Water Soluble Polymers for Pharmaceutical Applications

Abstract
: Advances in polymer science have led to the development of novel drug delivery systems. Some polymers are obtained from natural resources and then chemically modified for various applications, while others are chemically synthesized and used. A large number of natural and synthetic polymers are available. In the present paper, only water soluble polymers are described. They have been explained in two categories (1) synthetic and (2) natural. Drug polymer conjugates, block copolymers, hydrogels and other water soluble drug polymer complexes have also been explained. The general properties and applications of different water soluble polymers in the formulation of different dosage forms, novel delivery systems and biomedical applications will be discussed.1. Introduction
Advances in polymer science have led to the development of novel delivery systems. The introduction of new polymers has resulted in development of polymers with unique properties. Initially polymers were used as solubilisers, stabilizers and mechanical supports for sustained release of drugs. But over a period of time, the functionalities of polymers have changed. The polymers have been synthesized to suit specific needs or rather solve specific problems associated with development of drug delivery systems. So there is need to understand the role of polymers. Polymers can be classified based on any of the following categories: (1) source (Natural, semi synthetic, synthetic); (2) structure of polymer (Linear, Branched chain, Crosslinked or Network polymers); (3) type of polymerization (Addition, condensation polymers); (4) molecular forces (Elastomers, Fibres, Thermoplastic, Thermosetting); (5) Chain growth polymerization (Free radical governed); (6) degradability (biodegradable, non-biodegradable).
Water soluble polymers have a wide range of industrial applications like food, pharmaceuticals, paint, textiles, paper, constructions, adhesives, coatings, water treatment, etc. In this paper, the water soluble polymers have been divided into two categories (1) Synthetic and (2) Natural. This review describes water soluble polymers: their properties and applications in pharmaceutical and biomedical industries.
2. Synthetic Water Soluble Polymers
Synthetic water-soluble polymers are substances that dissolve, disperse or swell in water and, thus, modify the physical properties of aqueous systems in the form of gellation, thickening or emulsification/stabilization. These polymers usually have repeating units or blocks of units; the polymer chains contain hydrophilic groups that are substituents or are incorporated into the backbone. The hydrophilic groups may be nonionic, anionic, cationic or amphoteric [1].
2.1. Poly(ethylene glycol) (PEG)
In general, a low polydispersity index (PDI) is a prerequisite for the polymer to have pharmaceutical applications. A PDI value below 1.1 makes the polymer more homogenous so that it provides reliable residence time in the body [2].
All these prerequisites are fulfilled by PEG, since it has PDI of 1.01. This holds good for low molecular weight PEGs. Polyethylene glycol is synthesized by the interaction of ethylene oxide with water, ethylene glycol, or ethylene glycol oligomers. The starting materials used for synthesis of PEG polymers with low polydispersity index (narrow molecular weight distribution) are Ethylene glycol and its oligomers. Reactions catalyzed by anionic polymerization result in PEGs with low PDI.
In addition, PEG shows a high solubility in organic solvents and, therefore, end-group modifications are relatively easy. PEG is suitable for biological applications because it is soluble in water and has low intrinsic toxicity. The high hydrophilic nature of PEG enhances the solubility of hydrophobic drugs or carriers when conjugated with them. It enhances the physical and chemical stability of drugs and prevents aggregation of the drugs in vivo, as well as during storage, as a result of the steric hindrance and/or masking of charges provided through formation of a conformational cloud [3].
PEG helps in reducing the aggregation of red blood cells and so improves the blood compatibility of PEG copolymers that are implanted as cardiovascular devices such as stents. It is mainly used in storage of blood and organs.
Both temperature-responsive and chemically crosslinked hydrogels have been formed from PEG. Temperature-responsive systems have become increasingly attractive as injectable drug delivery systems [4]. Chemically crosslinked systems have also been studied for in situ photo polymerization [5].
The PEGylation technique was first introduced in the late 1970s. However, the applications of this concept to various carrier systems were widely explored in the 1990s (Figure 1) [6,7].
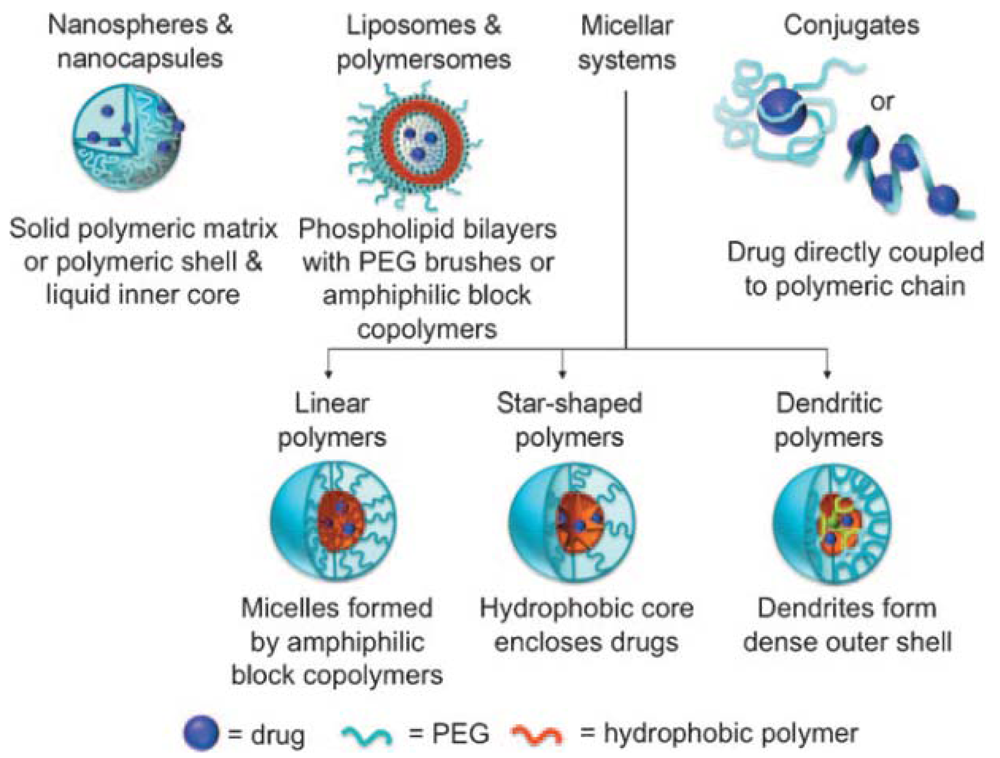
PEG-drug conjugates are being studied for a variety of molecules and drugs including insulin, daunorubicin camptothecin, peptides and lipids. The main advantages of PEG-drug conjugates are reduced protein immunogenicity, increased residence time in the body, reduced enzymatic degradation. All these features ensure that the drug reaches the site of action and prevents clearance from the body because it is not recognized as the foreign body. Therefore, the majority of conjugated drugs as well as liposomal and micellar formulations on the market or in advanced clinical trials are PEG-containing products [8]. Most of the polymer-based stealth drug-delivery systems that have reached the market are PEGylated products (Table 1) [8-11].
The conjugation of PEG with enzymes looks very promising in antitumoral therapy since several enzymes have proven to be active against various types of cancer by acting through different mechanisms. Enzymes that are able to reduce plasma levels of these tumor target amino acids (i.e., asparaginase, methioninase and arginine deiminase) are studied as therapeutic agents in cancer therapy. The advantage of enzymes is their great specificity.
Since the introduction of PEGylation, several antitumour agents, either proteins, peptides or low molecular weight drugs, have been considered for polymer conjugation but only a few entered clinical phase studies. the majority of the low molecular weight PEG drug conjugates which are in clinical phase are from the camptothecin family, namely camptothecin, SN38 and irinotecan [12]. Other PEG protein conjugation studies investigated include PEG-catalase, PEGuricase, PEG-honeybee venom, PEG-hemoglobin and PEG-modified ragweed pollen extract [13]. Following are some of the PEG conjugates with low molecular weight anti-cancer drugs investigated for different clinical applications.
2.1.1. PEG-Irinotecan (NKTR-102)
In a mouse model, the conjugate showed prolonged pharmacokinetic profiles with a half-life of 15 days when compared to 4 h with free irinotecan [14]. Currently the drug conjugate is being tested for its efficacy in breast cancer patients which is in phase 3 stage. Also phase 2 studies in ovarian and cervical cancer patients are in progress. Some of these studies have shown significant antitumor activity (reduction in tumor size) [15].
2.1.2. PEG-Docetaxel (NKTR-105)
NKTR-105 is a novel form of the anti-mitotic agent docetaxel, and was designed using Nektar's advanced polymer conjugate technology. NKTR-105 is in a Phase 1 clinical trial in patients with certain types of solid tumors including hormone-refractory prostate cancer. It is also being explored as a therapy for breast, non-small cell lung, gastric, head and neck cancers [16].
2.1.3. PEG-Camptothecin (PROTHECAN or Pegamotecan)
Pegamotecan (Enzon Pharmaceuticals, Inc.) is a prodrug obtained by coupling two molecules of camptothecin to a diol PEG of 40 kDa. The drug is linked through an ester bond involving the C-20 hydroxyl group and a carboxylic group of PEG. This PEGylation technique helped increasing the half-life of the drug in blood and stabilized camptothecin by acylation [17]. A phase II clinical study done in patients with gastric or gastro-esophageal adenocarcinoma is also documented [18].
2.1.4. PEG-SN38 (EZN-2208)
This is another advanced PEG conjugate which is in phase I clinical trials. This was synthesized by coupling a 4armPEG of 40 kDa with the camptothecin derivative SN38, through a glycine spacer. PEGSN38 showed an increase in drug loading with respect to Pegamotecan, reaching a value of 3.7 wt %. This conjugation resulted in a 1,000 fold increase in the solubility of SN38. The conjugated derrivative acts as a prodrug and showed some antitumour activity when studied using in vitro and in vivo techniques. When this conjugate was administered either as a single dose or multiple injections in mice, it showed better results than irinotecan [19].
Block copolymers containing PEG have also become very popular in recent years. Copolymers with poly(propylene oxide), poly(ethylene butylene), poly(caprolactone) are just a few examples. Copolymers containing PEG have been formed into thermo-sensitive gels, interconnected micelles, nanospheres, and films [20]. While the advantages of PEG in biomedical applications are lengthy, the primary disadvantage for its use in biomedical applications is its non-degradable structure.
Jeong et al. [20] have studied the thermo-reversible gelation of PEG-PLGA-PEG triblock copolymers. They have shown that an aqueous solution of PEG-PLGA-PEG triblock copolymers with a specific composition is a free flowing solution at room temperature but becomes a transparent gel at body temperature. The primary focus of this research was to use this thermo-reversible behavior as a long-term, injectable drug delivery device.
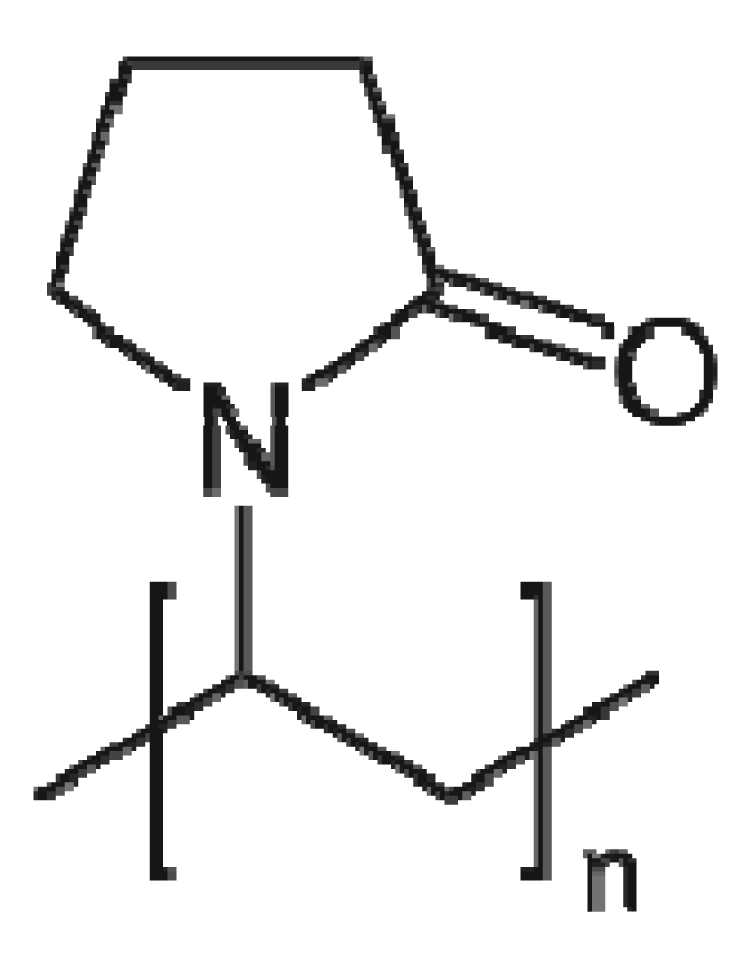
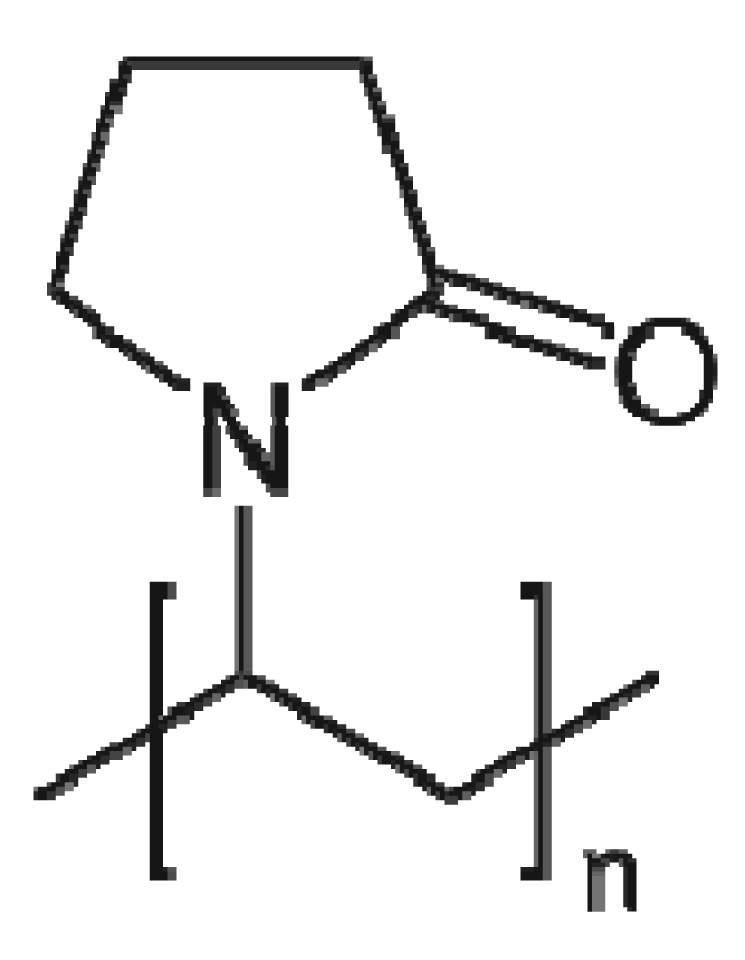
2.2. Polyvinyl pyrrolidone (PVP)
Polyvinyl pyrrolidone is a water soluble polymer having molecular weight ranging from 40,000 to 360,000. It is synthesized by polymerization of vinylpyrrolidone in water or isopropanol. The structure of PVP is given in Figure 2.
PVP is available in different grades based on molecular weights. It is mainly used as a binder in tablet formulations When compared to other binders, wet granulation with PVP having a molecular weight of 25,000 to 90,000 generally gives harder granulates with good flowability, higher binding and low friability [21,22]. In addition to enhancing the above properties, PVP also increases the dissolution of the active ingredient. Acetaminophen (paracetamol) tablets formulated with 4% PVP 90,000 as binder released the drug more quickly than tablets with gelatin or hydroxypropyl cellulose as binder, even though the povidone tablets were harder [23]. Similar results were obtained with 0.6 or 1.0% of PVP 90,000 or hydroxypropyl cellulose [24].
Many of the active substances have poor aqueous solubility due to which they have limited bioavailability. An easy way of enhancing the bioavailability of an active substance is to improve its dissolution by adding solubilizing agents, such as the soluble PVP grades. These form water-soluble complexes with many active substances and increase the bioavailability. The bioavailability of per oral gidazepam was increased by the addition of povidone.
The soluble PVP grades are also useful for preparing solid solutions and dispersions because of their good hydrophilization properties, universal solubility and ability to form water soluble complexes. More than 140 papers between 1960 and 1990 describe the preparation of drugs in solid solution, dispersions using PVP [25].
Soluble grades of PVP and polyvinyl pyrrolidone-vinyl acetate (PVP-VA) copolymer have been used to improve the bioavailability of many poorly water soluble drugs like indomethacin, tolbutamide, nifedipine [26]. These amorphous polymers can be used to formulate these drugs as glass solutions by hot melt extrusion (HME). The extrudates obtained by this process showed high dissolution of the drug depending on the chemical stability and temperature employed for the process. The use of PVP-VA as a polymer in improving the dissolution and bioavailability of these drugs by hot melt extrusion has been cited in other papers as well [27-29].
Povidone provides excellent stability to the tablet formulations (Ex; Phenytoin tablets) [30]. Lyophilisates are produced for parenteral and for oral preparations. Povidone is used to bind the lyophilisate together during freeze drying and to improve the solubility, stability and even the absorption of the active ingredient by virtue of its hydrophilic and complexing properties. Povidone and triesters of citric acid can be combined to obtain clear soft gelatin capsules of insoluble drug substances [31].
All grades of povidone can be used as hydrophilic polymers to physically stabilize suspensions. Their most important and primary function in all suspensions is as protective colloids, which hydrophilize the individual solid particles and sterically separate them. This increases the volume of any sediment and makes it easier to redisperse by shaking. Povidone also prevents the dissolved portion of the active substance from crystallizing out by forming soluble complexes with it. Table 2 lists the pharmaceutical applications of povidone [32].
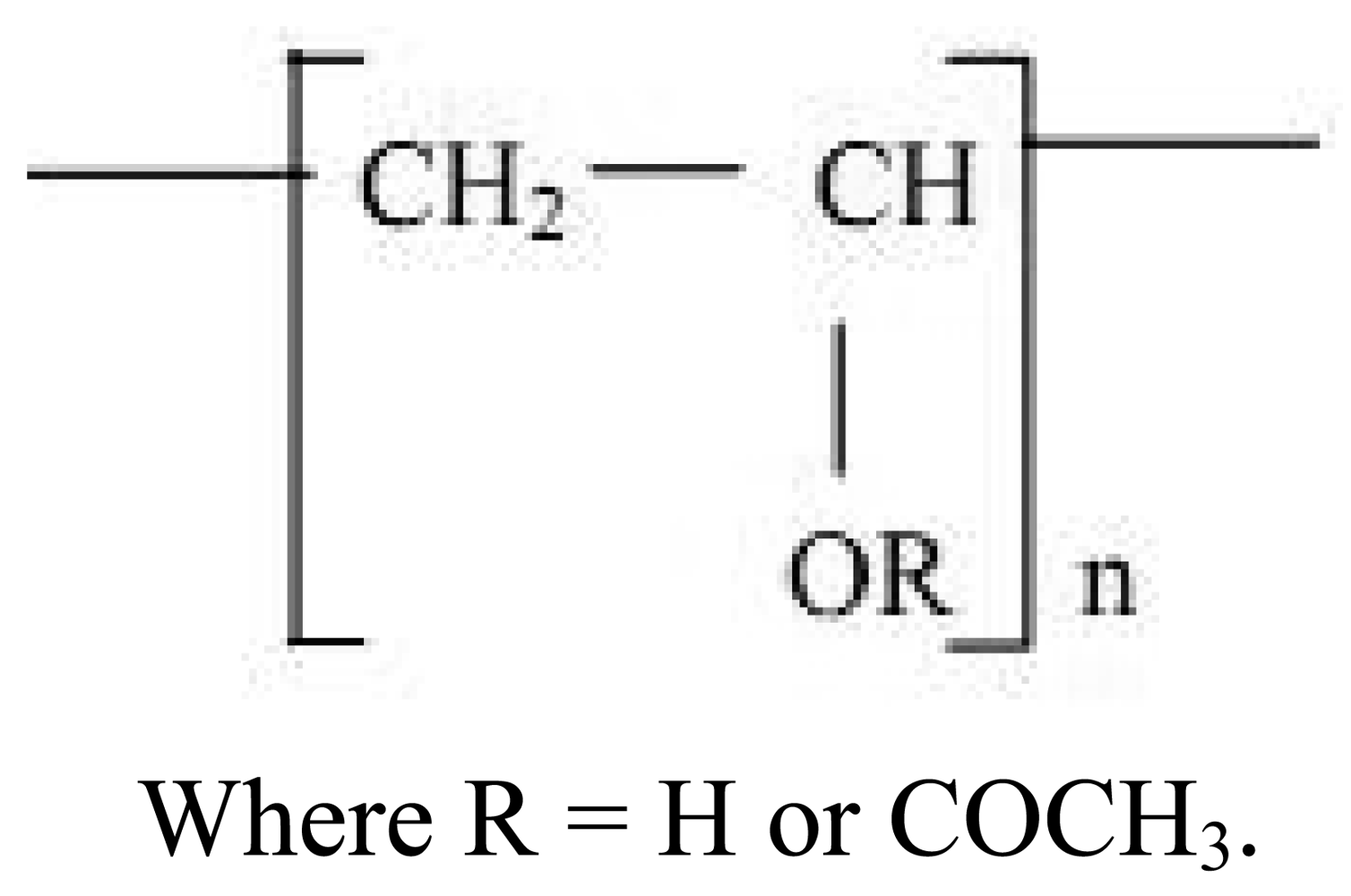
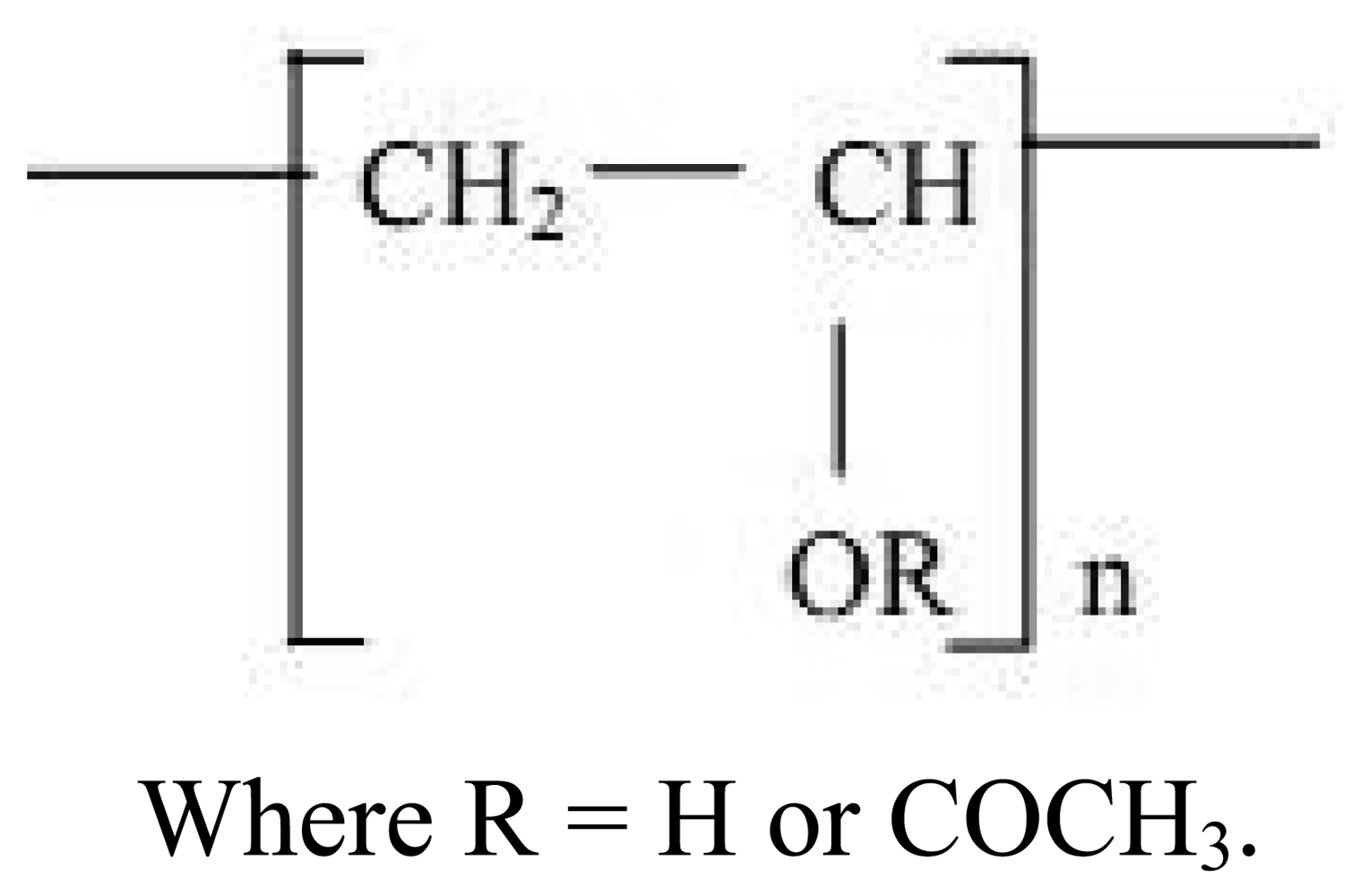
2.3. Polyvinyl alcohol (PVA)
Polyvinyl alcohol (PVA) has a hydroxyl group in its structure. It is synthesized by the polymerization of vinyl acetate to polyvinyl acetate (PVAc) which is then hydrolysed to get PVA. The structure of PVA is given in Figure 3. The extent of hydrolysis and content of acetate groups in PVA affect the crytallizability and solubility of PVA [33].
PVA is soluble in highly polar and hydrophilic solvents, such as water, Dimethyl Sulfoxide (DMSO), Ethylene Glycol (EG), and N-Methyl Pyrrolidone (NMP) [34]. Water is the most important solvent for PVA. The solubility of PVA in water depends on the degree of polymerization (DP), hydrolysis, and solution temperature. Any change in these three factors affects the degree and character of hydrogen bonding in the aqueous solutions, and hence the solubility of PVA.
It has been reported that PVA grades with high degrees of hydrolysis have low solubility in water. The solubility, viscosity, and surface tension of PVA depend on temperature, concentration, % hydrolysis and molecular weight of the material [35].
PVA hydrogels have been used for various biomedical and pharmaceutical applications [36]. PVA hydrogels have certain advantages which make them ideal candidates for biomaterials. Advantages of PVA hydrogels are that they are non-toxic, non-carcinogenic, and bioadhesive in nature. PVA also shows a high degree of swelling in water (or biological fluids) and a rubbery and elastic nature and therefore closely simulates natural tissue and can be readily accepted into the body. PVA gels have been used for contact lenses, the lining for artificial hearts, and drug- delivery applications.
PVA is mainly used in topical pharmaceutical and ophthalmic formulations (Table 3) [37,38]. It is used as a stabilizer in emulsions. PVA is used as a viscosity increasing agent for viscous formulations such as ophthalmic products. It is used as a lubricant for contact lens solutions, in sustained release oral formulations and transdermal patches [39].
2.4. Polyacrylic acid (PAA)
Polyacrylic acid is a biodegradable water soluble polymer with various industrial applications, including as a super adsorbent (e.g., in disposable nappies), in water treatment, etc. [40]. Poly(acrylic acid) (PAA) copolymers modified with block-copolymers of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) have a wide range of medicinal applications as their components are considered pharmaceutically safe [41].
The unique property of Polyacrylic acid is that it exists as a liquid at pH 5 and as a gel at pH 7. Permeation of cations into the gelled polymer converts the gel back to a liquid [42]. It is ideal for ocular delivery of ribozymes to the corneal epithelium as a drug delivery vehicle [43].
Hydrophobically modified poly(acrylic acid) (HMPAA) shows some interesting rheological properties in semidilute aqueous solutions, such as interchain aggregation followed by an increase in the apparent molecular weight and enhanced viscosity as well as shear sensitivity [44]. HMPAA is prepared by modification of PAA in its acidic form by alkylamines in an aprotic solvent in the presence of N,N′-dicyclohexylcarbodiimide (DCCD) [45].
Polyacrylic acid based polymers are mainly used for oral and mucosal contact applications such as controlled release tablets, oral suspensions and bioadhesives. It is also used as a thickening, suspending and emulsion stabilizing agent in low viscosity systems for topical applications. For bioadhesive applications, high molecular weight acrylic acid polymer crosslinked with divinyl glycol are extensively formulated in a variety of drug delivery systems for mucosal applications. Buccal, intestinal, nasal, vaginal and rectal bioadhesive products can all be formulated with such polymers [46].
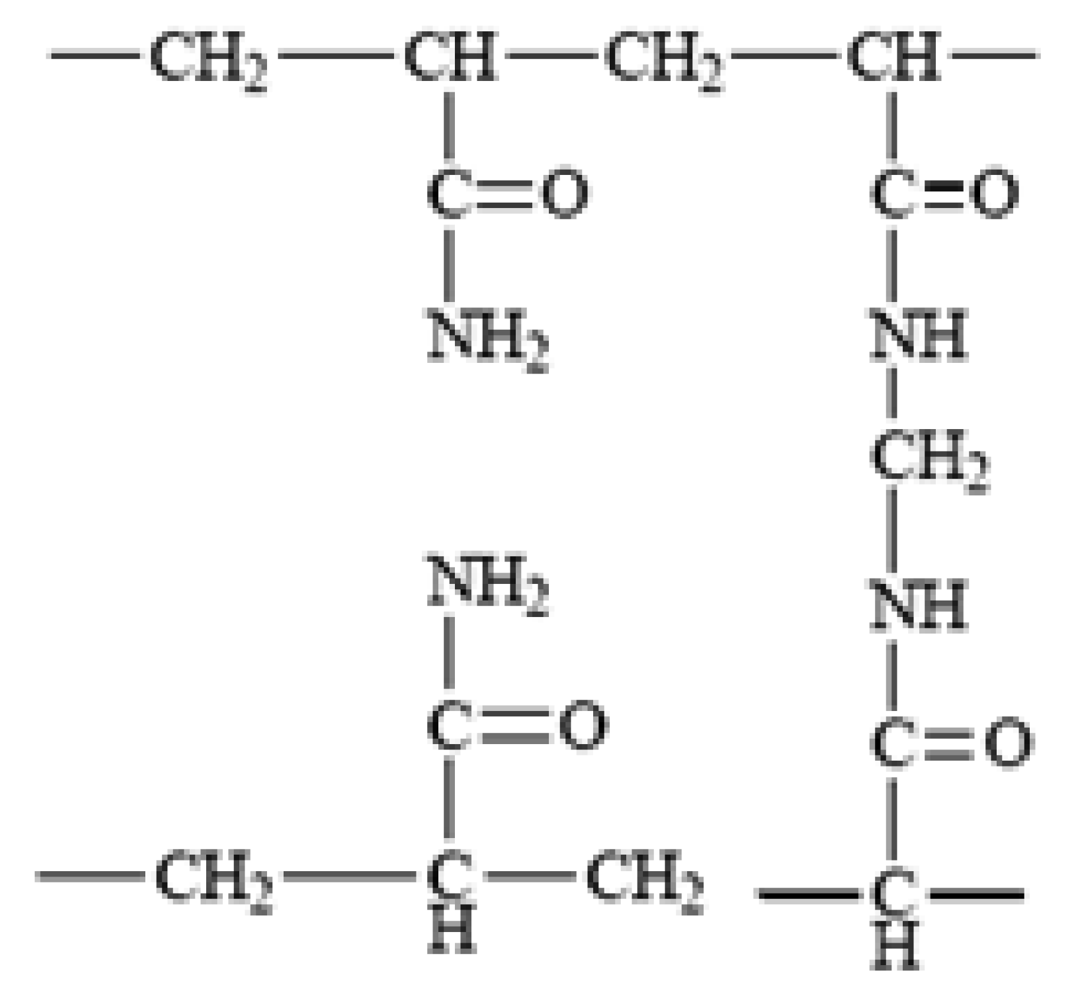
2.5. Polyacrylamides
Polyacrylamide, is a synthetic polymer derived from acrylamide monomer which was originally introduced for use as a support matrix for electrophoresis in 1959 [47]. Polyacrylamide gels result from polymerization of acrylamide with a suitable bifunctional crosslinking agent, most commonly, N,N′-methylenebisacrylamide (bisacrylamide) (Figure 4).
Gel polymerization is carried out using ammonium persulfate and the reaction rate is catalyzed by addition of N,N,′,N′-tetramethylethylenediamine (TEMED). Polyacrylamide gels with a range of pore size can be made to suite size fractionation of a variety of proteins for practical purposes by adjusting the total acrylamide concentration (% T), Polyacrylamide is stable over wide pH intervals (pH 3–11), as well as simple and economical. It has been widely used for a range of applications ranging from microanalysis to macro-fractionation for proteins, nucleic acid, and other biomolecules, and is nowadays the medium of choice in all electrophoretic techniques [48,49]. In addition to electrophoresis, polyacrylamides have also been used as carriers for delivery of drugs and bioactive molecules.
Polyacrylamide is a polymer that is formed from units of acrylamide, a known neurotoxin. However, polyacrylamide itself is non-toxic, but is a controversial ingredient because of its potential ability to secrete acrylamide. Polyacrylamide is used in wide range of cosmetic products (moisturizers, lotions, creams, self-tanning products, etc.). The Food and Drug Administration (FDA) allows Polyacrylamide (with less than 0.2% acrylamide monomer) to be used as a film former in the imprinting of soft-shell gelatin capsules. The Cosmetics Ingredient Review (CIR) Expert Panel allows the use of 5 ppm acrylamide residues in cosmetic products.
Polyacrylamides were first used as an implantable carrier for sustained delivery of insulin to lengthen the life of diabetic rats [50]. Since then, various drug delivery systems based on polyacrylamide have been developed [51,52]. It is also used as a carrier for other bioactive macromolecules and cells to produce the desired effects [53,54]. Polyacrylamide-chitosan hydrogels are biocompatible and are used for sustained antibiotic release [55].
Recently Tsung-Hua Yang [56] has reviewed several patents describing the use of polyacrylamide for drug delivery, biomedical and other applications. U.S. 5874095 patent describes pharmaceutical compositions comprising certain specific non-ionic polymers for topical application to the skin which increased transdermal penetration of the drugs through the skin.
The nonionic polymers used in the above invention are polyacrylamides and substituted polyacrylamides, branched or unbranched. These polymers are non-ionic water dispersible polymers which can be formed from a variety of monomers including acrylamide and methacrylamide which are unsubstituted or substituted with one or two alkyl groups (preferably C1–C5).
For example, polyacrylamide microgels derivatized by saponification of the –CONH2 group to the –COOH group are responsive to pH and ionic strength of the external medium [57]. Polyacrylamide that contains rationally designed single-strand DNA (ssDNA) as the cross-linker can shrink and swell in response to ssDNA samples and recognize a single base difference in the sample [58]. Among these polymers that can respond to external stimuli, poly-N-isopropylacrylamide (PNIPAA) has been widely examined as a smart drug delivery material because of its unique phase separation behavior upon external temperature change.
A device which is placed outside the body where total or partial blood diverted from the heart or arterial system is processed to remove unwanted toxic substances and subsequently returned to the circulation is called as extracorporeal toxin removal device. These devices are considered to be simple, efficient and economical to the patients. They have become popular because of their easy accessibility, lower infection and immune rejection probability, avoidance of a major surgical procedure [59]. The function of polyacrylamide in an extracorporeal toxin removal modality is to provide a support matrix for immobilization of the functional parts or ligands.
Because polyacrylamide is chemically inert and stable over various conditions, polyacrylamide has been employed, whether clinically or under development, to serve as a useful matrix for several types of extracorporeal toxin removal devices and has been described in the following patents: WO 02081006 [60], U.S. 7066900 [61].
2.6. N-(2-Hydroxypropyl) methacrylamide (HPMA)
The polymeric systems that have been very successfully used for passive drug targeting purposes are copolymers based on N-(2-hydroxypropyl) methacrylamide (HPMA). HPMA copolymers were initially developed as plasma expanders, they are highly hydrophilic, non-immunogenic and non-toxic, and reside in the circulation well.
Jindrich Kopecek and colleagues at the Czech (-oslovak) Academy of Sciences in Prague in the mid-1970s started using these long circulating synthetic macromolecules as carriers for low molecular weight drugs [62]. Rationale for using HPMA drug conjugates has been explained by Jindřich Kopeček &Pavla Kopečková [63] in one of their recent papers. HPMA copolymer-drug conjugates are nanosized (5–20 nm) water-soluble constructs. Their unique structural, physicochemical, and biological properties offer several advantages when compared to low molecular weight drugs. The concept of targeted polymer-drug conjugates was developed to address the lack of specificity of low molecular weight drugs for cancer cells. This approach was based on the work of De Duve, who realized that the endocytic pathway is suitable for lysosomotropic drug delivery [64].
The characteristic features needed to design an ideal conjugate are: a polymer-drug linker that is stable during transport and able to release the drug in the lysosomal compartment of the target cell at a controlled rate, solubility of the conjugate in the biological environment and the ability to target the diseased cell or tissue by an active (receptor ligand) or a passive (pathophysiological) mechanism. The first passively tumor-targeted polymeric prodrug to enter clinical trials was pHPMA-GFLG-doxorubicin [65,66]. The conjugate was named as PK1, i.e., Prague-Keele 1, Its average molecular weight is ~28 kDa and it contains on average 8% wt of doxorubicin [67]. The initial half-life time of PK1 was found to be 2.7 h, as compared to less than 10 min for free doxorubicin. Several other HPMA drug conjugates which are in various clinical phases are listed in Table 4.
2.7. Divinyl Ether-Maleic Anhydride (DIVEMA)
Divinyl ether-maleic anhydride (DIVEMA) is a water soluble polymer which has shown antitumor activity against various tumors [77]. The preparation of 1:2 divinyl ether-maleic anhydride copolymer was first reported by Butler. The biological activities of DIVEMA are due to its ability to activate immunocompetent cells, particularly macrophages and natural killer cells [78,79]. DIVEMA has been used as a drug carrier to superoxide dismutase [80] and anticancer agents such as Adriamycin and methotrexate.
Tumour necrosis factor (TNF-α) causes necrotic effect and has been shown to be effective against tumours induced in mice. The antitumor effect of TNF-α has been studied for murine tumors transplanted into mice and for human tumors transplanted into nude mice. However the clinical applications of TNF-α are limited because of the adverse effects. So water soluble polymer like DIVEMA was conjugated with TNF-α in order to increase the antitumor activity in vivo with reduced side effects.
The conjugate DIVEMA-TNF-α (+) showed a significant hemorrhagic necrotic effect on the tumor when compared to native TNF-α 24 h after i.v. injection into mice bearing Sarcoma-180 solid tumors. The antitumor effect was approximately 100 times greater than native TNF-α. The study proved that, upon administration of conjugated DIVEMA-TNF-α, the antitumor activity improved remarkably as compared to the activity observed when DIVEMA and TNF-α was given separately [81].
2.8. Polyoxazoline
Poly(2-alkyl-2-oxazolines) are gaining high interest in biomedical research as they are structurally similar to peptides Their physico chemical properties can be modulated by varying the alkyl substituent [82,83]. Their properties range from high hydrophilicity which enables synthesis of hydrophilic water soluble biocompatible polymers with good antibiofouling properties (alkyl = methyl or ethyl) through thermal sensitivity of thermoresponsive polymers (alkyl = isopropyl) to hydrophobicity typical for hydrophobic aromatic or aliphatic polymers (substituent = phenyl, butyl, nonyl, etc.) [82,84]. They act as versatile polymers and, as they have the ability to form nanostructures, they are being extensively investigated.
Poly(2-oxazolines) are used as adhesive and in coating formulations, and in various drug delivery applications [85]. Despite this, their commercial application is limited as the batch polymerisation times range from several hours to several days [86-89]. However, delayed polymerization reactions can be overcome by use of modern technology like microwave reactors. Commercially, however, only 2-methyl, 2-ethyl, 2-isopropyl and 2-phenyl oxazoline are currently available.
Woodle et al. reported the use of these polymers for the synthesis of lipo-polyoxazolines-poly(2-methyl-2-oxazoline) and poly(2-ethyl-2-oxazoline)-based lipid conjugates as an alternative to PEG-based materials [90]. The lipopolymers were used to prepare 67 Ga-labelled liposomes, which were subsequently injected into the bloodstream of rats. They behaved similar to the PEG based liposomes with long circulation time in blood, uptake by liver and spleen [91]. This similarity was attributed to their high mobility of chains and water binding ability both in turn contributing to the steric stabilization in polymer-lipid liposomes.
Apart from these, a number of other applications of polyoxazoline in preparation of nanoscale systems, thermo responsive systems, gene delivery applications have been reviewed by Nico Adams et al. [82].
2.9. Polyphosphates
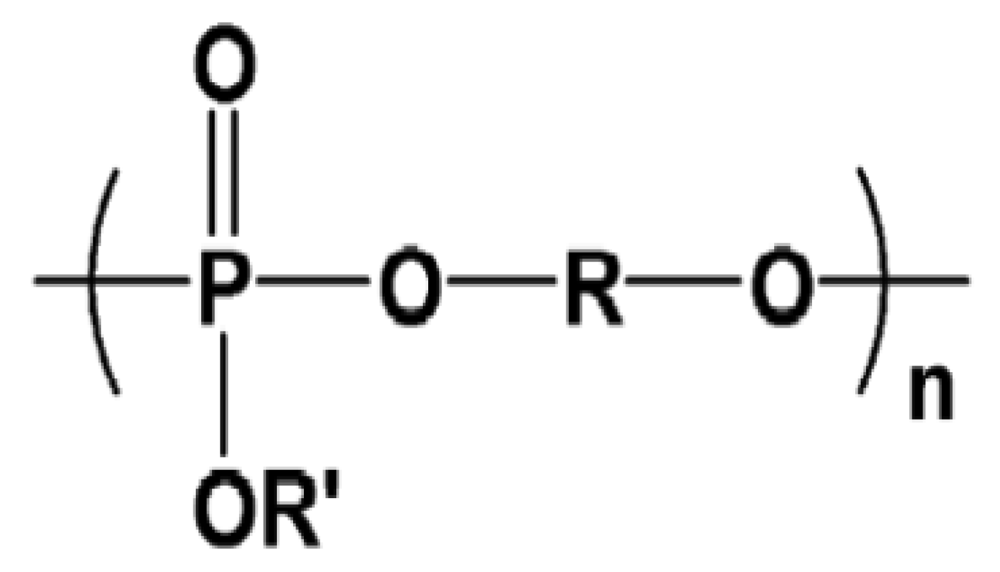
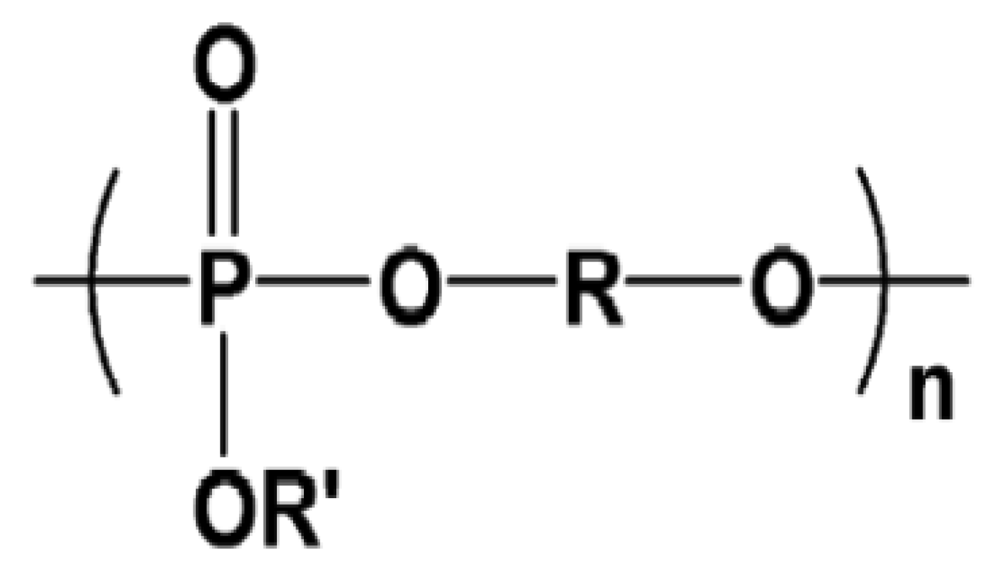
Biodegradable polyphosphoesters (PPE) like polyphosphates, polyphosphonates have been studied for their use in drug delivery, gene delivery and tissue engineering. These polymers have a backbone consisting of phosphorous atoms attached to either carbon or oxygen. The uniqueness of this class of polymer lies in the chemical reactivity of phosphorous, which enables attachment of side chains to alter the biodegradation rates and molecular weight of the polymer [92].
Penczek and others thought that these polymers, which are analogs of nucleic acid and and teichoic acids, would represent a large number of bio-macromolecules. They have reported the synthesis of many of these polymers by various methods [93-96]. The general structure of polyphosphoesters is given in Figure 5.
Water-soluble positively charged polymers are useful for gene delivery [97-101]. Positively charged polymer interacts with negatively charged DNA by electrostatic interactions resulting in the formation of complexes and thus providing protection to DNA from enzymatic attack. This also enables greater cellular uptake of DNA. The commonly used cationic polymers for gene delivery are the ones having amide bond like poly(L-lysine) and vinyl bonds like in polyethyleneimine (PEI) because they show excellent stability in aqueous solutions. Among a large number of cationic polymers reported in the literature, the most extensively studied polymeric gene carriers have either amide bonds (e.g., poly(L-lysine) and cationic PAMAM dendrimers) or vinyl bonds (e.g., poly-ethyleneimine, PEI). These bonds are very stable in aqueous solution and there is no direct evidence of their degradation in body fluid.
Wang et al. have reported the development of biodegradable polyphosphoesters including polyphosphates (PPEs) and polyphos-phoramidates (PPAs) as gene carriers [102-104]. They reported that these systems can show sustained release behavior both inside and outside the cell thus increasing the bioavailability of DNA in the cells. Further they showed that the controlled release behavior of these polymers can be controlled by changing the ratio of polycationic polymer to DNA ratio.
2.10. Polyphosphazenes
Polyphosphazene belongs to a class of polymers with inorganic moiety as the main chain and two active chloride groups on each repeat unit. Substitution of these chloride groups gives multifunctional polyphosphazenes with tunable physicochemical and biological properties [105]. These polymers have been used to formulate nano-fibers [106] and hydrogels [107]. Zhang et al. have synthesized and developed thermally sensitive amphiphilic phosphazenes for sustained local delivery [108,109]. They also synthesized methoxypoly(ethylene glycol) and ethyl-p-aminobenzoate side groups (PEG/EAB-PPPs) polyphosphazenes for delivery of water soluble anticancer agent like Doxorubicin HCl [110].
Some of the most important water-soluble polyphosphazenes such as poly[di(carboxylatophenoxy)phosphazene] (PCPP), poly[di(methoxyethoxyethoxy) phosphazene] (MEEP), and a number of others have been studied in pioneering works of H. Allcock and his coworkers [111,112]. Andrianov has reviewed about the advances in the synthesis of water-soluble polyphosphazene and their degradation pathways [113].
Some water-soluble polyphosphazenes containing ionic groups can be used to formulate hydrogel microspheres or nanospheres for controlled release and drug delivery applications. These methods are ideal for protein encapsulation as they do not use organic solutions or heat. Polymers include polyphosphazene immune adjuvants, which have been also formulated in microspheres and studied for mucosal immunization [114-117]. Water-soluble polyphosphazene containing amino aryloxy and methyl amino side groups has been synthesized and investigated as an inert polymeric carrier for the covalent attachment of biologically active agents.
3. Natural Water Soluble Polymers
3.1. Xanthan Gum
The primary structure of xanthan (Figure 6) consists of repeating pentasaccharide units consisting of two D-glucopyranosyl units, two D-mannopyranosyl units and one D-glucopyranosyluronic unit [118].
Xanthan is a free flowing powder soluble in both hot and cold water to give viscous solutions at low concentrations. Its industrial importance is based upon its ability to control the rheology of water based systems. It is a very effective thickener and stabilizer because it gives highly viscous solutions even at low concentrations as compared to other polysaccharide solutions. Xanthan gum solutions exhibit pseudoplastic behavior (viscosity is regained immediately even at high shear rates). Its pseudoplastic property enhances mouth feel effect and flavor release. Xanthan gum solutions offer very good stability. They are least affected by changes in pH and are stable in both alkaline and acidic conditions. The solution properties of xanthan are not affected in a pH range of 1–13. Xanthan is compatible with most commercially available thickeners such as sodium alginate, carboxymethyl cellulose and starch [118].
Xanthan gum is widely used in cosmetics and in toothpastes [119]. It can be easily extruded from the tube or dispenser because of the shear thinning flow behavior. It also ensures that toothpaste will keep a stable stand on the brush. The shear thinning characteristics also improve the dispersion on and the rinsing from the teeth. Toothpastes thickened with xanthan gum have a bright, shiny cord with short flow behavior. Xanthan gum is used as a thickener and stabilizer in personal care products like creams, eye gels. Typical xanthan gels feel very gentle and soft due to their shear thinning low behavior. In emulsions or suspensions for pharmaceutical use xanthan gum prevents the separation of insoluble ingredients, e.g., barium sulphate in X-ray contrast media. Most of the ready to eat, semi-prepared foods and convenience foods would not be possible without stabilizers and thickeners. In order to adjust the desired flow behavior, xanthan gum is often used in combination with other hydrocolloids [119].
3.2. Pectins
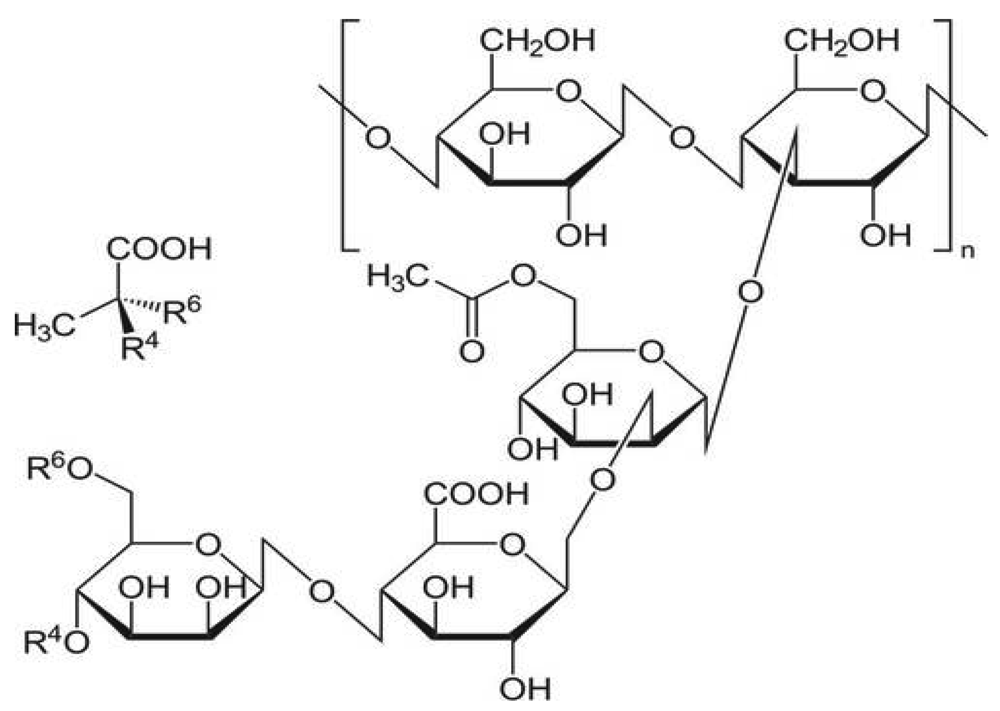
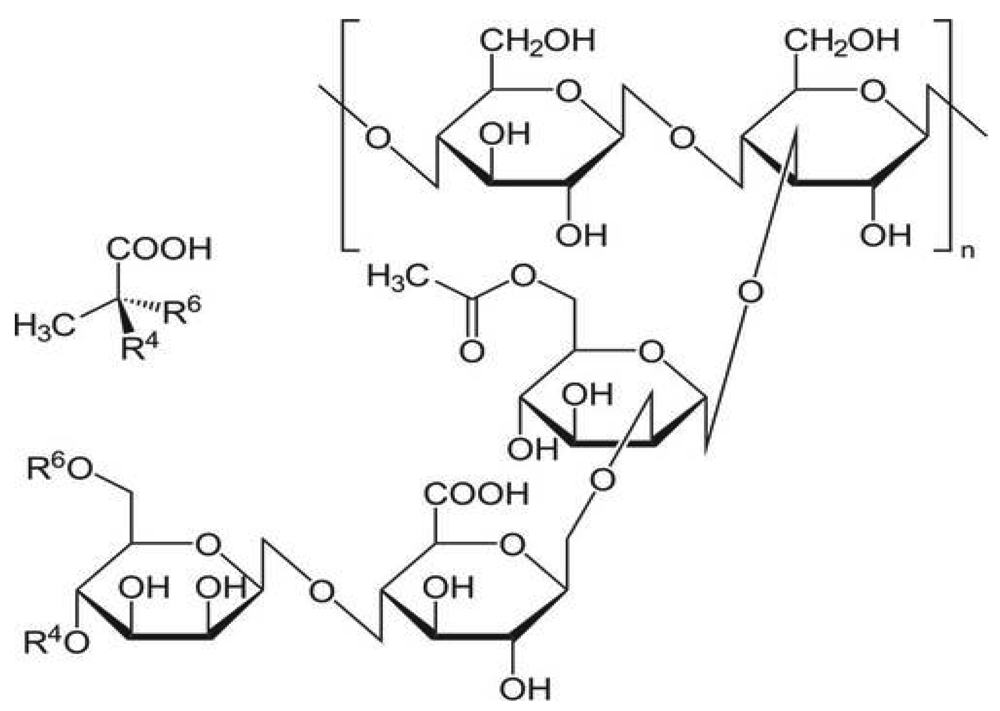
Pectin is a made up of mixture of polysaccharides. Pectins are mainly obtained from citrus peel or apple pomades, both of which are by-products of juice manufacturing process Apple pomade contains 10–15% of pectin on a dry matter basis while Citrus peel contains of 20–30%. Pectin is mainly composed of D-galacturonic acid (GalA) units [120] joined in chains by means of á-(1-4) glycosidic linkage. These uronic acids have carboxyl groups, some of which are naturally present as methyl esters and others which are commercially treated with ammonia to produce carboxamide groups (Figure 7).
Pectins are soluble in pure water. Monovalent cation (alkali metal) salts of pectinic and pectic acids are soluble in water; di- and tri-valent cations salts are weakly soluble or insoluble. Dry powdered pectin, when added to water, forms clumps. This clump formation can be prevented by dry mixing pectin powder with water-soluble carrier material or by the use of pectin having improved dispersibility [122]. Other properties like viscosity, solubility, and gelation are generally related. For instance, factors that increase gel strength will increase the tendency to gel, decrease solubility, and increase viscosity, and vice versa. These properties of pectins are a function of their structure, which is that of a linear polyanion (polycarboxylate). Monovalent cation salts of pectins are highly ionized in solution, and the distribution of ionic charges along the molecule keeps it in an extended form by coulombic repulsion [123].
Pectin has been used in the pharmaceutical industry for a wide range of applications [121]. Pure and standardized pectin has been used as a binding agent in tablets. High Methoxy (HM) pectin is used as monolithic bioerodible system, preparation of directly compressible tablets along with HPMC. Low Methoxy (LM) Pectin has been used to prepare beads by ionotropic gelation technique, sustained release drug delivery using calcium pectinate gel beads. Pectin based microspheres were also prepared by emulsification technique. Film coated tablets can also be prepared using combination of HM-pectin and ethyl cellulose aqueous dispersion, HM or LM pectin with chitosan mixtures.
Pectin also has several unique properties which have enabled it to be used as a matrix for the entrapment and/or delivery of a variety of drugs, proteins and cells. Pectin helps in reduction of blood cholesterol in a diverse group of subjects. At least 6 g/day of pectin is necessary to reduce cholesterol levels. Amounts less than 6 g/day of pectin are not effective [124]. Pectin has been used as a thickening stabilizing and gelling agent stabilizer in food and beverage industry. It effectively removes lead and mercury from the gastrointestinal tract and respiratory organs [125]. Intravenous administration of pectin shortens the coagulation time of drawn blood, thus helping in controlling hemorrhage or local bleeding.
Pectin hydrogels can be used as a binder in tablet formulations [126,127] and have been used in controlled-release matrix tablet formulations [128]. Using a extruder/spheronizer, spherical pellets containing calcium pectate were prepared. These were then coated in pectin solution resulting in the formation of insoluble calcium pectinate gel around the pellets. The use of pectin to develop other oral controlled release drug delivery systems has been reported by some authors.
3.3. Chitosan Derivatives
Chitin and chitosan have been used extensively in many areas ranging from food processing to waste management, medicine, biotechnology and pharmaceutical industries. Chitosan in particular has been used widely in pharmaceutical applications as a formulation excipient because it is biodegradable, biocompatible and less toxic. It has been used as a mucoadhesive, oral absorption enhancer and in protein and gene delivery [129].
The main drawback with chitin and chitosan is that it is difficult to dissolve them in water and in neutral pH. So, water soluble derivatives of chitosan and chitosan have been synthesized by various researchers by chemical modification. These chemical modifications result in the formation of hydrophilic chitin or chitosan which have more affinity to water or organic solvents [130]. Limited solubility of chitosan and chitin has been overcome by chemical modification. For example, carboxymethylation of chitosan results in formation of N-carboxymethylchitosan (N-CMC) which is soluble in wide range of pH [131].
Chitin and chitosan derivatives are also used in treatment of industrial effluents because of their affinity to metal ions. N-CMC has been used widely in pharmaceutical areas for achieving controlled release of drugs, orthopedic devices and connective tissue [132-137].
3.4. Dextran
Dextran can be produced by fermentation of media containing sucrose by Leuconostoc mesenteroides. B512F Dextran is an α-D-1,6-glucose-linked glucan with side-chains 1–3 linked to the backbone units of the Dextran biopolymer. A fragment of the Dextran structure is shown in Figure 8 [138].
Fractions of dextran are readily soluble in water to form clear, stable solutions. The solubility of dextran is not affected by pH. They are also soluble in other solvents like methyl sulfide, formamide, ethylene glycol, and glycerol. Dextran fractions are insoluble in alcohols like methanol, ethanol and isopropanol, and also most ketones, such as acetone and 2-propanone.
The research interest of past decades has focused on the use of dextran as macromolecular carriers, e.g., hydrogels, in which the drug can be incorporated. Dextran hydrogels can be obtained in various ways, based on either chemical or physical crosslinking. Dextran crosslinked with methacrylate (MA), hydroxyethylmethacrylate (HEMA) have been used as hydrogel implants, microspheres for scaffolds [139-144].
Thrombolytic enzymes are effective in treating myocardial infarction and other cardiac diseases. High costs, low availability due to low production, toxicity and antigenicity, lack of specificity to the affected area limit the clinical use of these enzymes. These problems can be overcome by using modified (immobilized) enzymes. Torchilin et al. [145] conjugated the enzyme with a water-soluble carrier like dextran, resulting in a stabilized enzyme preparation with longer circulation time and reduced immunogenicity [146]. They report the synthesis of streptokinase immobilized on activated dextran having a molecular weight of 35,000–50,000. This preparation is produced in Russia under the trademark Streptodekaza™ and is used for treatment of acute myocardial infarction, acute pulmonary artery thromboembolism, peripheral arterial and deep vein thrombosis [147,148]. In comparison with the native enzyme, Streptodekaza™ has a prolonged life-time in the circulation (increase in blood fibrinolytic activity can be observed even 80 h after administration) and causes few complications.
3.5. Carrageenan
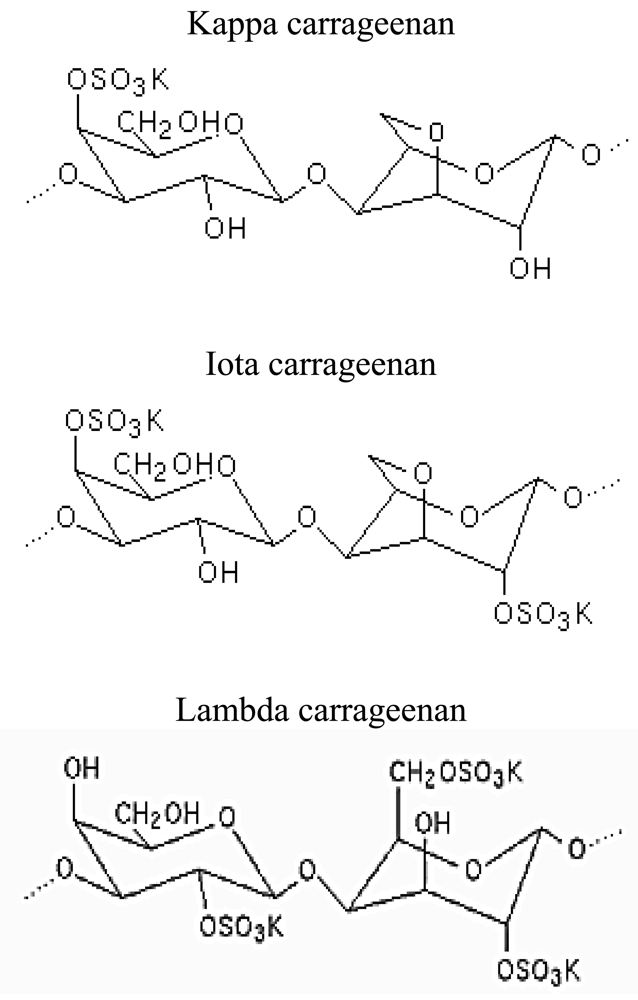
The main sources for carrageenan are the Chondrus crispus, Eucheuma cottonii and Eucheuma spinosum species. It is a natural ingredient obtained from certain species of the red seaweed, class Rhodophyceae [149,150]. Chemically, it comprises repeating galactose units and 3,6-anhydrogalactose (3,6-AG), sulfated and non-sulfated, joined by alternating α (1-)- and β (1-4)-glycosidic linkages [151,152]. Three main types of carrageenan which are widely used in food industry are called iota, kappa and lambda carrageenan. Table 5 summarizes the characteristic features of each type of carrageenan and Figure 9 represents their corresponding structures.
Carrageenan is considered to be a good substitute for gelatin (animal-based product) in hard and soft gel capsules. The incorporation in glycerin-water mixture masks the chalkiness of antacid gels. It can be used in both topical bases [153] and suppository bases [154]. The active ingredients can be trapped inside the fibres by spinning the insoluble carrageenan chitosan fibres. These systems help in wound healing by absorbing large quantities of water thereby keeping the wound clean. The texture of any formulation or polyols can be controlled by utilizing the property of unique interactions between carrageenan and polyols. Carrageenan is used as a thickening agent in hand lotions and shampoos thus promoting healthy skin and hair.
Contraceptive gels: Existing vaginal products have certain drawbacks like leakage because of their inability to maintain gel like structure when applied. Carrageenan gels can be modified suitably and can be used for quick rehealing and protection during intercourse.
Carrageenan has unique properties like viscosity, continuous phase gel formation and specific interactions with the abrasive. Combination of these properties helps in stabilizing the toothpaste preparations. The continuous phase gel matrix entraps the abrasive and flavor oil micelles within the gel matrix thereby enhances the emulsion stability. The gel structure also imparts short texture to the toothpaste providing a clean (non-stringy) break on extrusion from the tube or pump. Carrageenan helps in dispersing and stabilizing the solids thus preventing hardening, caking and drying out. This is because of interaction between carrageenan and surface of the abrasives. These distinct properties of carrageenan make it unique as compared to other binders used in dentrifice industry. It can be safely used with CMC as it does not contain enzymes while other binders like xanthan gum contain enzymes which attack CMC making it unsuitable for use in combinations. Apart from this, carrageenan is widely used in food industry as a thickening agent, stabilizer, gelling agents for a wide range of products like juices, dressings & sauces, beer and wine.
3.6. Guar Gum
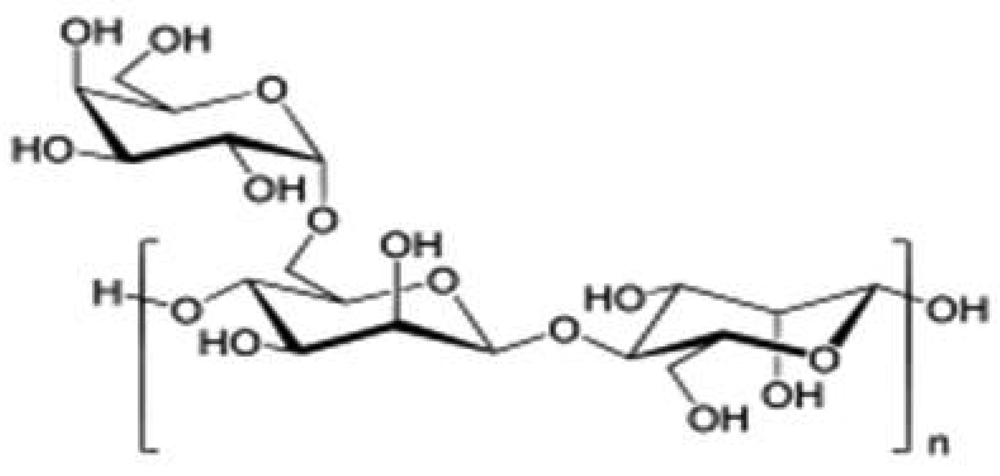
Guar Gum is derived from endosperm of the guar plant (Cyamopsis tetragonoloba). Chemically, guar gum is a polysaccharide composed of the sugars galactose and mannose. It consists chiefly of high molecular weight hydrocolloidal polysaccharide, composed of galactan and mannan units combined through glycosidic linkages and shows degradation in the large intestine due the presence of microbial enzymes [155-158]. The backbone is a linear chain of β 1,4-linked mannose residues to which galactose residues are 1,6-linked at every second mannose, forming short side-branches (Figure 10).
Guar gum is used as a binder, disintegrant in tablet formulations. It also acts as a stabilizers, emulsifier, thickening, and suspending agent in liquid formulations. It has been widely used for colonic drug delivery applications. The swelling ability of guar gum is used in the retardation of drug release from the dosage forms. Its utility as a carrier for colon specific drug delivery is based on its degradation by colonic bacteria [159-161].
3.7. Cellulose Ethers
A very wide range of products can be prepared using different cellulose ethers. They differ from each other with respect to type of substituents, substitution level, molecular weight (viscosity), and particle size. The most common types of cellulose ethers are:
Hydroxypropylmethyl cellulose (HPMC)
Hydroxypropyl cellulose (HPC)
Hydroxyethyl cellulose (HEC)
Sodium carboxy methyl cellulose (Na-CMC)
Pure cellulose as such is insoluble in hot or cold water due to strong intramolecular hydrogen bonding. So cellulose is converted to cellulose esters or cellulose ethers derivatives which are water soluble. These water soluble cellulose derivatives are used in wide range of applications. Thus, modified cellulose derivatives enhance water retention capacity, pseudoplastic behavior, film forming properties and complexation. The advantages of cellulose ethers are that they are biocompatible and hence can be used for pharmaceutical purposes; cosmetics and food [162]. They are mainly used as binders, coating agents, emulsifying, stabilizing, agents, and tablet disintegrants.
3.7.1. Sodium CMC
It is used as an emulsifying agent in pharmaceuticals, and in cosmetics. It is a preferred polymer because it has wide range of functional properties like binding, thickening, stabilizing agent. Also, NaCMC can be used in preparation of microspheres by using glutaraldehyde as a crosslinker. Ketorolac tromethamine, an anti-inflammatory and analgesic agent, was successfully encapsulated into these microspheres and drug encapsulation of up to 67% was achieved [163].
3.7.2. HPC
It is non-ionic water-soluble and pH insensitive cellulose ether. It can be used as thickening agent, tablet binding, modified release and film coating polymer. Buccal delivery formulations containing HPC and polyacrylic acid have been in use for many years [164,165], several researchers have reported the use of HPC in mucoadhesive delivery systems for several different drugs [166,167].
3.7.3. HPMC
It is a water soluble cellulose ether which is mainly used in the preparation of controlled release tablets. Viscosity is the main variable responsible for controlling the release. Ifat Katzhendler et al. studied the effect of molecular weight of HPMC on the mechanism of drug release of naproxen sodium (NS) and naproxen (N) [168]. The hydration and gel forming abilities of HPMC can be used to prolong the drug release of the active ingredient.
3.8. Hyaluronic acid (HA)
Hyaluronic acid (HA), a natural polyanionic polysaccharide distributed widely in the extracellular matrix and the joint liquid of mammalians and approved for injections by the Food and Drug Administration (FDA) [169]. It is non-toxic, biocompatible mucoadhesive polysaccharide having negative charge and is biodegradable. It is mainly distributed in the connective tissue, eyes, intestine and lungs. Above all, the overexpression of CD-44 receptor which is an endogenous ligand for HA, makes this a good candidate for drug targeting [170,171].
HA is composed of two sugar units-glucuronic acid and N-acetylglucosamine which is polymerized into large macromolecules of over 30,000 repeating units. It is readily soluble in water, and produces a gel. The high solubility of hyaluronic acid has proven to be problematic in the development of polymers for tissue engineering. Although this property of HA is more helpful in orthopaedic surgery, it also requires more chemical stabilization and structural stability. The length of the chain, degree of entanglement, cross linking, pH, chemical variations all effect the viscosity of the gel [172,173].
Hyaluronic acid polymers are used in the preparation of gels for delivery of drugs to eye and installation into other cavities. They are used along with other polymers like alginic acid, HPMC, poloxamers etc. for achieving the desired property in drug delivery systems (Bourlais et al. [174]. Combination of these polymers influences the biophysical properties and also alters the pharmacokinetics. HA-based corneal shields have demonstrated more prolonged steroid delivery than by direct application, with a consummate smoothing of dosage profile [175]. Insulin absorption from eye drops via the cornea is enhanced in the presence of HA. HA gel has been successfully studied as a carrier mechanism for antibiotics to the eye; the gel prevents tears from washing away the drug and gives a more prolonged release [176]. HA based nanosystems have been studied earlier for gene delivery, cancer and asthma [177-179]. Some of the commericial products containing HA are listed in Table 6.
Other applications of HA as reviewed by Price et al. [180] are as follows: (i) Wound healing by extracellular regeneration; (ii) Epithelial regeneration; (iii) Topical treatment of dry eye syndrome [181] and Sjögren's syndrome [182]; (iv) as a viscosity agent in pulmonary pathology for achieving alveolar patency [183]; (v) Commercial preparation (Synvist) available for intra-articular injection, (vi) as a filler in rejuvenative medicine for wrinkles and cutaneous lines.
It is also interesting to note that HA is used in the field of viscosurgery, viscosupplementation. In reproductive medicine, HA enhances the retention of the mobility of cryo-preserved and thawed spermatozoa. This property can be used to select spermatozoa which are viable and improve artificial insemination and other in vitro fertilization methods.
3.9. Albumin
Albumin has a molecular weight of 66.5 kDa and is the most abundant plasma protein (35–50 g/L human serum) synthesized in the liver. Human serum albumin (HSA) has a half-life of 19 days. It acts as a solubilising agent for long chain fatty acids and is therefore essential for the metabolism of lipids. It binds very well to penicillins, sulfonamides, indole compounds, and benzodiazepines, copper(II) and nickel(II) in a specific and calcium(II) and zinc(II) in a relatively nonspecific manner, it is responsible for osmotic pressure of the blood [185].
Albumin is an acidic and very soluble protein that is soluble in 40% ethanol. It is stable in the pH range of 4–9, soluble in 40% ethanol, and highly thermostable even when heated at 60 °C for up to 10 h. It is biodegradable in nature and lacks toxicity & immunogenicity. It is very well taken up by the tumor tissues. All these properties make it an ideal candidate for drug delivery. It is a versatile protein carrier which is used in drug targeting for achieving better pharmacokinetic profile of peptide or protein based drugs.
It is easy to purify, soluble in water which makes it convenient to delivery by injection and thus is considered as an ideal candidate for nanoparticle preparation [186,187]. Protein based nanoparticles have the advantage of greater stability during storage and are easy to scale up as compared to other delivery systems [188-191].
Covalent derivatization of albumin nanoparticles with drug targeting ligands is possible, due to the presence of functional groups (i.e., amino and carboxylic groups) on the nanoparticle surfaces [192,193]. HSA based formulations such as Abraxane [194,195] and Albunex [196] have shown good tolerability as evident from the clinical studies. So their efficacy of albumin formulations with minimum side effects is guaranteed. It is also suitable for gene delivery [197,198].
3.10. Starch or Starch Based Derivatives
Starch is a natural polymer which has widespread application ranging from a simple filler or binder [199] to a more functional ingredient in the formulation of capsules [200], coatings [201], subcutaneous implants [202], and tablets. In tablets, starch has been mainly used as a binder, diluents, disintegrant and also as a sustained release agent in matrix systems [203,204]. It is also used as a thickening and gelling agent in food industry.
It is synthesized from carbon dioxide and water by photosynthesis in plants [205]. Its low cost, biodegradability and renewability make it a suitable candidate for developing sustainable materials [206,207]. In order to conserve petrochemical resources and reduce environmental burden many efforts have been made to develop starch based polymers.
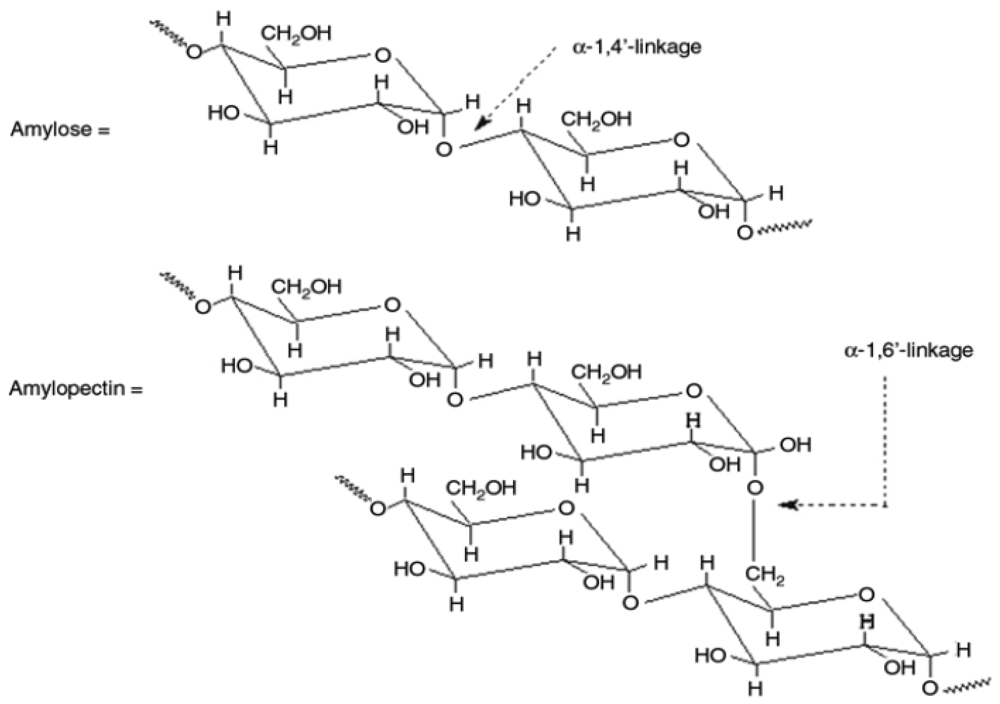
Starch is mainly composed of two homopolymers of D-glucose [208]: amylase, a mostly linear D (1, 4′)-glucan and branched amylopectin, having the same backbone structure as amylose but with many α-1, 6′-linked branch points (Figure 11). Starch has many hydroxyl functional groups in its structure and so it is hydrophilic in nature. Hydrophilicity of starch can be used to improve the degradation rate of some degradable hydrophobic polymers. Native starch is not used because of its poor processability, and poor mechanical properties of the end products [209].
Lu et al. have described in detail the preparation of starch based biodegradable polymers by physical blend, chemical modification and their applications in various fields [210]. Starch is either chemically or physically modified to improve the properties of starch. Such derivatives have physicochemical properties that are different from the parent while still maintaining the biodegradability.
Chemically, starch is modified by Hydroxypropylation to enhance starch clarity and cold-storage stability because the presence of hydroxypropyl groups increases water holding and reduces reassociation of starch chains. This results in formation of a more stable gel [211].
PCL and PLA are chemically bonded onto starch and can be used directly as thermoplastics or compatibilizer. Starch-g-PVA behaves exhibits properties of both components such as processability, hydrophilicity, biodegradability and gelation ability [212-216].
Starch-based biodegradeable polymers (SBBP) have good biocompatibility, its degradation products are non-toxic and have good mechanical properties [217-222]. These SBBPs have been widely used in bone tissue engineering scaffolds [223,224], in drug delivery as microspheres or hydrogels [225,226]. Modified starches have been studied as functional ingredients in sustained release applications because of their improved functionality over their native counterparts [227-231]. Among them, crosslinked high amylose corn starch is the most extensively studied one. The sustained release properties of crosslinked and substituted high amylose corn starch matrices and their swelling behavior in media with various pH and ionic strengths has been reported by Mulhbacher et al. [232]. The matrix characteristics of cross-linked high amylose starches have been studied by Dumoulin et al. [233] and Le Bail et al. [229]. The reasons for starch acting as a sustained release agent is due to its gel-forming ability, biodegradability, and biocompatibility [234].The molecular structure of the gel layer and the mechanical and physicochemical characteristics of the matrix such as gel strength and porosity contribute to the sustained release properties of the matrix.
Onofre and Wang [235] investigated the sustained release properties of hydroxypropylated corn starches with varying amounts of amylase. They characterized the matrices for water holding capacity, porosity, rheological properties, and morphology. Hydroxypropylation increased the water holding capacity and reduced the porosity of the tablets thus enhancing the sustained release ability of amylase containing starches.
4. Conclusions
Scientists around the globe are trying to find ways of improving therapeutic efficacy of drugs by modifying the formulation technique, polymeric systems, etc. The drawbacks associated with conventional dosage forms have been overcome by utilizing polymers synthesized specifically to solve the problems. The use of novel polymers not only offers benefits but also can prove to be harmful because of the toxicity and other incompatibilities associated with them. Care should be taken to properly select polymers while designing a delivery system. The ultimate goal is to introduce cost effective, biocompatible, multifunctional, less toxic polymers so that the delivery systems pass through the various phases of clinical trials and benefit the society. It is believed that the advances in polymer sciences will revolutionize the design, development and performance of polymer based drug delivery systems.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PEG drug candidate | Company | Indication | Year of approval |
|---|---|---|---|
| Adagen (11-17 × 5 kDa mPEG per adenosine deaminase) | Erzon Inc. (USA & Europe) | Immunodefficiency | 1990 (USA) |
| Oncospar (5 kDa mPEG-L-asparginase) | Erzon Inc. (USA/Rhone-Poulenc (Europe) | Acute lymphoblastic leukemia | 1994 (USA) |
| Doxil/Cadyx (SSL formulation of doxorubicin) | Alza Corp. (USA)/Schering Plough Corp. (Europe) | ovarian & breast cancer, multiple myeloma | 1995 (USA) 1999 (USA) 1996 (EU) |
| PEG-Intron (2 × 20 kDa mPEG-interferon-α-2a) | Schering Plough Corp. (USA & EU) | Chronic hepatitis C | 2000 (EU) 2001 (USA) |
| (Pegasys) 12 kDa interferon mPEG-interferon-α-2b) | Hoffmann-Laa-Roche (USA & EU) | Chronic hepatitis C | 2002 (USA & EU) |
| Neulasta (20 kDa mPEG-GCSF) | Amgen Inc. (USA & EU) | Febrile neutropenia | 2002 (USA & EU) |
| Somavert (4–6 × 5 kDa mPEG per structurally modified HG receptor antagonist) | Pfizer (USA & EU) | acromegaly | 2002 (EU) 2003 (USA) |
| Macugen (2 × 20 kDa mPEG anti-VEGF-aptamer) | Pfizer (EU/OSI Pharm Inc.) and Pfizer (USA) | Age related macular degeneration | 2004 (USA) 2006 (EU) |
| Cimzia (2 × 40 kDa MPEG anti TNFα) | CB S.A (USA & EU) | Crohns disease rheumatoid arthritis | 2008 (USA) 2009 (USA)2009 (EU) |
| Krystexxa (Pegloticase) PEGylated Uric acid | Savient Pharmaceutical Inc. (USA) | Chronic Gout | 2010 (USA) |
mPEG: methoxypoly(ethylene glycol); SSL: Sterically Stabilized Liposome; G-CSF: Granulocyte-Colony Stimulating Factor; HG: Human Growth; VEGF: Vascular Endothelial Growth Factor; TNF: Tumor Necrosis Factor.
| Function | Pharmaceutical form |
|---|---|
| Binder | Tablets, capsules, granules |
| Improved Bioavailability | Tablets, pellets, suppositories, transdermal systems |
| Film forming agent | Tablets, opthalmic solutions |
| Solubilising agent | Oral, parenteral and topical solutions |
| Taste masking | Oral solutions, chewing tablets |
| Lyophilizing agent | Injectables, oral lyophilisates |
| Stabiliser | Suspensions, dry syrups |
| Hydrophiliser | Sustained release forms of suspensions |
| Adhesive | Transdermal systems, adhesive gels |
| Stabilizer | Enzymes in diagnostics, different forms |
| Toxicity reducer | Injectables, oral preparations, etc. |
| Use | Concentration (%) |
|---|---|
| Emulsions | 0.5 |
| Ophthalmic formulations | 0.25–3.00 |
| Topical lotions | 2.5 |
| Acronym | Description | Phase | Ref. |
|---|---|---|---|
| PK1 | HPMA copolymer-bound doxorubicin; Prague-Keele-1; GFLG-spacer | II | [65-67] |
| PK2 | Galactosamine-modified PK1; GFLG-spacer; for liver targeting | I | [69-71] |
| PK3 | Tyrosinamide-modified PK1; for imaging purposes | I | [66] |
| AP5280 | Polymer-bound cisplatin-derivative; GFLG-spacer; well-tolerated; moderately active | I | [72,73] |
| AP5346 | Polymer-bound oxaliplatin; GGG-spacer; well-tolerated; moderately active | II | [74-76] |
| Iota | Kappa | Lambda |
|---|---|---|
| Gels most strongly with calcium salts | Gels most strongly with potassium salts | No gel formation |
| Elastic gel with no syneresis (draining out of water) | Brittle gel with some syneresis | forms highly viscoous solutions |
| Gel is freeze-thaw stable | Synergistic with locust bean gum | Fully soluble in cold |
| Completely soluble in hot water | Soluble in hot water | water |
| Company | Product/Application | ||
|---|---|---|---|
| Pharmacia & Upjohn Company | Healon surgical aid in cataract extraction | ||
| Fidia | Hyalgan—osteoarthritis | ||
| Amgen | Blend of HA with Interleukin-1 receptor antagonist | ||
| Anika | Incert®, Amvisc® for surgery | ||
| Orthovisc®, Hyvisc—osteoarthritis | |||
| Ossigel bone fracture recovery | |||
| BioCoat | Hydak—HA surface coating | ||
| Biomatrix | HA derivatives | ||
| Synvisc for viscosupplementation | |||
| Hylashiel for viscoprotection | |||
| Hylaform for viscoaugmentation | |||
| Clear Solutions Biotechnology | Halosol™, Halogel™, Halobeads™, HA-Quat™, Qualginate™, Halgin™ |  | cosmetic use |
| HA-Matricare™, Halosorb™—Medical applications | |||
| Hazomes-B2™, Cancept-HA™—drug delivery | |||
| HA-Bed™—Tissue engineering purposes | |||
| Collaborative Laboratories | HA products in the cosmetic area: liposomes (Micasomes™HOH) and specialty products (Botanigel™) | ||
| Genzyme | Hylucare—Cosmetic use (HyluMed®); Seprafilm®—Drug delivery | ||
| Seikagaku Corp. | HA-enzyme conjugates | ||
| Shiseido Company, Ltd. | HA products for cosmetics and drug delivery | ||
| SurModics Inc. | HA surface coating | ||
| Telios Pharmaceuticals, Inc. | HA hydrogels for tissue engineering | ||
Acknowledgments
The authors wish to thank the Department of Pharmaceutical sciences, Western University of Health Sciences, Pomona, CA, USA, for providing the facilities and necessary assistance.
References
- Will, R.; Loechner, U.; Yokose, K. Synthetic Water Soluble Polymers. Available online: http://www.sriconsulting.com/CEH/Public/Reports/582.0000/ (accessed on 28 June 2011).
- Veronese, F.M.; Pasut, G. PEGylation, Successful Approach to Drug Delivery. Drug Discovery Today 2005, 10, 1451–1458. [Google Scholar]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar]
- Ruel-Gariepy, E.; Leroux, J.-C. In Situ-Forming Hydrogels—Review of Temperature-Sensitive Systems. Eur. J. Pharm. Biopharm. 2004, 58, 409–426. [Google Scholar]
- Martens, P.; Holland, T.; Anseth, K.S. Synthesis and Characterization of Degradable Hydrogels Formed from Acrylate Modified Poly(vinyl alcohol) Macromers. Polymer 2002, 43, 6093–6100. [Google Scholar]
- Bhadra, D.; Bahdra, S.; Jain, P.; Jain, N.K. Pegnology: A Review of PEG-ylated Systems. Pharmazie 2002, 57, 5–29. [Google Scholar]
- Monfardini, C.; Veronese, F.M. Stabilization of Substances in Circulation. Bioconjugate Chem. 1998, 9, 418–450. [Google Scholar]
- Pasut, G.; Veronese, F.M. Polymer Drug Conjugation, Recent Achievements and General Strategies. Prog. Polym. Sci. 2007, 32, 933–961. [Google Scholar]
- Allen, T.M.; Cullis, P.R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303, 1818–1822. [Google Scholar]
- Duncan, R.; Vicent, M.J.; Greco, F.; Nicholson, R.I. Polymer-Drug Conjugates: Towards a Novel Approach for the Treatment of Endocrine-Related Cancer. Endocr. Relat. Cancer 2005, 12, S189–S199. [Google Scholar]
- Savient Pharmaceuticals, Inc. Product Pipeline—Overview. Available online: http://savientpharma.com/pipeline/ (accessed on 13 August 2011).
- Pasut, G.; Veronese, F.M. PEG Conjugates in Clinical Development or Use as Anticancer Agents: An Overview. Adv. Drug Deliv. Rev. 2009, 61, 1177–1188. [Google Scholar]
- Burnham, B. Polymers for Delivering Peptides and Proteins. Am. J. Hosp. Pharm. 1994, 51, 210–218. [Google Scholar]
- Eldon, M.A.; Staschen, C.M.; Viegas, T.; Bentley, M. NKTR-102, a Novel PEGylated Irinotecan Conjugate, Results in Sustained Tumor Growth Inhibition in Mouse Models of Human Colorectal and Lung Tumors that is Associated with Increased and Sustained Tumor SN38 Exposure. AACR-NCI-EORTC International Conference, San Francisco, CA, USA, 22–26 October 2007. poster C157.
- Von Hoff, D.D.; Jameson, G.S.; Borad, M.J.; Rosen, L.S.; Utz, J.; Basche, M.; Alemany, C.; Dhar, S.; Acosta, L.; Barker, T.; et al. First Phase I Trial of NKTR-102 (PEG-Irinotecan) Reveals Early Evidence of Broad Anti-Tumour Activity in Three Different Schedules. the 20th EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics, Geneva, Switzerland, 21–24 October 2008. poster#595.
- NKTR-105. Available online: http://www.nektar.com/product_pipeline/oncology_nktr-105.html (accessed on 12 August 2011).
- Zhao, H.; Lee, C.; Sai, P.; Choe, Y.H.; Boro, M.; Pendri, A.; Guan, S.; Greenwald, R.B. 20-Oacylcamptothecin Derivatives: Evidence for Lactone Stabilization. J. Org. Chem. 2000, 65, 4601–4606. [Google Scholar]
- Scott, L.C.; Yao, J.C.; Benson, A.B.; Thomas, A.L.; Falk, S.; Mena, R.R.; Picus, J.; Wright, J.; Mulcahy, M.F.; Ajani, J.A.; et al. A Phase II Study of Pegylatedcamptothecin (Pegamotecan) in the Treatment of Locally Advanced and Metastatic Gastric and Gastro-Oesophageal Junction Adenocarcinoma. Cancer Chemother. Pharmacol. 2009, 63, 363–370. [Google Scholar]
- Sapra, P.; Zhao, H.; Mehlig, M.; Malaby, J.; Kraft, P.; Longley, C.; Greenberger, L.M.; Horak, I.D. Novel Delivery of SN38 Markedly Inhibits Tumor Growth in Xenografts, Including a Camptothecin-11-Refractory Model. Clin. Cancer Res. 2008, 14, 1888–1896. [Google Scholar]
- Jeong, B.; Bae, Y.H.; Kim, S.W. Thermoreversible Gelation of PEG-PLGA-PEG Triblock Copolymer Aqueous Solutions. Macromolecules 1999, 32, 7064–7069. [Google Scholar]
- Chowhan, Z.T. Role of Binders in Moisture-Induced Hardness Increase in Compressed Tablets and Its Effect on in vitro Disintegration and Dissolution. J. Pharm. Sci. 1980, 69, 1–4. [Google Scholar]
- Chowhan, Z.T.; Amaro, A.A.; Ong, J.T.H. Punch Geometry and Formulation Considerations in Reducing Tablet Friability and Their Effect on in vitro Dissolution. J. Pharm. Sci. 1992, 81, 290–294. [Google Scholar]
- Jun, Y.B.; Min, B.H.; Kim, S.I.; Kim, Y.I.J. Preparation and Evaluation of Acetaminophen Tablets. Kor. Pharm. Sci. 1989, 19, 123–128. [Google Scholar]
- Sinchalpanid, N.; Mitrevej, A. Comparative Evaluation of Hydroxypropyl Cellulose and Povidone in Paracetamol Tablet Formulations. Mahidol J. Pharm. Sci. 1993, 20, 33–39. [Google Scholar]
- Stone, I.M. Water Dispersible Antibiotics U.S. Patent 3,089,818, 14 May 1963.
- Forster, A.; Hempenstall, J.; Rades, T. Characterization of Glass Solutions of Poorly Water-Soluble Drugs Produced by Melt Extrusion with Hydrophilic Amorphous Polymers. J. Pharm. Pharmacol. 2001, 53, 303–315. [Google Scholar]
- Jijun, F.; Lishuang, X.; Xiaoli, W.; Shu, Z.; Xiaoguang, T.; Xingna, Z.; Haibing, H.; Xing, T. Nimodipine (NM) Tablets with High Dissolution Containing NM Solid Dispersions Prepared by Hot-Melt Extrusion. Drug Dev. Ind. Pharm. 2011, 37, 934–944. [Google Scholar]
- He, H.; Yang, R.; Tang, X. In vitro and in vivo Evaluation of Fenofibrate Solid Dispersion Prepared by Hot-Melt Extrusion. Drug Dev. Ind. Pharm. 2010, 36, 681–687. [Google Scholar]
- Chokshi, R.J.; Sandhu, H.K.; Iyer, R.M.; Shah, N.H.; Malick, W.A.; Zia, H. Characterization of Physico-Mechanical Properties of Indomethacin and Polymers to Assess Their Suitability for Hot-Melt Extrusion Process as a Means to Manufacture Solid Dispersion/Solution. J. Pharm. Sci. 2005, 94, 2463–2474. [Google Scholar]
- Jachowicz, R. Dissolution Rates of Partially Water Soluble Drugs from Solid Dispersion Systems. II. Phenytoin. Int. J. Pharm. 1987, 35, 7–12. [Google Scholar]
- White, R.K. Pharmaceutical Compositions Containing Polyvinylpyrrolidone and a Tri-Ester and a Process of Manufacture Thereof. Internat. Patent WO/1994/025008, 10 November 1994. [Google Scholar]
- Bühler, V. Polyvinylpyrrolidone Excipients for Pharmaceuticals: Povidone, Crospovidone and Povidone, 1st ed.; Springer: Berlin, Germany, 2005. [Google Scholar]
- Tubbs, R.K. Sequence Distribution of Partially Hydrolyzed Poly(vinyl acetate). J. Polym. Sci. 1966, 4, 623–629. [Google Scholar]
- Tacx, J.C.J.F.; Schoffeleers, H.M.; Brands, A.G.M.; Teuwen, L. Dissolution Behavior and Solution Properties of Polyvinylalcohol as Determined by Viscometry and Light Scattering in DMSO, Ethylene Glycol and Water. Polymer 2000, 41, 947–957. [Google Scholar]
- Hassan, C.M.; Peppas, N.A. Structure and Applications of Poly(vinyl alcohol) Hydrogels Produced by Conventional Crosslinking or by Freezing/Thawing Methods. Adv. Polym. Sci. 2000, 153, 37–65. [Google Scholar]
- Reneker, D.H.; Yarin, A.L.; Fong, H.; Koombhongse, S. Bending Instability of Electrically Charged Liquid Jets of Polymer Solutions in Electrospinning. J. Appl. Phys. 2000, 87, 4531–4547. [Google Scholar]
- Krishna, N.; Brow, F. Polyvinyl Alcohol as an Ophthalmic Vehicle. Effect on Regeneration of Corneal Epithelium. Am. J. Opthalmol. 1964, 57, 99–106. [Google Scholar]
- Paton, T.F.; Robinson, J.R. Ocular Evaluation of Polyvinyl Alcohol Vehicle in Rabbits. J. Pharm. Sci. 1975, 64, 1312–1316. [Google Scholar]
- Wan, L.S.C.; Lim, L.Y. Drug Release from Heat Treated Polyvinyl Alcohol Films. Drug Dev. Ind. Pharm. 1992, 18, 1895–1906. [Google Scholar]
- Saunders, G.; MacCreath, B. Biodegradable Polymers Analysis of Biodegradable Polymers by GPC-SEC. Application Compendium; Agilent Technologies Inc.: Santa Clara, CA, USA, 2010. [Google Scholar]
- Bromberg, L. Polyether-Modified Poly(acrylic acid): Synthesis and Applications. Ind. Eng. Chem. Res. 1998, 37, 4267–4274. [Google Scholar]
- Craig, D.Q.M.; Tamburic, S.; Buckton, G.; Newton, J.M. An Investigation into the Structure and Properties of Carbopol® 934 Gels Using Dielectric Spectroscopy and Oscillatory Rheometry. J. Control. Release 1994, 30, 213–223. [Google Scholar]
- Ayers, D.; Cuthbertson, J.M.; Schroyer, K.; Sullivan, S.M. Polyacrylic Acid Mediated Ocular Delivery of Ribozymes. J. Control. Release 1996, 38, 167–175. [Google Scholar]
- Magny, B.; Iliopoulos, I.; Audebert, R. Aggregation of Hydrophobically Modified Polyelectrolytes in Dilute Solution: Tonic Strength Effects. In Macromolecular Complexes in Chemistry and Biology; Dubin, P., Bock, J., Davis, R., Schulz, D.N., Thies, C., Eds.; Springer-Verlag: Berlin, Germany, 1994; pp. 50–62. [Google Scholar]
- Wang, T.K.; Iliopoulos, I.; Audebert, R. Aqueous-Solution Behavior of Hydrophobically Modified Poly(acrylic acid). In Water Soluble Polymers; Shalaby, S.W., McCormick, C.L., Butler, G.B., Eds.; American Chemical Society: Washington, DC, USA, 1991. [Google Scholar]
- Polymers for Pharmaceutical Applications; Lubrizol Pharmaceutical Bulletin 1; Lubrizol: Wickliffe, OH, USA; 11; August; 2010.
- Weintraub, R.S. Acrylamide Gel as a Supporting Medium for Zone Electrophoresis. Science 1959, 130, 711–713. [Google Scholar]
- Raymond, S. Protein Purification by Elution Convection Electrophoresis. Science 1964, 146, 406–407. [Google Scholar]
- Chrambach, A.; Rodbard, D. Polyacrylamide Gel Electrophoresis. Science 1971, 172, 440–451. [Google Scholar]
- Davis, B.K. Control of Diabetes with Polyacrylamide Implants Containing Insulin. Experientia 1972, 28, 348. [Google Scholar]
- Hussain, M.D.; Rogers, J.A.; Mehvar, R.; Vudathala, G.K. Preparation and Release of Ibuprofen from Polyacrylamide Gels. Drug Dev. Ind. Pharm. 1999, 25, 265–271. [Google Scholar]
- Sairam, M.; Babu, V.R.; Vijaya, B.; Naidu, K.; Aminabhavi, T.M. Encapsulation Efficiency and Controlled Release Characteristics of Crosslinked Polyacrylamide Particles. Int. J. Pharm. 2006, 320, 131–136. [Google Scholar]
- Gao, D.; Xu, H.; Philbert, M.A.; Kopelman, R. Ultrafine Hydrogel Nanoparticles: Synthetic Approach and Therapeutic Application in Living Cells. Angew. Chem. Int. Ed. Engl. 2007, 46, 2224–2227. [Google Scholar]
- Patton, J.N.; Palmer, A.F. Physical Properties of Hemoglobinpoly(acrylamide) Hydrogel-Based Oxygen Carriers: Effect of Reaction pH. Langmuir 2006, 22, 2212–2221. [Google Scholar]
- Risbud, M.V.; Bhonde, R.R. Polyacrylamide-Chitosan Hydrogels: In Vitro Biocompatibility and Sustained Antibiotic Release Studies. Drug Deliv. 2000, 7, 69–75. [Google Scholar]
- Yang, T.H. Recent Applications of Polyacrylamide as Biomaterials. Recent Patents Mater. Sci. 2008, 1, 29–40. [Google Scholar]
- Soppimath, K.S.; Kulkarni, A.R.; Aminabhavi, T.M. Chemically Modified Polyacrylamide-g-Guar Gum-Based Crosslinked Anionic Microgels as pH-Sensitive Drug Delivery Systems: Preparation and Characterization. J. Control. Release 2001, 75, 331–345. [Google Scholar]
- Murakami, Y.; Mizuo, M. DNA-Responsive Hydrogels That can Shrink or Swell. Biomacromolecules 2005, 6, 2927–2929. [Google Scholar]
- Schmidt, B. Membranes in Artificial Organs. Artif. Organs 1996, 20, 375–380. [Google Scholar]
- Puetz, G.; Eckes, J. Method for Eliminating Potentially Toxic and/or Harmful Substances WO 02081006, 17 October 2002.
- Botto, S.A.; Roeth, P.J.; Faramus, E.L.; Nair, C.H. Removal of Metabolic Components from Blood U.S. Patent 7,066,900, 27 June 2006.
- Kopecek, J.; Bazilova, H. Poly[N-(2-hydroxypropyl)methacrylamide]. I. Radical Polymerization and Copolymerization. Eur. Polym. J. 1973, 9, 7–14. [Google Scholar]
- Kopeček, J.; Kopečková, P. HPMA Copolymers: Origins, Early Developments, Present and Future. Adv. Drug Deliv. Rev. 2010, 62, 122–149. [Google Scholar]
- De Duve, C.; De Barsy, T.; Poole, B.; Trouet, A.; Tulkens, P.; van Hoof, F. Lysosomotropic Agents. Biochem. Pharmacol. 1974, 23, 2495–2531. [Google Scholar]
- Vasey, P.A.; Duncan, R.; Kaye, S.B.; Cassidy, J. 929 Clinical Phase I Trial of PK1 (HPMA Co-Polymer Doxorubicin). Eur. J. Cancer 1995, 31A, S193. [Google Scholar]
- Vasey, P.A.; Kaye, S.B.; Morrison, R.; Twelves, C.; Wilson, P.; Duncan, R.; Thomson, L.S.; Murray, A.H.; Hilditch, T.E.; Murray, T.; et al. Cancer Research Campaign Phase I/II Committee, Phase I Clinical and Pharmacokinetic Study of PK1 [N-(2-hydroxypropyl)-methacrylamide Copolymer Doxorubicin]: First Member of a New Class of Chemotherapeutic Agents-Drug-Polymer Conjugates. Clin. Cancer Res. 1999, 5, 83–94. [Google Scholar]
- Bilim, V. Technology Evaluation: PK1, Pfizer/Cancer Research UK. Curr. Opin. Mol. Ther. 2003, 5, 326–330. [Google Scholar]
- Lammers, T. Improving the Efficacy of Combined Modality Anticancer Therapy Using HPMA Copolymer-Based Nanomedicine Formulations. Adv. Drug Deliv. Rev. 2010, 62, 203–230. [Google Scholar]
- Julyan, P.J.; Seymour, L.W.; Ferry, D.R.; Daryani, S.; Boivin, C.M.; Doran, J.; David, M.; Anderson, D.; Christodoulou, C.; Young, A.M.; et al. Preliminary Clinical Study of the Distribution of HPMA Copolymers Bearing Doxorubicin and Galactosamine. J. Control. Release 1999, 57, 281–290. [Google Scholar]
- Seymour, L.W.; Ferry, D.R.; Anderson, D.; Hesslewood, S.; Julyan, P.J.; Poyner, R.; Doran, J.; Young, A.M.; Burtles, S.; Kerr, D.J. Cancer Research Campaign Phase I/II Clinical Trials Committee, Hepatic Drug Targeting: Phase I Evaluation of Polymer-Bound Doxorubicin. J. Clin. Oncol. 2002, 20, 1668–1676. [Google Scholar]
- Seymour, L.W.; Ulbrich, K.; Wedge, S.R.; Hume, I.C.; Strohalm, J.; Duncan, R. N-(2-hydroxypropyl)methacrylamide Copolymers Targeted to the Hepatocyte Galactose-Receptor: Pharmacokinetics in DBA2 Mice. Br. J. Cancer 1991, 63, 859–866. [Google Scholar]
- Lin, X.; Zhang, Q.; Rice, J.R.; Stewart, D.R.; Nowotnik, D.P.; Howell, S.B. Improved Targeting of Platinum Chemotherapeutics. The Antitumour Activity of the HPMA Copolymer Platinum Agent AP5280 in Murine Tumour Models. Eur. J. Cancer 2004, 40, 291–297. [Google Scholar]
- Rademaker-Lakhai, J.M.; Terret, C.; Howell, S.B.; Baud, C.M.; De Boer, R.F.; Pluim, D.; Beijnen, J.H.; Schellens, J.H.; Droz, J.P. A Phase I and Pharmacological Study of the Platinum Polymer AP5280 Given as an Intravenous Infusion Once Every 3 Weeks in Patients with Solid Tumors. Clin. Cancer Res. 2004, 10, 3386–3395. [Google Scholar]
- Rice, J.R.; Gerberich, J.L.; Nowotnik, D.P.; Howell, S.B. Preclinical Efficacy and Pharmacokinetics of AP5346, a Novel Diaminocyclohexane-Platinum Tumor Targeting Drug Delivery System. Clin. Cancer Res. 2006, 12, 2248–2254. [Google Scholar]
- Campone, M.; Rademaker-Lakhai, J.M.; Bennouna, J.; Howell, S.B.; Nowotnik, D.P.; Beijnen, J.H.; Schellens, J.H. Phase I and Pharmacokinetic Trial of AP5346, a DACHplatinum-Polymer Conjugate, Administered Weekly for Three out of Every 4 Weeks to Advanced Solid Tumor Patients. Cancer Chemother. Pharmacol. 2007, 60, 523–533. [Google Scholar]
- Nowotnik, D.P.; Cvitkovic, E. ProLindac (AP5346): A Review of the Development of an HPMA DACH Platinum Polymer Therapeutic. Adv. Drug Deliv. Rev. 2009, 61, 1214–1219. [Google Scholar]
- Morahan, P.S.; Munson, J.A.; Baird, L.G.; Kaplan, A.M.; Regelson, W. Antitumor Action of Pyran Copolymer and Tilorone against Lewis Lung Carcinoma and B-l6 Melanoma. Cancer Res. 1974, 34, 506–511. [Google Scholar]
- Harmel, R.P., Jr.; Zbar, B. Tumor Suppression by Pyran Copolymer: Correlation with Production of Cytotoxic Macrophages. J. Natl. Cancer Inst. 1975, 54, 989–992. [Google Scholar]
- Santoni, A.; Puccetti, P.; Riccardi, C.; Herberman, R.B.; Bonmassar, E. Augmentation of Natural Killer Activity by Pyran Copolymer in Mice. Int. J. Cancer 1979, 24, 656–661. [Google Scholar]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen Radicals in Influenza-Induced Pathogenesis and Treatment with Pyran Polymer-Conjugated SOD. Science 1989, 244, 974–976. [Google Scholar]
- Kaneda, Y.; Yamamoto, Y.; Kamada, H.; Tsunoda, S.; Tsutsumi, Y.; Hirano, T.; Mayumi, T. Antitumor Activity of Tumor Necrosis Factor Conjugated with Divinyl Ether and Maleic Anhydride Copolymer on Solid Tumors in Mice. Cancer Res. 1998, 58, 290–295. [Google Scholar]
- Adams, N.; Schubert, U.S. Poly(2-oxazolines) in Biological and Biomedical Application Contexts. Adv. Drug Deliv. Rev. 2007, 59, 1504–1520. [Google Scholar]
- Park, J.S.; Akiyama, Y.; Winnik, F.M.; Kataoka, K. Versatile Synthesis of End-Functionalized Thermosensitive Poly(2-isopropyl-2-oxazolines). Macromolecules 2004, 37, 6786–6792. [Google Scholar]
- Persigehl, P.; Jordan, R.; Nuyken, O. Functionalization of Amphiphilic Poly(2-oxazoline) Block Copolymers: A Novel Class of Macroligands for Micellar Catalysis. Macromolecules 2000, 33, 6977–6981. [Google Scholar]
- Ansari, A.M.; Scaria, P.V.; Woodle, M.C. Polymers for Delivering Peptides and Small Molecules in vivo WO 2003/066069, 14 August 2003.
- Hoogenboom, R.; Fijten, M.W.M.; Schubert, U.S. Parallel Kinetic Investigation of 2-Oxazoline Polymerisations with Different Initiators as Basis for Designed Copolymer Synthesis. J. Polym. Sci. A: Polym. Chem. 2004, 42, 1830–1840. [Google Scholar]
- Wiesbrock, F.; Hoogenboom, R.; Leenen, M.; Van Nispen, S.F.G.M.; van der Loop, M.; Abeln, C.H.; van den Berg, A.M.J.; Schubert, U.S. Microwave-Assisted Synthesis of a 42-Membered Library of Diblock Copoly(2-oxazoline)s and Chain-Extended Homo Poly(2-oxazoline)s and Their Thermal Characterization. Macromolecules 2005, 38, 7957–7966. [Google Scholar]
- Huang, H.; Hoogenboom, R.; Leenen, M.A.M.; Guillet, P.; Jonas, A.M.; Schubert, U.S.; Gohy, J.F. Solvent-Induced Morphological Transition in Corecross-Linked Block Copolymer Micelles. J. Am. Chem. Soc. 2006, 128, 3784–3788. [Google Scholar]
- Hoogenboom, R.; Wiesbrock, F.; Huang, H.; Leenen, M.A.M.; Thijs, H.M.L.; van Nispen, S.F.G.M.; van der Loop, M.; Fustin, C.A.; Jonas, A.M.; Gohy, J.-F.; Schubert, U.S. Microwave-Assisted Cationic Ring-Opening Polymerization of 2-Oxazolines: A Powerful Method for the Synthesis of Amphiphilic Triblock Copolymers. Macromolecules 2006, 39, 4719–4725. [Google Scholar]
- Woodle, M.C. New Amphiphatic Polymer-Lipid Conjugates Forming Long Circulating Reticuloendothelial System-Evading Liposomes. Bioconjugate Chem. 1994, 5, 493–496. [Google Scholar]
- Lasic, D.D.; Martin, F.J.; Gabizon, A.; Huang, S.K.; Papahadjopoulosm, D. Sterically Stabilized Liposomes: A Hypothesis on the Molecular Origin of Extended Circulation Times. Biochim. Biophys. Acta 1991, 1070, 187–192. [Google Scholar]
- Mao, H.Q.; Kdaiyala, I.; Leong, K.W.; Zhao, Z.; Dang, W. Biodegradable Polymers: Poly (phosphoester)s. In Encyclopaedia of Controlled Drug Delivery; Mathowitz, E., Ed.; John Wiley and Sons: New York, NY, USA, 1999; Volume 1, pp. 45–60. [Google Scholar]
- Penczek, S.; Duda, A.; Kaluzynski, K.; Lapienis, G.; Nyk, A.; Szymanski, R. Thermodynamics and Kinetics of Ring-Opening Polymerization of Cyclic Alkylene Phosphates. Makromol. Chem. Macromol. Symp. 1993, 73, 91–101. [Google Scholar]
- Penczek, S.; Klosinski, P. Synthetic Polyphosphates Related to Nucleic and Teichoic Acids. In Models of Biopolymers by Ring-Opening Polymerization; Penczek, S., Ed.; CRC Press: Boca Raton, FL, USA, 1990; pp. 291–378. [Google Scholar]
- Penczek, S.; Lapienis, G.; Kaluzynski, K.; Nyk, A. Models of Biopolymers and Bioanalogous Polymers with Backbones of Poly(alkylene phosphate)s. Pol. J. Chem. 1994, 68, 2129–2142. [Google Scholar]
- Penczek, S.; Pretula, J.; Kaluzynski, K. Models of Biomacromolecules and Other Useful Structures Based on the Poly (alkylene phosphate) Chains. Pol. J. Chem. 2001, 75, 1171–1181. [Google Scholar]
- De Smedt, S.C.; Demeester, J.; Hennink, W.E. Cationic Polymer Based Gene Delivery Systems. Pharm. Res. 2000, 17, 113–126. [Google Scholar]
- Garnett, M.C. Gene-Delivery Systems Using Cationic Polymers. Crit. Rev. Ther. Drug Carrier Syst. 1999, 16, 147–207. [Google Scholar]
- Kabanov, A.V. Taking Polycation Gene Delivery Systems from in vitro to in vivo. Pharm. Sci. Technol. Today 1999, 2, 365–372. [Google Scholar]
- Pouton, C.W.; Seymour, L.W. Key Issues in Non-Viral Gene Delivery. Adv. Drug Deliv. Rev. 2001, 46, 187–203. [Google Scholar]
- Zhaob, Z.; Wanga, J.; Maoa, H.Q.C.; Leong, K.W. Polyphosphoesters in Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2003, 55, 483–499. [Google Scholar]
- Wang, J.; Mao, H.Q.; Leong, K.W. A Novel Biodegradable Gene Carrier Based on Polyphosphoester. J. Am. Chem. Soc. 2001, 123, 9480–9481. [Google Scholar]
- Wang, J.; Zhang, P.C.; Lu, H.F.; Ma, N.; Wang, S.; Mao, H.Q.; Leong, K.W.; Ong, L.; Leong, H.K.W. New Polyphosphoramidate with a Spermidine Side Chain as a Gene Carrier. J. Control. Release 2002, 83, 157–168. [Google Scholar]
- Wang, J.; Zhang, P.C.; Mao, H.Q.; Leong, K.W. Enhanced Gene Expression in Mouse Muscle by Sustained Release of Plasmid DNA Using PPE-EA as a Carrier. Gene Ther. 2002, 9, 1254–1261. [Google Scholar]
- Singh, A.; Krogman, N.R.; Sethuraman, S.; Nair, L.S.; Sturgeon, J.L.; Brown, P.W.; Laurencin, C.T.; Allcock, H.R. Effect of Side Group Chemistry on the Properties of Biodegradable L-Alanine Cosubstituted Polyphosphazenes. Biomacromolecules 2006, 7, 914–918. [Google Scholar]
- Nair, L.S.; Bhattacharyya, S.; Bender, J.D.; Greish, Y.E.; Brown, P.W.; Allcock, H.R.; Laurencin, C.T. Fabrication and Optimization of Methylphenoxy Substituted Polyphosphazene Nanofibers for Biomedical Applications. Biomacromolecules 2004, 5, 2212–2020. [Google Scholar]
- Seong, J.Y.; Jun, Y.J.; Kim, B.M.; Park, Y.M.; Sohn, Y.S. Synthesis and Characterization of Biocompatible Poly(organophosphazenes) Aiming for Local Delivery of Protein Drugs. Int. J. Pharm. 2006, 314, 90–96. [Google Scholar]
- Zhang, J.X.; Qiu, L.Y.; Zhu, K.J.; Jin, Y. Thermosensitive Micelles Self-Assembled by Novel N-Isopropylacrylamide Oligomer Grafted Polyphosphazene. Macromol. Rapid Commun. 2004, 25, 1563–1567. [Google Scholar]
- Zhang, J.X.; Li, X.J.; Qiu, L.Y.; Li, X.H.; Yan, M.Q.; Jin, Y.; Zhu, K.J. Indomethacin-Loaded Polymeric Nanocarriers Based on Amphiphilic Polyphosphazenes with Poly(N-isopropylacrylamide) and Ethyl Tryptophan as Side Groups: Preparation, in vitro and in vivo Evaluation. J. Control. Release 2006, 116, 322–329. [Google Scholar]
- Zheng, C.; Qiu, L.; Zhu, K. Novel Polymersomes Based on Amphiphilic Graft Polyphosphazenes and Their Encapsulation of Water-Soluble Anti-Cancer Drug. Polymer 2009, 50, 1173–1177. [Google Scholar]
- Allcock, H.R.; Kwon, S. An Ionically Crosslinkable Polyphosphazene: Poly[bis(carboxylatophenoxy)phosphazene] and Its Hydrogels and Membranes. Macromolecules 1989, 22, 75–79. [Google Scholar]
- Allcock, H.R.; Austin, P.E.; Neenan, T.X.; Sisko, J.T.; Blonsky, P.M.; Shriver, D.F. Polyphosphazenes with Etheric Side Groups: Prospective Biomedical and Solid Electrolyte Polymers. Macromolecules 1986, 19, 1508–1512. [Google Scholar]
- Andrianov, A.K. Water-Soluble Polyphosphazenes for Biomedical Applications. J. Inorg. Organomet. Polym. Mater. 2006, 16, 397–406. [Google Scholar]
- Andrianov, A.K.; Chen, J. Polyphosphazene Microspheres: Preparation by Ionic Complexation of Phosphazene Polyacids with Spermine. J. Appl. Polym. Sci. 2006, 101, 414–419. [Google Scholar]
- Andrianov, A.K.; Payne, L.G. Protein Release from Polyphosphazene Matrices. Adv. Drug Deliv. Rev. 1998, 31, 185–196. [Google Scholar]
- Andrianov, A.K.; Payne, L.G. Polymeric Carriers for Oral Uptake of Microparticulates. Adv. Drug Deliv. Rev. 1998, 34, 155–170. [Google Scholar]
- Andrianov, A.K.; Cohen, S.; Visscher, K.B.; Payne, L.G.; Allcock, H.R.; Langer, R. Controlled Release Using Ionotropic Polyphosphazene Hydrogels. J. Control. Release 1993, 27, 69–77. [Google Scholar]
- Sharma, B.R.; Naresh, L.; Dhuldhoya, N.C.; Merchant, S.U.; Merchant, U.C. Xanthan Gum—A Boon to Food Industry. Food Promot. Chron. 2006, 1, 27–30. [Google Scholar]
- Katzbauer, B. Properties and Applications of Xanthan Gum. Polym. Degrad. Stabil. 1998, 59, 81–84. [Google Scholar]
- Mukhiddinov, Z.K. Isolation and Structural Characterization of a Pectin Homo and Ramnogalacturonan. Talanta 2000, 53, 171–176. [Google Scholar]
- Sriamornsak, P. Chemistry of Pectin and Its Pharmaceutical Uses: A Review. Silpakorn Univ. Int. J. 2003, 3, 206–228. [Google Scholar]
- Rolin, C. Pectin. In Industrial Gums, 3rd ed.; Whistler, R.L., BeMiller, J.N., Eds.; Academic Press: New York, NY, USA, 1993. [Google Scholar]
- Paoletti, S. Chemistry and Function of Pectins; Fishman, M.L., Jen, J.J., Eds.; American Chemical Society: Washington, DC, USA, 1986. [Google Scholar]
- Ginter, E. Natural Hypocholesterolemic Agent: Pectin Plus Ascorbic Acid. Int. J. Vitic. Nat. Res. 1979, 49, 406–408. [Google Scholar]
- Kohn, R. Binding of Toxic Cations to Pectin, Its Oligomeric Fragment and Plant Tissues. Carbohydr. Polym. 1982, 2, 273–275. [Google Scholar]
- Slany, J. Study of Functional Action of Citrus Pectins in Tablets. Ceska a Slovenska Farmacie 1981, 30, 195–200. [Google Scholar]
- Slany, J. Evaluation of Tablets with Pectin as a Binding Agent. Farmaceuticky Obzor. 1981, 50, 491–498. [Google Scholar]
- Krusteva, S. Pharmaceutical Investigation of a Bioerodible Nystatin System. Pharmazie 1990, 45, 195–197. [Google Scholar]
- Khan, T.A.; Khiang, P.K. Reporting Degree of Deacetylation Values of Chitosan: The Influence of Analytical Methods. J. Pharm. Pharm. Sci. 2002, 5, 205–212. [Google Scholar]
- Masatoshi, S.; Minoru, M.; Hitoshi, S.; Hiroyuki, S.; Yoshihiro, S. Preparation and Characterization of Water-Soluble Chitin and Chitosan Derivatives. Carbohydr. Polym. 1998, 36, 49–59. [Google Scholar]
- TienAn, N.; Thien, D.T.; Dong, N.T.; Dung, P.L. Water-Soluble N-Carboxymethylchitosan Derivatives: Preparation, Characteristics and Its Application. Carbohydr. Polym. 2009, 75, 489–497. [Google Scholar]
- Chen, X.-G.; Park, H.-J. Chemical Characteristics of O-Carboxymethyl Chitosans Related to the Preparation Conditions. Carbohydr. Polym. 2003, 53, 355–359. [Google Scholar]
- de Abreu, F.R.; Campana-Filho, S.P. Preparation and Characterization of Carboxymethylation. Polímeros 2005, 15, 79–83. [Google Scholar]
- Ge, H.C.; Luo, D.K. Preparation of Carboxymethyl Chitosan in Aqueous Solution under Microwave Irradiation. Carbohydr. Res. 2005, 340, 1351–1356. [Google Scholar]
- Paulino, A.T.; Minasse, F.A.; Guilherme, M.R.; Reis, A.V.; Muniz, E.C.; Nozaki, J. Novel Adsorbent Based on Silkwormchrysalides for Removal of Heavy Metals froms Wastewaters. J. Colloid Interface Sci. 2006, 301, 479–487. [Google Scholar]
- Shengling, S.; Aiqin, W. Adsorption Properties and Mechanism of Crosslinked-Carboxymethyl Chitosan Resin with Zn(II) as Template Ion. React. Funct. Polym. 2006, 66, 819–826. [Google Scholar]
- Shengling, S.; Wang, L.; Wang, A. Adsorption Properties of Crosslinked-Carboxymethyl Chitosan Resin with Pb(II) as Template Ions. J. Hazard. Mater. 2006, 136, 930–937. [Google Scholar]
- Pharmacosmos. Dextran Structure. Available online: http://www.dextran.net/dextran-structure.html (accessed on 28 June 2011).
- van Dijk-Wolthuis, W.N.E.; Hoogeboom, J.A.M.; van Steenbergen, M.J.; Tsang, S.K.Y.; Hennink, W.E. Degradation and Release Behavior of Dextran-Based Hydrogels. Macromolecules 1997, 30, 4639–4645. [Google Scholar]
- Stenekes, R.J.H.; Franssen, O.; van Bommel, E.M.G.; Crommelin, D.J.A.; Hennink, W.E. The Preparation of Dextran Microspheres in an All-Aqueous System: Effects of the Formulation Parameters on Particle Characteristics. Pharm. Res. 1998, 15, 557–561. [Google Scholar]
- Franssen, O.; Vandervennet, L.; Roders, P.; Hennink, W.E. Degradable Dextran Hydrogels: Controlled Release of a Model Protein from Cylinders and Microspheres. J. Control. Release 1999, 60, 211–221. [Google Scholar]
- Lévesque, S.G.; Shoichet, M.S. Synthesis of Cell-Adhesive Dextran Hydrogels and Macroporous Scaffolds. Biomaterials 2006, 27, 5277–5285. [Google Scholar]
- Ferreira, L.; Gil, M.H.; Cabrita, A.M.S.; Dordick, J.S. Biocatalytic Synthesis of Highly Ordered Degradable Dextran-Based Hydrogels. Biomaterials 2005, 26, 4707–4716. [Google Scholar]
- De Jong, S.J.; De Smedt, S.C.; Demeester, J.; van Nostrum, C.F.; Kettenes-van den Bosch, J.J.; Hennink, W.E. Biodegradable Hydrogels Based on Stereocomplex Formation between Lactic Acid Oligomers Grafted to Dextran. J. Control. Release 2001, 72, 47–56. [Google Scholar]
- Torchilin, V.P.; Maksimenko, A.V.; Mazaev, A.V. Immobilized Thrombolytic Enzymes for Systemic and Local Application. Ann. N. Y. Acad. Sci. 2006, 501, 481–486. [Google Scholar]
- Torchilin, V.P. Immobilized Enzymes and the Use of Immobilization Principles for Drug Targeting. In Targeted Drugs; Goldberg, E.D., Ed.; Wiley: New York, NY, USA, 1983; pp. 127–152. [Google Scholar]
- Torchilin, V.P.; Voronkov, I.; Mazaev, A.V. Use of Immobilized Streptokinase (Streptodecase) for Treating Thromboses. Terapevt Arkh. 1982, 54, 21–25. [Google Scholar]
- Voronkov, Y.; Torchilinv, V.P. Immobilized Streptokinase (Streptodekaza) in Treatment of Intravitreal Haemorrhage. Vestnik Oftalmologii (Russ.) 1982, 4, 61–64. [Google Scholar]
- Kalia, A.N. Text Book of Industrial Pharmacognosy; CBS Publishers: New Delhi, India, 2005; p. 217. [Google Scholar]
- Evans, W.C.; Evans, D.; Trease, G.E. Trease and Evans Pharmacognosy, 15th ed.; Saunders/Elsevier: Edinburgh, UK, 2002; p. 206. [Google Scholar]
- Paul, C.S.; Richard, L.M. McGraw-Hill Encyclopedia of Science and Technology, 7th ed.; McGraw-Hill Publishing Company: New York, NY, USA, 1992; p. 285. [Google Scholar]
- Gennaro, A.R. Remington the Science and Practice of Pharmacy, 20th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; Volume 1, p. 1031. [Google Scholar]
- Lev, R.; Long, R.; Mallonga, L.; Schnaram, R.; Reily, W. Evaluation of Carrageenan as Base for Topical Gels. Pharm. Res. 1997, 14, 42. [Google Scholar]
- Lui, Y.; Schnaram, R.; Reily, W. Evaluation of Carrageenan as Suppository Base. Pharm. Res. 1997, 14, 41. [Google Scholar]
- Tomolin, J.; Taylor, J.S.; Read, N.W. The Effect of Mixed Faecal Bacteria on a Selection of Viscous Polysaccharide in vitro. Nutr. Rep. Int. 1989, 39, 121–135. [Google Scholar]
- Bayliss, C.E.; Houston, A.P. Degradation of Guar Gum by Faecal Bacteria. Appl. Environ. Microbiol. 1986, 48, 626–632. [Google Scholar]
- Macfarlane, G.T.; Hay, S.; Macfarlane, S.; Gibson, G.R. Effect of Different Carbohydrates on Growth Polysaccharidases and Glycosidase Production of Bacteroides Ovatus in Batch and Continuous Culture. J. Appl. Bacteriol 1990, 68, 179–187. [Google Scholar]
- Chourasia, M.K.; Jain, S.K. Potential of Guar Gum Microspheres for Target Specific Drug Release to Colon. J. Drug Target. 2004, 2, 1–8. [Google Scholar]
- Rama Prasad, Y.V.; Krishnaiah, Y.S.R.; Satyanarayana, S. In vitro Evaluation of Guar Gum as a Carrier for Colon-Specific Drug Delivery. J. Control. Release 1998, 51, 281–287. [Google Scholar]
- Krishnaiah, Y.S.R.; Satyanarayana, S.; Rama Prasad, Y.V.; Narasimha Rao, S. Gamma Scintigraphic Studies on Guar Gum Matrix Tablets for Colonic Drug Delivery in Healthy Subjects. J. Control. Release 1998, 55, 245–252. [Google Scholar]
- Krishnaiah, Y.S.R.; Satyanarayana, S.; Rama Prasad, Y.V.; Arasimha Rao, S. Evaluation of Guar Gum as a Compression Coat for Drug Targeting to Colon. Int. J. Pharm. 1998, 171, 137–146. [Google Scholar]
- Clasen, C.; Kulicke, W.M. Determination of Viscoelastic and Rheo-Optical Material Functions of Water Soluble Cellulose Derrivatives. Prog. Polym. Sci. 2001, 26, 1839–1919. [Google Scholar]
- Rokhade, A.P.; Agnihotri, S.A.; Patil, S.A.; Mallikarjuna, N.N.; Kulkarni, P.V.; Aminabhavi, T.M. Semi-Interpenetrating Polymer Network Microspheres of Gelatin and Sodium Carboxymethyl Cellulose for Controlled Release of Ketorolac Tromethamine. Carbohydr. Polym. 2006, 65, 243–252. [Google Scholar]
- Nagai, T.; Machida, Y. Advances in Drug Delivery: Mucosal Adhesive Dosage Forms. Pharm. Int. 1985, 6, 196–200. [Google Scholar]
- Satoh, K.; Takayama, K.; Machida, Y.; Suzuki, Y.; Nakagaki, M.; Nagai, T. Factors Affecting the Bioadhesive Property of Tablets Consisting of Hydroxypropyl Cellulose and Carboxyvinyl Polymer. Chem. Pharm. Bull. 1989, 37, 1366–1368. [Google Scholar]
- Senel, S.; Hincal, A.A. Drug Permeation Enhancement via Buccal Route: Possibilities and Limitations. J. Control. Release 2001, 72, 133–144. [Google Scholar]
- Okamoto, H.; Nakamori, T.; Arakawa, Y.; Iida, K.; Danjo, K. Development of Polymer Film Dosage Forms of Lidocaine for Buccal Administration. II. Comparison of Preparation Methods. J. Pharm. Sci. 2002, 91, 2424–2432. [Google Scholar]
- Katzhendler, I.; Mader, K.; Friedman, M. Structure and Hydration Properties of Hydroxypropyl Methylcellulose Matrices Containing Naproxen and Naproxen Sodium. Int. J. Pharm. 2000, 200, 161–179. [Google Scholar]
- Ito, T.; Iida-Tanaka, N.; Koyama, Y. Efficient in vivo Gene Transfection by Stable DNA/PEI Complexes Coated by Hyaluronic Acid. J. Drug Target. 2008, 16, 276–281. [Google Scholar]
- Abetamann, V.; Kern, H.F.; Elsasser, H.P. Differential Expression of the Hyaluronan Receptors CD44 and RHAMM in Human Pancreatic Cancer Cells. Clin. Cancer Res. 1996, 2, 1607–1618. [Google Scholar]
- Li, H.; Guo, L.; Li, J.W.; Liu, N.; Qi, R.; Liu, J. Expression of Hyaluronan Receptors CD44 and RHAMM in Stomach Cancers: Relevance with Tumor Progression. Int. J. Oncol. 2000, 17, 927–932. [Google Scholar]
- Kobayashi, Y.; Okamoto, A.; Nishinari, K. Viscoelasticity of Hyaluronic Acid with Different Molecular Weights. Biorheology 1994, 31, 235–244. [Google Scholar]
- Parka, J.W.; Chakrabartib, B. Conformational Transition of Hyaluronic Acid Carboxylic Group Participation and Thermal Effect. Biochim. Biophys. Acta 1978, 541, 263–269. [Google Scholar]
- Bourlais, C.L.; Acar, L.; Zia, H.; Sado, P.A.; Needham, T.; Leverge, R. Ophthalmic Drug Delivery Systems—Recent Advances. Prog. Retin. Eye Res. 1998, 17, 33–58. [Google Scholar]
- Bucolo, C.; Mangiafico, S.; Spadaro, A. Methylprednisolone Delivery by Hyalobend Corneal Shields and Its Effects on Rabbit Ocular Inflammation. J. Ocul. Pharmacol. Ther. 1996, 12, 141–149. [Google Scholar]
- Cho, K.Y.; Chung, T.W.; Kim, B.C.; Kim, M.K.; Lee, J.H.; Wee, W.R.; Cho, C.S. Release of Ciprofloxacin from Poloxamer-graft-Hyaluronic Acid Hydrogels in vitro. Int. J. Pharm. 2003, 260, 83–91. [Google Scholar]
- de la Fuente, M.; Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. Nanoparticle as Protein and Gene Carriers to Mucosal Surfaces. Nanomedicine 2008, 3, 845–857. [Google Scholar]
- Platt, V.M.; Szoka, F.C., Jr. Anticancer Therapeutics: Targeting Macromolecules and Nanocarriers to Hyaluronan or CD44, a Hyaluronan Receptor. Mol. Pharm. 2008, 5, 474–486. [Google Scholar]
- Oyarzun-Ampuero, F.A.; Brea, J.; Loza, M.I.; Torres, D.; Alonso, M.J. Chitosan Hyaluronic Acid Nanoparticles Loaded with Heparin for the Treatment of Asthma. Int. J. Pharm. 2009, 381, 122–129. [Google Scholar]
- Price, R.D.; Berry, M.G.; Navsaria, H.A. Hyaluronic Acid: The Scientific and Clinical Evidence. J. Plast. Reconstruct. Aesth. Surg. 2007, 60, 1110–1119. [Google Scholar]
- McDonald, C.C.; Kaye, S.B.; Figueiredo, F.C.; Macintosh, G.; Lockett, C. A Randomised, Crossover, Multicentre Study to Compare the Performance of 0.1% (w/v) Sodium Hyaluronate with 1.4% (w/v) Polyvinyl Alcohol in the Alleviation of Symptoms Associated with Dry Eye Syndrome. Eye 2002, 16, 601–607. [Google Scholar]
- Aragona, P.; Di Stefano, G.; Ferreri, F.; Spinella, R.; Stilo, A. Sodium Hyaluronate Eye Drops of Different Osmolarity for the Treatment of Dry Eye in Sjogren's Syndrome Patients. Br. J. Ophthalmol. 2002, 86, 879–884. [Google Scholar]
- Bray, B.A. The Role of Hyaluronan in the Pulmonary Alveolus. J. Theor. Biol. 2001, 210, 121–130. [Google Scholar]
- Prestwich, G.D.; Vercruysse, K.P. Profiles Therapeutic Applications of Hyaluronic Acid and Hyaluronan Derivatives. Pharm. Sci. Technol. Today 1998, 1, 42–43. [Google Scholar]
- Peters, T. Serum Albumin. Adv. Protein Chem. 1985, 37, 161–245. [Google Scholar]
- Kratz, F.; Fichtner, I.; Schumarcher, P.; Roth, T.; Feibig, H.H.; Unger, C. Antitumor Activity of Acid Labile Transferrin and Albumin Doxorubicin Conjugates in vitro and in vivo Human Tumor Xerograft Models. Eur. J. Cancer 1997, 33, S175. [Google Scholar]
- Rahimnejad, M.; Jahanshahi, M.; Najafpour, G.D. Production of Biological Nanoparticles from Bovine Serum Albumin for Drug Delivery. Afr. J. Biotechnol. 2006, 5, 1918–1923. [Google Scholar]
- Langer, K.; Balthasar, S.; Vogel, V.; Dinauer, N.; Von Briesen, H.; Schubert, D. Optimization of the Preparation Process for Human Serum Albumin (HSA) Nanoparticles. Int. J. Pharm. 2003, 257, 169–180. [Google Scholar]
- Rubino, O.P.; Kowalsky, R.; Swarbrick, J. Albumin Microspheres as a Drug Delivery System: Relation among Turbidity Ratio, Degree of Cross-Linking and Drug Release. Pharm. Res. 1993, 10, 1059–1065. [Google Scholar]
- Kommareddy, S.; Amiji, M. Preparation and Evaluation of Thiol-Modified Gelatin Nanoparticles for Intracellular DNA Delivery in Response to Glutathione. Bioconjugate Chem. 2005, 16, 1423–1432. [Google Scholar]
- Azarmi, S.; Tao, X.; Chen, H.; Wang, Z.; Finlay, W.H.; Löbenberg, R.; Roa, W.H. Formulation and Cytotoxicity of Doxorubicin Nanoparticles Carried by Dry Powder Aerosol Particles. Int. J. Pharm. 2006, 319, 155–161. [Google Scholar]
- Ulbrich, K.; Hekmatara, T.; Herbert, E.; Kreuter, J. Transferrin- and Transferrin-Receptor-Antibody-Modified Nanoparticles Enable Drug Delivery across the Blood-Brain Barrier (BBB). Eur. J. Pharm. Biopharm. 2009, 71, 251–256. [Google Scholar]
- Steinhauser, I.M.; Langer, K.; Strebhardt, K.M.; Spänkuch, B. Effect of Trastuzumab-Modified Antisense Oligonucleotide-Loaded Human Serum Albumin Nanoparticles Prepared by Heat Denaturation. Biomaterials 2008, 29, 4022–4028. [Google Scholar]
- Damascelli, B.; Cantù, G.; Mattavelli, F.; Tamplenizza, P.; Bidoli, P.; Leo, E.; Dosio, F.; Cerrotta, A.M.; di Tolla, G.; Frigerio, L.F.; et al. Intraarterial Chemotherapy with Polyoxyethylated Castor Oil Free Paclitaxel, Incorporated in Albumin Nanoparticles (ABI-007): Phase II Study of Patients with Squamous Cell Carcinoma of the Head and Neck and Anal Canal: Preliminary Evidence of Clinical Activity. Cancer 2001, 92, 2592–2602. [Google Scholar]
- Ibrahim, N.K.; Desai, N.; Legha, S.; Soon-Shiong, P.; Theriault, R.L.; Rivera, E.; Esmaeli, B.; Ring, S.E.; Bedikian, A.; Hortobagyi, G.N.; Ellerhorst, J.A. Phase I and Pharmacokinetic Study of ABI-007, a Cremophor-Free, Protein-Stabilized, Nanoparticle Formulation of Paclitaxel. Clin. Cancer. Res. 2002, 8, 1038–1044. [Google Scholar]
- Geny, B.; Mettauer, B.; Muan, B.; Bischoff, P.; Epailly, E.; Piquard, F.; Eisenmann, B.; Haberey, P. Safety and Efficacy of a New Transpulmonary Echo Contrast Agent in Echocardiographic Studies in Patients. J. Am. Coll. Cardiol. 1993, 22, 1193–1198. [Google Scholar]
- Brzoska, M.; Langer, K.; Coester, C.; Loitsch, S.; Wagner, T.O.; Mallinckrodt, C. Incorporation of Biodegradable Nanoparticles into Human Airway Epithelium Cells—In Vitro Study of the Suitability as a Vehicle for Drug or Gene Delivery in Pulmonary Diseases. Biochem. Biophys. Res. Commun. 2004, 318, 562–570. [Google Scholar]
- Simoes, S.; Slepushkin, V.; Pires, P.; Gaspar, R.; de Lima, P.M.C.; Duzgunes, N. Human Serum Albumin Enhances DNA Transfection by Lipoplexes and Confers Resistance to Inhibition by Serum. Biochim. Biophys. Acta 2004, 1463, 459–469. [Google Scholar]
- Roper, H. Applications of Starch and Its Derivatives. Carbohydr. Eur. 1996, 15, 14–21. [Google Scholar]
- Vilivalam, V.D.; Illum, L.; Iqbal, K. Starch Capsules: An Alternative System for Oral Drug Delivery. Pharm. Sci. Technol. Today 2000, 3, 64–69. [Google Scholar]
- Milojevic, S.; Newton, J.M.; Cummings, J.H.; Gibson, G.R.; Botham, R.L.; Ring, S.G.; Stockham, M.; Allwood, M.C. Amylose as a Coating for Drug Delivery to the Colon: Preparation and in vitro Evaluation Using 5-Aminosalicylic Acid Pellets. J. Control. Release 1996, 38, 75–84. [Google Scholar]
- Désévaux, C.; Dubreuil, P.; Lenaerts, V.; Girard, C. Tissue Reaction and Biodegradation of Implanted Cross-Linked High Amylose Starch in Rats. J. Biomed. Mater. Res. 2002, 63, 772–779. [Google Scholar]
- Mulhbacher, J.; Ispas-Szabo, P.; Lenaerts, V.; Mateescu, M.A. Cross-Linked High Amylose Starch Derivatives as Matrices for Controlled Release of High Drug Loadings. J. Control. Rel. 2001, 76, 51–58. [Google Scholar]
- Nabais, T.; Brouillet, F.; Kyriacos, S.; Mroueh, M.; Amores da Silva, P.; Bataille, B.; Chebli, C.; Cartilier, L. High-Amylose Carboxymethyl Starch Matrices for Oral Sustained Drug-Release: In vitro and in vivo Evaluation. Eur. J. Pharm. Biopharm. 2007, 65, 371–378. [Google Scholar]
- Teramoto, N.; Motoyama, T.; Yosomiya, R.; Shibata, M. Synthesis, Thermal Properties, and Biodegradability of Propyl-Etherified Starch. Eur. Polym. J. 2003, 39, 255–261. [Google Scholar]
- Araújo, M.A.; Cunha, A.; Mota, M. Enzymatic Degradation of Starch-Based Thermoplastic Compounds Used in Protheses: Identification of the Degradation Products in Solution. Biomaterials 2004, 25, 2687–2693. [Google Scholar]
- Zhang, J.F.; Sun, X.Z. Mechanical Properties of PLA/Starch Composites Compatibilized by Maleic Anhydride. Biomacromolecules 2004, 5, 1446–1451. [Google Scholar]
- Pareta, R.; Edirisinghe, M.J. A Novel Method for the Preparation of Starch Films and Coatings. Carbohydr. Polym. 2006, 63, 425–431. [Google Scholar]
- Choi, E.J.; Kim, C.H.; Park, J.K. Synthesis and Characterization of Starch-g-Polycaprolactone Copolymer. Macromolecules 1999, 32, 7402–7408. [Google Scholar]
- Lu, D.R.; Xiao, C.M.; Xu, S.J. Starch-Based Completely Biodegradable Polymer Materials. eXPRESS Polym. Lett. 2009, 3, 366–375. [Google Scholar]
- Pal, J.; Singhal, R.S.; Kulkarni, P.R. Physicochemical Properties of Hydroxypropyl Derivative from Corn and Amaranth Starch. Carbohydr. Polym. 2002, 48, 49–53. [Google Scholar]
- Fanta, G.F.; Burr, R.C.; Doane, W.M.; Russell, C.R. Graft Polymerization of Vinyl Acetate onto Starch. Saponification to Starch-g-Poly(vinyl alcohol). J. Appl. Polym. Sci. 1979, 23, 229–240. [Google Scholar]
- Simi, C.K.; Abraham, T.E. Hydrophobic Grafted and Crosslinked Starch Nanoparticles for Drug Delivery. Bioprocess Biosyst. Eng. 2007, 30, 173–180. [Google Scholar]
- Samaha, S.H.; Nasr, H.E.; Hebeish, A. Synthesis and Characterization of Starch-Poly(vinyl acetate) Graft Copolymer and Their Saponified Form. J. Polym. Res. 2005, 12, 343–353. [Google Scholar]
- Xiao, C.M.; Yang, M.L. Controlled Preparation of Physical Cross-Linked Starch-g-PVA Hydrogel. Carbohydr. Polym. 2006, 64, 37–40. [Google Scholar]
- Zhu, Z.F.; Zhuo, R.X. Slow Release Behavior of Starch-g-Poly(vinyl alcohol) Matrix for 2,4,5-Trichlorophenoxyacetic Acid Herbicide. Eur. Polym. J. 2001, 37, 1913–1919. [Google Scholar]
- Marques, A.P.; Reis, R.L.; Hunt, J.A. The Biocompatibility of Novel Starch-Based Polymers and Composites: In Vitro Studies. Biomaterials 2002, 23, 1471–1478. [Google Scholar]
- Mendes, S.C.; Reis, R.L.; Bovell, Y.P.; Cunha, A.M.; van Blitterswijk, C.A.; de Bruijn, J.D. Biocompatibility Testing of Novel Starch-Based Materials with Potential Application in Orthopaedic Surgery: A Preliminary Study. Biomaterials 2001, 22, 2057–2064. [Google Scholar]
- Azevedo, H.S.; Gama, F.M.; Reis, R.L. In vitro Assessment of the Enzymatic Degradation of Several Starch Based Biomaterials. Biomacromolecules 2003, 4, 1703–1712. [Google Scholar]
- Defaye, J.; Wong, E. Structural Studies of Gum Arabic, the Exudate Polysaccharide from Acacia Senegal. Carbohydr. Res. 1986, 150, 221–231. [Google Scholar]
- Reddy, S.M.; Sinha, V.R.; Reddy, D.S. Novel Oral Colon-Specific Drug Delivery Systems for Pharmacotherapy of Peptides and Nonpeptide Drugs. Drugs Today 1999, 35, 537–580. [Google Scholar]
- Sinha, V.R.; Kumria, R. Polysaccharides in Colon-Specific Drug Delivery. Int. J. Pharm. 2001, 224, 19–38. [Google Scholar]
- Boesel, L.F.; Mano, J.F.; Reis, R.L. Optimization of the Formulation and Mechanical Properties of Starch Based Partially Degradable Bone Cements. J. Mater. Sci. Mater. Med. 2004, 15, 73–83. [Google Scholar]
- Gomes, M.E.; Sikavitsas, V.I.; Behravesh, E.; Reis, R.L.; Mikos, A.G. Effect of Flow Perfusion on the Osteogenic Differentiation of Bone Marrow Stromal Cells Cultured on Starch-Based Three-Dimensional Scaffolds. J. Biomed. Mater. Res. Part A 2003, 67, 87–95. [Google Scholar]
- Balmayor, E.R.; Tuzlakoglu, K.; Marques, A.P.; Azevedo, H.S.; Reis, R.L. A Novel Enzymatically Mediated Drug Delivery Carrier for Bone Tissue Engineering Applications: Combining Biodegradable Starch Based Microparticles and Differentiation Agents. J. Mater. Sci. Mater. Med. 2008, 19, 1617–1623. [Google Scholar]
- Reis, A.V.; Guilherme, M.R.; Moia, T.A.; Mattoso, L.H.C.; Muniz, E.C.; Tambourgi, E.B. Synthesis and Characterization of a Starch-Modified Hydrogel as Potential Carrier for Drug Delivery System. J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 2567–2574. [Google Scholar]
- Herman, J.; Remon, J.P. Modified Starches as Hydrophilic Matrices for Controlled Oral Delivery. II. In Vitro Drug Release Evaluation of Thermally Modified Starches. Int. J. Pharm. 1989, 56, 65–70. [Google Scholar]
- Te Wierik, G.H.P.; Eissens, A.C.; Bergsma, J.; Arends-Scholte, A.W.; Lerk, C.F. A New Generation of Starch Products as Excipient in Pharmaceutical Tablets. II. High Surface Area Retrograded Pregelatinized Potato Starch Products in Sustained Release Tablets. J. Control. Release 1997, 45, 25–33. [Google Scholar]
- Le Bail, P.; Morin, F.G.; Marchessault, R.H. Characterization of a Crosslinked High Amylose Starch Excipient. Int. J. Biol. Macromol. 1999, 26, 193–200. [Google Scholar]
- Chebli, C.; Cartilier, L.; Hartman, N.G. Substituted Amylose as a Matrix for Sustained-Drug Release: A Biodegradation Study. Int. J. Pharm. 2001, 222, 183–189. [Google Scholar]
- Yoon, H.-S.; Kweon, D.-K.; Lim, S.-T. Effects of Drying Process for Amorphous Waxy Maize Starch on Theophylline Release from Starch-Based Tablets. J. Appl. Polym. Sci. 2007, 105, 1908–1913. [Google Scholar]
- Mulhbacher, J.; Ispas-Szabo, P.; Mateescu, M.A. Cross-Linked High Amylase Starch Derivatives for Drug Release. II. Swelling Properties and Mechanistic Study. Int. J. Pharm. 2004, 278, 231–238. [Google Scholar]
- Dumoulin, Y.; Alex, S.; Szabo, P.; Cartilier, L.; Mateescu, M.A. Cross-Linked Amylase as Matrix for Drug Controlled Release. X-Ray and FT-IR Structural Analysis. Carbohydr. Polym. 1998, 37, 361–370. [Google Scholar]
- Zhang, L.M.; Yang, C.; Yan, L. Perspectives on: Strategies to Fabricate Starch Based Hydrogels with Potential Biomedical Applications. J. Bioact. Compat. Polym. 2005, 20, 297–314. [Google Scholar]
- Onofre, F.O.; Wang, Y.J. Hydroxypropylated Starches of Varying Amylose Contents as Sustained Release Matrices in Tablets. Int. J. Pharm. 2010, 385, 104–112. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kadajji, V.G.; Betageri, G.V. Water Soluble Polymers for Pharmaceutical Applications. Polymers 2011, 3, 1972-2009. https://doi.org/10.3390/polym3041972
Kadajji VG, Betageri GV. Water Soluble Polymers for Pharmaceutical Applications. Polymers. 2011; 3(4):1972-2009. https://doi.org/10.3390/polym3041972
Chicago/Turabian StyleKadajji, Veeran Gowda, and Guru V. Betageri. 2011. "Water Soluble Polymers for Pharmaceutical Applications" Polymers 3, no. 4: 1972-2009. https://doi.org/10.3390/polym3041972
APA StyleKadajji, V. G., & Betageri, G. V. (2011). Water Soluble Polymers for Pharmaceutical Applications. Polymers, 3(4), 1972-2009. https://doi.org/10.3390/polym3041972