Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3


Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Synthesis
2.2.1. Synthesis of N-Carboxyanhydride (NCA) of γ-Benzyl-L-glutamate
2.2.2. Synthesis of N-Carboxyanhydride (NCA) of Nε-Benzylcarbonyl-L-lysine
2.2.3. Synthesis of N-carboxyanhydride (NCA) of L-alanine
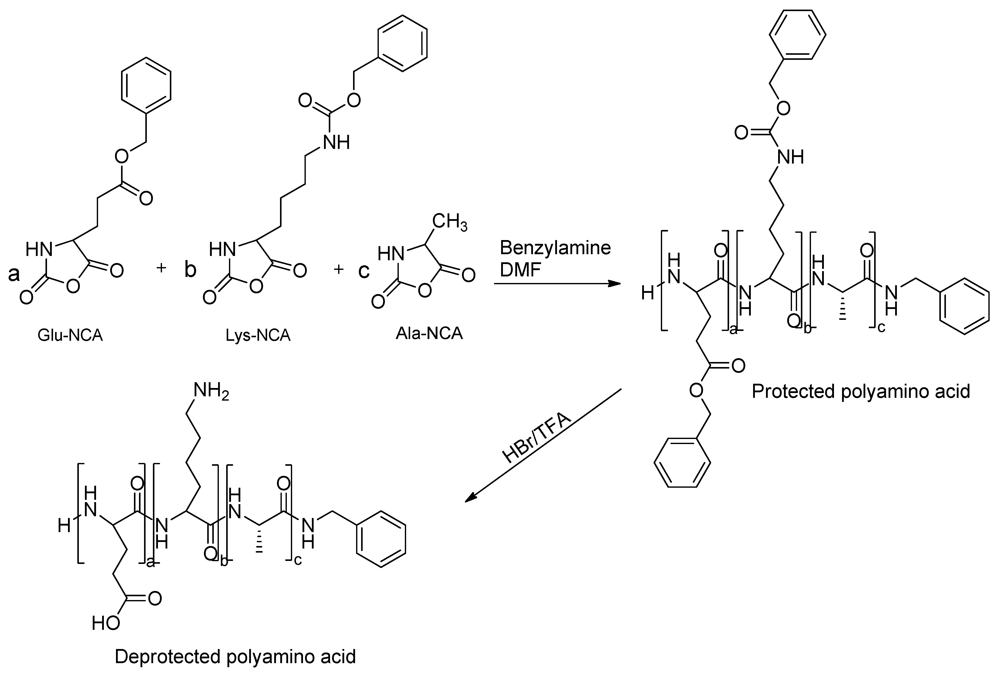
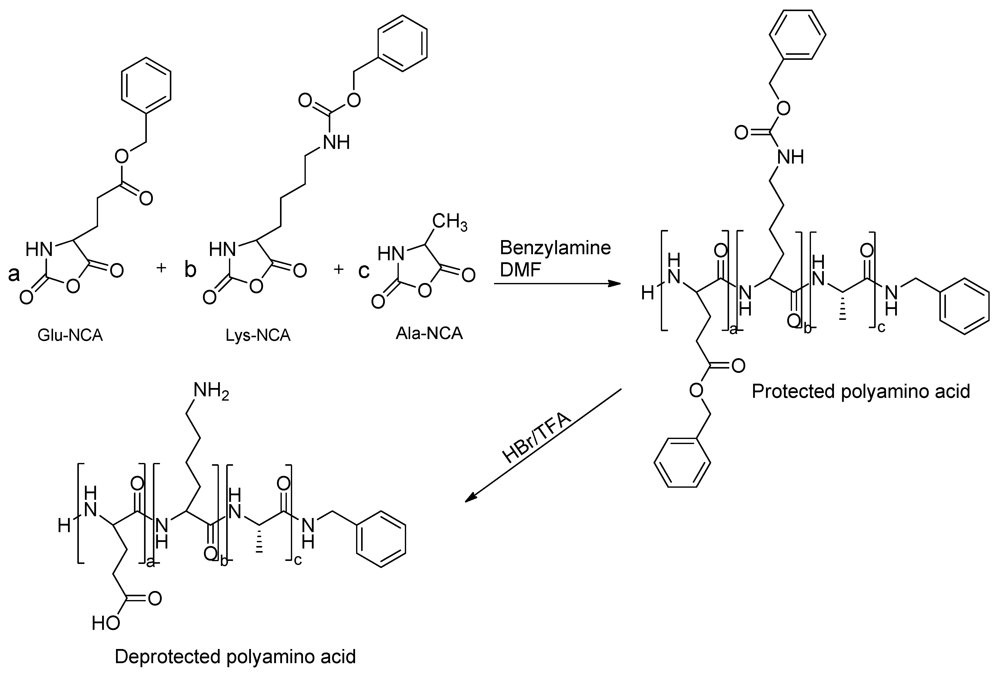
2.2.4. Random NCA Copolymerization
2.2.5. Deprotection of Polyamino Acids
2.3. Characterization
2.3.1. Size Exclusion Chromatography (SEC)
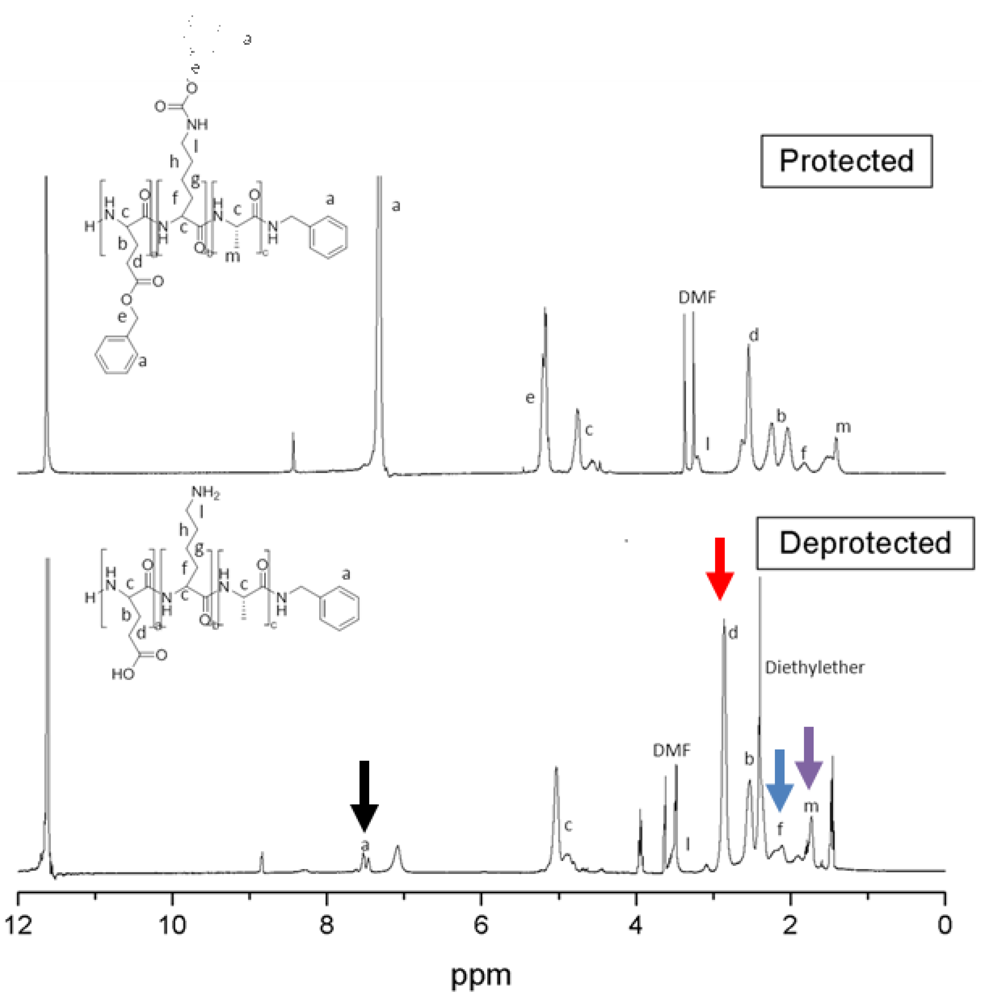
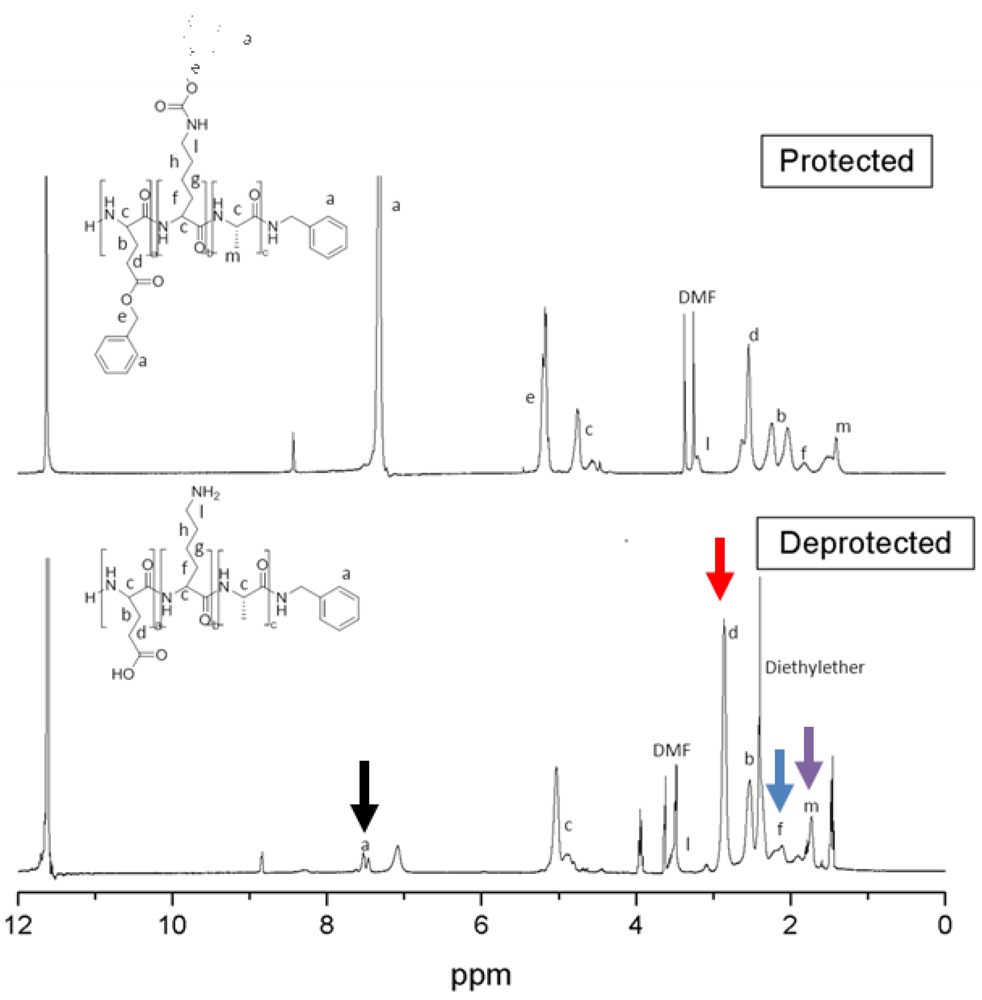
2.3.2. 1H-NMR
2.3.3. Titration Experiments
2.3.4. Scanning Electron Microscopy (SEM)
2.3.5. Powder X-ray Diffraction (PXRD)
2.3.6. Raman Microscopy
3. Results and discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target composition | DP (a) | Mn (g/mol) (a) | PDI (a) | Experimental composition (b) |
|---|---|---|---|---|
| E14K14A14 | 56 | 10400 | 1.20 | E11K25A18 |
| E18K12A12 | 50 | 9600 | 1.60 | E13K20A15 |
| E22K10A10 | 50 | 9700 | 1.57 | E16K17A13 |
| E26K9A9 | 47 | 9400 | 1.45 | E21K19A13 |
| E34K4A4 | 45 | 9500 | 1.61 | E28K9A7 |
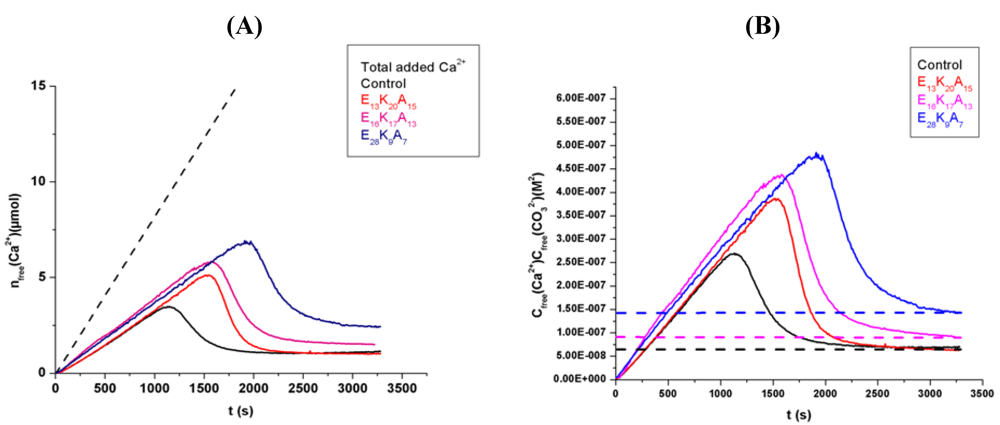
3.1. Random Poly(Amino Acid)s in CaCO3 Mineralization
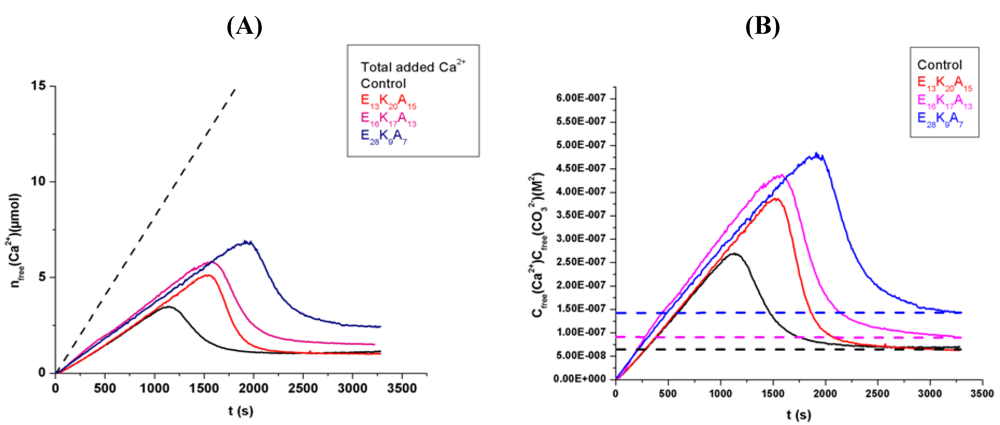
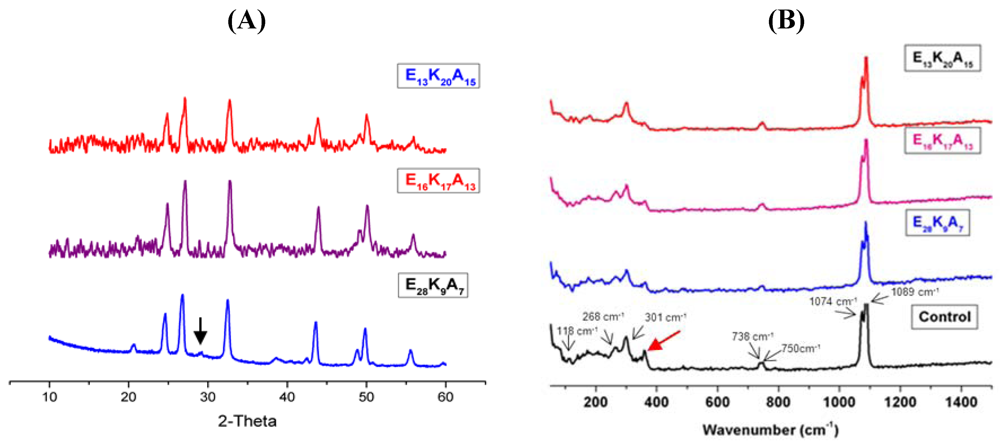

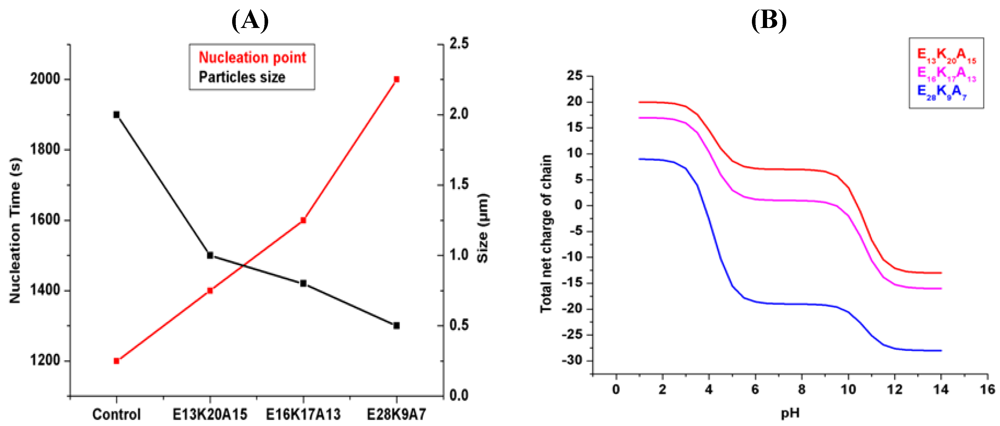
4. Conclusions
Acknowledgements
References
- Addadi, L.; Joester, D.; Nudelman, F.; Weiner, S. Mollusk shell formation: A source of new concepts for understanding biomineralization processes. Chem. Eur. J. 2006, 12, 981–987. [Google Scholar]
- Addadi, L.; Weiner, S. Control and design principles in biological mineralization. Angew. Chem. Int. Edit. Engl. 1992, 31, 153–169. [Google Scholar]
- Bauerlein, E. Progress in biology, molecular biology, and application. In Biomineralization, 2nd; Bauerlein, E., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Mann, S. Biomineralization: Principles and Concepts in Bioinorganic Materials Chemistry; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Delak, K.; Giocondi, J.; Orme, C.; Evans, J.S. Modulation of crystal growth by the terminal sequences of the prismatic-associated asprich protein. Cryst. Growth Des. 2008, 8, 4481–4486. [Google Scholar]
- Gotliv, B.A.; Addadi, L.; Weiner, S. Mollusk shell acidic proteins: In search of individual functions. Chem. Biochem. 2003, 4, 522–529. [Google Scholar]
- Gotliv, B.A.; Kessler, N.; Sumerel, J.L.; Morse, D.E.; Tuross, N.; Addadi, L.; Weiner, S. Asprich: A novel aspartic acid-rich protein family from the prismatic shell matrix of the bivalve atrina rigida. Chem. Biochem. 2005, 6, 304–314. [Google Scholar]
- Politi, Y.; Mahamid, J.; Goldberg, H.; Weiner, S.; Addadi, L. Asprich mollusk shell protein: In vitro experiments aimed at elucidating function in CaCO3 crystallization. Cryst. Eng. Comm. 2007, 9, 1171–1177. [Google Scholar]
- Belcher, A.M.; Wu, X.H.; Christensen, R.J.; Hansma, P.K.; Stucky, G.D.; Morse, D.E. Control of crystal phase switching and orientation by soluble mollusc-shell proteins. Nature 1996, 381, 56–58. [Google Scholar]
- Falini, G.; Albeck, S.; Weiner, S.; Addadi, L. Control of aragonite or calcite polymorphism by mollusk shell macromolecules. Science 1996, 271, 67–69. [Google Scholar]
- Metzler, R.A.; Tribello, G.A.; Parrinello, M.; Gilbert, P.U.P.A. Asprich peptides are occluded in calcite and permanently disorder biomineral crystals. J. Am. Chem. Soc. 2010, 132, 11585–11591. [Google Scholar]
- Metzler, R.A.; Evans, J.S.; Killian, C.E.; Zhou, D.; Churchill, T.H.; Appathurai, N.P.; Coppersmith, S.N.; Gilbert, P.U.P.A. Nacre protein fragment templates lamellar aragonite growth. J. Am. Chem. Soc. 2010, 132, 6329–6334. [Google Scholar]
- Berman, A.; Addadi, L.; Kvick, A.; Leiserowitz, L.; Nelson, M.; Weiner, S. Intercalation of sea-urchin proteins in calcite-study of a crystalline composite-material. Science 1990, 250, 664–667. [Google Scholar]
- Berman, A.; Addadi, L.; Weiner, S. Interactions of sea-urchin skeleton macromolecules with growing calcite crystals—A study of intracrystalline proteins. Nature 1988, 331, 546–548. [Google Scholar]
- Colfen, H. Bio-inspired mineralization using hydrophilic polymers. In Biomineralization II: Mineralization Using Synthetic Polymers and Templates; Springer-Verlag Berlin: Berlin, Germany, 2007; Volume 271, pp. 1–77. [Google Scholar]
- Imai, H. Self-organized formation of hierarchical structures. In Biomineralization I: Crystallization and Self-organization Process; Springer: Berlin, Germay, 2007; Volume 270, pp. 43–72. [Google Scholar]
- Song, R.Q.; Coelfen, H.; Xu, A.W.; Hartmann, J.; Antonietti, M. Polyelectrolyte-directed nanoparticle aggregation: Systematic morphogenesis of calcium carbonate by nonclassical crystallization. ACS Nano 2009, 3, 1966–1978. [Google Scholar]
- Ye, G.; Nam, N.H.; Kumar, A.; Saleh, A.; Shenoy, D.B.; Amiji, M.M.; Lin, X.; Sun, G.; Parang, K. Synthesis and evaluation of tripodal peptide analogues for cellular delivery of phosphopeptides. J. Med. Chem. 2007, 50, 3604–3617. [Google Scholar]
- Walton, A.G.; Blackwell, J. Biopolymers; Academic press: New York, NY, USA, 1973. [Google Scholar]
- Deber, C.M. Peptides: Structure and Function; Pierce Chem. Co: Rockford, IL, USA, 1985. [Google Scholar]
- Stahmann, M.A. Polyamino Acids, Poly Peptides and Proteins; University of Wisconsin Press: Wisconsin, WI, USA, 1962. [Google Scholar]
- Block, H. Poly(g-benzyl-l-glutamate) and Other Glutamicacid Containing Polymers; Gordon and Breach: London, UK, 1983. [Google Scholar]
- Kricheldorf, H.R. A-amino Acid-N-Carboxyanhydrides and Related Heterocycles; Springer Verlag: New York, NY, USA, 1987. [Google Scholar]
- Kricheldorf, H.R. Polypeptides and 100 years of chemistry of alpha-amino acid n-carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef]
- Adams, D.J.; Young, I. Oligopeptide-based amide functional initiators for atrp. J. Polym. Sci. A Polym. Chem. 2008, 46, 6082–6090. [Google Scholar]
- Aliferis, T.; Iatrou, H.; Hadjichristidis, N. Living polypeptides. Biomacromolecules 2004, 5, 1653–1656. [Google Scholar]
- Deming, T.J. Facile synthesis of block copolypeptides of defined architecture. Nature 1997, 390, 386–389. [Google Scholar] [CrossRef]
- Deming, T.J. Amino acid derived nickelacycles: Intermediates in nickel-mediated polypeptide synthesis. J. Am. Chem. Soc. 1998, 120, 4240–4241. [Google Scholar]
- Deming, T.J. Cobalt and iron initiators for the controlled polymerization of alpha-amino acid-N-carboxyanhydrides. Macromolecules 1999, 32, 4500–4502. [Google Scholar]
- Dimitrov, I.; Schlaad, H. Synthesis of nearly monodisperse polystyrene-polypeptide block copolymers via polymerisation of N-carboxyanhydrides. Chem. Commun. 2003, 23, 2944–2945. [Google Scholar]
- Kricheldorf, H.R.; Hauser, K. Polylactones. 55. A-B-A triblock copolymers of various polypeptides. Syntheses involving 4-aminobenzoyl-terminated poly(epsilon-caprolactone) as B block. Biomacromolecules 2001, 2, 1110–1115. [Google Scholar] [CrossRef]
- Yang, J.X.; Zhao, K.; Gong, Y.X.; Vologodskii, A.; Kallenbach, N.R. Alpha-helix nucleation constant in copolypeptides of alanine and ornithine or lysine. J. Am. Chem. Soc. 1998, 120, 10646–10652. [Google Scholar]
- Lu, H.; Cheng, J. Hexamethyldisilazane-mediated controlled polymerization of alfa-amino acid N-carboxyanhydrides. J. Am. Chem. Soc. 2008, 129, 14114–14115. [Google Scholar]
- Huang, J.; Habraken, G.; Audouin, F.; Heise, A. Hydrolytically stable bioactive synthetic glycopeptide homo- and copolymers by combination of NCA polymerization and click reaction. Macromolecules 2010, 43, 6050–6057. [Google Scholar]
- Huang, J.; Bonduelle, C.; Thévenot, J.; Lecommandoux, S.; Heise, A. Biologically active polymersomes from amphiphilic glycopeptides. J. Am. Chem. Soc. 2012, 134, 119–122. [Google Scholar]
- Vayaboury, W.; Giani, O.; Cottet, H.; Deratani, A.; Schue, F. Living polymerization of alpha-amino acid N-carboxyanhydrides (NCA) upon decreasing the reaction temperature. Macromol. Rapid Commun. 2004, 25, 1221–1224. [Google Scholar]
- Deng, Z.; Habraken, G.J.M.; Peeters, M.; Heise, A.; de With, G.; Sommerdijk, N.A.J.M. Fluorescein functionalized random amino acid copolymers in the biomimetic synthesis of CaCO3. Soft Matter 2011, 7, 9685–9694. [Google Scholar]
- Davies, C.W. Ion Association; Butterworths: London, UK, 1962. [Google Scholar]
- Corneille, F.; Copier, J.L.; Senet, J.P.; Robin, Y. Procédé de Préparation des N-carboxyanhydrides. Eur. Pat. Appl. 1201659, 2 May 2002. [Google Scholar]
- Gibson, M.I.; Cameron, N.R. Experimentally facile controlled polymerization of N-carboxyan hydrides (NCAS), including O-benzyl-L-threonine NCA. J. Polym. Sci. A Polym. Chem. 2009, 47, 2882–2891. [Google Scholar]
- Habraken, G.J.M.; Peeters, M.; Dietz, C.H.J.T.; Koning, C.E.; Heise, A. How controlled and versatile is N-carboxy anhydride (NCA) polymerization at 0 °C? Effect of temperature on homo-, block- and graft (co)polymerization. Polym. Chem. 2010, 1, 514–524. [Google Scholar]
- Habraken, G.J.M.; Wilsens, K.H.R.M.; Koning, C.E.; Heise, A. Optimization of N-carboxyanhydride (NCA) polymerization by variation of reaction temperature and pressure. Polym. Chem. 2011, 2, 1322–1330. [Google Scholar]
- Gebauer, D.; Voelkel, A.; Coelfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar]
- Dandeu, A.; Humbert, B.; Carteret, C.; Muhr, H.; Plasari, E.; Bossoutrot, J.M. Raman spectroscopy—A powerful tool for the quantitative determination of the composition of polymorph mixtures: Application to CaCO3 polymorph mixtures. Chem. Eng. Technol. 2006, 29, 221–225. [Google Scholar]
- Lin, Y.P.; Singer, P.C. Inhibition of calcite crystal growth by polyphosphates. Water Res. 2005, 39, 4835–4843. [Google Scholar]
- Sommerdijk, N.A.J.M.; de With, G. Biomimetic CaCO3 mineralization using designer molecules and interfaces. Chem. Rev. 2008, 108, 4499–4550. [Google Scholar]
- Malkaj, P.; Dalas, E. Calcium carbonate crystallization in the presence of aspartic acid. Cryst. Growth Des. 2004, 4, 721–723. [Google Scholar] [CrossRef]
- Manoli, F.; Dalas, E. Calcium carbonate crystallization in the presence of glutamic acid. J. Cryst. Growth 2001, 222, 293–297. [Google Scholar]
- Cantaert, B.; Kim, Y.Y.; Ludwig, H.; Nudelman, F.; Sommerdijk, N.A.J.M.; Meldrum, F.C. Think positive: Phase separation enables a positively charged additive to induce dramatic changes in calcium carbonate morphology. Adv. Funct. Mater. 2012, 22, 907–915. [Google Scholar]
- Evans, J.S. Tuning in to mollusk shell nacre- and prismatic-associated protein terminal sequences. Implications for biomineralization and the construction of high performance inorganic-organic composites. Chem. Rev. 2008, 108, 4455–4462. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dmitrovic, V.; Habraken, G.J.M.; Hendrix, M.M.R.M.; Habraken, W.J.E.M.; Heise, A.; De With, G.; Sommerdijk, N.A.J.M. Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3. Polymers 2012, 4, 1195-1210. https://doi.org/10.3390/polym4021195
Dmitrovic V, Habraken GJM, Hendrix MMRM, Habraken WJEM, Heise A, De With G, Sommerdijk NAJM. Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3. Polymers. 2012; 4(2):1195-1210. https://doi.org/10.3390/polym4021195
Chicago/Turabian StyleDmitrovic, Vladimir, Gijs J.M. Habraken, Marco M.R.M. Hendrix, Wouter J.E.M. Habraken, Andreas Heise, Gijsbertus De With, and Nico A.J.M Sommerdijk. 2012. "Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3" Polymers 4, no. 2: 1195-1210. https://doi.org/10.3390/polym4021195
APA StyleDmitrovic, V., Habraken, G. J. M., Hendrix, M. M. R. M., Habraken, W. J. E. M., Heise, A., De With, G., & Sommerdijk, N. A. J. M. (2012). Random Poly(Amino Acid)s Synthesized by Ring Opening Polymerization as Additives in the Biomimetic Mineralization of CaCO3. Polymers, 4(2), 1195-1210. https://doi.org/10.3390/polym4021195