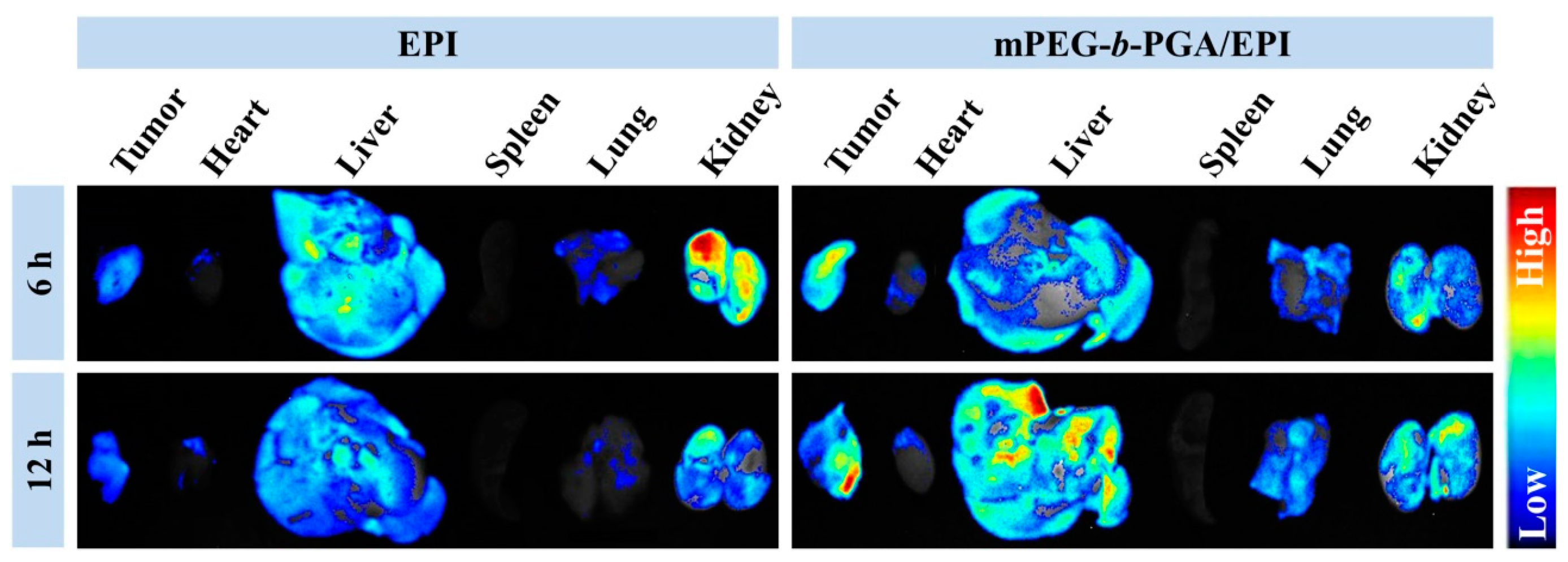
3.4. Improved Tissue Distribution and Enhanced Antitumor Efficacy of mPEG-b-PGA/EPI in Vivo
The tissue biodistribution of antitumor pharmaceuticals is a crucial factor on therapy efficacy and side effects. To detect the influence of mPEG-
b-PGA on the biodistribution of EPI, the
ex vivo fluorescence imaging tests of isolated tumors and major organs (
i.e., heart, liver, spleen, lung, and kidney) were performed at 6 or 12 h post-injection into H22 HCC-xenografted BALB/c mice. As shown in
Figure 8, liver and kidney appeared strong EPI fluorescence after the administration of EPI formulations, suggesting that both of them were mainly metabolized by liver and kidney [
17,
30]. It is worth mentioning that the fluorescence intensities of tumor and test organs were observed to fall in the free EPI group when the time extended from 6 to 12 h, while that in the mPEG-
b-PGA/EPI group increased somewhat. The different changes indicated that the micellization was able to improve the pharmacokinetic character of free drug and made them metabolize longer [
16,
35]. More meaningfully, the tumor fluorescence intensity of mouse treated with mPEG-
b-PGA/EPI for 6 h is stronger than that with free EPI, and the distinction was more obvious at 12 h post-injection.
Figure 8.
Ex vivo EPI fluorescence images showing tissue distribution of free EPI and mPEG-b-PGA/EPI in tumors and major organs (i.e., heart, liver, spleen, lung, and kidney) of H22 HCC-xenografted BALB/c mice at 6 or 12 h post-injection (n = 3).
Figure 8.
Ex vivo EPI fluorescence images showing tissue distribution of free EPI and mPEG-b-PGA/EPI in tumors and major organs (i.e., heart, liver, spleen, lung, and kidney) of H22 HCC-xenografted BALB/c mice at 6 or 12 h post-injection (n = 3).
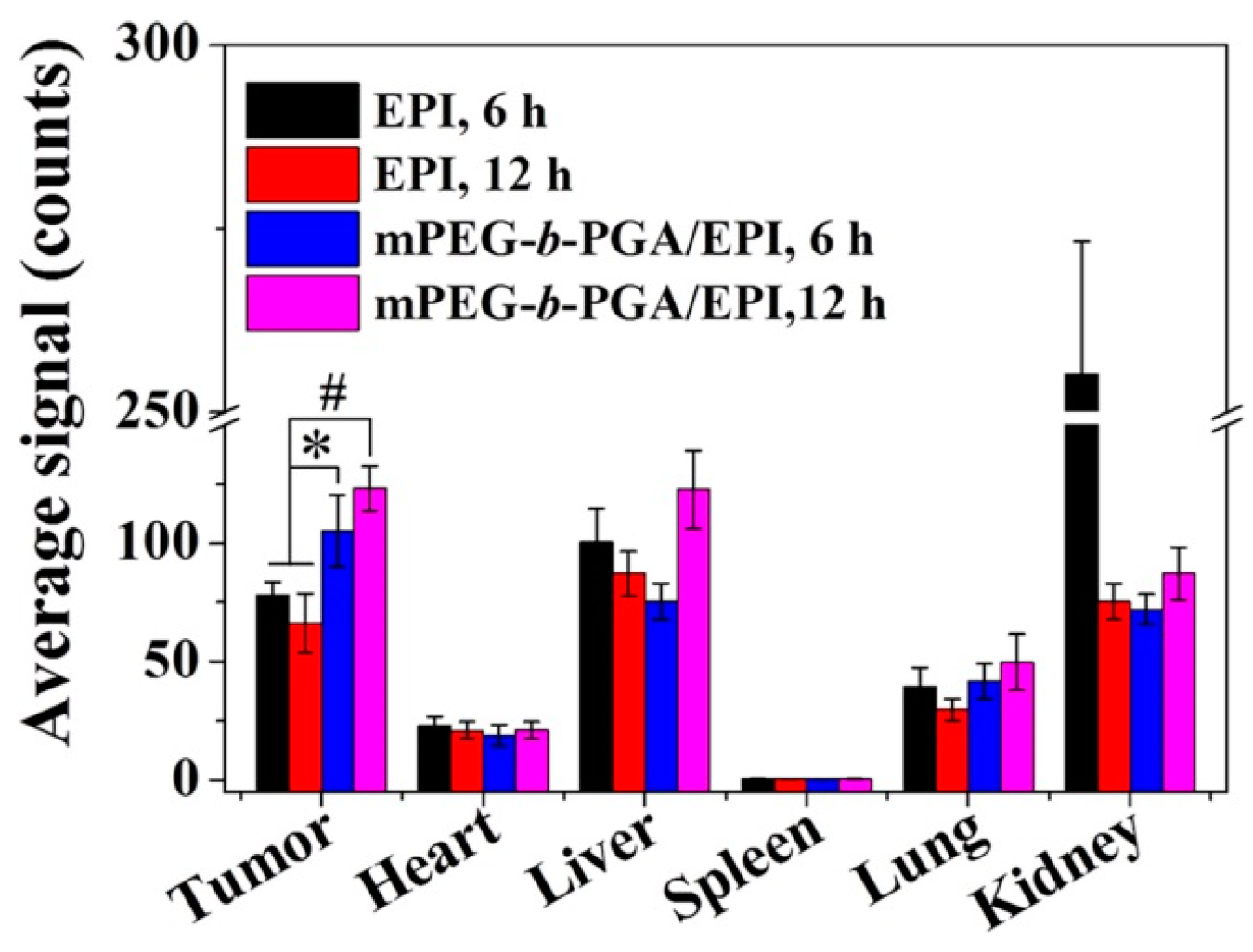
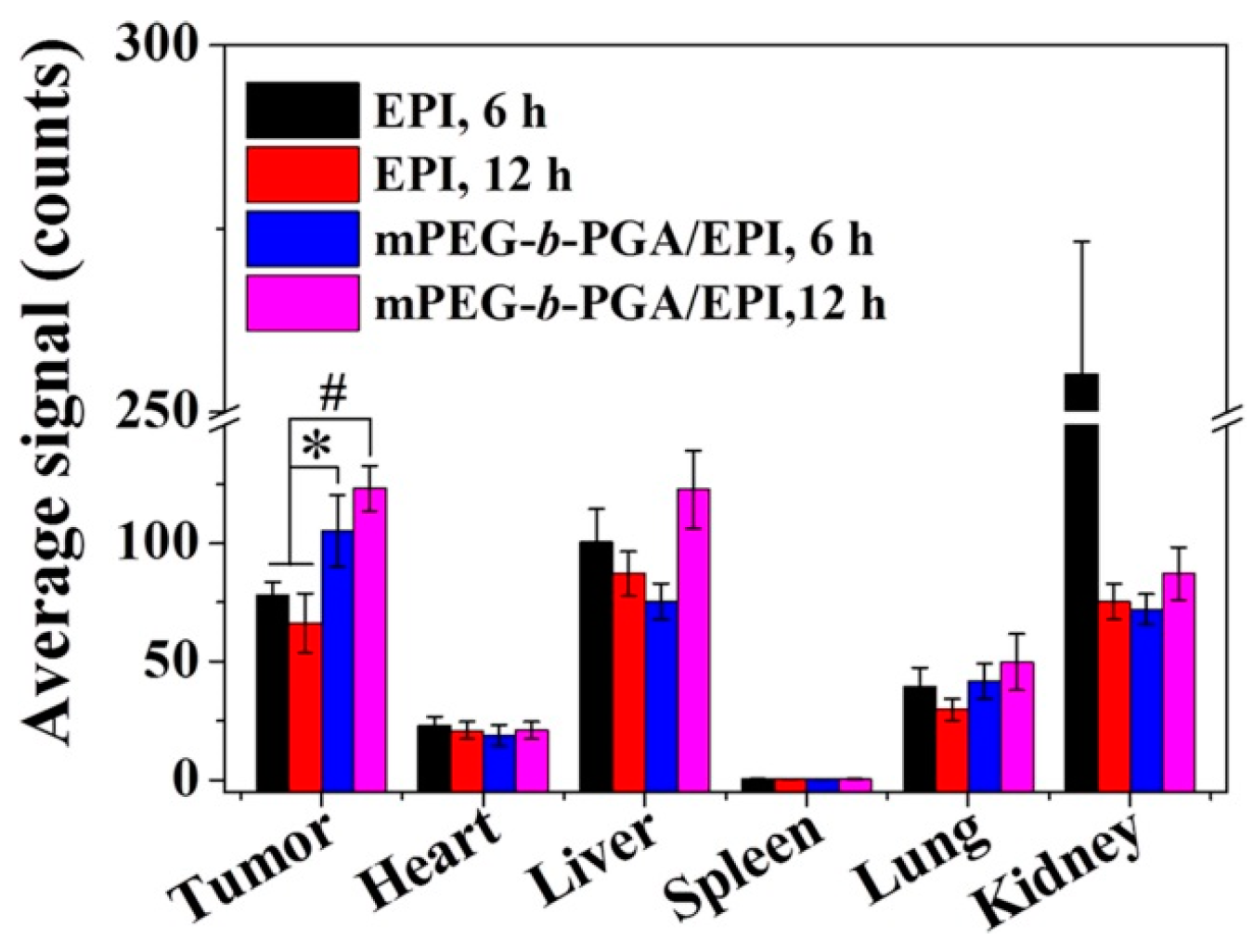
The semi-quantitative analysis of fluorescence intensity was carried out by Maestro™ 2.4 software (CRi Inc., Woburn, MA, USA). As shown in
Figure 9, the average signals,
i.e., photon numbers per unit area, were calculated. Generally, the average signals of organs in the free EPI group were relative higher than those of mPEG-
b-PGA/EPI group at 6 h post-injection. As the time extended from 6 to 12 h, the variation tendency of average signals in both free EPI and mPEG-
b-PGA/EPI groups were opposite as general observation above. More fascinatingly, the average signals of tumors in the mPEG-
b-PGA/EPI group were 105.3 ± 15.1 and 123.2 ± 9.5 at 6 and 12 h post-injection, respectively, which were significantly higher than 78.3 ± 5.4 and 66.3 ± 12.4 of free EPI group (
p < 0.05 for 6 h, and
p < 0.01 for 12 h). The increased agent accumulation in tumor may depend on the EPR effect and predict the increased anticancer efficacy [
36].
Figure 9.
Average signals collected from the tumors and major organs (i.e., heart, liver, spleen, lung, and kidney) in H22 HCC-xenografted BALB/c mice at 6 or 12 h after intravenous injection of free EPI or mPEG-b-PGA/EPI. Data were presented as a mean ± SD (n =3; * p < 0.05, # p < 0.01).
Figure 9.
Average signals collected from the tumors and major organs (i.e., heart, liver, spleen, lung, and kidney) in H22 HCC-xenografted BALB/c mice at 6 or 12 h after intravenous injection of free EPI or mPEG-b-PGA/EPI. Data were presented as a mean ± SD (n =3; * p < 0.05, # p < 0.01).
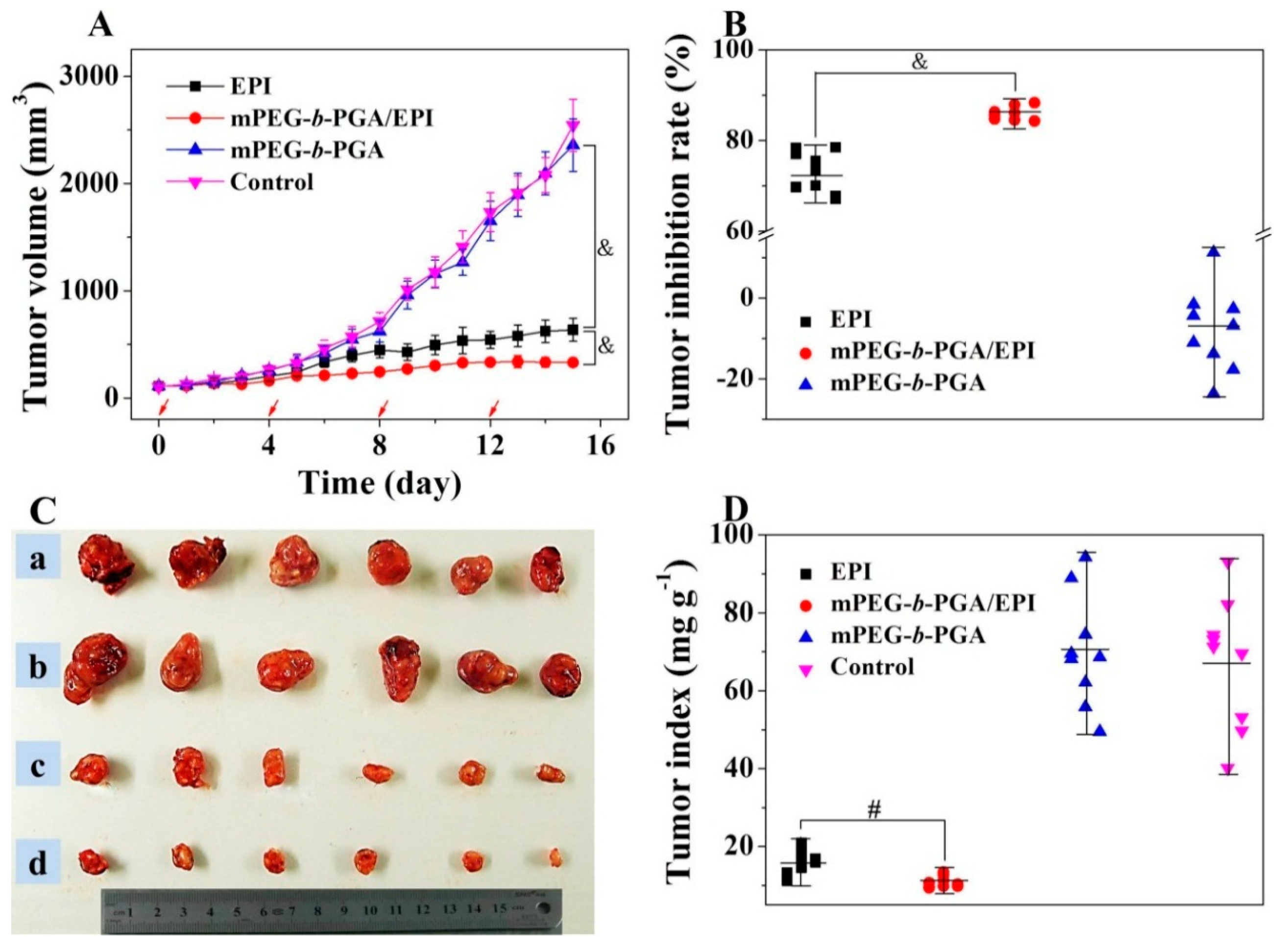
The improvement of antitumor efficacy and reduction of side effects from traditional chemotherapy agents are the ultimate goals of polymer nanomedicines. Due to the targeted delivery and increased tolerability of antitumor drugs, mPEG-
b-PGA/EPI might possess superior antitumor efficacy with decreased side effects. In this study, H22 cells were chosen as the tumor source to establish the subcutaneous HCC murine model, and then the chemotherapy efficacies of free EPI and mPEG-
b-PGA/EPI were assessed. Based on the results of MTD tests, free EPI and mPEG-
b-PGA/EPI at EPI equivalent dose of 8.0 mg·(kg·BW)
−1, or mPEG-
b-PGA at the corresponding material dose in the mPEG-
b-PGA/EPI group were administrated through intravenous injection. As shown in
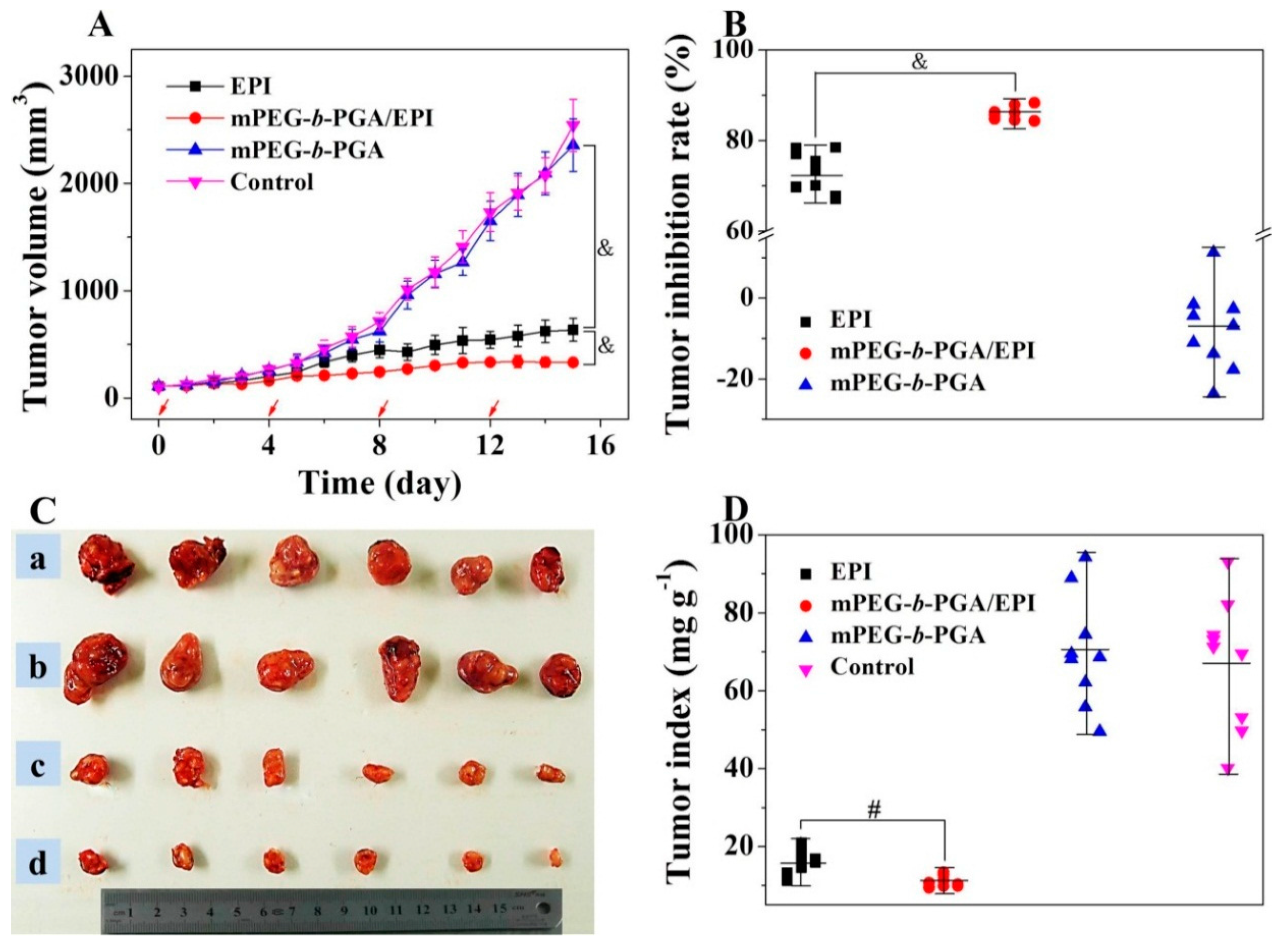
Figure 10, the growth of tumors in the mPEG-
b-PGA and control groups was extremely fast, and both of the average tumor volumes exceeded 2000 mm
3 within 16 days, indicating the copolymer is not effective against tumor. Although the two EPI formulations both inhibited the growth of tumor compared with NS, the treatment of mPEG-
b-PGA/EPI showed a significantly better efficacy than that of free EPI. The average tumor volume in the mPEG-
b-PGA/EPI group (
i.e., 330.1 ± 35.5) was just half of that in the free EPI group (
i.e., 635.1 ± 106.5) at the end of experiment (
p < 0.001;
Figure 10A). As shown in
Figure 10B, the tumor inhibitory rate of mPEG-
b-PGA/EPI is about 13% higher than that of free EPI (73.1 ± 4.5
vs. 86.0 ±1.5,
p < 0.001). At the end of all treatments, mice were sacrificed, and the tumors were isolated and weighted to observe the tumor suppression effects generally, and tumor indices were calculated. As shown in
Figure 10C, the tumor volumes of mPEG-
b-PGA and control groups were obviously bigger than those of drug-treated groups, and the mPEG-
b-PGA/EPI group had the smallest tumor volume. The results of tumor indices showed that the mass rate of tumor and mouse (mg·kg
−1) in the mPEG-
b-PGA/EPI group was 10.8 ± 1.4, which was significantly lower than 15.6 ± 3.1 of free EPI group (
Figure 10D,
p < 0.01). The enhanced antitumor efficacy of mPEG-
b-PGA/EPI could be explained with the following two reasons. First, the slow and pH-sensitive drug release could be realized with the protection of mPEG-
b-PGA, so the blood circulation time and effective action time to tumor were prolonged [
37]. Second, micellization created a higher agent concentration in tumor site by the EPR effect, and the tumor toxicity was enhanced.
Figure 10.
Tumor volumes (A); tumor inhibitory rates (B); tumor macroscopical images (C); and tumor indices (D) of H22 HCC-xenografted mice after treatment with free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. The red arrows in (A) meant the treatment times. Tumors in a, b, c, and d of (C) were from control, mPEG-b-PGA, free EPI, and mPEG-b-PGA/EPI groups, respectively. Data were presented as mean ± SD (n = 10; # p < 0.01, & p < 0.001).
Figure 10.
Tumor volumes (A); tumor inhibitory rates (B); tumor macroscopical images (C); and tumor indices (D) of H22 HCC-xenografted mice after treatment with free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. The red arrows in (A) meant the treatment times. Tumors in a, b, c, and d of (C) were from control, mPEG-b-PGA, free EPI, and mPEG-b-PGA/EPI groups, respectively. Data were presented as mean ± SD (n = 10; # p < 0.01, & p < 0.001).
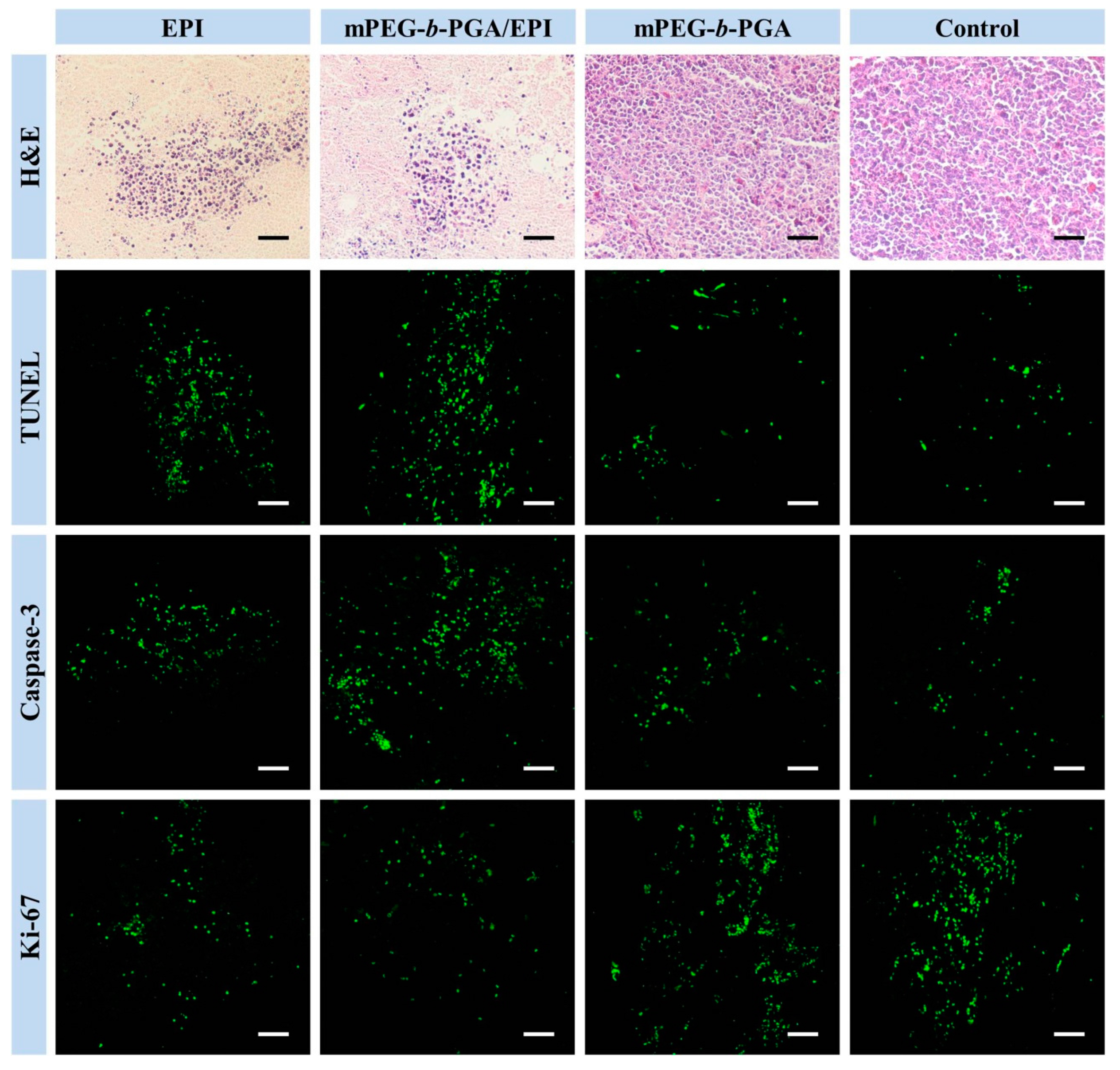
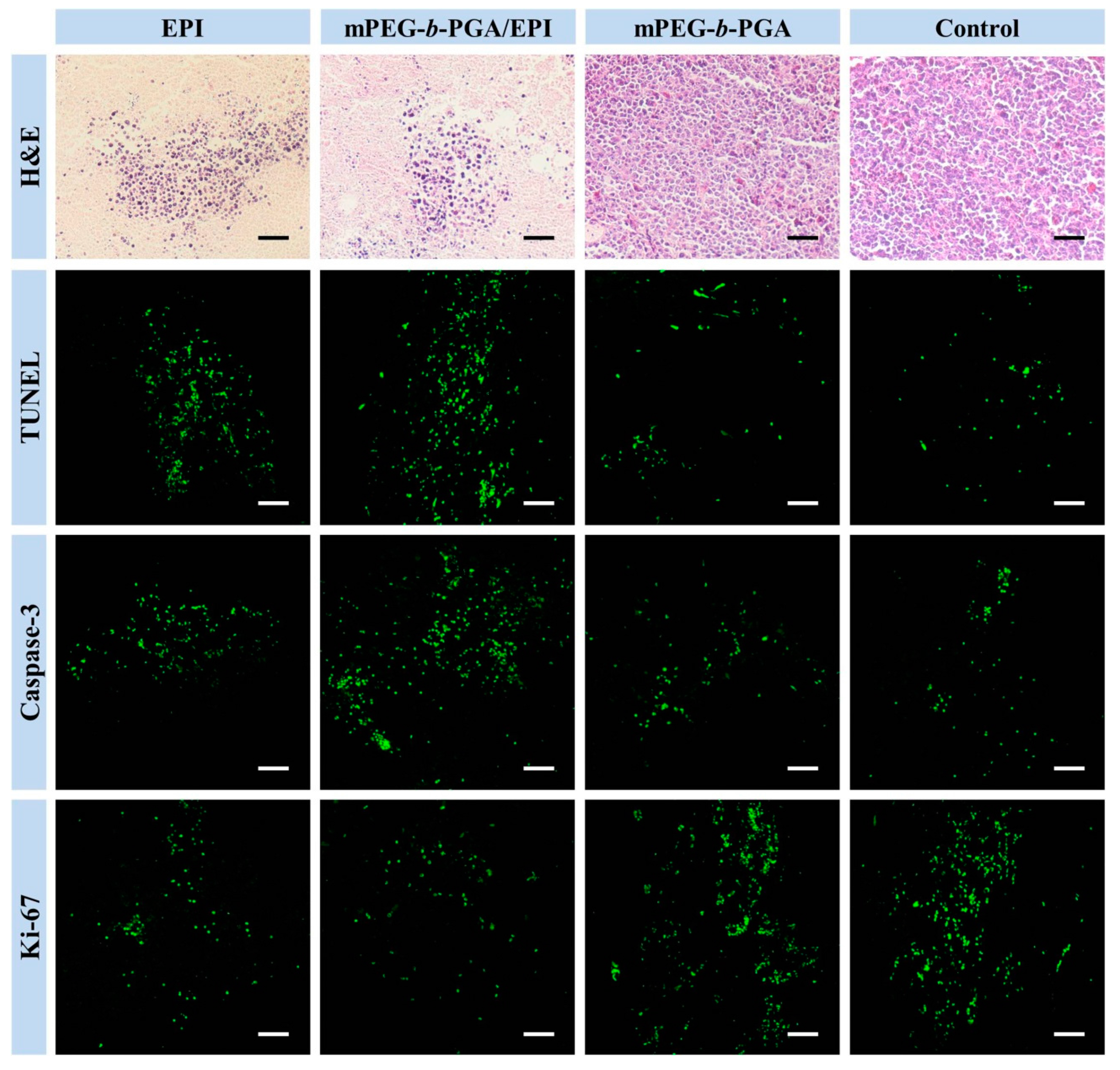
Besides the macroscopic observation, the microscopic examinations were also necessary to evaluate the antitumor efficacies. For this purpose, the histopathological assay,
in situ apoptosis, and immunohistochemical assays of isolated tumors were carried out at the end of all treatments (
Figure 11).
Figure 11.
Histopathological (i.e., H&E), in situ cell apoptosis (i.e., TUNEL), and immunohistochemical analyses (i.e., cleaved caspase-3 and Ki-67) of tumor sections after treatment with free EPI, mPEG-b-PGA/EPI, mPEG-b-PGA, or NS as control. Scale bar: 50.0 μm.
Figure 11.
Histopathological (i.e., H&E), in situ cell apoptosis (i.e., TUNEL), and immunohistochemical analyses (i.e., cleaved caspase-3 and Ki-67) of tumor sections after treatment with free EPI, mPEG-b-PGA/EPI, mPEG-b-PGA, or NS as control. Scale bar: 50.0 μm.
H&E staining, a gold standard for diagnoses of many diseases, including malignancy,
etc., is widely used in the observation and identification of cell apoptosis and tissue necrosis. In normal tissues, the cell nuclei will be stained bluish violet by alkaline hematoxylin, while the extracellular matrix and cytoplasm present varying degrees of pink for the reaction between protein and acidic eosin. On contrast, necrotic cells loss their normal cellular morphologies, and nuclear pyknosis, fragmentation, and even disappearance appeared. The results of H&E staining in this study showed that there almost no pathological changes in the tumors of both mPEG-
b-PGA and control groups, while large areas of amorphous substance could be seen in the groups of therapeutic agents (
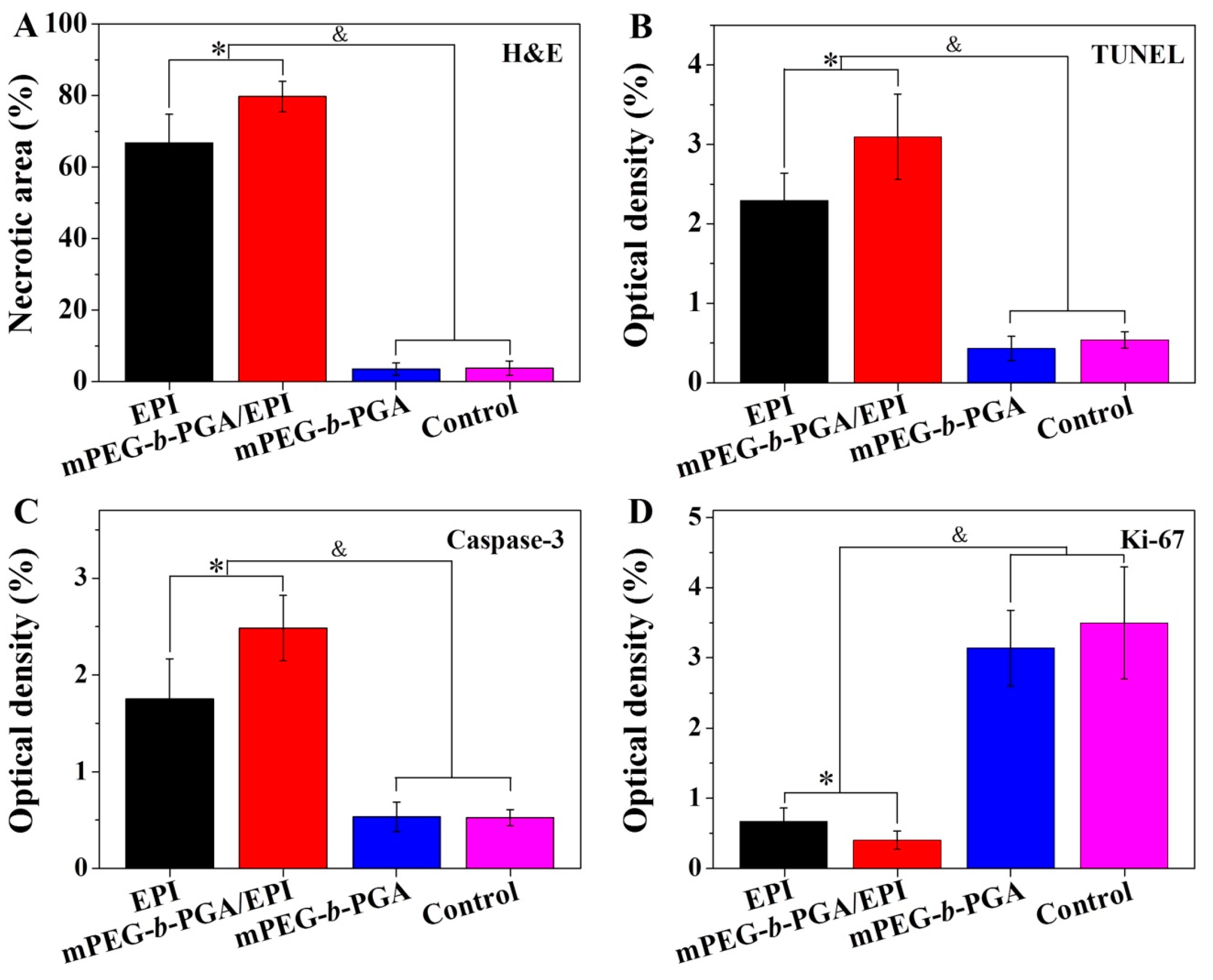
Figure 11). The semi-quantitative calculation of tumor necrosis areas with NIS-Elements BR Imaging Software (Nikon Instruments SpA, Florence, Italy) showed that the mPEG-
b-PGA/EPI group exhibited the highest necrosis degree among all the test groups, and its relative tumor necrosis area,
i.e., 79.7% ± 4.2%, was about 13% higher than that of free EPI group (
Figure 12A). All these results are consistent with those from the macroscopic observation and conformed again a better therapeutic efficacy of mPEG-
b-PGA/EPI.
Apoptosis is an important pathway of cell death and can be induced by chemotherapy effectively. During the process of apoptosis, the activated endogenous nucleic acid enzyme can cut genomic DNA into fragments of different sizes, which can be labeled by green fluorescein isothiocyanate (FITC) in TUNELkit. The results of TUNEL staining toward the tumor sections of different treatment groups are similar to those of histopathological tests (
Figure 11). There are only a few scattered fluorescent signals in the tumor sections of both mPEG-
b-PGA and control groups, indicating a small number of physiologic apoptosis. In contrast, the fluorescent signals in the groups with the injections of both EPI formulations obviously increased. The sorting of average relative optical densities in various groups calculated with ImageJ software was as follow: mPEG-
b-PGA/EPI > free EPI > mPEG-
b-PGA ≈ NS as control (
p < 0.05;
Figure 12B). In details, the signal of mPEG-
b-PGA/EPI group was 3.1% ± 0.5%, about 1.3-fold higher than that of free EPI group,
i.e., 2.3% ± 0.3%. The results of TUNEL tests declared that free EPI could inhibit the growth of HCC though the manner of inducting apoptosis, and this manner could be enhanced by mPEG-
b-PGA/EPI.
Figure 12.
Relative necrotic areas of tumor sections from H&E staining (A); and relative optical densities of tumor sections from TUNEL assay (B); cleaved caspase-3 (C); and Ki-67 analyses (D) after treatment of free EPI, mPEG-b-PGA/EPI, mPEG-b-PGA, or NS as control. Data were presented as mean ± SD (n =3; * p < 0.05, & p < 0.001).
Figure 12.
Relative necrotic areas of tumor sections from H&E staining (A); and relative optical densities of tumor sections from TUNEL assay (B); cleaved caspase-3 (C); and Ki-67 analyses (D) after treatment of free EPI, mPEG-b-PGA/EPI, mPEG-b-PGA, or NS as control. Data were presented as mean ± SD (n =3; * p < 0.05, & p < 0.001).
Caspase 3 plays an important role in the apoptosis pathway and is the executor of cell apoptosis. A lot of key enzymes for keeping cellular activity were the substrates of cleaved caspase-3, that is, an activated form of caspase-3 [
38]. The expression level of cleaved caspase-3 is an index of cellular apoptosis. Cytotoxic drugs can activate caspase-3 through multiple signaling pathways and further induce the apoptosis of tumor cells [
39]. To further confirm a higher apoptosis-inducing potentiation of mPEG-
b-PGA/EPI compared to that of free EPI, the cleaved caspase-3 levels in tumors of all groups were detected by immunohistochemistry. The point-like or flake-like green particles under CLSM represent the expression of this activated protein. Compared with control, both of the EPI formulations could obviously increase the expression of cleaved caspase-3, and the effect of mPEG-
b-PGA/EPI was stronger (
Figure 11). The situation of average relative optical densities in various groups was the same as those of TUNEL tests (
Figure 12C). More specifically, the mPEG-
b-PGA/EPI group had the highest relative optical density, and the value was 2.6% ± 0.5%, which was about 1.5-fold higher than that of free EPI group,
i.e., 1.7% ± 0.4%. These results confirmed the advantage of mPEG-
b-PGA/EPI on another side.
Ki-67 is a kind of nuclear nonhistone protein, which is expressed widely in proliferating cells but lack in quiescent ones. The level of cellular proliferation is able to be illustrated by the Ki-67 detection. It is suggested that Ki-67 may be a predictive factor for the development and prognosis of malignant tumors, and high levels of Ki-67 are often associated with poor prognosis [
40]. As shown in
Figure 11, there was vast Ki-67 fluorescence signal in the tumor sections of both mPEG-
b-PGA and control groups, indicating the vigorous proliferation of tumor cells. As expected, the administration of agents could reverse this situation and the lowest fluorescence signal appeared in the mPEG-
b-PGA/EPI group (
Figure 11). To explain this situation in more details, the relative optical density of survivin was counted with a similarly above-mentioned embodiment as TUNEL and caspase 3. As shown in
Figure 12D, the sorting of apoptosis area was as follow: mPEG-
b-PGA ≈ NS (as control) > free EPI > PEG-
b-PGA/EPI (
p < 0.05). The relative optical densities of tumors treated with free EPI and PEG-
b-PGA/EPI were 0.6% ± 0.2% and 0.4% ± 0.1%, respectively, and the rate was about 0.7. Therefore, the fact was verified that mPEG-
b-PGA/EPI had a better tumor inhibition effect than free EPI.
3.5. Excellent Security of mPEG-b-PGA/EPI in Vivo
The problem of side effects cannot be ignored in the applications of various drugs, and the security test must be considered in the development of new antitumor drug formulations [
41]. EPI possesses the reduced side effects comparing with DOX, but its dose-dependent cardiotoxicity, hepatotoxicity, and myelosuppression are still principal elements to limit its application seriously. To detect the impact of mPEG-
b-PGA on EPI side effects, the security assessments
in vivo were designed in this study. The contents included body weight changes, organ index detections, and organ pathology assays.
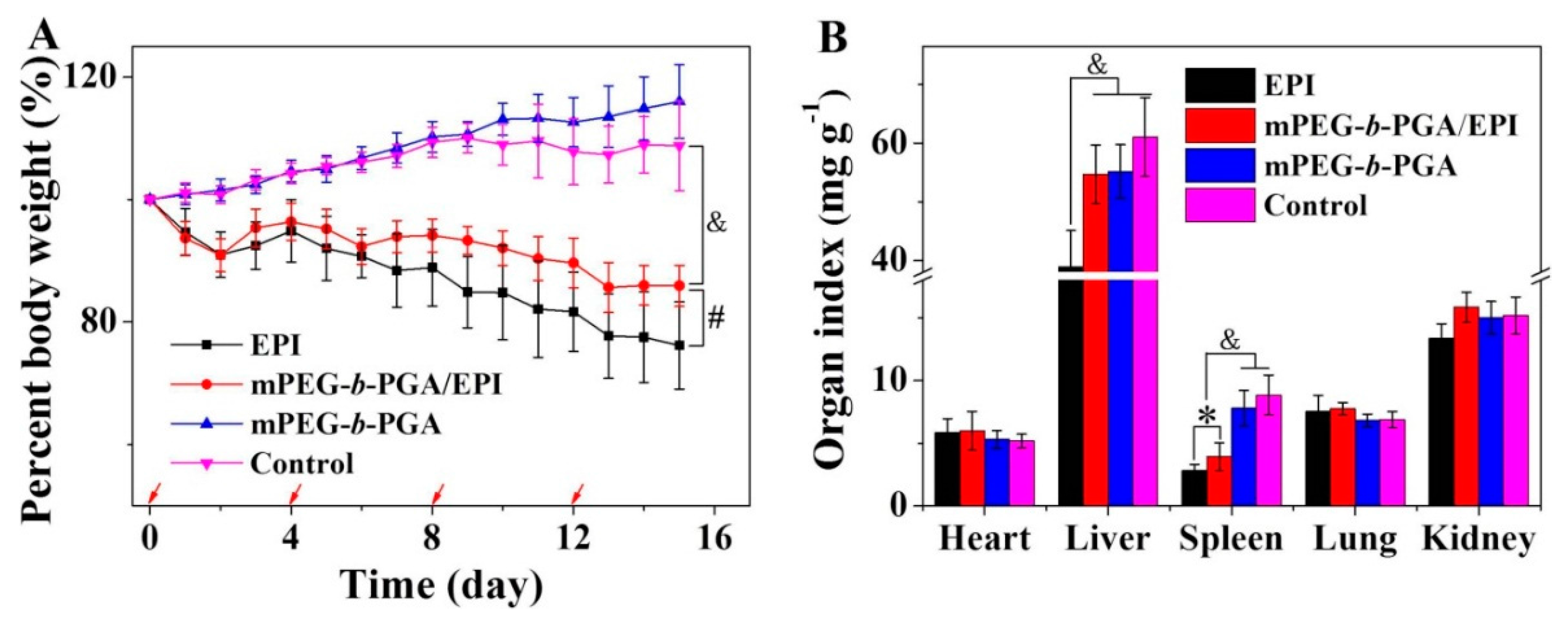
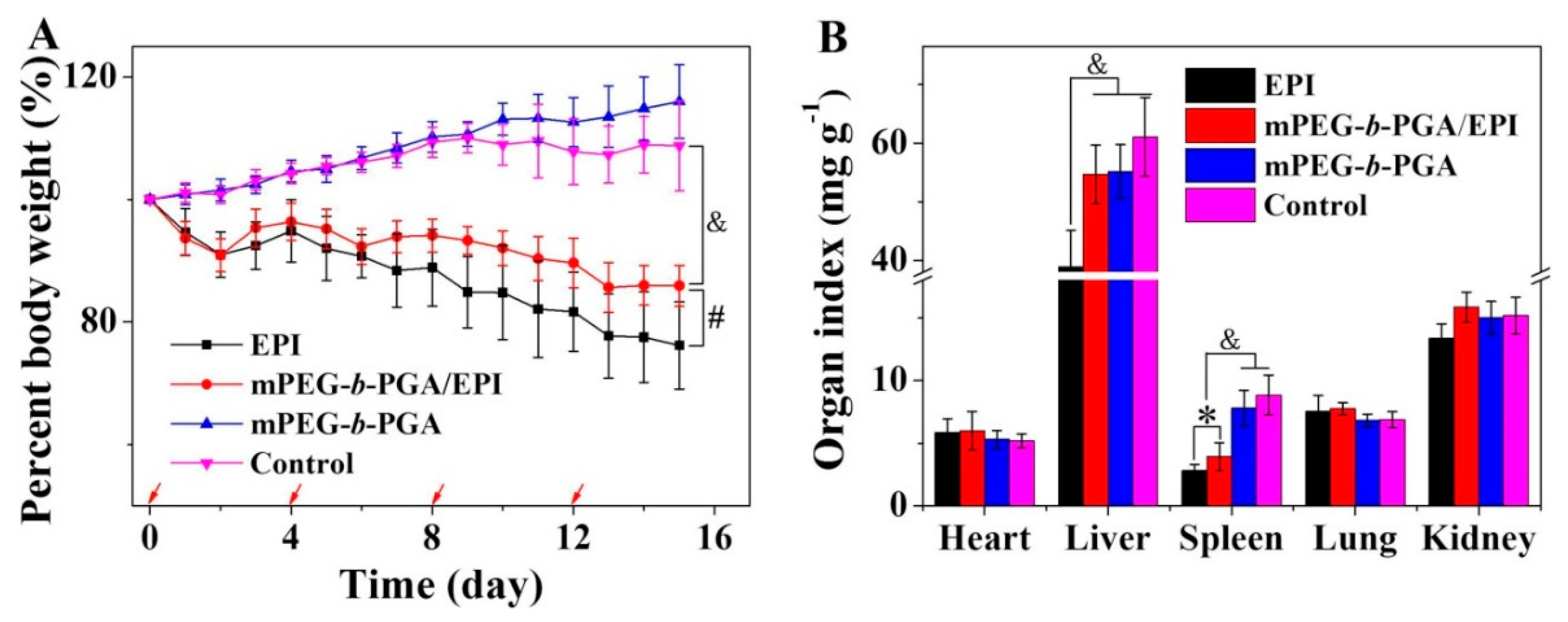
The body weight change is a visualized and important index, which can reflect the systemic toxicity of drugs.
Figure 13A depicted the changes of mouse body weights during treatments. Similarly as that of control group, the repeated injection of mPEG-
b-PGA did not influence the growth of mouse body weight, indicating its excellent biocompatibility and safety for
in vivo application again. More meaningful, the use of mPEG-
b-PGA/EPI significantly relieved the body weight loss situation caused by free EPI. The percentage of body weight decrease in the free EPI group was about 23.9%, while that in the mPEG-
b-PGA/EPI group was only 14.1% (
p < 0.01). This result suggested that this kind of micellar EPI might be a more secure formulation.
Organ indices, the ratios of organs (mg) and body weight (g), is a common index in toxicology experiment and can reflect the organ toxicity of agents partially. In this study, the major organs (
i.e., heart, liver, spleen, lung, and kidney) were isolated at the end of all treatments, and organ indices were obtained. As shown in
Figure 13B, the major organ indices of mPEG-
b-PGA group were nearly same as that of control group. Meanwhile, the dramatically decreased spleen index of mice treated with mPEG-
b-PGA/EPI or free EPI was observed, and the value was 3.9 ± 1.1 or 2.8 ± 0.5 mg·kg
−1, respectively. However, the former degree is significantly slighter by comparing (
p < 0.05). In addition, mPEG-
b-PGA/EPI almost completely reversed the liver index decrease in the free EPI group. The liver index in the mPEG-
b-PGA/EPI group was 54.7 ± 5.0 mg·kg
−1, which was very close to that in the control group (
i.e., 61.0 ± 6.7 mg·kg
−1) and was significantly higher than that in the free EPI group (
i.e., 38.9 ± 6.2 mg·kg
−1,
p < 0.001). All these results prompted that the protection of mPEG-
b-PGA could reduce the organ side effects of free drug.
Figure 13.
Body weight changes (A) and organ indices of H22 HCC-xenografted mice (B) after treatment of free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. The red arrows of (A) meant the treatment times. Data were presented as mean ± SD (n = 10; * p < 0.05, # p < 0.01, & p < 0.001).
Figure 13.
Body weight changes (A) and organ indices of H22 HCC-xenografted mice (B) after treatment of free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. The red arrows of (A) meant the treatment times. Data were presented as mean ± SD (n = 10; * p < 0.05, # p < 0.01, & p < 0.001).
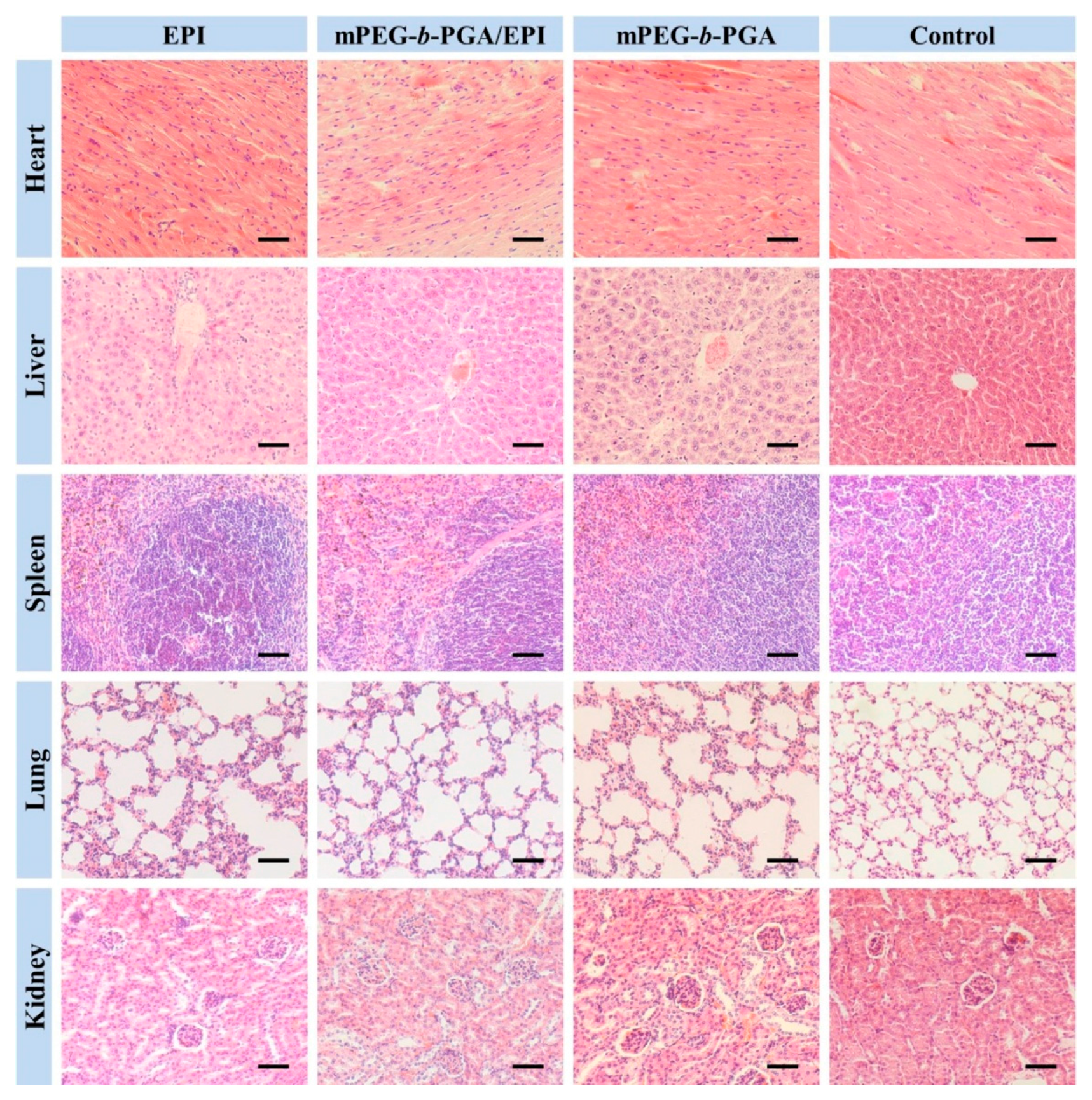
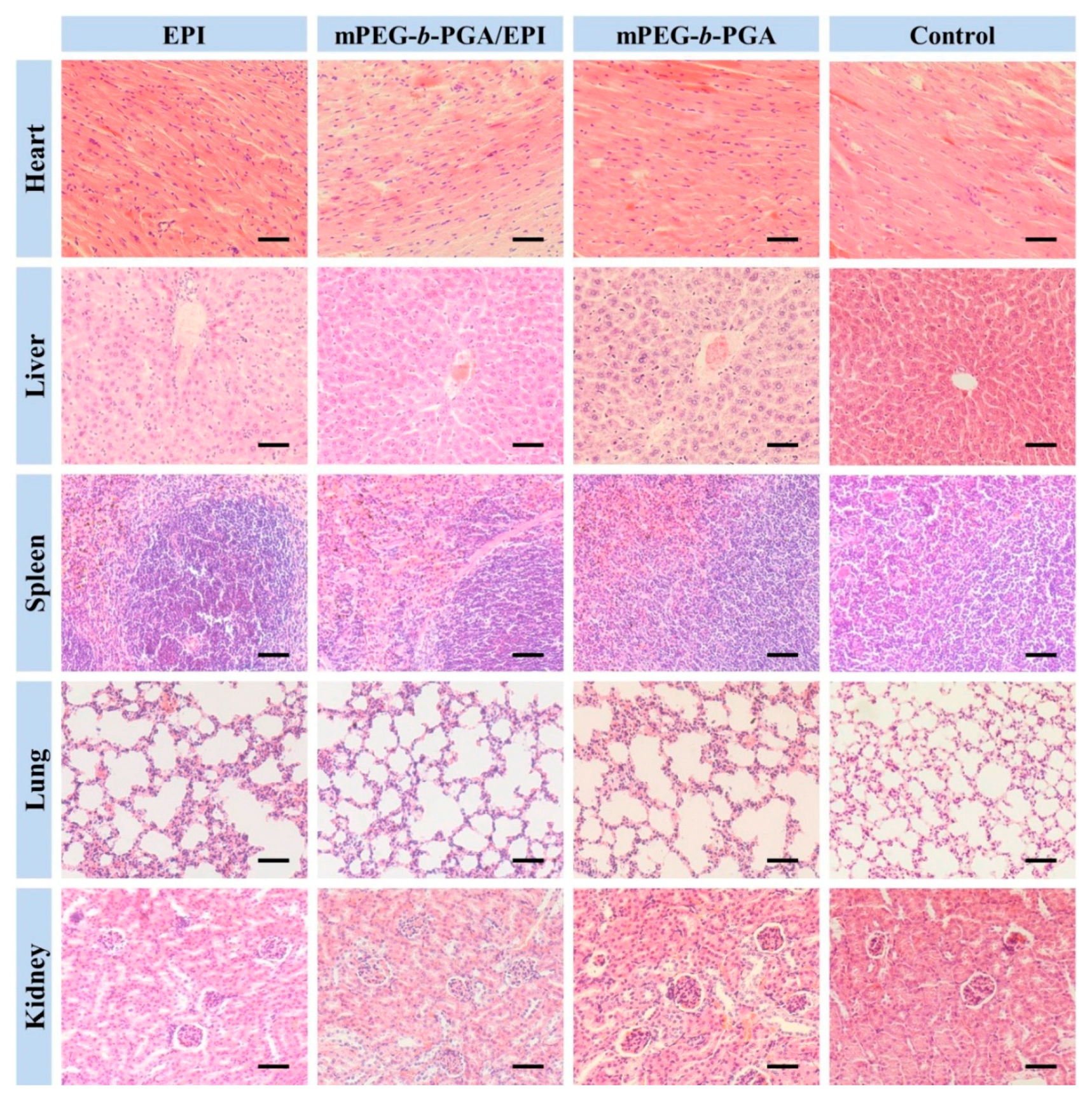
To further verify the advantages of mPEG-
b-PGA/EPI on security, the H&E staining of major organs were carried out. The results were showed in
Figure 14. There were almost no histopathological changes in the organs of both mPEG-
b-PGA and control groups, while varying degree of organ micro-morphological changes, especially in heart, liver, and kidney, could be observed in the EPI formulation groups. Acting as anthracycline antitumor drug, the organ damages of EPI in this study were very similar to those of DOX [
42]. The main pathological features included: (i) partial myocardial cells disordered and degenerated with inflammatory cell infiltration more or less; (ii) liver tissue appeared congestion with scattered inflammatory cells, and atrophic and eosinophilic hepatocytes could be seen; and (iii) renal interstitial hyperemia occurred, and some renal capsule shrank or even disappeared. Fortunately, all these changes in the mPEG-
b-PGA/EPI group were more moderate compared with those in the free EPI group.
Figure 14.
Histological analyses of main organs (i.e., heart, liver, spleen, lung, and kidney) from H22 HCC-xenografted mice after treatment of free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. Scale bar: 50.0 μm.
Figure 14.
Histological analyses of main organs (i.e., heart, liver, spleen, lung, and kidney) from H22 HCC-xenografted mice after treatment of free EPI, mPEG-b-PGA/EPI, or mPEG-b-PGA with NS as control. Scale bar: 50.0 μm.
In summary, mPEG-
b-PGA/EPI possessed better security than free EPI and could be safely applied
in vivo. The improved security might benefit from the structure of micelle, which improved the pharmacokinetic and biodistribution characteristics of free drug [
43]. Besides, the pH-sensitive micelle could obviously reduce the payload release in normal tissue, which was also an important reason.



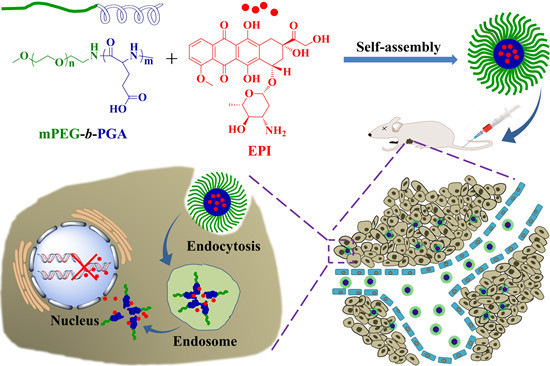
{kind=link}
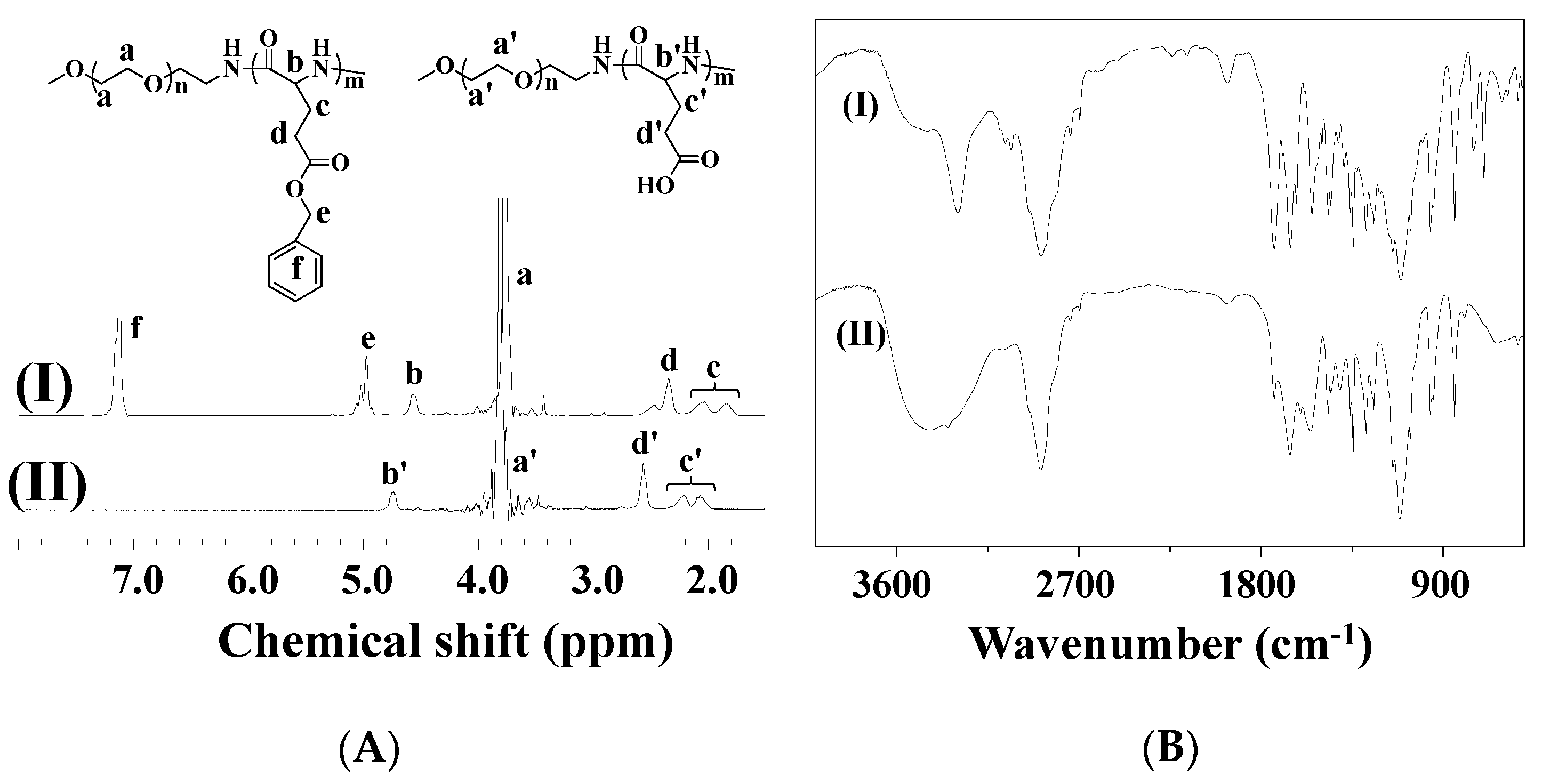{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}