Surface Initiated Polymerizations via e-ATRP in Pure Water

1
Chemical Process Engineering, RWTH Aachen University, Forckenbeckstrasse 50, Aachen D-52056, Germany
2
Department of Mechanical & Industrial Engineering, Electrocatalytic Green Engineering Group, Concordia University, 1455 de Maisonneuve Blvd. West, Montreal, PQ H3G 1M8, Canada
3
DWI an der RWTH, IPC Macromolecular Materials and Surfaces, RWTH Aachen University, Forckenbeckstrasse 50, Aachen D-52056, Germany
4
Department of Biomedical Engineering-FB40, W.J. Kolff Institute for Biomedical Engineering and Materials Science, University of Groningen, University Medical Center Groningen, Antonius Deusinglaan 1, Groningen 9713 AV, The Netherlands
*
Author to whom correspondence should be addressed.
Polymers 2013, 5(4), 1229-1240; https://doi.org/10.3390/polym5041229
Submission received: 24 August 2013
/
Revised: 11 October 2013
/
Accepted: 21 October 2013
/
Published: 25 October 2013
Abstract
:Here we describe the combined process of surface modification with electrochemical atom transfer radical polymerization (e-ATRP) initiated from the surface of a modified gold-electrode in a pure aqueous solution without any additional supporting electrolyte. This approach allows for a very controlled growth of the polymer chains leading towards a steady increase in film thickness. Electrochemical quartz crystal microbalance displayed a highly regular increase in surface confined mass only after the addition of the pre-copper catalyst which is reduced in situ and transformed into the catalyst. Even after isolation and washing of the modified electrode surface, reinitiation was achieved with retention of the controlled electrochemical ATRP reaction. This reinitiation after isolation proves the livingness of the polymerization. This approach has interesting potential for smart thin film materials and offers also the possibility of post-modification via additional electrochemical induced reactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Atom transfer radical polymerization (ATRP) is one of the most widely used reaction to create well defined polymers, linear as well as more complex architectures which has been used also for functionalization of various surfaces composed of different materials (e.g., Au, Ag, SiO2, etc.) [1,2,3,4,5,6,7]. The controlled/living character of the polymerization allows for the creation of well-defined films even providing block-copolymer compositions with a controlled thickness for a widespread of applications e.g., in antimicrobial [8,9] and protective coatings preventing adsorption of plasma proteins and platelets [10,11], protein affinity layers [12,13] and responsive films [14,15]. In these applications, precise control over the polymerization properties is imperative since delicate changes can result in repulsion layers or affinity layers [16]. The application of ATRP in aqueous systems tremendously increased the importance of it for functionalization of biocompatible systems [17,18,19] or bio-surfaces [20] but also for biomedical applications [21,22]. It allowed for the modification of proteins in order to tailor new hybrid materials [23,24,25]. Recently a new impulse has been given to ATRP by controlling the reaction using electrochemistry, so-called e-ATRP which had was previously also attempted in combination with reversible addition-fragmentation chain transfer (RAFT) polymerization [26]. By adjusting the electrochemical potential, this method reduces Cu(II) to Cu(I) in-situ and thereby controlling the amount of Cu(I) in the system, and hence control over the polymerization reaction even in combination with the presence of oxygen which was previously only possible with activators regenerated by electron transfer atom transfer radical polymerization (ARGET-ATRP) [27,28]. This technique was quickly developed to function effectively also in aqueous buffered solutions, increasing the biocompatibility of e-ATRP [29]. The newly developed approach would benefit greatly when combined with surface modification. While normally this would be initiation of a modified surface from solution, direct modification of electrode surfaces is much more convenient. This is common for the formation of conjugated polymers directly from the surface of the electrode via oxidative electrochemical processes but an absolute novelty via e-ATRP [30,31,32,33].
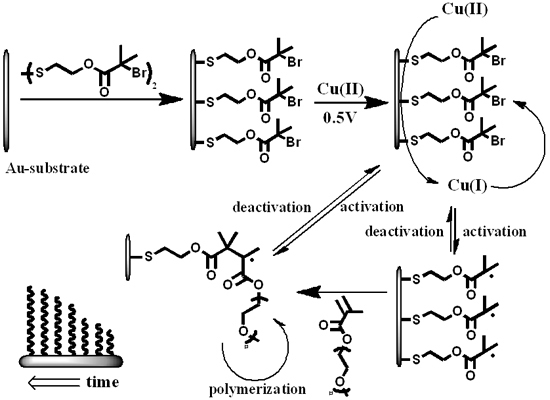
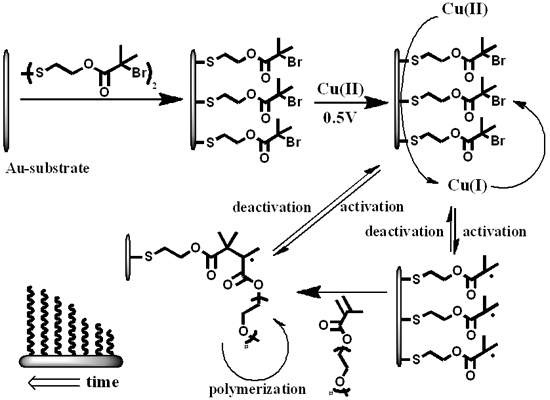
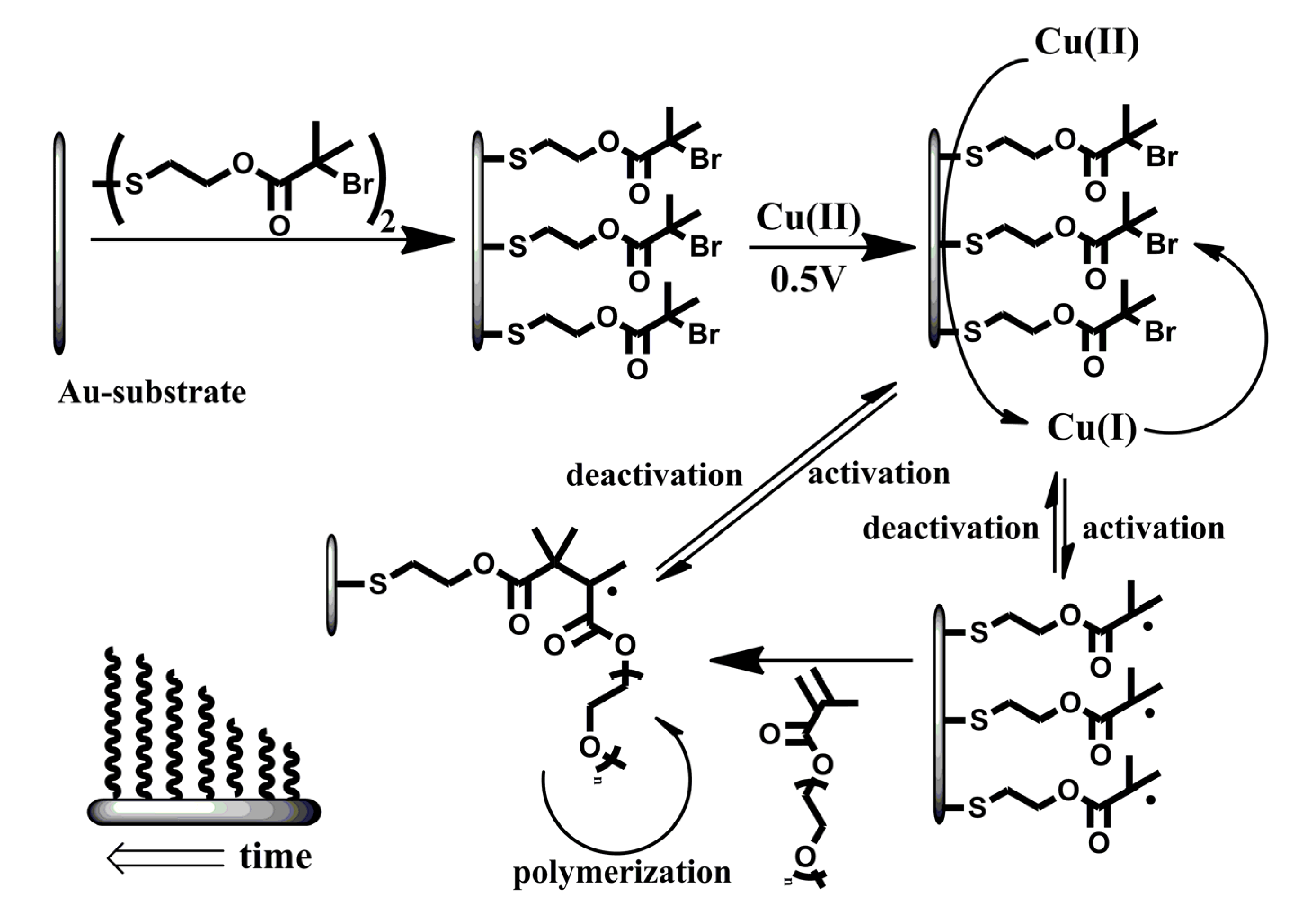
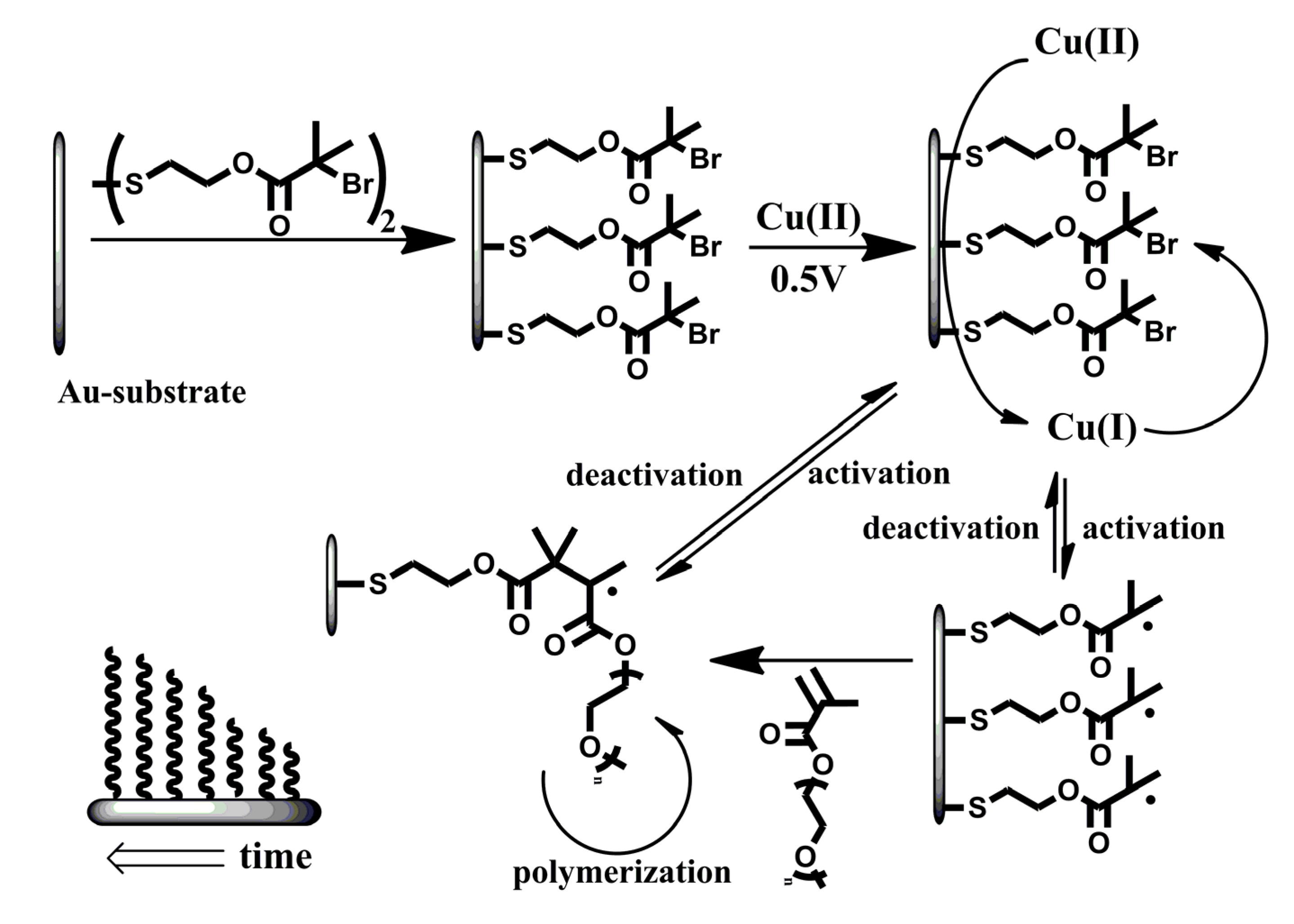
Here, we describe the combined process of surface modification with electrochemical ATRP initiated from the surface of a modified gold-electrode in a pure aqueous solution without any supporting electrolyte (Scheme 1). The surface of the electrode is modified with an ATRP-initiator (bis[2-(2′-bromoisobutyryloxy)ethyl]disulfide) and placed in an aqueous solution containing Cu(II)Br2/Me6TREN (tris(2-dimethyl-aminoethyl)amine) (1.0 mM, 1:1) and oligo(ethylene glycol)methyl ether methacrylate (OEGMA475) (2.5% w/w, 52.6 mM) or 2-hydroxethyl methacrylate (2.5% w/w, 192.3 mM). The system initially lacks the presence of activator [Cu(I)] and has a high amount of deactivator [Cu(II)]. The working gold electrode, covered with ATRP initiator, is able to reduce Cu(II) to Cu(I) and hence initiate the polymerization reaction. The formation of activator-species occurs in the vicinity of the initiator-layer which enhances the likelihood of activation. Upon activation, the polymerization proceeds until deactivation by the high amount of deactivator in the system. This reversible and electrochemically controlled process allows the formation of polymer layers with controllable dry thicknesses of up to 47 nm within two hours. The conditions presented are the most challenging for the system since complete (100%) surface coverage by initiator conventionally induces limited degree of polymerization and omitting additional electrolyte as well as any co-solvents would influence electro-chemical activity and solubility, respectively [34]. These operating conditions reflect the strength of the electro-chemical ATRP approach for surface modification.
Scheme 1.
Schematic representation of the proposed system. A monolayer of atom transfer radical polymerization (ATRP)-initiator on a flat gold substrate which is used as the working electrode in the electrochemical setup. Cu(II) is reduced to Cu(I) which initiates the reaction on the electrode surface. The activation and deactivation is a reversible process and maintains the control over the polymerization-reaction.
Scheme 1.
Schematic representation of the proposed system. A monolayer of atom transfer radical polymerization (ATRP)-initiator on a flat gold substrate which is used as the working electrode in the electrochemical setup. Cu(II) is reduced to Cu(I) which initiates the reaction on the electrode surface. The activation and deactivation is a reversible process and maintains the control over the polymerization-reaction.
2. Experimental Section
2.1. Materials
Oligo(ethylene glycol) methyl ether methacrylate (OEGMA475, average Mw: 475 g/mol) and 2-hydroxyethyl methacrylate (HEMA) were purchased from Sigma Aldrich (Munich, Germany) and passed over basic alumina to remove inhibitor. Copper(II)Br2 (99%, Aldrich), bis[2-(2′-bromoisobutyryloxy)ethyl]disulfide and ethanol (HPLC grade) were purchased from Sigma Aldrich and used without further purifications. Gold coated silicon wafer was also obtained from Sigma Aldrich and before use rinsed with water, ethanol and cleaned with CO2 snow cleaning. All reactions were performed in Milli-Q water (Millipore GmbH, Schwalbach am Taunus, Germany). Me6TREN was synthesized as described by Ciampolini [35].
Scanning force microscopy images were taken on a Veeco Instruments Scanning Force Microscope (Veeco, Mannheim, Germany) via tapping mode, operating on Nanoscope software (version 1.10, Veeco).
2.2. Electrochemical Polymerization
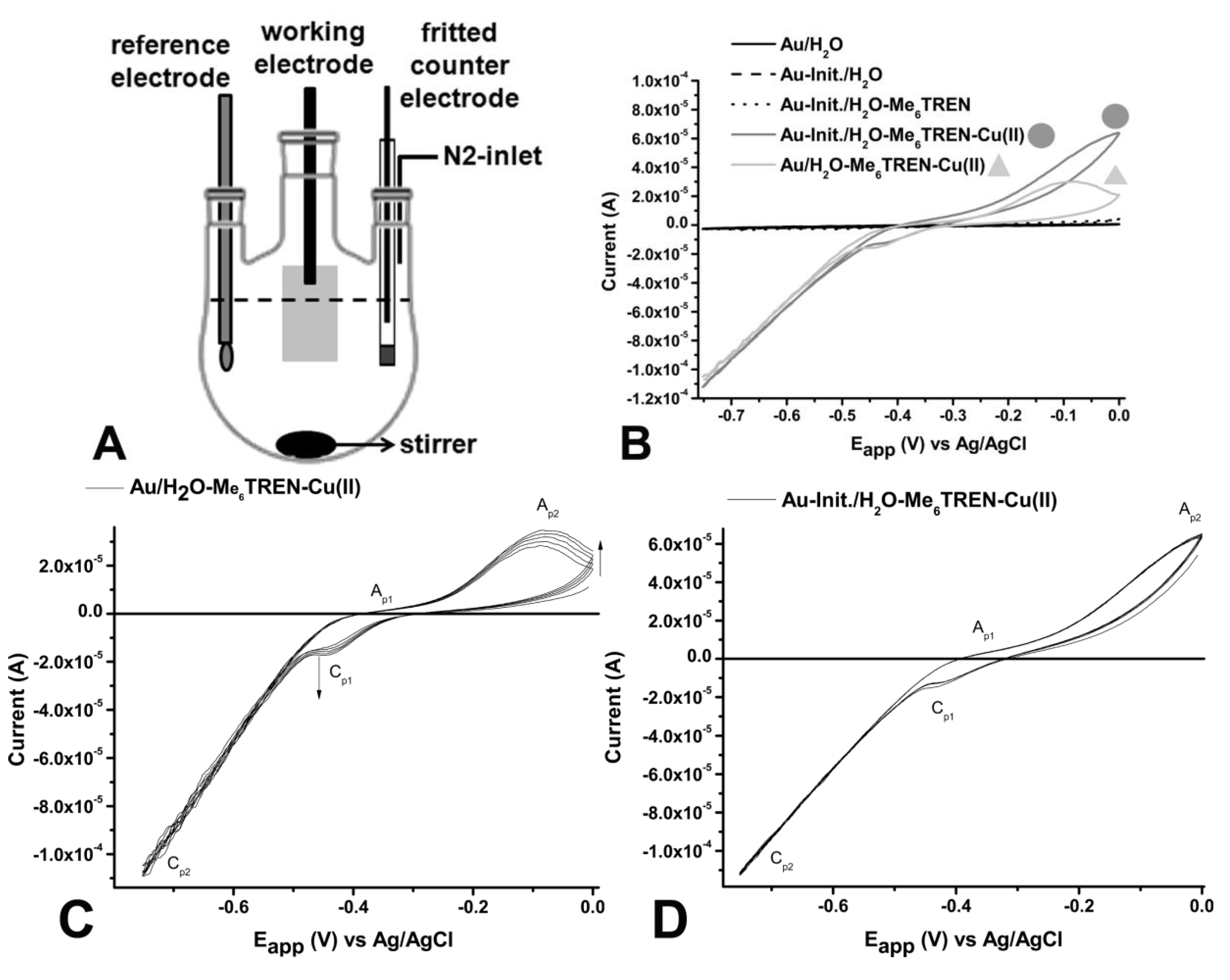
Cyclic voltammetry (CV) and controlled-potential electrolysis were carried out on an AutoLab potentiostat (Autolab PGSTAT302N, Metrohm Autolab b.v., Filderstadt, Germany) operated with NOVA 1.7 software in a three-electrode cell under a nitrogen atmosphere, using platinum-wire isolated in a glass-fritted tube as a counter electrode. The gold electrode of about 1 cm2 acting as the working electrode was connected via a clamp to a platinum wire and as a Ag/AgCl reference electrode was used (See schematic setup Figure 1A).
All experiments were performed at ambient temperature and under inert atmosphere (N2). The working solution was first bubbled with nitrogen for 30 min before the reaction was initiated. During the polymerization the solution was gently stirred not to create air bubbles which would adhere to the electrode surface or create turbulence at the liquid-air interface to see a clear effect between surface which was in contact with the solution and the area which was not. After the polymerization the samples were washed immediately with water and dried under a nitrogen-flow. Operating potential for polymerizations was −0.5 V vs. Ag/AgCl. Scan rate for CV was 0.02 V∙s−1. Electrochemical ATRP via QCM was performed on a Quartz Crystal Microbalance—QCM200, Standford Research Systems, Sunnyvale, CA, USA—from Stanford research systems. The reaction conditions as well as the used potentials and electrodes were identical to the conventional polymerization reaction as described above.
Figure 1.
(A) Schematic representation of the reaction setup; (B) Cyclic voltammograms of flat gold-electrodes non-covered (—, black) and modified with the ATRP-initiator (Init.) (– –) in degassed MilliQ-water and modified gold-electrode in the presence of 1 mM Me6TREN (- - -), Cu(II)Br2/Me6TREN (1:1) (—, ![Polymers 05 01229 i001]() ) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—, ![Polymers 05 01229 i002]() ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
 ) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,  ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
Figure 1.
(A) Schematic representation of the reaction setup; (B) Cyclic voltammograms of flat gold-electrodes non-covered (—, black) and modified with the ATRP-initiator (Init.) (– –) in degassed MilliQ-water and modified gold-electrode in the presence of 1 mM Me6TREN (- - -), Cu(II)Br2/Me6TREN (1:1) (—, ![Polymers 05 01229 i001]() ) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—,
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—, ![Polymers 05 01229 i002]() ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.
) and Cu(II)Br2/Me6TREN (1:1) on unmodified gold-electrode (—, ) recorded at scan speed (v) v = 0.02 V∙s−1; (C) Cyclic voltammograms of flat unmodified gold-electrodes in degassed MilliQ-water in the presence of 1mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. Arrows indicate the direction to how changes in signals progress with each cycle; and (D) Cyclic voltammograms of flat with initiator modified gold-electrodes in degassed MilliQ-water in the presence of 1 mM Cu(II)Br2/Me6TREN (1:1) recorded at v = 0.02 V∙s−1. The cyclic voltammogram does not display any changes over multiple runs.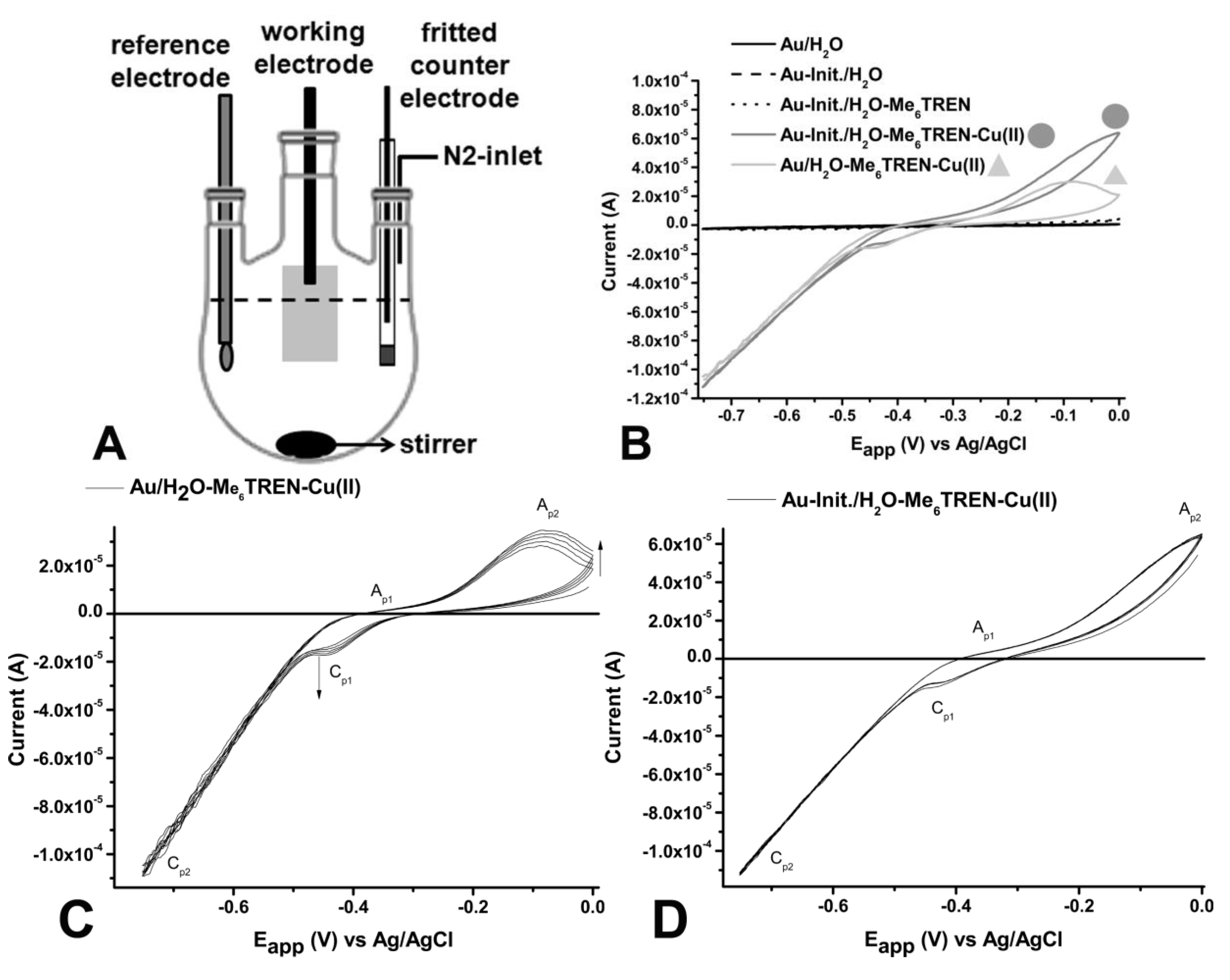
3. Results and Discussion
To determine the electrochemical behavior of the Cu(II)Br2/Ligand complex in combination with the modified gold-electrode, cyclic-voltammetry (CV) measurements were performed using a flat gold (modified and unmodified) covered silicon-wafer as the working electrode connected to a platinum-wire. A platinum-wire inside a glass fritted tube and a Ag/AgCl electrode were used as the counter electrode and the reference electrode, respectively. From the CV-measurements it was observed that neither the unmodified gold electrode nor the initiator-functionalized gold electrode, gives any appreciable signal in pure degassed water. Sequentially, the ligand Me6TREN was added to ensure that any signal in the system is solely due to the presence of the copper. Also after addition of ligand no deviating behavior was observed. However, upon addition of the ligand/Cu(II)Br2 complex an increased cathodic and anodic signal was observed (Figure 1B). The cathodic signal results from the reduction of Cu(II) to Cu(I).
Upon comparing the cyclo-voltammograms of the Cu(II)Br2/Me6TREN with the unmodified electrode and the electrode containing surface-tethered initiator moieties it was observed that they behave differently with respect to the cathodic and anodic peaks (Figure 1C). Here the cathodic peaks (Cp), Cp1 and Cp2 reflect the reduction of Cu(II) to Cu(I) and Cu(I) to Cu(0), respectively (Figure 1C). The anodic peaks Ap1 and Ap2 depict the oxidation of Cu(0) to Cu(I) and Cu(I) to Cu(II), respectively. In accordance with other electrochemical investigations, Ap1 is very weak and not visible at low scanning rates. In contrast, for continuous cycling for the system lacking the initiator, an increase in signal intensity is observed, indicating an irreversible adsorption of CuBr2/Me6TREN, as reported previously [36]. The increase of the current density in each cycle might result from the growth of a Cu/Me6TREN film. The initiator-modified electrode shows no adsorption behavior (Figure 1D). This could be due to the lower accessibility of the surface, which is covered by the initiator.
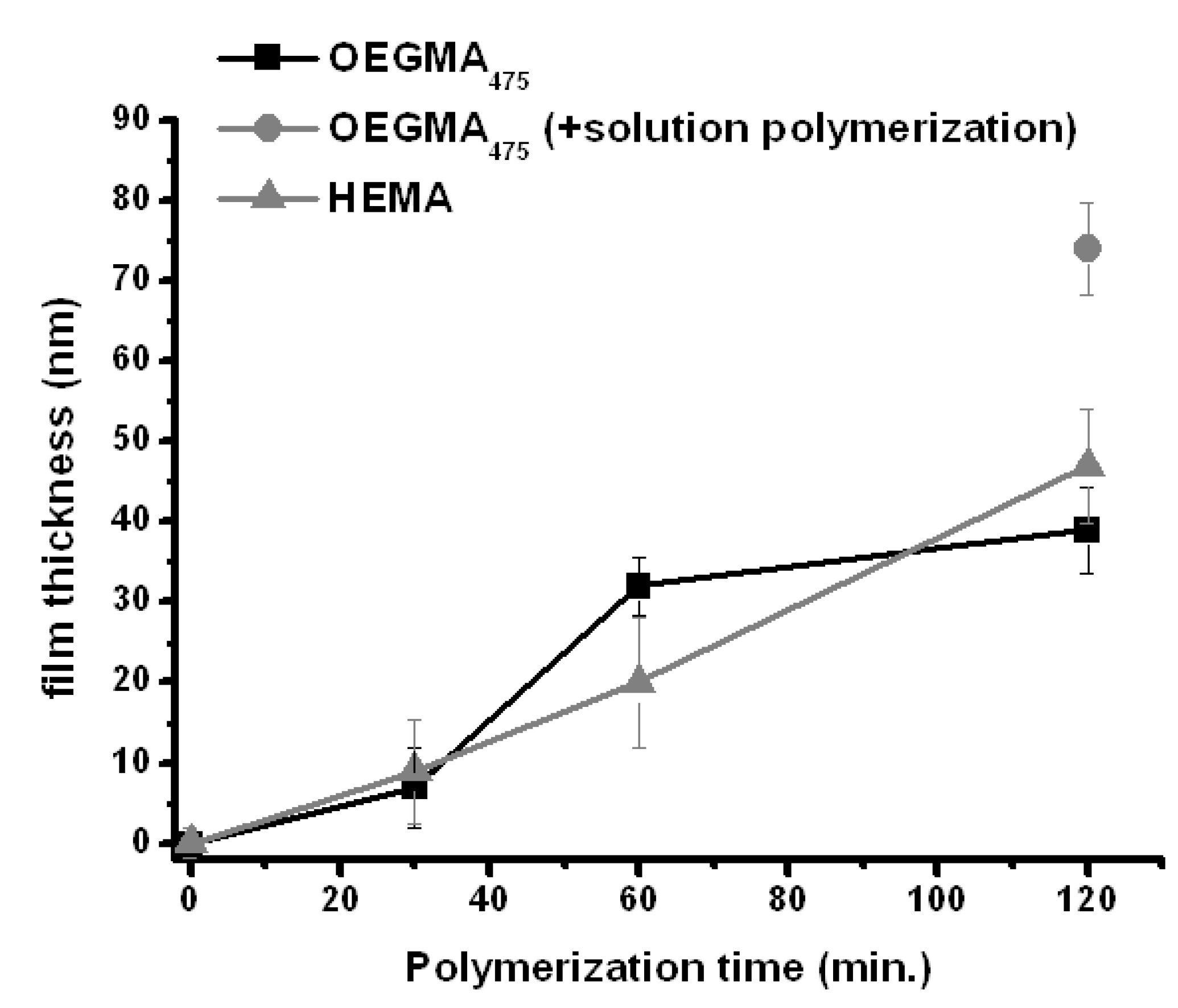
The gold surface was modified by placing the squared gold substrates in an ethanolic solution of bis[2-(2′-bromoisobutyryloxy)ethyl]disulfide (1 mg∙mL−1, 2.2 mM) overnight. Afterwards the electrodes were rinsed with ethanol and dried over a stream of nitrogen. The modified gold-electrode was connected to a Pt-wire via a small clamp and placed into the polymerization solution in such a way that the Pt-wire was not in contact with the solution. This prevents solution-based processes adding to the polymerization reaction, as Cu(II) can also be transformed on the Pt-wire surface. The applied potential (Eapp) was set to −0.5 V vs. Ag/AgCl and polymerization times were varied from 30 to 120 min. Upon applying the potential, or “switching on” the reaction, the surface slowly displays a slight discoloration over time. After the polymerization reaction, the surfaces were extensively rinsed with water and ethanol. The dry film thickness was analyzed by scanning force microscopy (SFM). The surface morphology and height-profile, obtained by scratching the surface with a sharp cannula down to the SiO2-support, was compared to untreated gold (all SFM images shown in Figures S1–S10). The surface of the untreated gold is smooth and the gold-layer thickness was found to be 81 nm (Figure S1). This thickness was used for the correction of the height determination of the modified substrates. Both layer-thickness and surface roughness of the modified electrodes increase during the polymerization process (Figure 2 and Figures S1–S10). After only 30 min a polymer layer of about 7 and 9 nm was found for OEGMA475 and HEMA, respectively. With longer polymerization times the thickness gradually increases reaching up to 39 nm for OEGMA475 and 47 nm for HEMA after 120 min. The elemental composition of the surface was analyzed by X-ray photoelectron spectroscopy (XPS). The main components are indeed carbon and oxygen (Figure S8) with an elevated carbon-content with respect to oxygen. This can be attributed to the surface morphology as more non-polar segments will be on the air-polymer interface. The absence of a Cu(I/II) signal indicates that it is present in amounts smaller than 0.1%, which is the detection limit of the XPS instrument used.
Figure 2.
Layer-thickness evolution with increasing polymerization times. Thickness obtained by scanning force microscopy (Figures S2–S9). There is a steady increase in polymer layer thickness with increasing reaction times for OEGMA475 (■) and HEMA ( ![Polymers 05 01229 i003]() ). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation ( ![Polymers 05 01229 i004]() ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
 ). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (  ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
Figure 2.
Layer-thickness evolution with increasing polymerization times. Thickness obtained by scanning force microscopy (Figures S2–S9). There is a steady increase in polymer layer thickness with increasing reaction times for OEGMA475 (■) and HEMA ( ![Polymers 05 01229 i003]() ). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation (
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation ( ![Polymers 05 01229 i004]() ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.
). Additionally OEGMA475 was polymerized with the connecting electrode positioned also in solution to see the effect on film thickness of additional solution-based Cu(I)-formation ( ). Error bars depict the standard deviation for the analyzed sample which is obtained from large area analysis.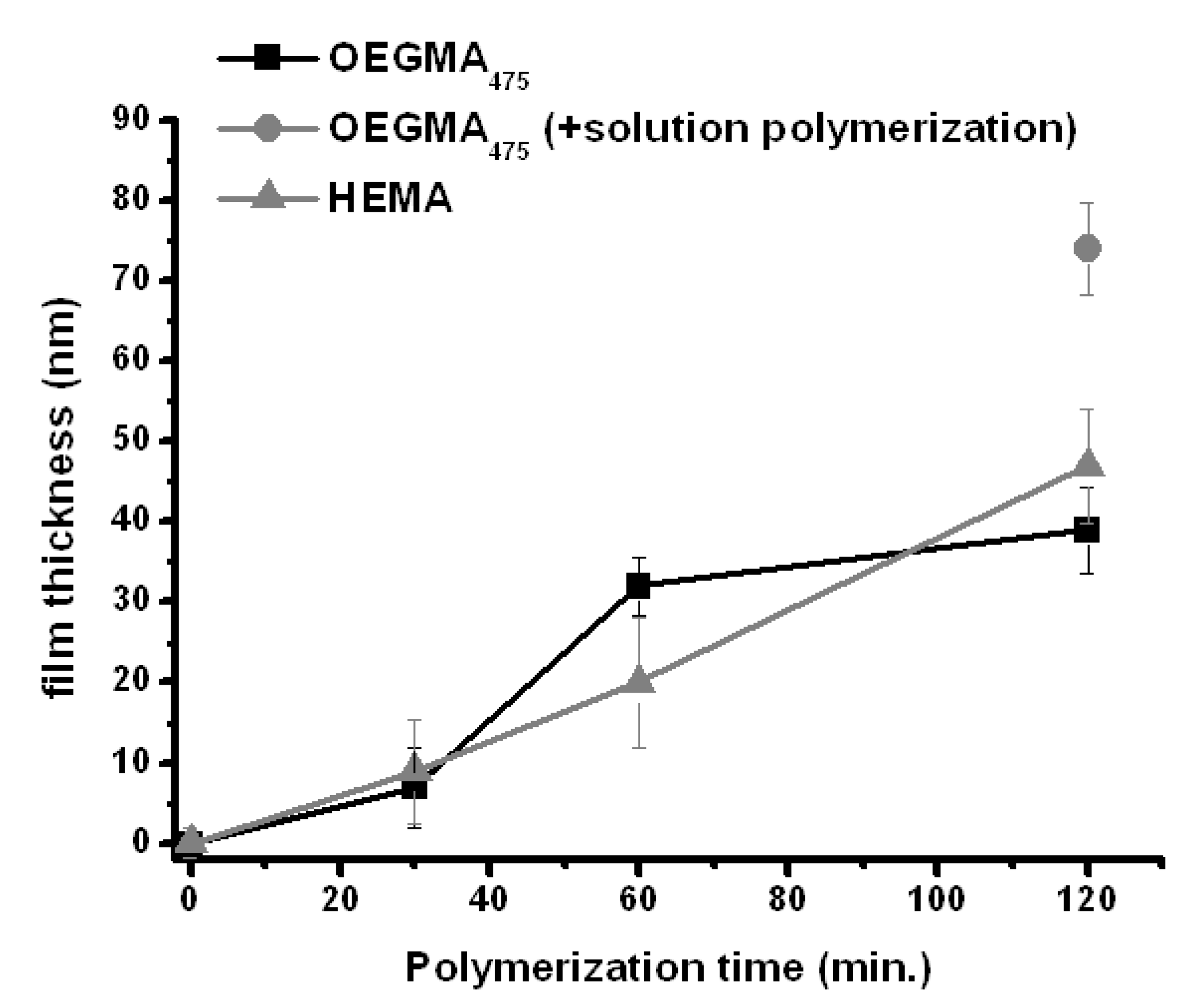
The controlled polymerization process provided by ATRP is also reflected to a certain degree in the controlled increase in layer thickness. While for the OEGMA475-film formation the curve progression is more irregular, it is very close to being linear for the HEMA-film. It must be noted that an average thickness was taken over several micrometers of polymer film and due to the rather large variations in the local thickness; the standard deviation for each point is relatively high.
Control experiments revealed several interesting aspects of the surface initiated e-ATRP reaction. First, in all cases for the time-progressive polymerization experiments, the Pt-wire connection was always separated from the solution and while the thickness of the OEGMA475-layer reached 39 nm after 120 min it significantly increased when the Pt-wire was also placed inside the polymerization solution (Figure 2 and Figure S9) reaching a thickness of about 74 nm. Consequently, also Cu(I) from solution contributes to the polymerization reaction. Secondly, the initiation of the polymerization is very much surface confined. Since the transformation of the Cu(II) into Cu(I) occurs at the electrode surface which contains the ATRP initiator, the subsequent initiation quickly commences after the formation of Cu(I) without expelling any significant amounts into the solution. This is supported by using electrodes of which parts of the surface are not directly connected and therefore unable to reduce Cu(II). No polymerization is observed in these areas (Figure 3), which would be the case when Cu(I) was present in solution. Also omitting the Cu(II) from solution did not provide any polymer layer which means that the polymer is not grown via a homolytic cleavage of the ATRP initiator. It has recently been shown that this is also a possibility induced via an electrochemical reductive process [37]. Though, the XPS-data indicates that the amount of copper in the polymer layer is lower than 0.1%, the presence of it is obvious from a discoloration, which would not result solely from the polymer. Therefore, the substrate was immersed in a THF-Me6TREN solution over night to attempt to extract residual Cu-ions from the polymer layer. Indeed, the color disappears and the height of the polymer layer only slightly decreases as shown by SFM (Figure S10).
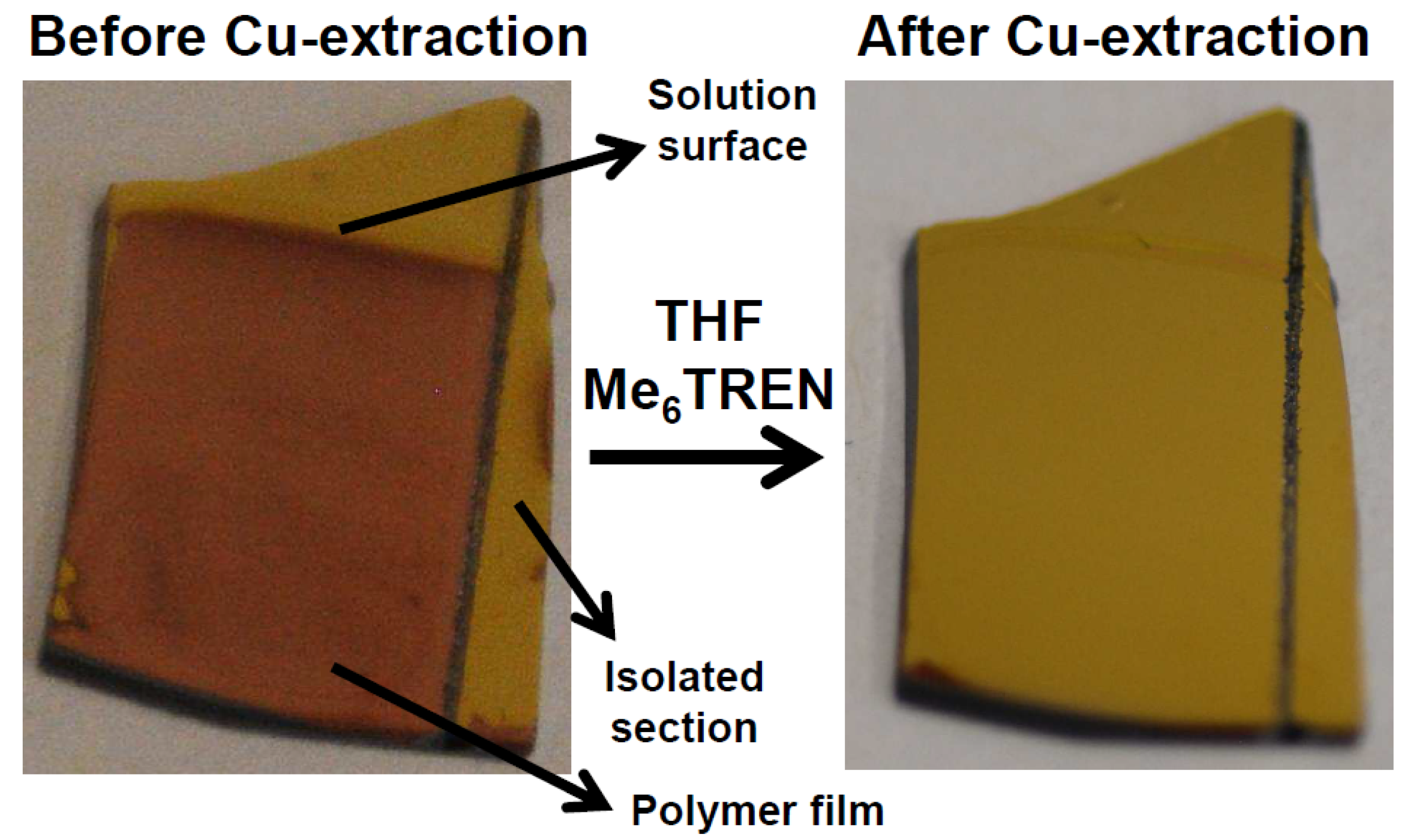
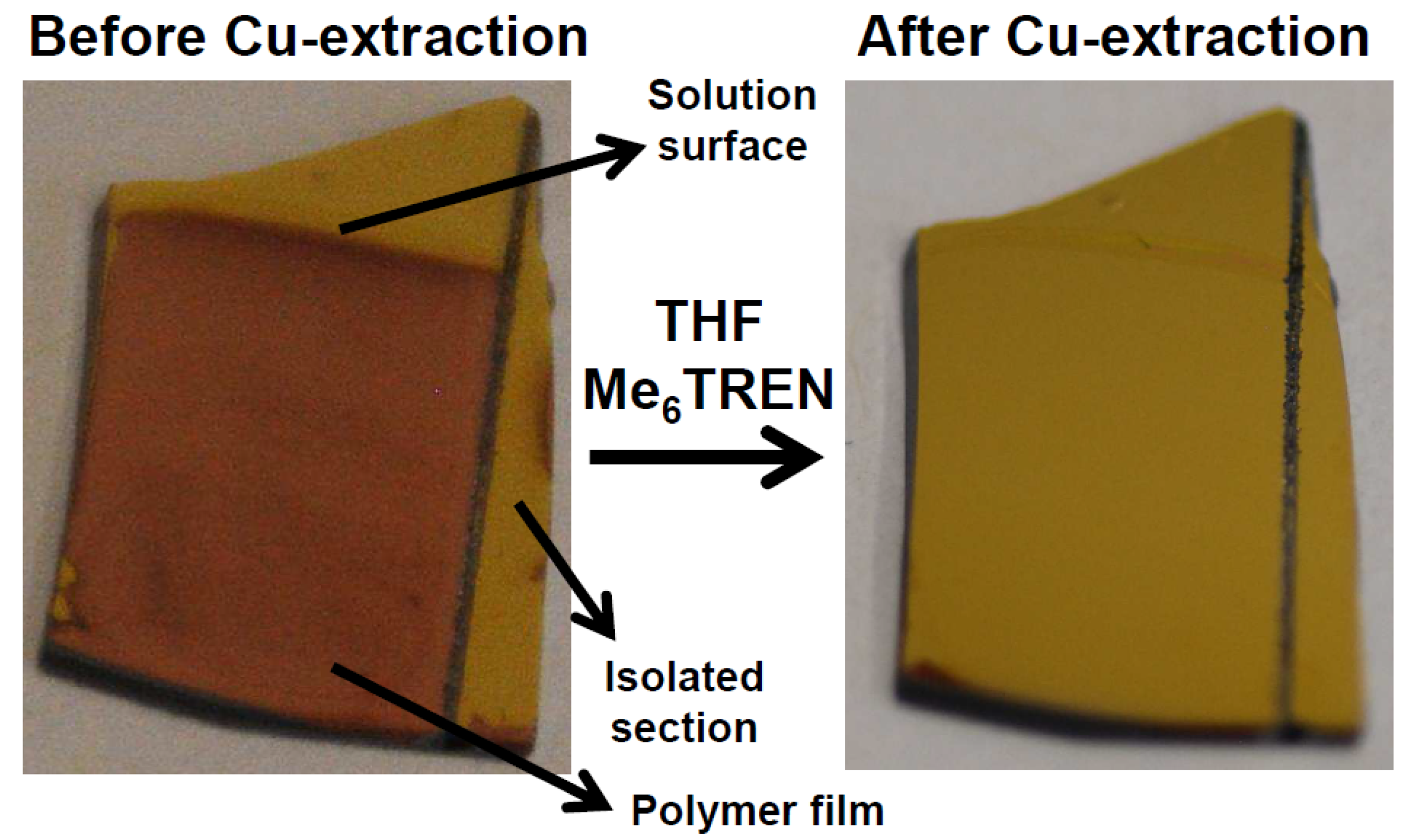
Figure 3.
Photograph of a modified gold electrode via e-ATRP, a colored area is observed which is still from residual Cu-ions. The color can be mostly washed away by immersing the substrate in THF with Me6TREN. The surface only shows modification on the areas which were immersed in the solution and are connected to the electrode. The isolated area provided by a scratch through the conductive surface is not modified even though immersed in the solution.
Figure 3.
Photograph of a modified gold electrode via e-ATRP, a colored area is observed which is still from residual Cu-ions. The color can be mostly washed away by immersing the substrate in THF with Me6TREN. The surface only shows modification on the areas which were immersed in the solution and are connected to the electrode. The isolated area provided by a scratch through the conductive surface is not modified even though immersed in the solution.
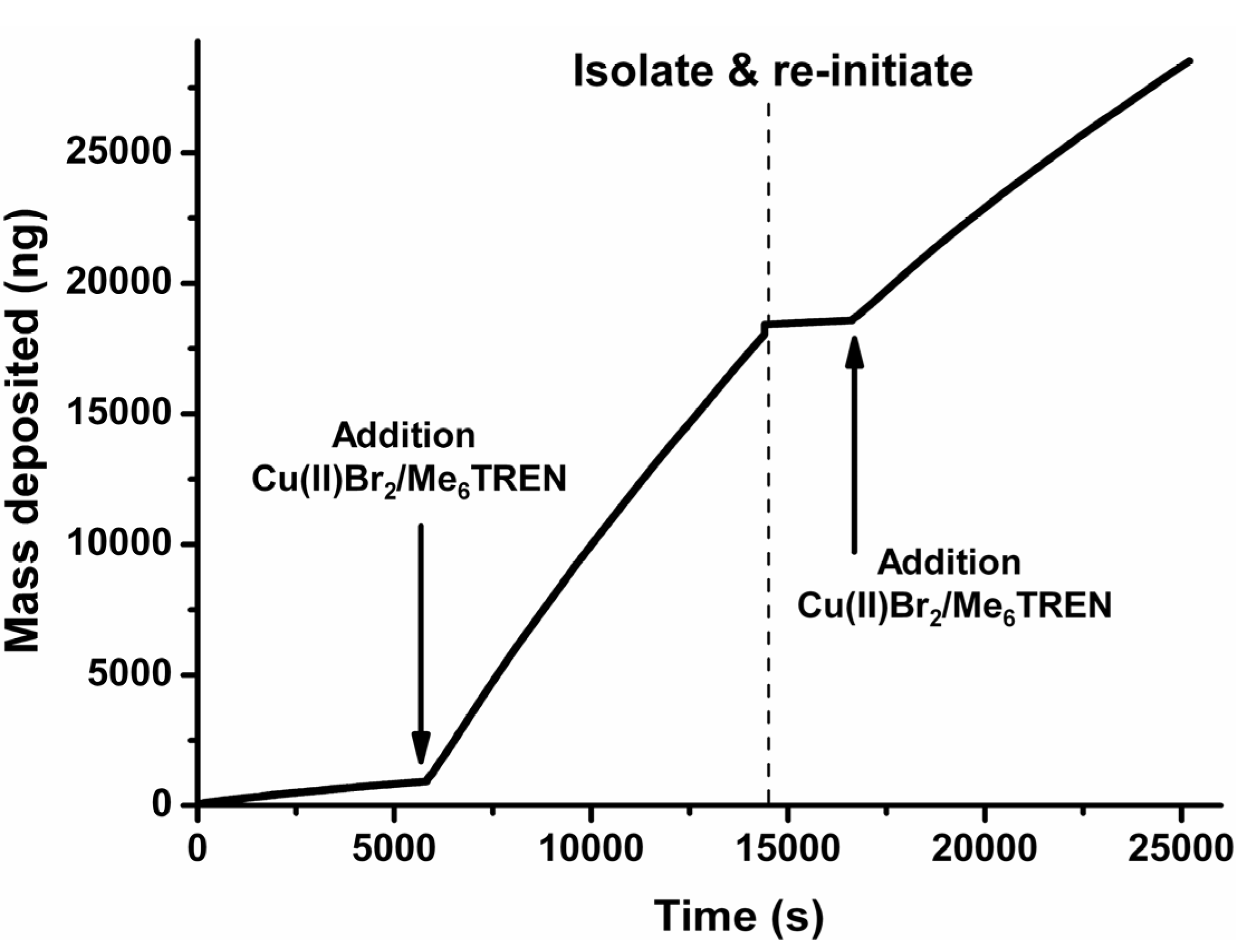
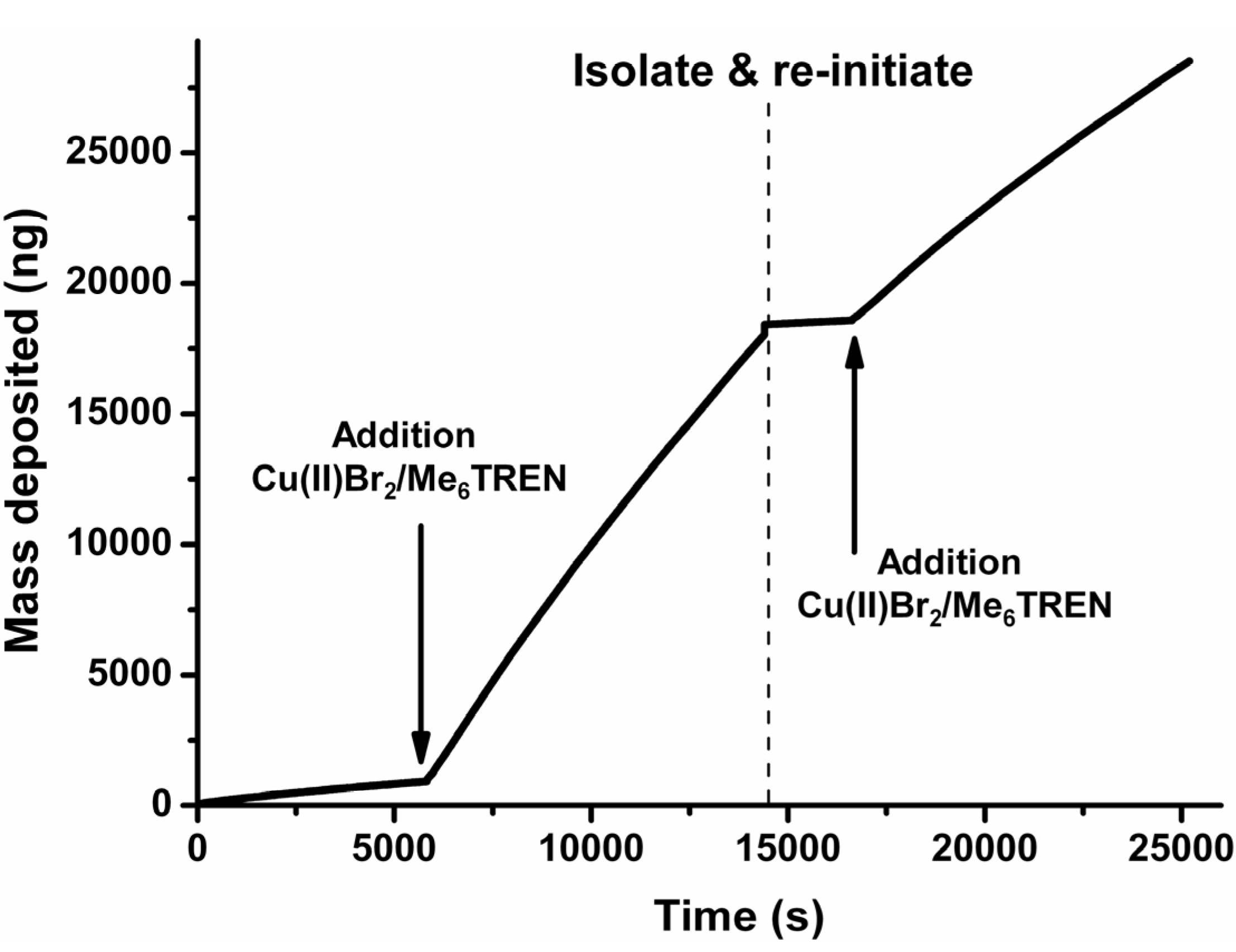
The living character as well as the possibility for removal of the thiol-tethered polymers was investigated using E-QCM (electrochemical quartz crystal microbalance). Identical experiments as described earlier in the SFM-analysis were repeated and the polymerization was followed in situ by determining the increase in surface-confined mass. Initially, a gold-coated quartz crystal was modified with ATRP initiator and mixed with HEMA (Figure 4). Upon applying a negative potential of −0.5 V vs. Ag/AgCl, there was no significant increase in mass. Once the Cu(II)Br2/Me6TREN was added, a drastic increase in mass deposition was observed, indicating polymer growth. The electrode was isolated and rinsed after 4 h of reaction time and put into a fresh solution of monomer and the same experiment was repeated. Without the Cu(II)Br2/Me6TREN complex there was again no significant increase in mass but once added again a drastic increase was observed. The mass accumulation in the re-initiation reaction was less strong than in the first experiment (Figure 4 and Figure S11) 1.85 ng∙s−1 for the first polymerization vs. 1.09 ng∙s−1 for the re-initiated polymerization. This can be attributed to the formed polymer layer which inhibits the transport of Cu(II) due to an inhibited diffusion towards the electrode surface as well as possible termination of the polymer chains. Diffusion plays an important role since introducing a surface which is partially uncoated increases the layer thickness substantially as indicated in Figure 2. The accumulation of mass during the polymerization process is highly regular indicating almost a linear increase in mass deposition i.e., surface confined polymer evolution (linear regression with R2 ≈ 0.998). This linearity indicate the lack of free radical formation which would show a fast polymerization in combination with a quick leveling off with respect to the mass increase and also not having the possibility of reinitiation. While similar behavior with respect to difference between initial polymerization and re-initiation as well as for the average increase in mass over time (0.54–4.32 ng∙s−1 (values given recalculated to achieve ng∙s−1) depending on the monomer/catalyst ratio with OEGMA475 as the monomer) was found for conventional ATRP [38], the development of mass deposition was shown to be much less controlled and displays a decreased mass accumulation over time [38,39]. It has to be noted that the mass deposition is composed of a combination of polymer with additional components such as water and other solubilized species confined in the polymer layer. The increase in mass was shown not to be a result of deposition of Cu(I/II) species (Figure S12) as the relative increase in mass (same reaction conditions without monomer present) is negligible with respect to the polymerization reaction which is in accordance with the continuous cyclo-voltammogram (Figure 1D) which also indicated no appreciable adsorption of Cu-species onto the surface.
An additional advantage of surface initiated e-ATRP is that in one single experiment, based on electrochemical induced reactions, both surface modifications as well as cleavage of the polymers from the surface are possible. Due to the nature of the thiol-linkage to gold, it can be reduced by applying a relatively large negative potential and hence cleaving the bond between the polymer and the electrode surface [40]. The removal of the polymers from the electrode would be convenient for preparing cross-linked films and removing these from the electrode to produce self-supporting thin polymer films. Cleaving the thiol-connection via reduction at a higher negative potential is feasible which was shown by applying a potential of −2.0 V vs. Ag/AgCl and investigating the decrease in surface confined mass. Initially a decrease in mass was observed which leveled after about 1 h. Though, the decrease in mass indicates the removal of surface-bound polymer, the detachment is not efficient. Most likely the presence of residual Cu-species inside the polymer film forms a solid-Cu(0) layer which embeds the thiol-linked polymers and prevents further polymer detachment (Figure S13). For a more efficient detachment either complete removal of residual copper or a different functional group is necessary which is not conflicting with the metal/ligand complex.
Figure 4.
Cumulative E-QCM profile of HEMA polymerization via surface initiated e-ATRP. The profile shows a large increase in mass only after the addition of Cu(II)Br2/Me6TREN complex. It is shown that the polymerization reaction can be re-initiated after isolating and cleaning the electrode which indicates the “living” character of the ATRP-reaction.
Figure 4.
Cumulative E-QCM profile of HEMA polymerization via surface initiated e-ATRP. The profile shows a large increase in mass only after the addition of Cu(II)Br2/Me6TREN complex. It is shown that the polymerization reaction can be re-initiated after isolating and cleaning the electrode which indicates the “living” character of the ATRP-reaction.
4. Conclusions
Electrochemical ATRP for the direct modification of electrode material surfaces was shown to function as well as for regular modification for non-electrochemical ATRP but with a more linear trend in growth comparing the mass deposition over time. The polymerization was performed in a completely aqueous environment without any additional supporting electrolyte which simplifies the system and omits any possible interference of electrolyte with monomer/polymer functionalities. Currently we are investigating various methods of cross-linking as well as additional modifications via electrochemically induced reactions, allowing the complete formation of a responsive thin polymer film to be a “one pot” process. Further investigations will also focus on using this newly surface initiated e-ATRP process to produce more complicated polymer architectures like block-copolymer systems and the incorporation of other functional moieties like protein structures and other nanostructures [41]. The development of surface initiated e-ATRP would provide a new approach to obtain more complex polymer films potentially useful for photo-voltaic devices, sensing structures and functional film-development, strengthening the importance and wide applicability of this newly developed approach. Especially, when translated also to other substrates which could be used as electrode materials, e.g., doped silicon, platinum, or transparent materials like ITO. Obviously, this will only work when there is the ability to modify the surface as well as finding the conditions to reduce the Cu(II)-species with the respective electrode.
Acknowledgments
Roes, J. is kindly acknowledged for the XPS measurements and Merle, G., Kühne, A. and Pester, C. for helpful discussions. The authors thank Böker, A., Wessling, M. and Wüthrich, R. for support and access to instrumentation required for this project. This project is financially supported by the Alexander von Humboldt-Stiftung (PvR) and the PDRF scholarship (SSH) from the Department of Foreign Affairs and Intentional Trade Canada (DFAIT).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cunningham, M.F. Controlled/living radical polymerization in aqueous dispersed systems. Prog. Polym. Sci. 2008, 33, 365–398. [Google Scholar] [CrossRef]
- Qiu, J.; Charleux, B.; Matyjaszewski, K.; Pierre, Â.; Curie, M. Controlled/living radical polymerization in aqueous media: Homogeneous and heterogeneous systems. Prog. Polym. Sci. 2001, 26, 2083–2134. [Google Scholar]
- Tsarevsky, N.V.; Matyjaszewski, K. “Green” atom transfer radical polymerization: From process design to preparation of well-defined environmentally friendly polymeric materials. Chem. Rev. 2007, 107, 2270–2299. [Google Scholar]
- Matyjaszewski, K.; Tsarevsky, N.V. Nanostructured functional materials prepared by atom transfer radical polymerization. Nat. Chem. 2009, 1, 276–288. [Google Scholar] [CrossRef]
- Edmondson, S.; Osborne, V.L.; Huck, W.T.S. Polymer brushes via surface-initiated polymerizations. Chem. Soc. Rev. 2004, 33, 14–22. [Google Scholar]
- Stuart, M.A.C.; Huck, W.T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef]
- Minko, S. Responsive polymer brushes. J. Macromol. Sci. Polym. Rev. 2006, 46, 397–420. [Google Scholar] [CrossRef]
- Ramstedt, M.; Cheng, N.; Azzaroni, O.; Mossialos, D.; Mathieu, H.J.; Huck, W.T. Synthesis and characterization of poly(3-sulfopropylmethacrylate) brushes for potential antibacterial applications. Langmuir 2007, 23, 3314–3321. [Google Scholar]
- Kenawy, E.-R.; Worley, S.D.; Broughton, R. The chemistry and applications of antimicrobial polymers: A state-of-the-art review. Biomacromolecules 2007, 8, 1359–1384. [Google Scholar]
- Ma, H.; Hyun, J.; Stiller, P.; Chilkoti, A. “Non-fouling” oligo(ethylene glycol)-functionalized polymer brushes synthesized by surface-initiated atom transfer radical polymerization. Adv. Mater. 2004, 16, 338–341. [Google Scholar] [CrossRef]
- Feng, W.; Gao, X.; McClung, G.; Zhu, S.; Ishihara, K.; Brash, J.L. Methacrylate polymer layers bearing poly(ethylene oxide) and phosphorylcholine side chains as non-fouling surfaces: In vitro interactions with plasma proteins and platelets. Acta Biomater. 2011, 7, 3692–3699. [Google Scholar]
- Zhang, Z.; Chen, S.; Jiang, S. Dual-functional biomimetic materials: Nonfouling poly(carboxybetaine) with active functional groups for protein immobilization. Biomacromolecules 2006, 7, 3311–3315. [Google Scholar]
- Ladd, J.; Zhang, Z.; Chen, S.; Hower, J.C.; Jiang, S. Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 2008, 9, 1357–1361. [Google Scholar] [CrossRef]
- Sun, L.; Baker, G.L.; Bruening, M.L. Polymer brush membranes for pervaporation of organic solvents from water. Macromolecules 2005, 38, 2307–2314. [Google Scholar]
- Zhou, F.; Biesheuvel, P.M.; Choi, E.Y.; Shu, W.; Poetes, R.; Steiner, U.; Huck, W.T. Polyelectrolyte brush amplified electroactuation of microcantilevers. Nano Lett. 2008, 8, 725–730. [Google Scholar] [CrossRef]
- Ma, H.; He, J.; Liu, X.; Gan, J.; Jin, G.; Zhou, J. Surface initiated polymerization from substrates of low initiator density and its applications in biosensors. ACS Appl. Mater. Interfaces 2010, 2, 3223–3230. [Google Scholar]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/living radical polymerization: Features, developments, and perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Grover, G.N.; Maynard, H.D. Protein-polymer conjugates: Synthetic approaches by controlled radical polymerizations and interesting applications. Curr. Opin. Chem. Biol. 2010, 14, 818–827. [Google Scholar] [CrossRef]
- Wang, X.-S.; Lascelles, S.F.; Jackson, R.A.; Armes, S.P. Facile synthesis of well-defined water-soluble polymers via atom transfer radical polymerization in aqueous media at ambient temperature. Chem. Commun. 1999, 1817–1818. [Google Scholar]
- Slavin, S.; Khoshdel, E.; Haddleton, D.M. Biological surface modification by “thiol-ene” addition of polymers synthesised by catalytic chain transfer polymerisation (CCTP). Polym. Chem. 2012, 3, 1461–1466. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.J.; Neoh, K.G.; Kang, E.T. Bioactive surfaces and biomaterials via atom transfer radical polymerization. Prog. Polym. Sci. 2009, 34, 719–761. [Google Scholar]
- Zeng, Q.; Li, T.; Cash, B.; Li, S.; Xie, F.; Wang, Q. Chemoselective derivatization of a bionanoparticle by click reaction and ATRP reaction. Chem. Commun. 2007, 1453–1455. [Google Scholar]
- Hu, Y.; Samanta, D.; Parelkar, S.S.; Hong, S.W.; Wang, Q.; Russell, T.P.; Emrick, T. Ferritin-polymer conjugates: Grafting chemistry and integration into nanoscale assemblies. Adv. Funct. Mater. 2010, 20, 3603–3612. [Google Scholar]
- Mougin, N.C.; van Rijn, P.; Park, H.; Müller, A.H.E.; Böker, A. Hybrid capsules via self-assembly of thermoresponsive and interfacially active bionanoparticle-polymer conjugates. Adv. Funct. Mater. 2011, 21, 2470–2476. [Google Scholar]
- Bünsow, J.; Mänz, M.; Vana, P.; Johannsmann, D. Electrochemically induced RAFT polymerization of thermoresponsive hydrogel films: Impact on film thickness and surface morphology. Macromol. Chem. Phys. 2010, 211, 761–767. [Google Scholar]
- Magenau, A.J.D.; Strandwitz, N.C.; Gennaro, A.; Matyjaszewski, K. Electrochemically mediated atom transfer radical polymerization. Science 2011, 332, 81–84. [Google Scholar]
- Matyjaszewski, K.; Dong, H.; Jakubowski, W.; Pietrasik, J.; Kusumo, A. Grafting from surfaces for “everyone”: ARGET ATRP in the presence of air. Langmuir 2007, 23, 4528–4531. [Google Scholar]
- Bortolamei, N.; Isse, A.A.; Magenau, A.J.D.; Gennaro, A.; Matyjaszewski, K. Controlled aqueous atom transfer radical polymerization with electrochemical generation of the active catalyst. Angew. Chem. Int. Ed. 2011, 50, 11391–11394. [Google Scholar]
- Gurunathan, K.; Murugan, A.V.; Marimuthu, R.; Mulik, U.P.; Amalnerkar, D.P. Electrochemically synthesised conducting polymeric materials for applications towards technology in electronics, optoelectronics and energy storage devices. Mater. Chem. Phys. 1999, 61, 173–191. [Google Scholar]
- Li, B.; Yu, B.; Huck, W.T.S.; Zhou, F.; Liu, W. Electrochemically induced surface-initiated atom-transfer radical polymerization. Angew. Chem. Int. Ed. 2012, 51, 5092–5095. [Google Scholar]
- Ibrahim, M.M.; Mersal, G.A.M. Solution studies of tris(2-aminoethyl)amine with metal ions of biological interest: The electroanalytical determination of the phosphate triester hydrolysis. J. Inorg. Organomet. Polym. Mater. 2009, 19, 549–557. [Google Scholar] [CrossRef]
- Li, B.; Yu, B.; Huck, W.T.S.; Liu, W.; Zhou, F. Electrochemically mediated atom transfer radical polymerization on nonconducting substrates: Controlled brush growth through catalyst diffusion. J. Am. Chem. Soc. 2013, 135, 1708–1710. [Google Scholar]
- Jones, D.M.; Brown, A.A.; Huck, W.T.S. Surface-initiated polymerizations in aqueous media: Effect of initiator density. Langmuir 2002, 18, 1265–1269. [Google Scholar]
- Ciampolini, M.; Nardi, N. Five-coordinated high-spin complexes of bivalent cobalt, nickel, and copper with tris(2-dimethylaminoethyl)amine. Inorg. Chem. 1966, 5, 41–44. [Google Scholar] [CrossRef]
- Cosnier, S.; Holzinger, M. Electrosynthesized polymers for biosensing. Chem. Soc. Rev. 2011, 40, 2146–2156. [Google Scholar] [CrossRef]
- Isse, A.A.; Gennaro, A.; Lin, C.Y.; Hodgson, J.L.; Coote, M.L.; Guliashvili, T. Mechanism of carbon-halogen bond reductive cleavage in activated alkyl halide initiators relevant to living radical polymerization: Theoretical and experimental study. J. Am. Chem. Soc. 2011, 133, 6254–6264. [Google Scholar]
- He, J.; Wu, Y.; Wu, J.; Mao, X.; Fu, L.; Qian, T.; Fang, J.; Xiong, C.; Xie, J.; Ma, H. Study and application of a linear frequency–thickness relation for surface-initiated atom transfer radical polymerization in a quartz crystal microbalance. Macromolecules 2007, 40, 3090–3096. [Google Scholar] [CrossRef]
- Moya, S.E.; Brown, A.A.; Azzaroni, O.; Huck, W.T.S. Following polymer brush growth using the quartz crystal microbalance technique. Macromol. Rapid Commun. 2005, 26, 1117–1121. [Google Scholar] [CrossRef]
- Buchmann, M.B.; Fyles, T.M.; Mischki, T.; Sutherland, T.; Tong, C.C.; Yip, V.L.Y. Intramolecular thiolysis of 4-mercaptobutyrate esters: Developing a “traceless” linker for alcohol release from self-assembled monolayers on gold. Arch. Org. Chem. 2011, 2011, 19–26. [Google Scholar]
- Broyer, R.M.; Grover, G.N.; Maynard, H.D. Emerging synthetic approaches for protein–polymer conjugations. Chem. Commun. 2011, 47, 2212–2226. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Hosseiny, S.S.; Van Rijn, P. Surface Initiated Polymerizations via e-ATRP in Pure Water. Polymers 2013, 5, 1229-1240. https://doi.org/10.3390/polym5041229
AMA Style
Hosseiny SS, Van Rijn P. Surface Initiated Polymerizations via e-ATRP in Pure Water. Polymers. 2013; 5(4):1229-1240. https://doi.org/10.3390/polym5041229
Chicago/Turabian StyleHosseiny, Seyed Schwan, and Patrick Van Rijn. 2013. "Surface Initiated Polymerizations via e-ATRP in Pure Water" Polymers 5, no. 4: 1229-1240. https://doi.org/10.3390/polym5041229