Synthesis and Polymerizability of Atom-Bridged Bicyclic Monomers

Abstract
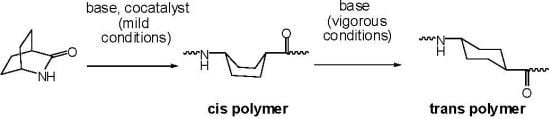
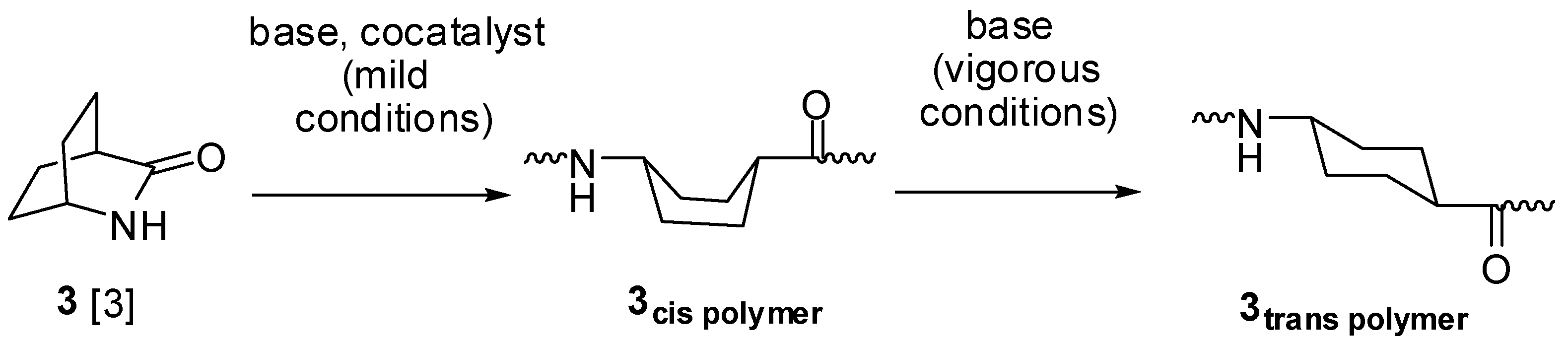
:
1. Introduction
- UNCOORDINATED ANIONIC POLYMERIZATIONS of lactams, ureas, and urethanes
- COORDINATED ANIONIC POLYMERIZATIONS of lactones and carbonates
- CATIONIC POLYMERIZATIONS of ethers, acetals, orthoesters and amines
2. Discussion
2.1. Lactams (1–15)

| Bicyclo[2.2.1]heptanes | ||||
 |   | |||
| Bicyclo[2.2.2]octanes | ||||
 | ||||
| Bicyclo[3.2.1]octanes | Bicyclo[4.1.1]octane | Bicyclo[3.2.2]nonane | ||
 |  |  |  | |
| Bicyclo[3.3.1]nonanes | Bicyclo[4.2.1]nonane | Bicyclo[4.3.1]decane | ||
 | ||||
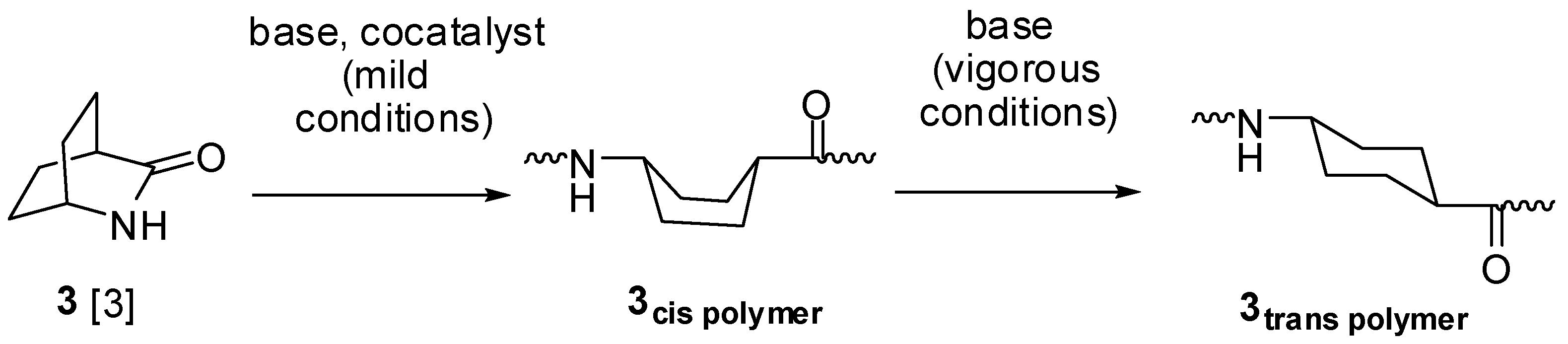

2.2. Ureas (17–23)







2.3. Urethanes(24–29)






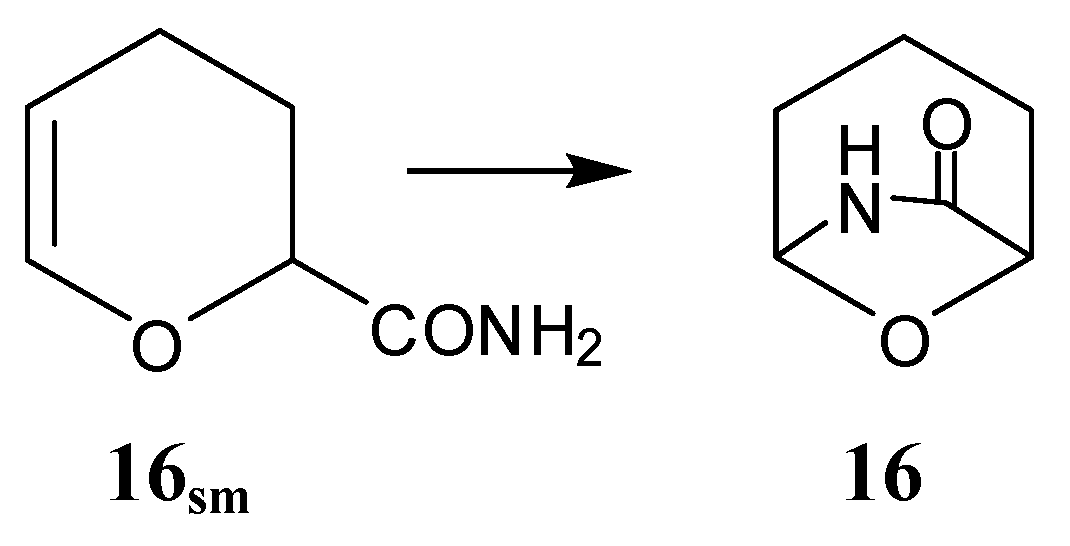
2.4. Lactones (30–37) and Carbonates(38–39)
| Bicyclo[2.1.1]hexane | Bicyclo[2.2.2]octanes | ||||
 |  |  |  |  | |
| Bicyclo[3.2.1]octanes | Bicyclo[3.3.1]nonane | Bicyclo[3.2.2]nonane | |||
 |  |  |  |  | |
2.5. Ethers (40–43)




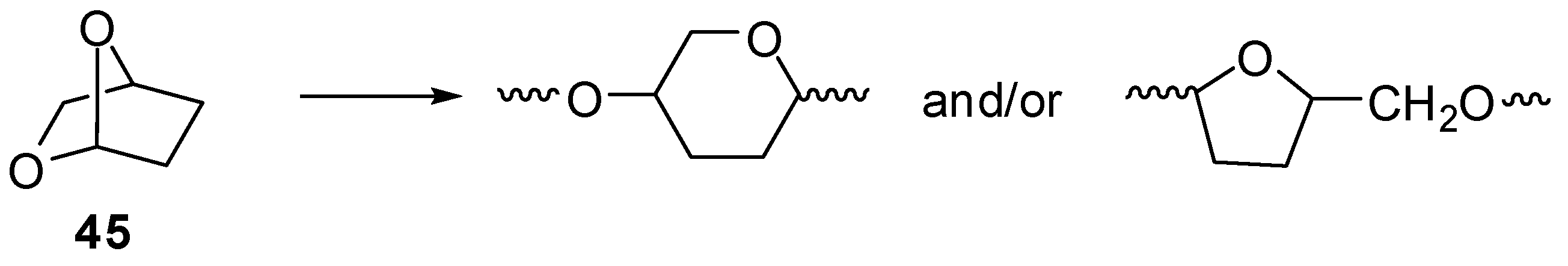
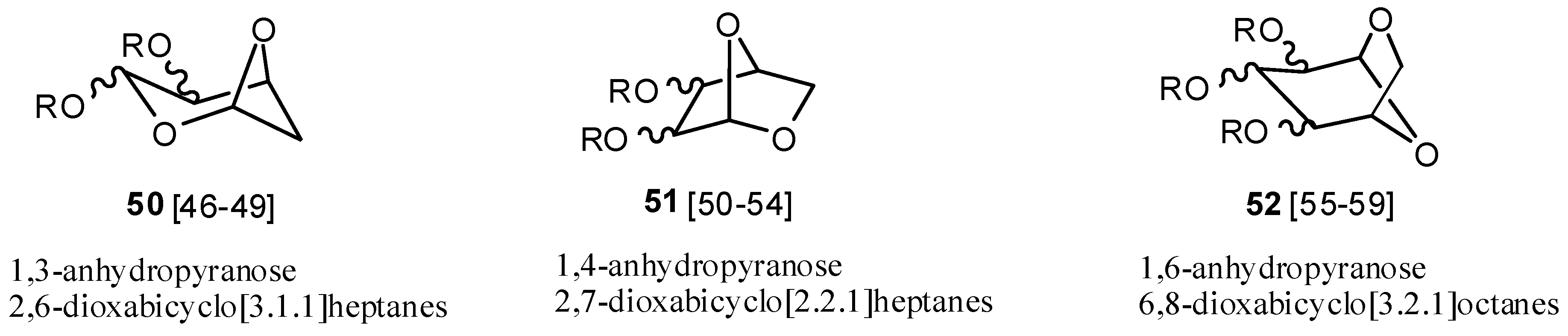
2.6. Acetals (44–52)
| Bicyclo[2.2.1]heptanes | Bicyclo[2.2.2]octane | ||
 |  |  | |
| Bicyclo[3.1.1]heptane | Bicyclo[3.2.1]octanes | ||
 |  |  | |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.7. Orthoesters (53–56)
| Bicyclo[2.2.1]heptane | Bicyclo[3.2.1]octane | Bicyclo[3.3.1]nonane | |
 |  |  |  |
2.8. Amines (57–58)
| Bicyclo[2.2.2]octane | Bicyclo[3.2.2]nonane |
 |  |
2.9. Attempted Correlations with Monomer Properties
2.9.1. Infrared
2.9.2. Saponification Rates
2.9.3. Dipole Moments
2.9.4. Polymer Properties and Uses
3. Conclusions
| Monomer Structure | Polymerizability |
|---|---|
| Bicyclohexane [2.1.1] | + |
| Bicycloheptane [2.2.1] | + |
| Bicyclooctanes [2.2.2] | + |
| [3.2.1] | + |
| [4.1.1] | + |
| Bicyclononanes [3.3.1] | ± |
| [3.2.2] | + |
| [4.2.1] | + |
| Bicyclodecane [4.3.1] | + |
Acknowledgments
References
- Dubois, P.; Coulembier, O.; Raquez, J.-M. Handbook of Ring-Opening Polymerization; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Cho, H.N.; Choi, K.Y.; Choi, S.K. Polymerization of 2-azabicyclo[2.2.1]heptane-3-one. J. Polym. Sci. Polym. Chem. Ed. 1985, 23, 623–634. [Google Scholar] [CrossRef]
- Hall, H.K., Jr. Polymerization and ring strain in bridged bicyclic compounds. J. Am. Chem. Soc. 1958, 80, 6412–6420. [Google Scholar] [CrossRef]
- Al-Obeidi, F.A.; Micheli, B.J.M.; Barfield, M.; Padias, A.B.; Wei, Y.; Hall, H.K., Jr. Synthesis and NMR studies of activated derivatives of cis- and trans-5-amino-6-oxopiperidine-2-carboxylic acid and the corresponding bicyclic dilactam DBO: Potential building blocks for stereoregular polyamides and peptides. Macromolecules 1999, 32, 6507–6516. [Google Scholar] [CrossRef]
- Lukes, R. A new application of Bredt’s rule. Coll. Czech. Chem. Comm. 1938, 10, 148–152. [Google Scholar]
- Yakhontov, L.N.; Rubstov, M.V. Synthesis of 2-quinuclidone. Z. Obsh. Khimi 1957, 27, 72–77. [Google Scholar]
- Pracejus, H. 2,2-Dimethyl-6-quinuclidone, a resonance-free amide. Chem. Ber. 1959, 92, 988–989. [Google Scholar] [CrossRef]
- Claydon, J.; Moran, W.J. The twisted amide 2-quinuclidone. Angew. Chem. Int. Ed. 2006, 45, 7118–7120. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Mori, H.; Hall, H.K., Jr.; Chan, J.H.; Bruck, M. Synthesis and ring-opening polymerization of novel bicyclic oxalactams: 2-Oxa-5-azabicyclo[2.2.2]octan-6-one. J. Polym. Sci. Polym. Chem. Ed. 1990, 8, 3251–3260. [Google Scholar]
- Okada, M.; Sumitomo, H.; Sassa, T.; Takai, M.; Hall, H.K., Jr.; Bruck, M. Synthesis and ring-opening polymerization of novel bicyclic oxalactams, 2-oxa-6-azabicyclo[2.2.2]octan-5-one. Macromolecules 1990, 23, 2427–2432. [Google Scholar] [CrossRef]
- Hall, H.K., Jr. Synthesis and polymerization of atom-bridged bicyclic lactams. J. Am. Chem. Soc. 1960, 82, 1209–1215. [Google Scholar] [CrossRef]
- Hall, H.K., Jr. Synthesis and polymerization of 3-azabicyclo[4.3.1]decan-4-one and 7,7-dimethyl-2-azabicyclo[4.1.1]octan-3-one. J. Org. Chem. 1963, 28, 3213–3214. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Shaw, R.A.; Deutschmann, A., Jr. Anti-bredt molecules. 2. 1-azabicyclo[3.3.1]nonan-2-one, a new bicyclic lactam containing bridgehead nitrogen. J. Org. Chem. 1980, 45, 3722–3724. [Google Scholar] [CrossRef]
- Okada, M. Ring-opening polymerization of bicyclic and spiro compounds. Reactivities and polymerization mechanisms. Adv. Polym. Sci. 1992, 102, 1–46. [Google Scholar] [CrossRef]
- Sumitomo, H.; Okada, M. Ring-opening polymerization of bicyclic acetals, lactones, and lactams. Adv. Polym. Sci. 1978, 28, 47–82. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Atsumi, M.; Hall, H.K., Jr. Ring-opening polymerization of bicyclic oxalactones and oxalactams. Makromol. Chem. Macromol. Symp. 1991, 42-43, 355–364. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Johnson, R.C. 3-Isopropyl-1,3-diazabicyclo[3.3.1]nonan-2-one. A simple bicyclic urea with a bridgehead nitrogen atom. J. Org. Chem. 1972, 37, 697–699. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Ekuchukwu, O.E.; Deutschmann, A., Jr.; Rose, C. Anti-bredt molecules. 6. Synthesis and polymerization of 1,3-diazabicyclo[3.3.1]nonan-2-one. Polym. Bull. 1980, 3, 375–382. [Google Scholar]
- Hall, H.K., Jr.; El-Shekeil, A. Anti-bredt molecules. 3. Synthesis of two bicyclic urethanes possessing bridgehead nitrogen. J. Org. Chem. 1980, 45, 5325–5328. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; El-Shekeil, A. Anti-bredt molecules. 5. Synthesis and polymerization of 1-aza-7-oxabicyclo[3.2.1]octan-7-one. Polym. Bull. 1980, 3, 233–239. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; El-Shekeil, A. Anti-bredt molecules. 4. Polymerization of 1-aza-3-oxabicyclo[3.3.1]nonan-2-one. Polym. Bull. 1980, 2, 829–836. [Google Scholar]
- Hall, H.K., Jr.; El-Shekeil, A. Anti-bredt bridgehead nitrogen compounds in ring-opening polymerization. Chem. Rev. 1983, 83, 549–555. [Google Scholar]
- Hall, H.K., Jr.; Blanchard, E.P., Jr.; Martin, E.L. Synthesis and polymerization of 2-oxabicyclo[2.1.1] hexan-3-ones (cyclobutane1,3-lactones). Macromolecules 1971, 4, 142–146. [Google Scholar] [CrossRef]
- Ceccarelli, C.; Andruzzi, F.; Paci, M. NMR spectroscopy of polyesters from 2-oxabicyclo- [2.2.2]octan-3-one. Polymer 1979, 20, 605–610. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Yamada, S.; Atsumi, M.; Hall, H.K., Jr.; Chan, R.J.H.; Ortega, R.B. Synthesis and ring-opening polymerization of bicyclic lactones containing a tetrahydropyran ring. 2,5-Dioxabicyclo[2.2.2]octan-3-one. Macromolecules 1986, 19, 953–959. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Atsumi, M.; Hall, H.K., Jr.; Ortega, R.B. Synthesis and ring-opening polymerization of bicyclic lactones containing a tetrahydropyran ring. 2,6-Dioxabicyclo[2.2.2]octan-3-one. Macromolecules 1986, 19, 503–509. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Atsumi, M.; Hall, H.K., Jr. Synthesis of polyesters having pendant ester groups by ring-opening polymerization of 4-methoxycarbonyl-2,6-dioxabicyclo[2.2.2]octan-3-one. Macromolecules 1987, 20, 1199–1205. [Google Scholar] [CrossRef]
- Sandin, R.B.; Rebel, W.J.; Levine, S. Ring closure of 2,5-dibromoadipic acids. J. Org. Chem. 1966, 31, 3879–3880. [Google Scholar] [CrossRef]
- Drumright, R.E.; Harmann, M.; Wolf, R. Copolymers of Cyclic Esters and Carbonates and Methods for Making Same. PCT. U.S. Patent 2002018443, 7 June 2002. [Google Scholar]
- Moore, J.A.; Kelly, J.E. Synthesis and polymerization of 2-oxo-3,8-dioxabicyclo[3.2.1]-octane. J. Polym. Sci. Polym. Lett. Ed. 1975, 13, 333–336. [Google Scholar] [CrossRef]
- Wittbecker, E.L.; Hall, H.K., Jr.; Campbell, T.W. Synthesis and polymerization of bridged bicyclic ethers. J. Am. Chem. Soc. 1960, 82, 1218–1222. [Google Scholar]
- Saegusa, T.; Motoi, M.; Matsumoto, S.; Fujei, H. Stereochemistry of the ring-opening polymerization of 2-methyl-7-oxabicyclo[2.2.1]heptane. Macromolecules 1972, 5, 233–236. [Google Scholar] [CrossRef]
- Baccaredda, M.; Giusti, P.; Andruzzi, F.; Cerrai, D.; DiMaina, N. PF5-catalyzed polymer of exo-2-methyl-7-oxabicyclo[2.2.1]heptane. J. Polym. Sci. Polym. Symp. 1970, 31, 159–176. [Google Scholar]
- Kops, J.; Spanggard, D. Polymerization of 2-methyl-7-oxabicyclo[2.2.1]heptane. J. Macromol. Sci. Chem. 1973, 7, 1455–1469. [Google Scholar] [CrossRef]
- Saegusa, T.; Hadaka, T.; Fujii, H. Polymer of 2-oxabicyclo[2.2.2]octane. Polym. J. 1971, 2, 670–671. [Google Scholar]
- Andruzzi, F.; Ceccarelli, G.; Paci, M. NMR spectra of poly-3-oxabicyclo[3.2.2]nonane. Polymer 1980, 21, 1180–1184. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; de Blauwe, F. 2,6- And 2,7-dioxabicyclo[2.2.1]heptanes. J. Am. Chem. Soc. 1975, 97, 655–656. [Google Scholar]
- Hall, H.K., Jr.; Carr, L.J.; Kellman, R.; de Blauwe, F. New ring system. 2,6-Dioxabicyclo[2.2.2]octane, a highly reactive bicyclic acetal. J. Am. Chem. Soc. 1974, 96, 7265–7269. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; de Blauwe, F.; Carr, L.J.; Rao, V.S.; Reddy, G.S. Synthesis, hydrolytic reactivity, and polymerization of 2,6- and 2,7-dioxabicyclo[2.2.1]heptanes. J. Polym. Sci. Symp. 1976, 56, 101–115. [Google Scholar]
- Hall, H.K., Jr.; Steuck, M.J. Polymerization of 6,8-dioxabicyclo[3.2.1]octane and 3,6,8-trioxabicyclo[3.2.1]octane. J. Polym. Sci. Polym. Chem. Ed. 1973, 11, 1035–1042. [Google Scholar] [CrossRef]
- Padias, A.B.; Szymanski, R.; Hall, H.K., Jr. Synthesis and polymerization of atom-bridged bicyclic acetals and orthoesters; A dioxacarbenium ion mechanism for orthoester polymerization. In Ring Opening Polymerization; McGrath, J.E., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1985; pp. 313–333, Chapter 286. [Google Scholar]
- Padias, A.B.; Szymanski, R.; Hall, H.K., Jr. Synthesis and polymerization of atom-bridged bicyclic acetals and orthoesters: A new mechanism. ACS Polym. Prepr. 1984, 25, 258–259. [Google Scholar]
- Yokoyama, Y.; Hall, H.K., Jr. Ring-opening polymerization of atom-bridged and bond-bridged bicyclic ethers, acetals and orthoester. Adv. Polym. Sci. 1982, 42, 107–138. [Google Scholar] [CrossRef]
- Reich, W.; Schwalm, R.; Haussling, L.; Nuyken, O.; Raether, R.B. Preparation Method for 2,7-Dioxabicyclo[3.2.1]octane. Ger. Patent DE19710992A1, 30 October 1971. [Google Scholar]
- Reich, W.; Schwalm, R.; Haussling, L.; Nuyken, O.; Raether, R.B. Polymers From 2,7-Dioxa-bicyclo[3.2.1]octane. Ger. Patent DE19614635A1, 16 October 1961. [Google Scholar]
- Good, F.J., Jr.; Schuerch, C. Synthesis of (1→3)-α-D-glucopyranan by stereoregular cationic polymerization of substituted 2,6-dioxabicyclo[3.1.1]heptanes: 1,3-anhydrotri-(p-substituted-benzyl)-β-D-glucopyranoses. Macromolecules 1985, 18, 595–599. [Google Scholar] [CrossRef]
- Kong, F.; Schuerch, C. Synthesis of (1→3)-α-D-mannopyranan by stereoregular cationic polymerization of substituted 2,6-dioxabicyclo[3.1.1]heptanes. Macromolecules 1984, 17, 983–989. [Google Scholar] [CrossRef]
- Good, F.; Schuerch, C. Improved synthesis of substituted 2,6-dioxabicyclo[3.1.1]heptanes: 1,3-anhydro-2,4,6-tri-O-benzyl-2,4,6-tri-O-p-bromobenzyl- and -2,4,6-tri-O-p-methylbenzyl-β-D-glucopyranose. Carbohydr. Res. 1984, 125, 165–171. [Google Scholar] [CrossRef]
- Kong, F.; Schuerch, C. Improved synthesis of substituted 2,6-dioxabicyclo[3.1.1]heptanes: 1,3-anhydro-2,4,6-tri-O-benzyl- and 1,3-anhydro-2,4,6-tri-O-p-bromobenzyl-β-D-mannopyranose. Carbohydr. Res. 1983, 112, 141–147. [Google Scholar] [CrossRef]
- Kops, J.; Schuerch, C. Polymerization of 1,4-Anhydro Sugar Derivative. J. Polym. Sci. C 1965, 11, 119–138. [Google Scholar] [CrossRef]
- Hagino, A.; Yoshida, S.; Shinpuku, T.; Matsuzaki, K.; Uryu, T. Selective ring-opening polymerization of 1,4-anhydro-α-D-lyxopyranose derivatives and synthesis of stereoregular (1→5)-α-D-lyxofuranan. Macromolecules 1986, 19, 1–7. [Google Scholar] [CrossRef]
- Ogawa, M.; Hatanaka, K.; Uryu, T. Synthesis of a novel cellulose-type hexopyranan 6-deoxy-(1→4)-α-L-talopyranan by selective ring opening polymerization of 1,4-anhydro sugar derivatives. Macromolecules 1991, 24, 987–992. [Google Scholar] [CrossRef]
- Uryu, T.; Yamanouchi, J.; Hayashi, S.; Tamaki, H.; Matsuzaki, K. Selective ring-opening polymerization of 1,4-anhydro-2,3-di-O-benzyl-α-D-xylopyranose and synthesis of stereoregular (1→5)-alpha-D-xylofuranan. Macromolecules 1983, 16, 320–326. [Google Scholar] [CrossRef]
- Uryu, T.; Kitano, K.; Ito, K.; Yamanouchi, J.; Matsuzaki, K. Selective ring-opening polymerization of 1,4-anhydro-α-D-ribopyranose derivatives and synthesis of stereoregular (1→4)-beta-D-ribopyranan. Macromolecules 1981, 14, 1–9. [Google Scholar] [CrossRef]
- Kobayashi, K.; Ichikawa, H.; Sumitomo, H.; Schuerch, C. Sterically controlled ring-opening polymerization of a 1,6-anhydro-beta-D-galactopyranose derivative by neighboring group participation. (1→6)-beta-D-Galactopyranan. Macromolecules 1988, 21, 542–543. [Google Scholar] [CrossRef]
- Uryu, T.; Katsuhiro, I.; Kobazashi, K.-I.; Matsuzahi, K. Spectroscopic studies on key-opening polymerization of 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose. Macromol. Chem. Phys. 1979, 180, 1509–1519. [Google Scholar] [CrossRef]
- Uryu, T.; Tachikawa, H.; Ohaku, K.-I.; Terui, K.; Matsuzahi, K. Synthesis of 2,3,4-tri-O-benzyl-[1→6]-α-D-glucopyranan. Macromol. Chem. Phys. 1977, 178, 1929–1940. [Google Scholar] [CrossRef]
- Uryu, T.; Ito, K.; Kobayashi, K.I.; Matsuzaki, K. Ring opening polymerization of 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose. Makromol. Chem. 1979, 180, 1509–1519. [Google Scholar] [CrossRef]
- Uryu, T.; Tachikawa, H.; Ohaku, K.I.; Terui, K.; Matsuzaki, K. Polymerization of 1,6-anhydro-2,3,4-tri-O-benzyl-β-D-glucopyranose. Makromol. Chem. 1977, 178, 1929–1940. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; de Blauwe, F.; Pyriadi, T. 2,6,7-Trioxabicyclo[2.2.1]heptane. J. Am. Chem. Soc. 1975, 97, 3854. [Google Scholar]
- Hall, H.K., Jr.; Yokoyama, Y. Polymerization of 2,6,7-trioxabicyclo[2.2.1]heptane with either contraction or expansion. Polym. Bull. 1980, 2, 281–287. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Padias, A.B.; de Blauwe, F.; Hall, H.K., Jr. Synthesis and polymerization of 2,6,7-trioxabicyclo[2.2.1]heptane and 1-methyl-2,6,7-trioxabicyclo[2.2.1]heptane. Macromolecules 1980, 13, 252–261. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Padias, A.B.; Bratoeff, E.A.; Hall, H.K., Jr. Synthesis and polymerization of 2,6,7-trioxabicyclo[2.2.2]octane and its derivatives. Macromolecules 1982, 15, 11–17. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Hall, H.K., Jr. Polymerization of 2,7,8-trioxabicyclo[3.2.1]octane and 2,8,9-trioxabicyclo[3.3.1]nonane. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 3133–3147. [Google Scholar] [CrossRef]
- Crank, G.; Eastwood, F.W. Derivatives of Orthoesters. 1. Bicyclic Orthoesters. Aust. J. Chem. 1964, 17, 1385–1391. [Google Scholar] [CrossRef]
- Heller, J.; Barr, J.; Ng, S.Y.; Abdellanci, K.S.; Gurry, R. Poly(orthoesters). Adv. Drug Deliv. Rev. 2002, 547, 1015–1039. [Google Scholar]
- Burt, R.A.; Chiang, Y.; Hall, H.K., Jr.; Kresge, A.J. The hydrolysis of bicyclic orthoesters in the 2,6,7-trioxabicyclo[2.2.1]heptane series. Confirmation of the absence of strain-relief rate acceleration. J. Am. Chem. Soc. 1982, 104, 3687–3690. [Google Scholar]
- Hall, H.K., Jr. Polymerization of two atom-bridged bicyclic amines. J. Org. Chem. 1963, 28, 223–224. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Zbinden, R. Infrared spectra and strain in cyclic carbonyl compounds. J. Am. Chem. Soc. 1958, 80, 6428–6432. [Google Scholar] [CrossRef]
- Zbinden, R.; Hall, H.K., Jr. Infrared carbonyl and carbon-hydrogen frequencies in bridged bicyclic ketones. J. Am. Chem. Soc. 1960, 82, 1215–1218. [Google Scholar] [CrossRef]
- Hall, H.K., Jr.; Brandt, M.K.; Mason, R.M. Hydrolysis rates and mechanisms of cyclic monomers. J. Am. Chem. Soc. 1958, 80, 6420–6427. [Google Scholar]
- Hall, H.K., Jr. Mechanisms of hydrolysis of several atom-bridged bicyclic anhydrides, N-methylimides and lactone. J. Org. Chem. 1963, 28, 2027–2029. [Google Scholar] [CrossRef]
- Lee, C.M.; Kumler, W.D. Dipole moments and structure of cyclic compounds: Lactones, lactams, anhydrides, carbonates, carbamates, ureides, and imides. J. Org. Chem. 1963, 28, 1438–1439. [Google Scholar] [CrossRef]
- Berti, C.; Binassi, E.; Celli, A.; Colonna, M.; Fiorini, M.; Marchese, P.; Marianucci, E.; Gazzano, M.; Di Credico, F.; Brunelle, D. Poly(1,4-cyclohexylenedimethylene 1,4-cyclohexanedicarboxylate): Influence of stereochemistry of 1,4-cyclohexylene units on the thermal properties. J. Polym. Sci. B Polym. Phys. 2008, 46, 619–630. [Google Scholar] [CrossRef]
- Liu, Y.; Turner, S.R. Synthesis and properties of cyclic diester-based aliphatic copolyesters. J. Polym. Sci. A Polym. Chem. 2010, 48, 2162–2169. [Google Scholar] [CrossRef]
- Schuerch, C. Biomedical applications of synthetic polysaccharides. In Polymer and Fiber Science: Recent Advances; Fornes, R.E., Gilbert, R.D., Mark, H.F., Eds.; Wiley-VCH: New York, NY, USA, 1992; pp. 9–16. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hall, H.K. Synthesis and Polymerizability of Atom-Bridged Bicyclic Monomers. Polymers 2012, 4, 1674-1686. https://doi.org/10.3390/polym4041674
Hall HK. Synthesis and Polymerizability of Atom-Bridged Bicyclic Monomers. Polymers. 2012; 4(4):1674-1686. https://doi.org/10.3390/polym4041674
Chicago/Turabian StyleHall, Henry K. 2012. "Synthesis and Polymerizability of Atom-Bridged Bicyclic Monomers" Polymers 4, no. 4: 1674-1686. https://doi.org/10.3390/polym4041674
APA StyleHall, H. K. (2012). Synthesis and Polymerizability of Atom-Bridged Bicyclic Monomers. Polymers, 4(4), 1674-1686. https://doi.org/10.3390/polym4041674