Multiscale Modeling for Host-Guest Chemistry of Dendrimers in Solution

Abstract
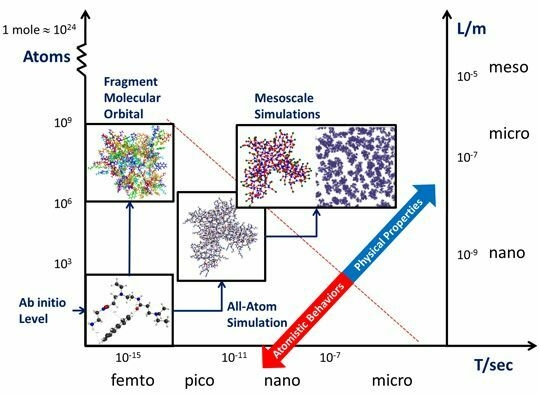
:
{kind=link}
{kind=link}
{kind=link}
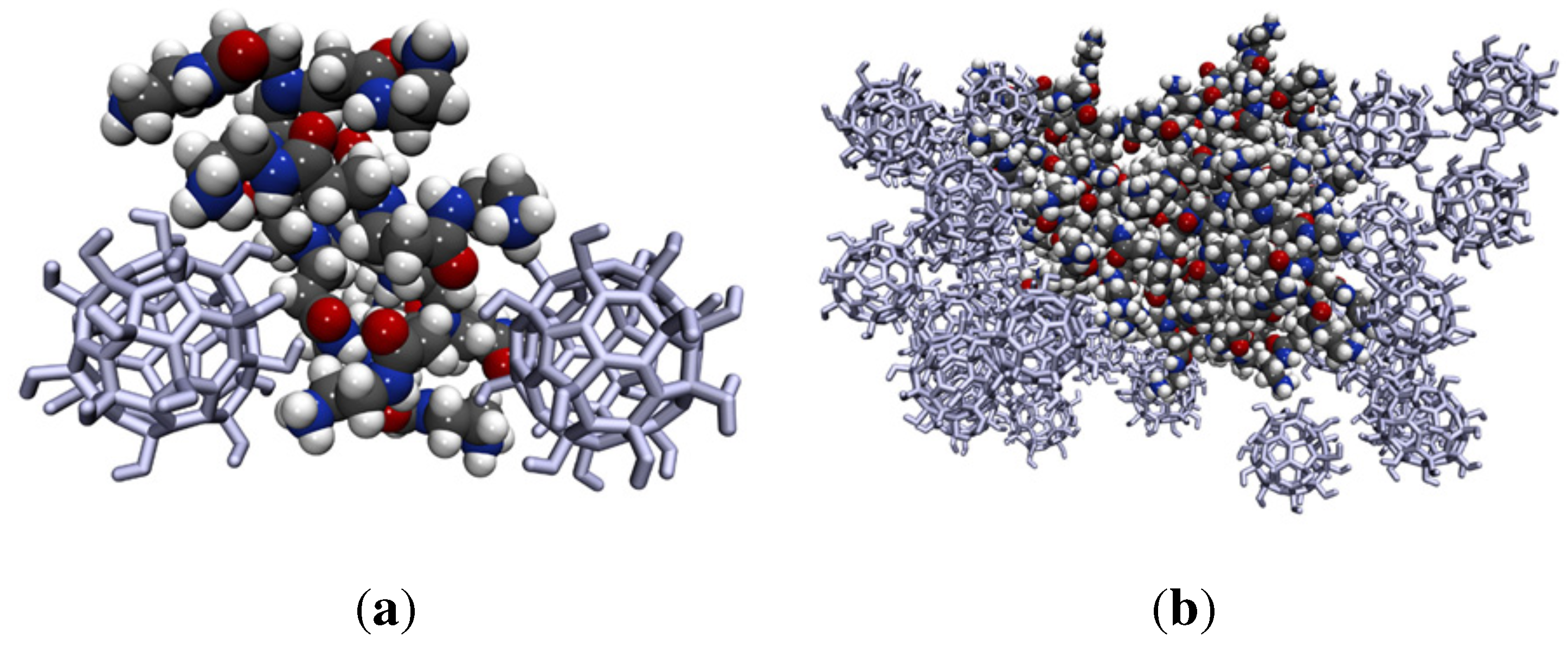{kind=link}
{kind=link}
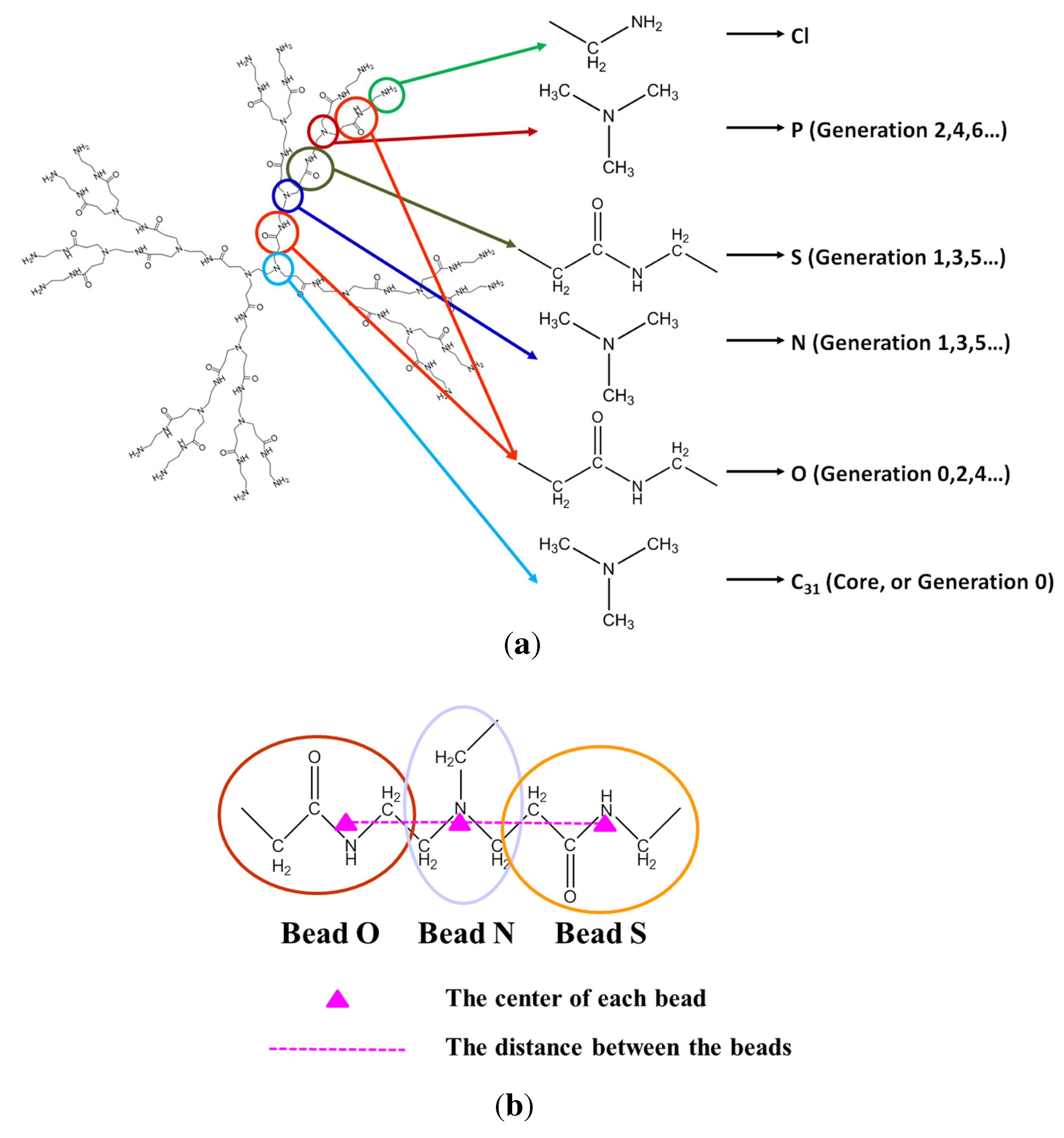{kind=link}
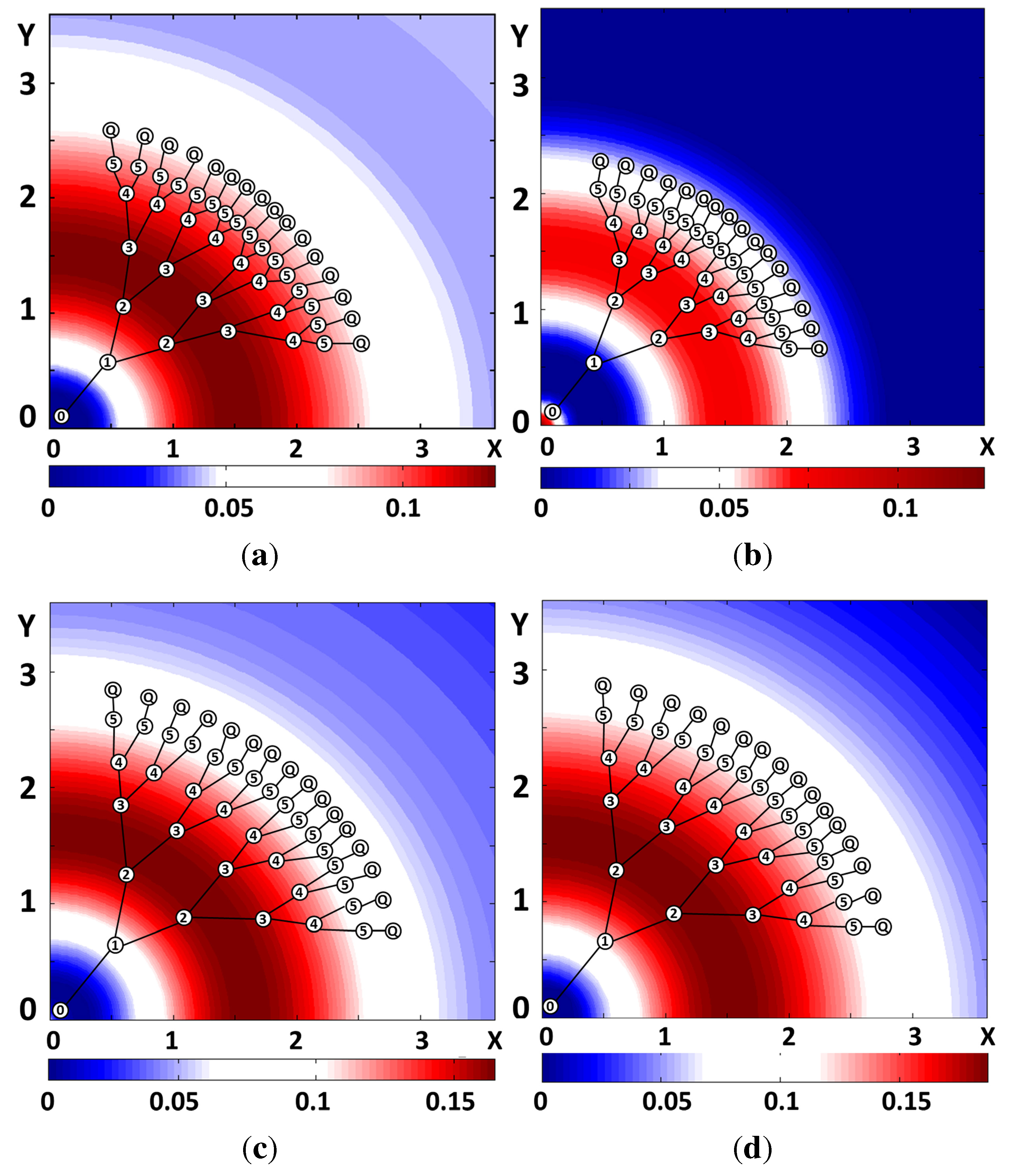{kind=link}
{kind=link}
1. Introduction
2. All-Atom Molecular Dynamics Simulations
2.1. All-Atom Force Fields for Modeling Dendrimers
2.2. The Physical Properties of Dendrimers in Solution
2.3. The Physical Properties of Complex Systems of Dendrimers with Guest Molecules in Solution
3. Coarse-Grained Simulations
3.1. The Physical Properties of Dendrimers in Solution
3.2. The Physical Properties of Complex Systems of Dendrimers with Guest Molecules in Solution
4. Statistical Field Theories
5. Conclusions
Acknowledgements
References
- Evans, H.M.; Ahmad, A.; Ewert, K.; Pfohl, T.; Martin-Herranz, A.; Bruinsma, R.F.; Safinya, C.R. Structural polymorphism of DNA-dendrimer complexes. Phys. Rev. Lett. 2003, 91, 075501:1–075501:4. [Google Scholar] [CrossRef] [PubMed]
- Boas, U.; Christensen, J.B.; Heegaard, P.M.H. Dendrimers in Medicine and Biotechnology: New Molecular Tools; Royal Society of Chemistry: Cambridge, UK, 2006. [Google Scholar]
- Licata, N.A.; Tkachenko, A.V. Kinetic limitations of cooperativity-based drug delivery systems. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef] [PubMed]
- Menjoge, A.R.; Kannan, R.M.; Tomalia, D.A. Dendrimer-based drug and imaging conjugates: Design considerations for nanomedical applications. Drug Discov. Today 2010, 15, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Navath, R.S.; Menjoge, A.R.; Wang, B.; Romero, R.; Kannan, S.; Kannan, R.M. Amino acid-functionalized dendrimers with heterobifunctional chemoselective peripheral groups for drug delivery applications. Biomacromolecules 2010, 11, 1544–1563. [Google Scholar] [CrossRef] [PubMed]
- Turrin, C.; Caminade, A. Dendrimers for imaging. In Dendrimers: Towards Catalytic, Material and Biomedical Uses; Caminade, A., Turrin, C., Laurent, R., Ouali, A., Delavaux-Nicot, B., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 393–412. [Google Scholar]
- Olson, E.S.; Jiang, T.; Aguilera, T.A.; Nguyen, Q.T.; Ellies, L.G.; Scadeng, M.; Tsien, R.Y. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and mr imaging of proteases. Proc. Natl. Acad. Sci. USA 2010, 107, 4311–4316. [Google Scholar] [CrossRef] [PubMed]
- Lang, H.; May, R.A.; Iversen, B.L.; Chandler, B.D. Dendrimer-encapsulated nanoparticle precursors to supported platinum catalysts. J. Am. Chem. Soc. 2003, 125, 14832–14836. [Google Scholar] [CrossRef]
- Yancey, D.F.; Carino, E.V.; Crooks, R.M. Electrochemical synthesis and electrocatalytic properties of Au@Pt dendrimer-encapsulated nanoparticles. J. Am. Chem. Soc. 2010, 132, 10988–10989. [Google Scholar] [CrossRef]
- Albiter, M.A.; Morales, R.; Zaera, F. Dendrimer-based synthesis of Pt catalysts for hydrocarbon conversion. Appl. Catal. A Gen. 2011, 391, 386–393. [Google Scholar] [CrossRef]
- Diallo, M.S.; Christie, S.; Swaminathan, P.; Balogh, L.; Shi, X.; Um, W.; Papelis, C.; Goddard, W.A., III.; Johnson, J.H. Dendritic chelating agents. 1. Cu(II) binding to ethylene diamine core poly(amidoamine) dendrimers in aqueous solutions. Langmuir 2004, 20, 2640–2651. [Google Scholar] [CrossRef]
- Diallo, M.S.; Christie, S.; Swaminathan, P.; Johnson, J.H.; Goddard, W.A., III. Dendrimer enhanced ultrafiltration. 1. Recovery of Cu(II) from aqueous solutions using pamam dendrimers with ethylene diamine core and terminal NH2 groups. Environ. Sci. Technol. 2005, 39, 1366–1377. [Google Scholar] [CrossRef]
- Diallo, M.S.; Falconer, K.; Johnson, J.H. Dendritic anion hosts: Perchlorate uptake by G5-NH2 poly(propyleneimine) dendrimer in water and model electrolyte solutions. Environ. Sci. Technol. 2007, 41, 6521–6527. [Google Scholar] [CrossRef] [PubMed]
- Diallo, M.S.; Arasho, W.; Johnson, J.H.; Goddard, W.A., III. Dendritic chelating agents. 2. U(VI) binding to poly(amidoamine) and poly(propyleneimine) dendrimers in aqueous solutions. Environ. Sci. Technol. 2008, 42, 1572–1579. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A. Dendrons/dendrimers: Quantized, nano-element like building blocks for soft-soft and soft-hard nano-compound synthesis. Soft Matter 2010, 6, 456–474. [Google Scholar] [CrossRef]
- Newkome, G.R.; Moorefield, C.N.; Vogtle, F. Dendritic Molecules: Concepts, Syntheses, Perspectives; John Wiley & Sons: Hoboken, NJ, USA, 1996. [Google Scholar]
- Ke, W.; Shao, K.; Huang, R.; Han, L.; Liu, Y.; Li, J.; Kuang, Y.; Ye, L.; Lou, J.; Jiang, C. Gene delivery targeted to the brain using an angiopep-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. Biomaterials 2009, 30, 6976–6985. [Google Scholar] [CrossRef]
- Peng, S.F.; Su, C.J.; Wei, M.C.; Chen, C.Y.; Liao, Z.X.; Lee, P.W.; Chen, H.L.; Sung, H.W. Effects of the nanostructure of dendrimer/DNA complexes on their endocytosis and gene expression. Biomaterials 2010, 31, 5660–5670. [Google Scholar] [CrossRef]
- Shcharbin, D.; Dzmitruk, V.; Shakhbazau, A.; Goncharova, N.; Seviaryn, I.; Kosmacheva, S.; Potapnev, M.; Pedziwiatr-Werbicka, E.; Bryszewska, M.; Talabaev, M.; et al. Fourth generation phosphorus-containing dendrimers: Prospective drug and gene delivery carrier. Pharmaceutics 2011, 3, 458–473. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Olson, E.S.; Aguilera, T.A.; Jiang, T.; Scadeng, M.; Ellies, L.G.; Tsien, R.Y. Surgery with molecular fluorescence imaging using activatable cell-penetrating peptides decreases residual cancer and improves survival. Proc. Natl. Acad. Sci. USA 2010, 107, 4317–4322. [Google Scholar] [CrossRef]
- Han, L.; Li, J.; Huang, S.; Huang, R.; Liu, S.; Hu, X.; Yi, P.; Shan, D.; Wang, X.; Lei, H.; et al. Peptide-conjugated polyamidoamine dendrimer as a nanoscale tumor-targeted T1 magnetic resonance imaging contrast agent. Biomaterials 2011, 32, 2989–2998. [Google Scholar] [CrossRef]
- Jasmine, M.J.; Prasad, E. Fractal growth of PAMAM dendrimer aggregates and its impact on the intrinsic emission properties. J. Phys. Chem. B 2010, 114, 7735–7742. [Google Scholar] [CrossRef]
- Arkas, M.; Tsiourvas, D.; Paleos, C.M. Functional dendrimeric “nanosponges" for the removal of polycyclic aromatic hydrocarbons from water. Chem. Mater. 2003, 15, 2844–2847. [Google Scholar] [CrossRef]
- Lard, M.; Kim, S.H.; Lin, S.; Bhattacharya, P.; Ke, P.C.; Lamm, M.H. Fluorescence resonance energy transfer between phenanthrene and PAMAM dendrimers. Phys. Chem. Chem. Phys. 2010, 12, 9285–9291. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Boisselier, E.; Ornelas, C. Dendrimers designed for functions: From physical, photophysical, and supramolecular properties to applications in sensing, catalysis, molecular electronics, photonics, and nanomedicine. Chem. Rev. 2010, 110, 1857–1959. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.V.; Liroff, M.G.; Triplett, L.D.; Leroueil, P.R.; Mullen, D.G.; Wallace, J.M.; Meshinchi, S.; Baker, J.R.; Orr, B.G.; Holl, M.M.B. Stoichiometry and structure of poly(amidoamine) dendrimer-lipid complexes. ACS Nano 2009, 3, 1886–1896. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Cheng, Y.; Wu, Q.; Wen, L.; Xu, T. Evaluation of phenylbutazone and poly(amidoamine) dendrimers interactions by a combination of solubility, 2D-NOESY NMR, and isothermal titration calorimetry studies. J. Pharm. Sci. 2009, 98, 1075–1085. [Google Scholar] [CrossRef]
- Jensen, L.B.; Mortensen, K.; Pavan, G.M.; Kasimova, M.R.; Jensen, D.K.; Gadzhyeva, V.; Nielsen, H.M.; Foged, C. Molecular characterization of the interaction between siRNA and PAMAM G7 dendrimers by SAXS, ITC, and Molecular dynamics simulations. Biomacromolecules 2010, 11, 3571–3577. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, P.; Kim, S.H.; Chen, P.; Chen, R.; Spuches, A.M.; Brown, J.M.; Lamm, M.H.; Ke, P.C. Complexation of PAMAM dendrimer and fullerenol for drug delivery and environmental remediation. unpublished.
- Shi, X.; Bi, X.; Ganser, T.R.; Hong, S.; Myc, L.A.; Desai, A.; Holl, M.M.B.; Baker, J.R. HPLC analysis of functionalized poly(amidoamine) dendrimers and the interaction between a folate-dendrimer conjugate and folate binding protein. Analyst 2006, 131, 842–848. [Google Scholar] [CrossRef]
- Orberg, M.; Schillen, K.; Nylander, T. Dynamic light scattering and fluorescence study of the interaction between double-stranded DNA and poly(amido amine) dendrimers. Biomacromolecules 2007, 8, 1557–1563. [Google Scholar] [CrossRef]
- Froehlich, E.; Mandeville, J.S.; Jennings, C.J.; Sedaghat-Herati, R.; Tajmir-Riahi, H.A. Dendrimers bind human serum albumin. J. Phys. Chem. B 2009, 113, 6986–6993. [Google Scholar] [CrossRef]
- Mandeville, J.S.; Tajmir-Riahi, H.A. Complexes of dendrimers with bovine serum albumin. Biomacromolecules 2010, 11, 465–472. [Google Scholar] [CrossRef]
- Hu, J.; Cheng, Y.; Ma, Y.; Wu, Q.; Xu, T. Host-guest chemistry and physicochemical properties of the dendrimer-mycophenolic acid complex. J. Phys. Chem. B 2009, 113, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Cheng, Y.; Wu, Q.; Zhao, L.; Xu, T. Host-guest chemistry of dendrimer-drug complexes. 2. Effects of molecular properties of guests and surface functionalities of dendrimers. J. Phys. Chem. B 2009, 113, 10650–10659. [Google Scholar] [CrossRef] [PubMed]
- Montanez, M.I.; Najera, F.; Perez-Inestrosa, E. NMR studies and molecular dynamic simulation of synthetic dendritic antigens. Polymers 2011, 3, 1533–1553. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R. Multiscale modeling of dendrimers and their interactions with bilayers and polyelectrolytes. Molecules 2009, 14, 423–438. [Google Scholar] [CrossRef]
- Nandy, B.; Maiti, P.K. DNA compaction by a dendrimer. J. Phys. Chem. B 2011, 115, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Jayamurugan, G.; Vasu, K.S.; Rajesh, Y.B.R.D.; Kumar, S.; Vasumathi, V.; Maiti, P.K.; Sood, A.K.; Jayaraman, N. Interaction of single-walled carbon nanotubes with poly(propyl ether imine) dendrimers. J. Chem. Phys. 2011, 134, 104507:1–104507:6. [Google Scholar] [CrossRef] [PubMed]
- Voth, G.A. Coarse-Graining of Condensed Phase and Biomolecular Systems, 1st ed.; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar]
- Lin, C.; Wu, K.; Sa, R.; Mang, C.; Liu, P.; Zhuang, B. Density functional theory studies on the potential energy surface and hyperpolarizability of polyamidoamide dendrimer. Chem. Phys. Lett. 2002, 363, 343–348. [Google Scholar] [CrossRef]
- Tarazona-Vasquez, F.; Balbuena, P.B. Complexation of the lowest generation poly(amidoamine)-NH2 dendrimers with metal ions, metal atoms, and Cu(II) hydrates: An ab initio study. J. Phys. Chem. B 2004, 108, 15992–16001. [Google Scholar] [CrossRef]
- Wan, H.; Li, S.; Konovalova, T.A.; Shuler, S.F.; Dixon, D.A.; Street, S.C. Experimental and theoretical studies of the photoreduction of copper(II)-dendrimer complexes. J. Phys. Chem. C 2008, 112, 1335–1344. [Google Scholar] [CrossRef]
- Kitaura, K.; Ikeo, E.; Asada, T.; Nakano, T.; Uebayasi, M. Fragment molecular orbital method: An approximate computational method for large molecules. Chem. Phys. Lett. 1999, 313, 701–706. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Extending the power of quantum chemistry to large systems with the fragment molecular orbital method. J. Phys. Chem. A 2007, 111, 6904–6914. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, D.G.; Kitaura, K. Pair interaction energy decomposition analysis. J. Comput. Chem. 2007, 28, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, D.G.; Kitaura, K.; Li, H.; Jensen, J.H.; Gordon, M.S. The polarizable continuum model (PCM) interfaced with the fragment molecular orbital method (FMO). J. Comput. Chem. 2006, 27, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.K.; Messina, R. Counterion distribution and ζ-potential in PAMAM dendrimer. Macromolecules 2008, 41, 5002–5006. [Google Scholar] [CrossRef]
- Kim, S.H.; Lamm, M.H. Reintroducing explicit solvent to a solvent-free coarse-grained model. Phys. Rev. E 2011, 84, 025701:1–025701:4. [Google Scholar] [CrossRef]
- Tanis, I.; Karatasos, K. Association of a weakly acidic anti-inflammatory drug (ibuprofen) with a poly(amidoamine) dendrimer as studied by molecular dynamics simulations. J. Phys. Chem. B 2009, 113, 10984–10993. [Google Scholar] [CrossRef]
- Shi, X.; Lee, I.; Chen, X.; Shen, M.; Xiao, S.; Zhu, M.; Baker, J.R.; Wang, S.H. Influence of dendrimer surface charge on the bioactivity of 2-methoxyestradiol complexed with dendrimers. Soft Matter 2010, 6, 2539–2545. [Google Scholar] [CrossRef]
- Maiti, P.K.; Cagin, T.; Lin, S.; Goddard, W.A., III. Effect of solvent and pH on the structure of PAMAM dendrimers. Macromolecules 2005, 38, 979–991. [Google Scholar] [CrossRef]
- Han, M.; Chen, P.; Yang, X. Molecular dynamics simulation of PAMAM dendrimer in aqueous solution. Polymer 2005, 46, 3481–3488. [Google Scholar] [CrossRef]
- Lee, H.; Baker, J.R.; Larson, R.G. Molecular dynamics studies of the size, shape, and internal structure of 0% and 90% acetylated fifth-generation polyamidoamine dendrimers in water and methanol. J. Phys. Chem. B 2006, 110, 4014–4019. [Google Scholar] [CrossRef]
- Giri, J.; Diallo, M.S.; Goddard, W.A., III.; Dalleska, N.F.; Fang, X.; Tang, Y. Partitioning of poly(amidoamine) dendrimers between n-octanol and water. Environ. Sci. Technol. 2009, 43, 5123–5129. [Google Scholar] [CrossRef] [PubMed]
- Weiner, P.K.; Kollman, P.A. AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem. 1981, 2, 287–303. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Pissurlenkar, R.R.; Shaikh, M.S.; Iyer, R.P.; Coutinho, E.C. Molecular mechanics force fields and their applications in drug design. Anti-Infect. Agents Med. Chem. 2009, 8, 128–150. [Google Scholar] [CrossRef]
- Dauber-Osguthorpe, P.; Roberts, V.A.; Osguthorpe, D.J.; Wolff, J.; Genest, M.; Hagler, A.T. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins Struct. Funct. Bioinform. 1988, 4, 31–47. [Google Scholar] [CrossRef]
- Mayo, S.L.; Olafson, B.D.; Goddard, W.A., III. DREIDING: A generic force field for molecular simulations. J. Phys. Chem. 1990, 94, 8897–8909. [Google Scholar] [CrossRef]
- Maiti, P.K.; Goddard, W.A., III. Solvent quality changes the structure of G8 pamam dendrimer, a disagreement with some experimental interpretations. J. Phys. Chem. B 2006, 110, 25628–25632. [Google Scholar] [CrossRef]
- Liu, Y.; Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A., III. Pamam dendrimers undergo pH responsive conformational changes without swelling. J. Am. Chem. Soc. 2009, 131, 2798–2799. [Google Scholar] [CrossRef]
- Welch, P.; Muthukumar, M. Dendrimer-polyelectrolyte complexation: A model guest-host system. Macromolecules 2000, 33, 6159–6167. [Google Scholar] [CrossRef]
- Lee, I.; Athey, B.D.; Wetzel, A.W.; Meixner, W.; Baker, J.R. Structural molecular dynamics studies on polyamidoamine dendrimers for a therapeutic application: Effects of pH and generation. Macromolecules 2002, 35, 4510–4520. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Evers, L.J.; van der Schoot, P.; Darinskii, A.A.; Lyulin, A.V.; Michels, M.A.J. Effect of solvent quality and electrostatic interactions on size and structure of dendrimers. Brownian dynamics simulation and mean-field theory. Macromlecules 2004, 37, 3049–3063. [Google Scholar] [CrossRef]
- Maiti, P.K.; Bagchi, B. Diffusion of flexible, charged, nanoscopic molecules in solution: Size and pH dependence for PAMAM dendrimer. J. Chem. Phys. 2009, 131, 214901. [Google Scholar] [CrossRef] [PubMed]
- Pranami, G.; Lamm, M.H.; Vigil, R.D. Molecular dynamics simulation of fractal aggregate diffusion. Phys. Rev. E 2010, 82, 051402:1–051402:10. [Google Scholar] [CrossRef]
- Maiti, P.K.; Bagchi, B. Structure and dynamics of DNA-dendrimer complexation: Role of counterions, water, base pair sequence. Nano Lett. 2006, 6, 2478–2485. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.; Orr, B.; Holl, M.M.B.; Andricioaei, I. Microscopic basis for the mesoscopic extensibility of dendrimer-compacted DNA. Biophys. J 2010, 98, 834–842. [Google Scholar] [CrossRef]
- Ouyang, D.; Zhang, H.; Parekh, H.S.; Smith, S.C. Structure and dynamics of multiple cationic vectors-siRNA complexation by all-atomic molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 9231–9237. [Google Scholar] [CrossRef] [PubMed]
- Vasumathi, V.; Maiti, P.K. Complexation of siRNA with dendrimer: A molecular modeling approach. Macromolecules 2010, 43, 8264–8274. [Google Scholar] [CrossRef]
- Ouyang, D.; Zhang, H.; Parekh, H.S.; Smith, S.C. The effect of pH on PAMAM dendrimer-siRNA complexation - Endosomal considerations as determined by molecular dynamics simulation. Biophys. Chem. 2011, 158, 126–133. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparision of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Darinskii, A.A.; Lyulin, A.V. Computer simulation of complexes of dendrimers with linear polyelectrolytes. Macromlecules 2005, 38, 3990–3998. [Google Scholar] [CrossRef]
- Pavan, G.M.; Albertazzi, L.; Danani, A. Ability to adapt: Different generations of PAMAM dendrimers show different behaviors in binding siRNA. J. Phys. Chem. B 2010, 114, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- Suek, N.W.; Lamm, M.H. Effect of terminal group modification on the solution properties of dendrimers: A molecular dynamics simulation study. Macromolecules 2006, 39, 4247–4255. [Google Scholar] [CrossRef]
- Lyulin, S.V.; Vattulainen, I.; Gurtovenko, A.A. Complexes comprised of charged dendrimers, linear polyelectrolytes, and counterions: Insight through coarse-grained molecular dynamics simulations. Macromolecules 2008, 41, 4961–4968. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Complexation of a linear polyelectrolyte with a charged dendrimer: Polyelectrolyte stiffness effects. Macromolecules 2010, 43, 1575–1582. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Effects of valences of salt ions at various concentrations on charged dendrimers. Soft Matter 2010, 6, 1308–1316. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The martini force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef]
- Izvekov, S.; Voth, G.A. Multiscale coarse graining of liquid-state systems. J. Chem. Phys. 2005, 123, 134105–134113. [Google Scholar] [CrossRef]
- Izvekov, S.; Voth, G.A. Solvent-free lipid bilayer model using multiscale coarse-graining. J. Phys. Chem. B 2009, 113, 4443–4455. [Google Scholar] [CrossRef]
- Lu, L.; Izvekov, S.; Das, A.; Andersen, H.C.; Voth, G.A. Efficient, regularized, and scalable algorithms for multiscale coarse-graining. J. Chem. Theory Comput. 2010, 6, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Larin, S.; Lyulin, S.; Lyulin, A.; Darinskii, A. Charge inversion of dendrimers in complexes with linear polyelectrolytes in the solutions with low pH. Polym. Sci. Ser. A 2009, 51, 459–468. [Google Scholar] [CrossRef]
- Larin, S.V.; Darinskii, A.A.; Lyulin, A.V.; Lyulin, S.V. Linker formation in an overcharged complex of two dendrimers and linear polyelectrolyte. J. Phys. Chem. B 2010, 114, 2910–2919. [Google Scholar] [CrossRef]
- Yan, L.; Yu, X. Charged dendrimers on lipid bilayer membranes: Insight through dissipative particle dynamics simulations. Macromolecules 2009, 42, 6277–6283. [Google Scholar] [CrossRef]
- Maiti, P.K.; Li, Y.; Cagin, T.; Goddard, W.A., III. Structure of polyamidoamide dendrimers up to limiting generations: A mesoscale description. J. Chem. Phys. 2009, 130, 144902. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Larson, R.G. Molecular dynamics simulations of PAMAM dendrimer-induced pore formation in DPPC bilayers with a coarse-grained model. J. Phys. Chem. B 2006, 110, 18204–18211. [Google Scholar] [CrossRef]
- Maiti, P.K.; Cagin, T.; Wang, G.; Goddard, W.A., III. Structure of pamam dendrimers: Generations 1 through 11. Macromolecules 2004, 37, 6236–6254. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Coarse-grained molecular simulation of interacting dendrimers. Soft Matter 2011, 7, 500–505. [Google Scholar] [CrossRef]
- Fredrickson, G.H. The Equilibrium Theory of Inhomogeneous Polymers; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Boris, D.; Rubinstein, M. A self-consistent mean field model of a starburst dendrimer: Dense core vs. dense shell. Macromolecules 1996, 29, 7251–7260. [Google Scholar] [CrossRef]
- Giupponi, G.; Buzza, D.M.A.; Adolf, D.B. Are polyelectrolyte dendrimers stimuli responsive? Macromolecules 2007, 40, 5959–5965. [Google Scholar] [CrossRef]
- Kim, S.H.; Cochran, E.W. Localization of spherical nanoparticles within lamellar AB diblock copolymer melts through self-consistent field theory. Polymer 2011, 52, 2328–2339. [Google Scholar] [CrossRef]
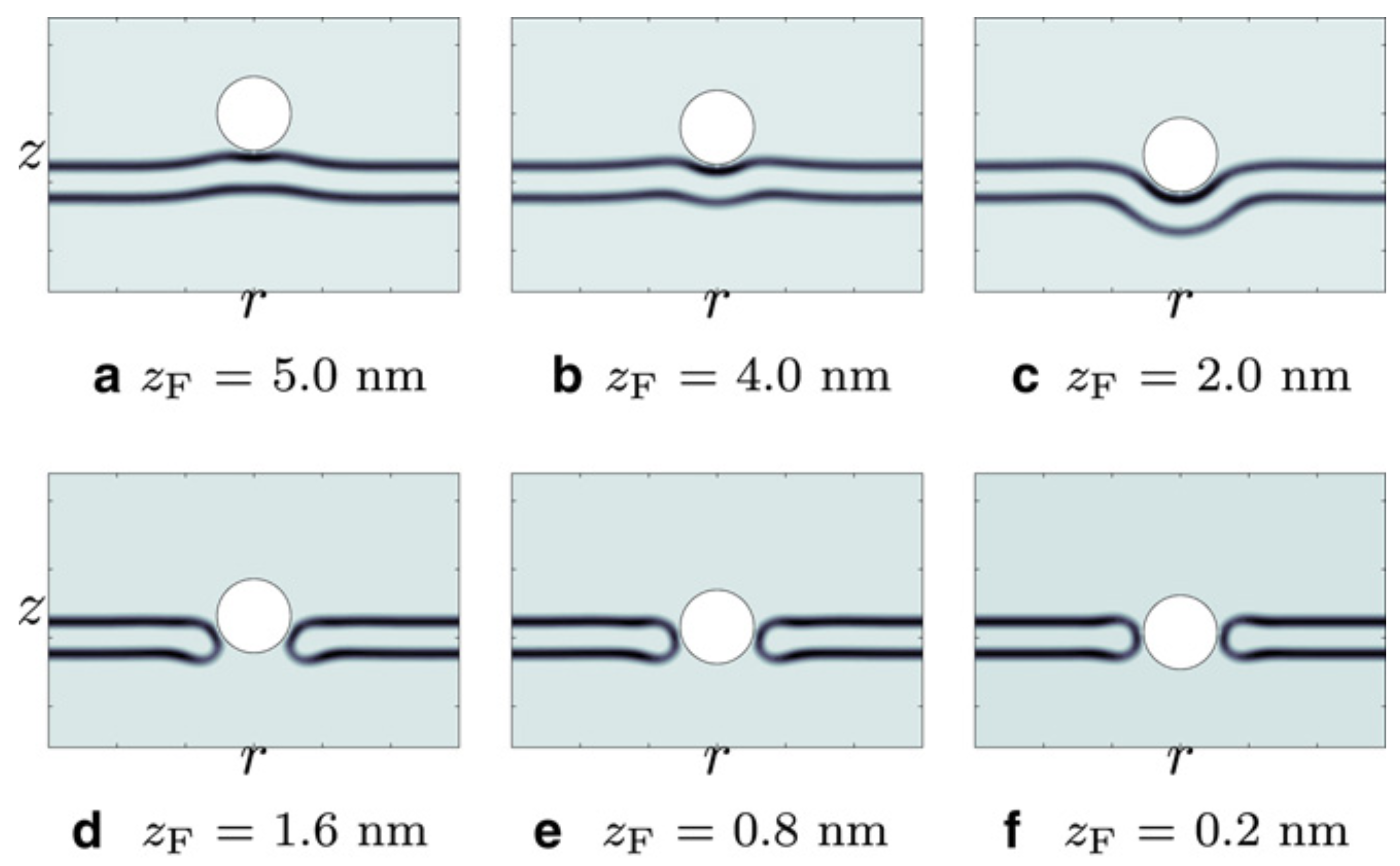
- Ting, C.L.; Wang, Z. Interactions of a charged nanoparticle with a lipid membrane: Implications for gene delivery. Biophys. J. 2011, 100, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Ting, C.L.; Appelo, D.; Wang, Z. Minimum energy path to membrane pore formation and rupture. Phys. Rev. Lett. 2011, 106, 168101:1–168101:4. [Google Scholar] [CrossRef]
- Lee, H.; Larson, R.G. Lipid bilayer curvature and pore formation induced by charged linear polymers and dendrimers: The effect of molecular shape. J. Phys. Chem. B 2008, 112, 12279–12285. [Google Scholar] [CrossRef]
- E, W.; Ren, W.; Vanden-Eijnden, E. String method for the study of rare events. Phys. Rev. B 2002, 66, 052301. [Google Scholar] [CrossRef]







© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, S.H.; Lamm, M.H. Multiscale Modeling for Host-Guest Chemistry of Dendrimers in Solution. Polymers 2012, 4, 463-485. https://doi.org/10.3390/polym4010463
Kim SH, Lamm MH. Multiscale Modeling for Host-Guest Chemistry of Dendrimers in Solution. Polymers. 2012; 4(1):463-485. https://doi.org/10.3390/polym4010463
Chicago/Turabian StyleKim, Seung Ha, and Monica H. Lamm. 2012. "Multiscale Modeling for Host-Guest Chemistry of Dendrimers in Solution" Polymers 4, no. 1: 463-485. https://doi.org/10.3390/polym4010463