Historical Perspective of Advances in the Science and Technology of Polymer Blends

Abstract
:1. Introduction
2. Theory: Review of Historical Developments
2.1. Flory-Huggins Equation
2.2. Solubility Parameter Concept
2.3. Equation of State (EOS)
2.4. Mean Field Approach
 and
and  are the volume fractions of b and c repeat units in the copolymer with
are the volume fractions of b and c repeat units in the copolymer with  . If Bbc is much larger than Bab and Bac, compositions will exist where miscibility is observed. This window of miscibility will exist if the expression below is satisfied:
. If Bbc is much larger than Bab and Bac, compositions will exist where miscibility is observed. This window of miscibility will exist if the expression below is satisfied:
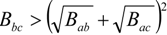
2.5. Hydrogen Bonding Fundamentals
3. Experimental Methods
3.1. Determination of χ12, B12, ΔHm
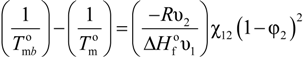
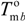 and
and 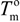 are the equilibrium melting points of the blend and the undiluted crystalline polymer. υ1 and υ2 are the molar volumes of the miscible polymer diluents and the crystalline polymer respectively.
are the equilibrium melting points of the blend and the undiluted crystalline polymer. υ1 and υ2 are the molar volumes of the miscible polymer diluents and the crystalline polymer respectively. 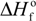 is the heat of fusion of the crystalline polymer at 100% crystallinity and φ2 is the volume fraction of the crystalline polymer. This method has shown good qualitative agreement with other methods employed to determine χ12 for the same blends.
is the heat of fusion of the crystalline polymer at 100% crystallinity and φ2 is the volume fraction of the crystalline polymer. This method has shown good qualitative agreement with other methods employed to determine χ12 for the same blends.3.2. Experimental (Other)
4. Compatibilization of Immiscible and Incompatible Polymer Blends
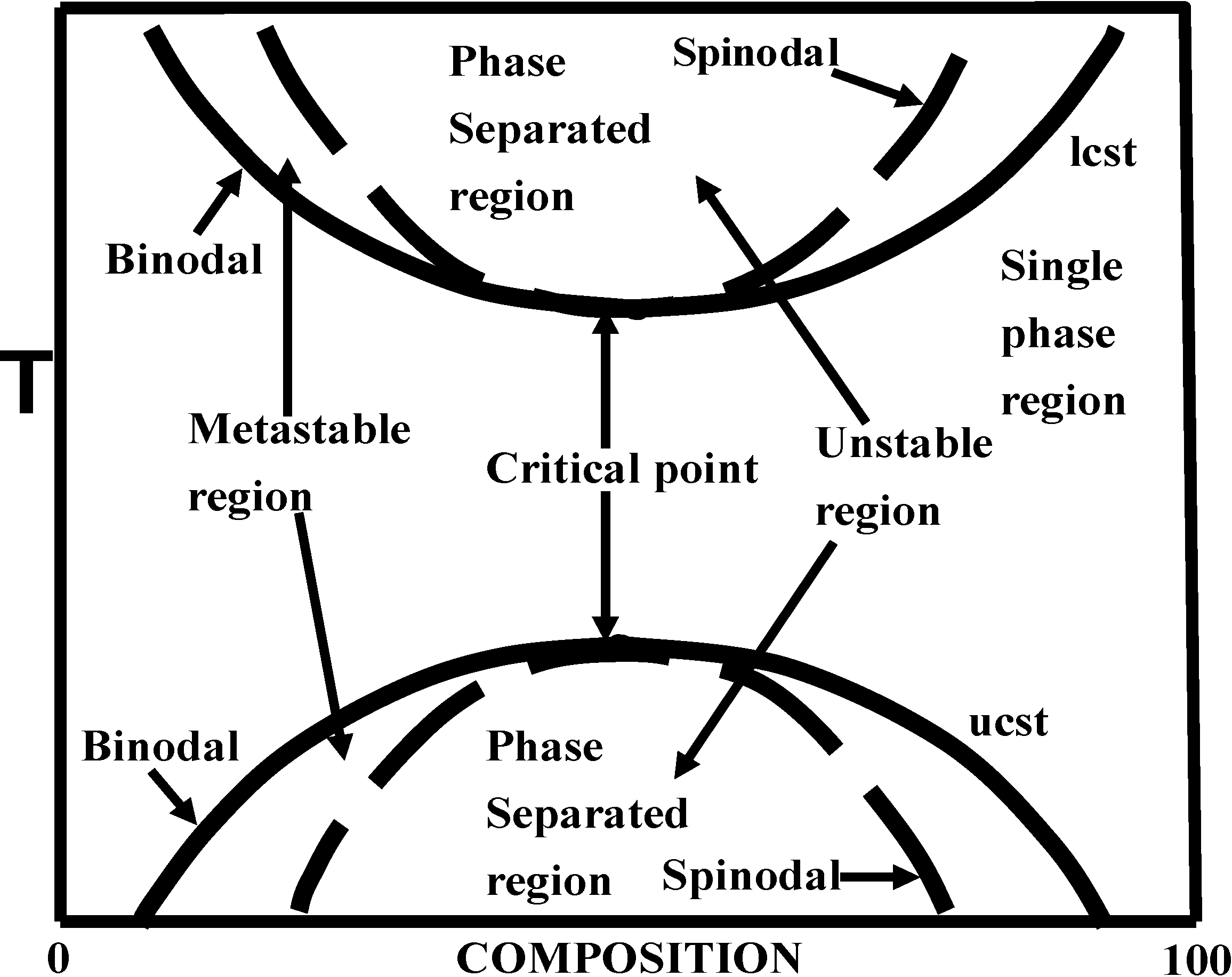
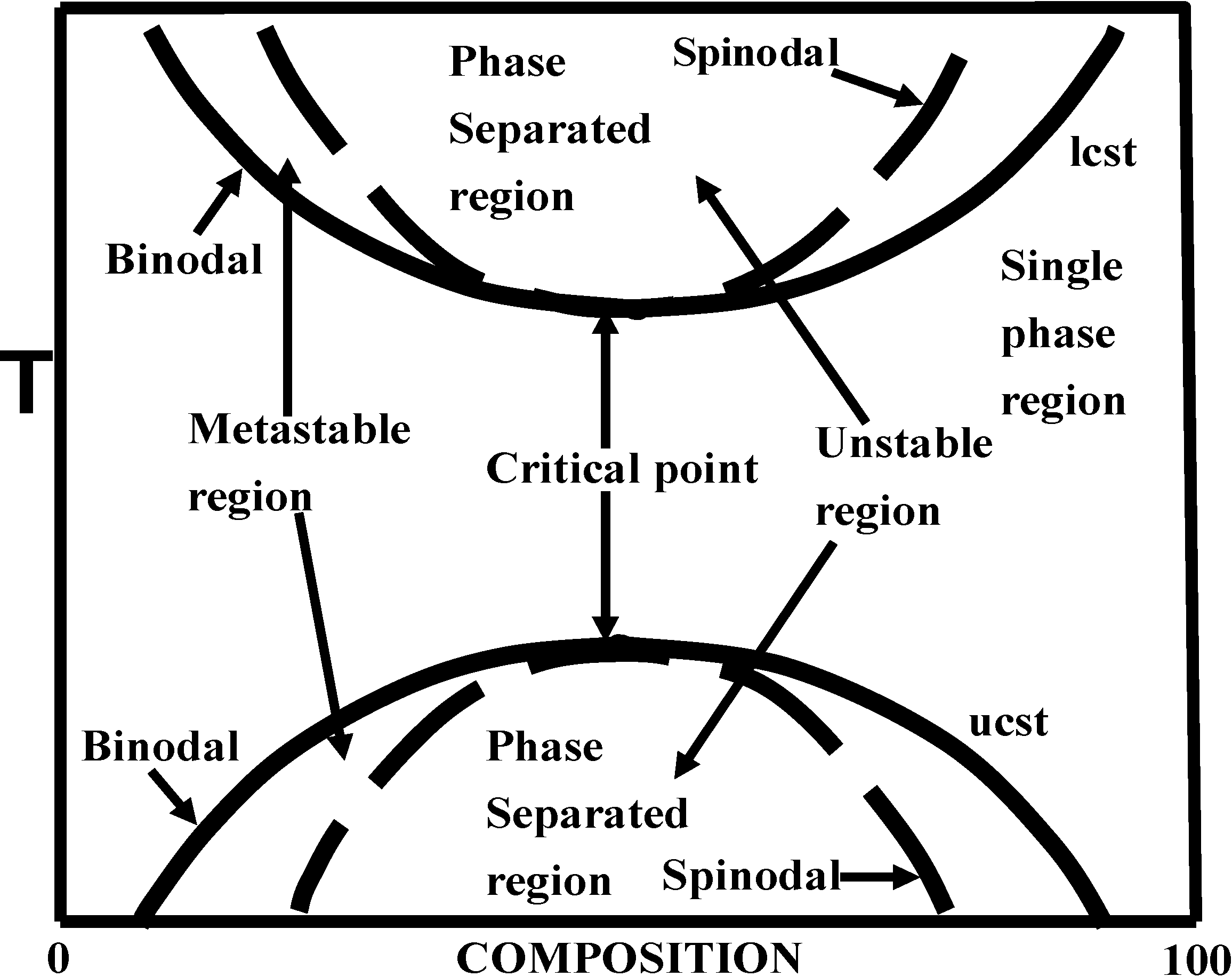
5. Phase Separation: Spinodal Decomposition versus Nucleation and Growth

{kind=link}
| Property | Nucleation and Growth | Spinodal Decomposition |
|---|---|---|
| Size of phase separated region | increases with time | size constant |
| Concentration of phase separated region | constant with time | increases with time |
| Diffusion coefficient | positive | negative |
| Phase structure | separated | interconnected |
| Activation energy | required | not required |
| Region of phase diagram | metastable or unstable region | only unstable region |
6. Commercial Developments
7. Conclusions
Conflicts of Interest
References
- Cizek, E.P. Blend of a Polyphenylene Ether and a Styrene Resin. U.S. Patent 3,383,435, 14 May 1968. [Google Scholar]
- Henton, D.E.; Bubeck, R.A. The manufacture and physical properties of rubber-toughened styrenics. In Polymer Toughening; Arends, C.B., Ed.; Marcel Dekker: New York, NY, USA, 1996; pp. 237–291. [Google Scholar]
- Badum, E. Ozone Resistant Cable Construction. U.S. Patent 2,297,194, 29 September 1942. [Google Scholar]
- Henderson, D.E. Mixture of Polymerized Materials. U.S. Patent 2,330,353, 28 September 1943. [Google Scholar]
- Gibbs, J.W. Transactions of the Connecticut Academy of Arts and Sciences; Connecticut Academy of Arts and Sciences: New Haven, CT, USA, 1873; pp. 382–404. [Google Scholar]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1941, 9, 660–661. [Google Scholar] [CrossRef]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Huggins, M.L. Solutions of long chain compounds. J. Chem. Phys. 1941, 9. [Google Scholar] [CrossRef]
- Huggins, M.L. Some properties of solutions of long-chain compounds. J. Phys. Chem. 1942, 46, 151–158. [Google Scholar] [CrossRef]
- Hildebrand, J.H. Solubility. J. Am. Chem. Soc. 1916, 38, 1452–1473. [Google Scholar] [CrossRef]
- Scatchard, G. Equilibrium in non-electrolyte solutions in relation to the vapor pressure and densities of the compounds. Chem. Revs. 1931, 8, 321–333. [Google Scholar] [CrossRef]
- Hildebrand, J.H.; Scott, R.L. The Solubility of Non-Electrolytes; Reinhold: New York, NY, USA, 1936. [Google Scholar]
- Hansen, C.M. The three dimensional solubility parameter-key to paint component affinities. I. Solvents, plasticizers, polymers and resins. J. Paint. Technol. 1967, 39, 104–117. [Google Scholar]
- Small, P.A. Some factors affecting the solubility of polymers. J. Appl. Chem. 1953, 3, 71–80. [Google Scholar] [CrossRef]
- Van Krevelan, D.W. Properties of Polymers: Correlations with Chemical Structure; Elsevier: Amsterdam, The Netherlands, 1972; pp. 135–142. [Google Scholar]
- Hoy, K.L. New values of the solubility parameter from vapor pressure data. J. Paint Technol. 1970, 42, 76–118. [Google Scholar]
- Coleman, M.M.; Graf, J.F.; Painter, P.C. Specific Interactions and the Miscibility of Polymer Blends; Technomic Publishing Company Inc.: Lancaster, PA, USA, 1991. [Google Scholar]
- Prigogine, I. The Molecular Theory of Solutions; North-Holland Pub.: Amsterdam, The Netherlands, 1957. [Google Scholar]
- Flory, P.J.; Orwoll, R.A.; Vrij, A.J. Statistical thermodynamics of chain molecule liquids. I. An equation of state for normal paraffin hydrocarbons. J. Am. Chem. Soc. 1964, 86, 3507–3514. [Google Scholar] [CrossRef]
- Flory, P.J.; Orwoll, R.A.; Vrij, A.J. Statistical thermodynamics of chain molecule liquids. II. Liquid mixtures of normal paraffin hydrocarbons. J. Am. Chem. Soc. 1964, 86, 3515–3520. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical thermodynamics of mixtures. J. Am. Chem. Soc. 1965, 87, 1833–1838. [Google Scholar] [CrossRef]
- McMaster, L.P. Aspects of polymer-polymer thermodynamics. Macromolecules 1973, 6, 760–773. [Google Scholar] [CrossRef]
- Shaw, M.T. Studies of polymer-polymer solubility using a two-dimensional solubility parameter approach. J. Appl. Polym. Sci. 1974, 18, 449–472. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. An elementary molecular theory of classical fluids. Pure fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
- Lacombe, R.H.; Sanchez, I.C. Statistical thermodynamics of fluid mixtures. J. Phys. Chem. 1976, 80, 2568–2580. [Google Scholar] [CrossRef]
- Somcynsky, T.; Simha, R. On the statistical thermodynamics of spherical and chain molecule fluids. Macromolecules 1969, 2, 343–350. [Google Scholar]
- Jain, R.K.; Simha, R. On the statistical thermodynamics of multicomponent fluids: Equation of state. Macromolecules 1980, 13, 1501–1508. [Google Scholar] [CrossRef]
- Jain, R.K.; Simha, R. On the equation of state of argon and organic liquids. J. Chem. Phys. 1980, 72, 4909–4912. [Google Scholar] [CrossRef]
- Dee, G.T.; Walsh, D.J. Equations of state for polymer liquids. Macromolecules 1988, 21, 811–814. [Google Scholar] [CrossRef]
- Dee, G.T.; Walsh, D.J. A modified cell model equation of state for polymer liquids. Macromolecules 1988, 21, 815–817. [Google Scholar] [CrossRef]
- Delmas, G.; Patterson, D. The molecular weight dependence of lower and upper critical solution temperatures. J. Polym. Sci. 1970, 30, 1–8. [Google Scholar]
- Klotz, H.C.; Mathias, P.M.; Robeson, L.M. An equation of state for polymer/polymer systems. Fluid Phase Equilib. 1989, 53, 311–322. [Google Scholar] [CrossRef]
- Paul, D.R.; Barlow, J.W. A binary interaction model for miscibility of copolymers in blends. Polymer 1984, 25, 487–494. [Google Scholar] [CrossRef]
- Kambour, R.P.; Bendler, J.T.; Bopp, R.C. Phase behavior of polystyrene, poly(2,6-dimethyl-1,4-phenylene oxide) and their brominated derivatives. Macromolecules 1983, 16, 753–757. [Google Scholar] [CrossRef]
- ten Brinke, G.; Karasz, F.E.; MacKnight, W.J. Phase behavior in copolymer blends: Poly(2,6-dimethyl-1,4-phenylene oxide) and halogen substituted styrene copolymers. Macromolecules 1983, 16, 1827–1832. [Google Scholar] [CrossRef]
- Cruz, C.A.; Barlow, J.W.; Paul, D.R. The basis for miscibility in polyester-polycarbonate blends. Macromolecules 1979, 12, 726–731. [Google Scholar]
- Wang, T.T.; Nishi, T. Spherulitic crystallization in compatible blends of poly(vinylidene fluoride) and poly(methyl methacrylate). Macromolecules 1977, 10, 421–425. [Google Scholar] [CrossRef]
- Kruse, W.A.; Kriste, R.G.; Haas, J.; Schmitt, B.J.; Stein, D.J. Experimentter nachweis des molekular dispersen charakters der nischung von zwei polymeren und bestimmung des chemischen potentials in diesen mischungen. Die Makromol. Chem. 1976, 177, 1145–1160. (In German) [Google Scholar] [CrossRef]
- Kaplan, D.J. Structure-property relationships in copolymers to composites: Molecular interpretation of the glass transition phenomenon. J. Appl. Polym. Sci. 1976, 20, 2615–2629. [Google Scholar] [CrossRef]
- Utrachi, L.A. Thermodynamics of polymer blends. In Polymer Blends Handbook; Utrachi, L.A., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; Volume 1, pp. 123–201. [Google Scholar]
- Harris, J.E.; Paul, D.R.; Barlow, J.W. A comparison of miscible binary blend interaction parameters measured by different methods. Polym. Eng. Sci. 1983, 23, 676–681. [Google Scholar] [CrossRef]
- Olabisi, O. Polymer compatibility by gas-liquid chromatography. Macromolecules 1975, 8, 316–322. [Google Scholar]
- Kwei, T.K.; Pearce, E.M.; Ren, F.; Chen, J.P. Hydrogen bonding in polymer mixtures. J. Polym. Sci. 1986, 24, 1597–1609. [Google Scholar] [CrossRef]
- Morawetz, H. Fluorescence studies of conformational mobility and the mutual interpenetration of flexible chain molecules. Pure Appl. Chem. 1980, 52, 277–284. [Google Scholar] [CrossRef]
- Amrani, F.; Hung, J.M.; Morawetz, H. Studies of polymer compatibility by nonradiative energy transfer. Macromolecules 1980, 13, 649–653. [Google Scholar] [CrossRef]
- Frank, C.W.; Gashgari, M.A. Excimer fluorescence as a molecular probe of polymer compatibility. I. Blends of poly(2-vinyl naphthalene) with poly(alkyl methacrylates). Macromolecules 1979, 12, 163–165. [Google Scholar] [CrossRef]
- Semerak, S.M.; Frank, C.W. Excimer fluorescence as a molecular probe of blend miscibility. 3. Effect of molecular weight of the host matrix. Macromolecules 1981, 14, 443–449. [Google Scholar] [CrossRef]
- Robeson, L.M. Polymer Blends: A Comprehensive Review; Hanser Publishers: Munich, Germany, 2007. [Google Scholar]
- Simon, G.P. Polymer Characterization Techniques and Their Application to Blends; Oxford University Press: New York, NY, USA, 2003. [Google Scholar]
- Hobbs, S.Y.; Dekkers, M.E.J.; Watkins, V.H. Effect of interfacial forces on polymer blend morphologies. Polymer 1988, 29, 1598–1602. [Google Scholar] [CrossRef]
- Epstein, B.N. Tough Thermoplastic Nylon Compositions. U.S. Patent 4,174,358, 13 November 1979. [Google Scholar]
- Ide, F.; Hasegawa, A. Studies on polymer blend of nylon 6 and polypropylene or nylon 6 and polystyrene using the reaction of polymer. J. Appl. Polym. Sci. 1974, 18, 963–974. [Google Scholar] [CrossRef]
- McGrath, J.E.; Robeson, L.M.; Matzner, M. Polysulfone-nylon 6 block copolymers and alloys. In Recent Advances in Polymer Blends, Grafts, and Blocks; Sperling, L.H., Ed.; Plenum Press: New York, NY, USA, 1974; pp. 195–211. [Google Scholar]
- Robeson, L.M.; Famili, A.; Nangeroni, J.F. Reactive extrusion compatibilization of poly(vinyl alcohol)-polyolefin blends. In Science and Technology of Polymers and Advanced Materials; Prasad, P.N., Ed.; Plenum Press: New York, NY, USA, 1998; pp. 9–23. [Google Scholar]
- Xanthos, M. Reactive Extrusion: Principles and Practice; Hanser Publishers: Munich, Germany, 1992. [Google Scholar]
- Baker, W.; Scott, C.; Hu, G.H. Reactive Polymer Blending; Hanser Publishers: Munich, Germany, 2001. [Google Scholar]
- McMaster, L.P. Aspects of liquid-liquid phase transition phenomena in multicomponent polymeric systems. Adv. Chem. Ser. 1975, 142, 43–65. [Google Scholar] [CrossRef]
- Cahn, J.W.; Hilliard, J.E. Free energy of a nonuniform system 1. Interfacial free energy. J. Chem. Phys. 1958, 28, 258–267. [Google Scholar]
- Cahn, J.W. Phase separation by spinodal decomposition in isotropic systems. J. Chem. Phys. 1965, 42, 93–99. [Google Scholar] [CrossRef]
- Nishi, T.; Wang, T.T.; Kewi, T.K. Thermally induced phase separation behavior of compatible polymer mixtures. Macromolecules 1975, 8, 227–234. [Google Scholar] [CrossRef]
- Alysworth, J.W. Plastic Composition. U.S. Patent 1,111,284, 22 September 1914. [Google Scholar]
- Wagner, E.R.; Robeson, L.M. Impact polystyrene: Factors controlling the rubber efficiency. Rubber Chem. Technol. 1970, 43, 1129–1137. [Google Scholar]
- Robeson, L.M. Applications of polymer blends: Emphasis on recent advances. Polym. Eng. Sci. 1984, 24, 587–597. [Google Scholar] [CrossRef]
- Robeson, L.M. Recent advances in polymer blend technology. In Multiphase Macromolecular Systems; Culbertson, B.M., Ed.; Plenum Publishing Corporation: New York, NY, USA, 1989; pp. 177–212. [Google Scholar]
- Polymers Blends; Paul, D.R.; Newman, S. (Eds.) Academic Press: New York, NY, USA, 1978; Volumes 1 and 2.
- Olabisi, O.; Robeson, L.M.; Shaw, M.T. Polymer-Polymer Miscibility; Academic Press: New York, NY, USA, 1979. [Google Scholar]
- Utracki, L.A. Polymer Blends and Alloys: Thermodynamics and Rheology; Hanser Publishers: New York, NY, USA, 1989. [Google Scholar]
- Shonaike, G.O.; Simon, G.P. Polymer Blends and Alloys; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Paul, D.R.; Bucknall, C.B. Polymer Blends; John Wiley & Sons: New York, NY, USA, 2000; Volume 1 (Formulation) and Volume 2 (Performance). [Google Scholar]
- Utracki, L.A. Polymer Blends Handbook; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002; Volumes 1 and 2. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Robeson, L. Historical Perspective of Advances in the Science and Technology of Polymer Blends. Polymers 2014, 6, 1251-1265. https://doi.org/10.3390/polym6051251
Robeson L. Historical Perspective of Advances in the Science and Technology of Polymer Blends. Polymers. 2014; 6(5):1251-1265. https://doi.org/10.3390/polym6051251
Chicago/Turabian StyleRobeson, Lloyd. 2014. "Historical Perspective of Advances in the Science and Technology of Polymer Blends" Polymers 6, no. 5: 1251-1265. https://doi.org/10.3390/polym6051251