Structural and Mechanical Hysteresis at the Order-Order Transition of Block Copolymer Micellar Crystals

Abstract
: Concentrated solutions of a water-soluble block copolymer (PEO)20-(PPO)70-(PEO)20 show a thermoreversible transition from a liquid to a gel. Over a range of concentration there also exists an order-order transition (OOT) between cubically-packed spherical micelles and hexagonally-packed cylindrical micelles. This OOT displays a hysteresis between the heating and cooling transitions that is observed at both the macroscale through rheology and nanoscale through small angle neutron scattering (SANS). The hysteresis is caused by the persistence of the cubically-packed spherical micelle phase into the hexagonally-packed cylindrical micelle phase likely due to the hindered realignment of the spherical micelles into cylindrical micelles and then packing of the cylindrical micelles into a hexagonally-packed cylindrical micelle phase. This type of hysteresis must be fully characterized, and possibly avoided, for these block copolymer systems to be used as templates in nanocomposites.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Amphiphilic block copolymers self assemble into micelles when dispersed in a selective solvent, or a solvent that has preferential solubility of one of the blocks. At higher polymer concentrations, this selectivity leads to the formation of micelles and spontaneous packing of micelles into crystal lattices. In some amphiphilic block copolymers solutions, solvent selectivity is brought on by changing the temperature and, in that case, ordered packed micelle phases exhibit the property of thermoreversibility between a disordered liquid phase and an ordered micelle phase. In thermoreversible systems, the amphiphilic block copolymer solutions easily transition between the disordered and ordered phases [1,2].
Amphiphilic block copolymers of poly(ethylene oxide)n-poly(propylene oxide)m-poly(ethylene oxide)n are known by the trade names Pluronic®, Synperonic® or Polyoxamer® and are widely studied. The thermodynamics, phase behavior, and thermoreversible gelation of this set of block copolymers are well known [3-12]. At temperatures below about 15 °C both blocks are soluble in water. As the temperature is increased, the middle poly(propylene oxide) block becomes dehydrated and the block copolymers self assemble into micelles. At high enough concentrations, these micelles will pack into ordered micelle phases [7]. This ordered phase formation is a thermoreversible disorder-order transition (ODT) between a disordered micelle liquid phase and an ordered micelle, or crystal, phase. The details of the crystal packing depend on the relative block molecular weights of the block copolymer [6]. Many of these systems also exhibit order-order transitions (OOT) between different micellar crystal phases; transitions induced by changes in temperature (solvent quality) or concentration.
One potential application of these nanostructured materials is for the spatial control of nanoparticulate material [13]. A simple and general method to template hydrophilic nanoparticles and proteins into the interstitial spaces of aqueous block copolymer micelle crystal phases to form nanocomposite materials has been presented previously for the cubically-packed micelle phase and the hexagonally-packed cylindrical micelle phase of different block copolymer architectures [14-17]. A specific architecture of Pluronic®, denoted P123, is used as a template for nanoparticles in both its cubically-packed spherical micelle and hexagonally-packed cylindrical micelle phases [18]. Block copolymers often have more than one phase and each OOT must be fully understood before they can be used effectively as templates. The Pluronic® P123 phase diagram with ordered phases of cubically-packed spherical micelles and hexagonally-packed cylindrical micelles was shown previously, but the dynamics and reversibility of the OOT were not studied in detail [6]. A recent report of the hysteresis in this system using viscosity and grazing-incidence SANS shows persistent structures near a solid surface and the complexity of the phase diagram [19]. A thermoreversible order-order transition (OOT) between face-centered cubic (FCC) and body-centered cubic (BCC) phases has been observed previously for block copolymer solutions of Pluronics® [6,20,21] and other block copolymer solutions and melts [22-26].
In this work, we focus on the OOT between the cubically-packed spherical micelle and hexagonally-packed cylindrical micelle phases of the block copolymer Pluronic® P123, (PEO)20-(PPO)70-(PEO)20, at both the macroscale and the nanoscale. This OOT exhibits a hysteresis between the heating and cooling which is characterized with both rheology and small angle neutron scattering (SANS). Many approaches are used to alter the micellar crystals and phase behavior of Pluronic® systems, including variation of the selective solvent or mixtures of different triblocks [27-29]. Here, we will base our work on the neat system of Pluronic® P123 and water.
2. Materials and Methods
Pluronic® P123 has a reported chemical formula of (PEO)20-(PPO)70-(PEO)20 and was donated by BASF (Mount Olive, NJ). The Pluronic® P123 has less than 0.4 wt% water content and was used as received with no further purification. All block copolymer samples were dissolved in de-ionized water (conductivity of greater than 18 MΩ). For SANS experiments, the block copolymer samples were dissolved in 99.8% deuterium oxide purchased from Cambridge Isotopes (Andover, MA). Previous work shows minimal to no difference in the position or behavior of the gelation transition between samples prepared with either H2O or D2O. Experiments performed as part of a contrast variation study showed minimal to no change in micelle spacing or dimensions for these samples as the H2O to D2O ratio was changed [30]. Based on these observations, we assume that there is no practical different between block copolymer solutions prepared in light and heavy water. To prepare samples, concentrated block copolymer solution was diluted with de-ionized water or deuterium oxide and the samples gently shaken to promote mixing. Samples were allowed to equilibrate for several days at 10 °C to a homogenous solution. This process has been well characterized and documented previously [14-16,18].
Rheology experiments were performed on a stress-controlled SR-5 rheometer (T.A. Instruments, New Castle, DE). Parallel plates with a diameter of 25 mm and a 0.8 mm gap were utilized for all samples. Oscillatory rheology experiments were performed with stress amplitudes in the linear regime (verified at several frequencies). The temperature was varied using a Peltier heating element to control the temperature of the bottom plate, the top plate is not thermally controlled. All samples were loaded cold (T ˜10 °C) and brought to the experimental conditions. Rheology experiments covering the temperature range from 5–45 °C were conducted at a variety of heating/cooling rates ranging from 1 °C/min to 0.1 °C/min. Rheological samples were loaded and then equilibrated for half an hour at the temperature of interest for thermal history experiments prior to testing unless otherwise stated. A thin layer of mineral oil from Sigma Aldrich (St. Louis, MO) was applied to the outer edge of the samples to prevent evaporation of the sample at the edge. Control samples were run with and without the oil at equivalent conditions to ensure that the oil did not impact the measurements.
Birefringence was probed qualitatively by observing back lit samples between linear sheet polarizers with perpendicularly aligned polarization directions, or crossed polarizers. Samples were heated in a water bath for 1 hour prior to birefringence characterization to achieve thermal equilibration. All thermally equilibrated samples were visually inspected for clarity and birefringence.
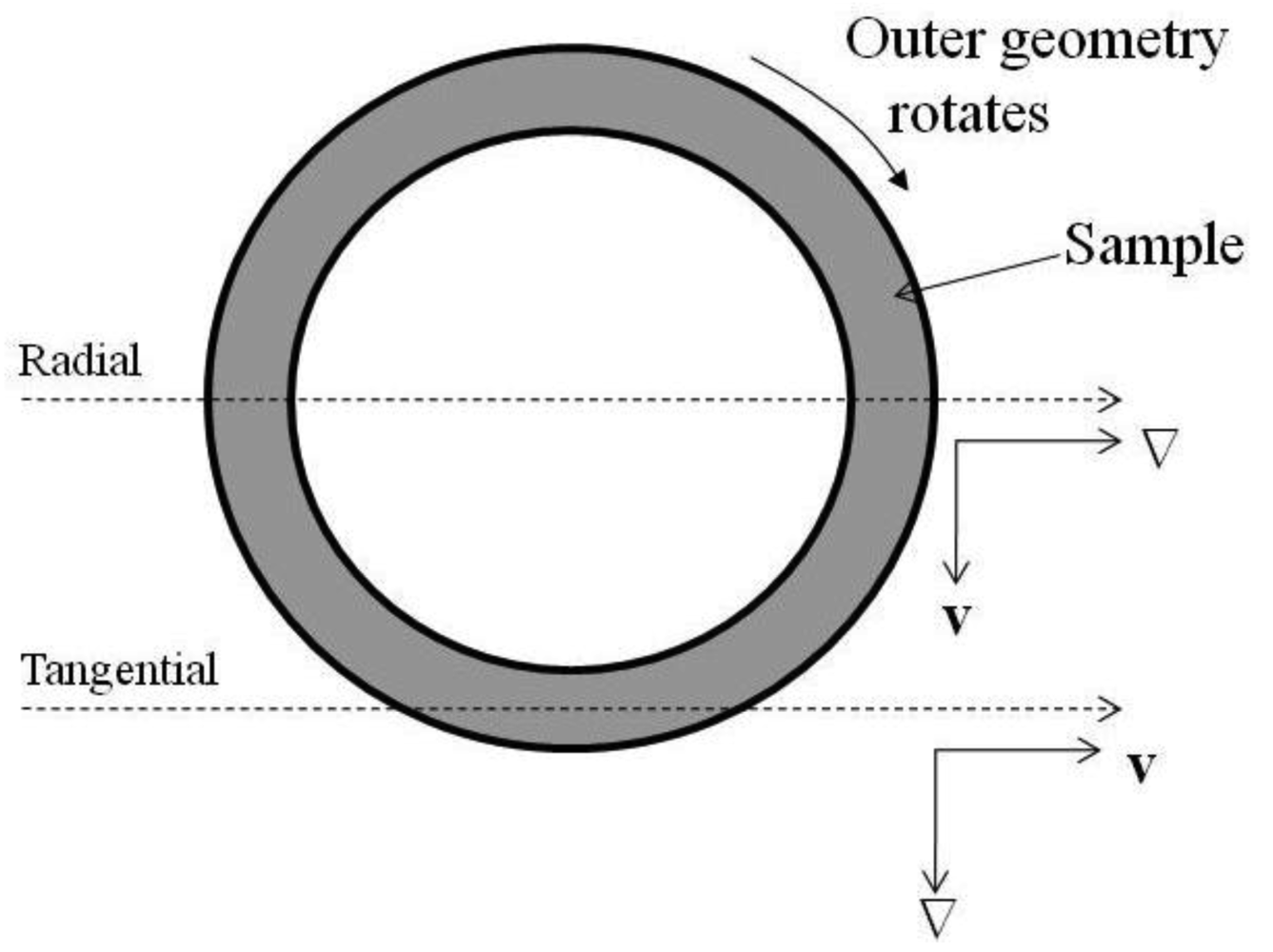
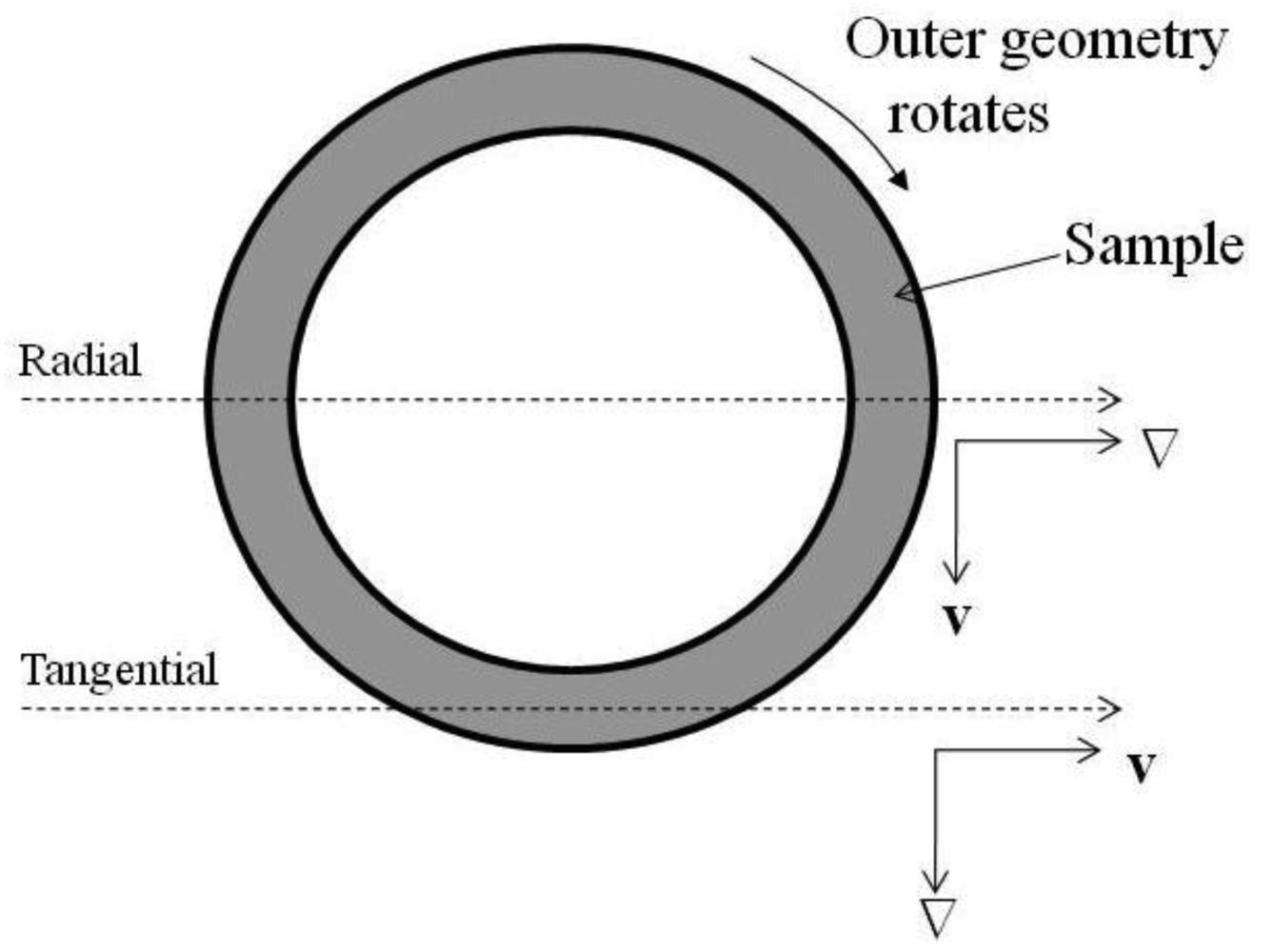
Small angle neutron scattering (SANS) experiments were performed on the NG3 30 m SANS instrument at the NIST Center for Neutron Research (NIST, Gaithersburg, MD) [31]. The mean incident neutron wavelength, λ, was 6 Å with a spread Δλ/λ of 0.15 and sample-to-detector distances of 1.3 m and 3.5 m were used, providing a scattering vector ( , where θ denotes the scattering angle between the incident beam and radial position on the detector) range of 0.022–0.135 Å−1. A Couette geometry shear cell was used in the SANS experiments to align the block copolymer micelle gel [32]. The Couette shear cell consisted of an inner quartz cylindrical stator (O.D. = 59mm) and an outer quartz cylindrical rotor (I.D. = 61 mm), resulting in a 1 mm sample thickness. The samples were studied at temperatures between 5–45 °C under static conditions, under oscillation (triangular wave function, 5 Hz and a peak strain of 100%) and under simple shear (γ˙ =1 s−1 or 5 s−1). A top view schematic of the cell is shown in Figure 1 which indicates the two scattering projections that were used.
The radial geometry probes the scattering in the plane defined by the velocity vector (v) and the vorticity vector (e). The tangential geometry is defined by a thin slit in the gap of the cell and probes scattering in the plane defined by the vorticity vector (e) and the shear gradient vector (∇). The scattering data is reduced to an absolute scale by correcting for absorption, background scattering, detector sensitivity and the empty cell scattering and compared to open beam intensity using the analysis software (version 6.01) in Igor Pro® (version 6.0.3.1) described elsewhere [33].
Small angle X-ray scattering (SAXS) experiments are preformed under vacuum using a Rigaku S-Max3000 with a two-dimensional multi-wire detector (Rigaku Americas, The Woodlands, TX). The sample to detector distance is approximately 3 m, the wavelength, λ, of the radiation source is 1.54 Å (Cu Kα) and the scattering vector (defined similarly to the SANS work) is calibrated using a silver behenate standard. Samples are loaded into a static sample cell with Kapton® windows and thermally equilibrated using a Linkam LTS 350 high temperature control stage. The two-dimensional scattering patterns are averaged to obtain plots of scattered intensity (normalized to the open beam, arbitrary units) versus the magnitude of the scattering vector, Q.
3. Results
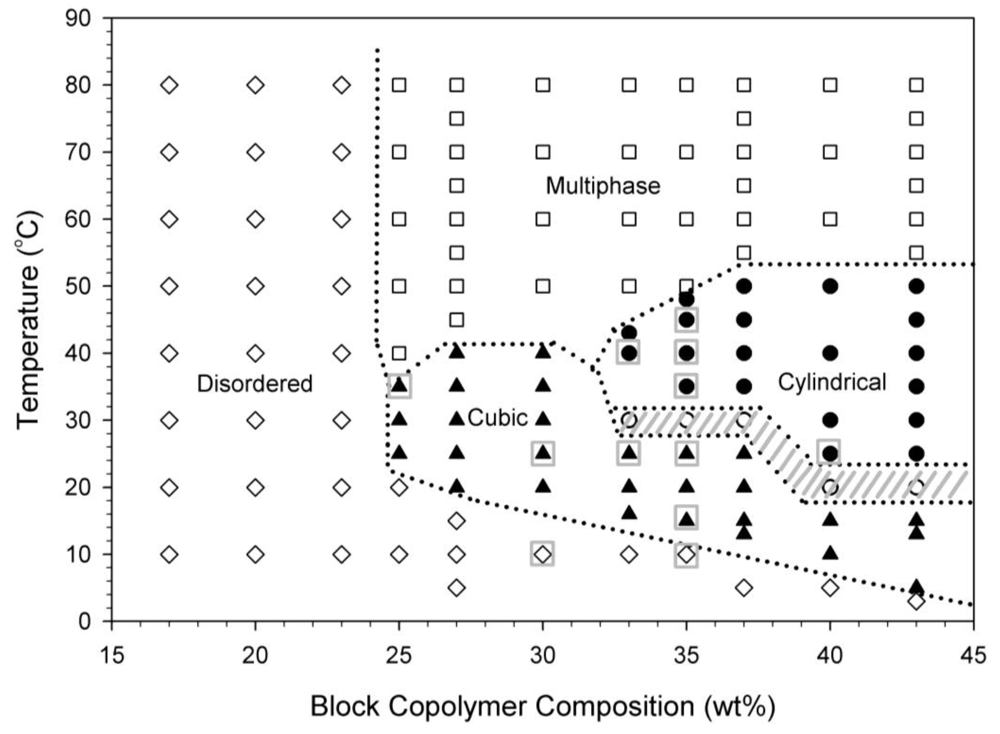
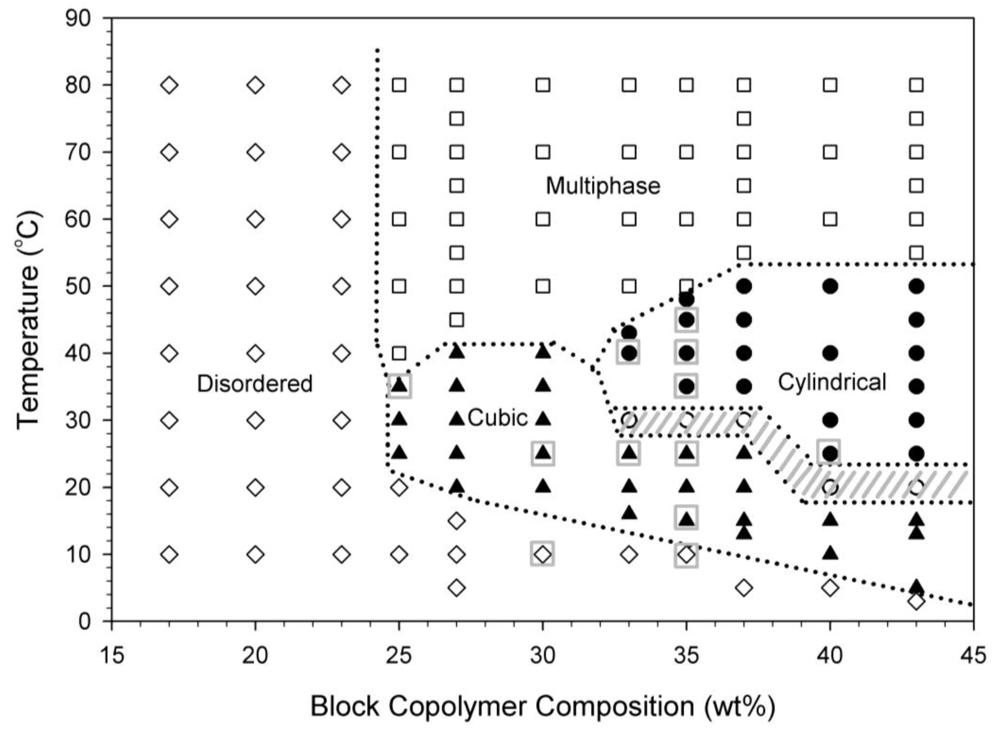
Block copolymer solutions between 5–43 wt% consist of a disordered viscous liquid region and an ordered packed micelle phase which is initially determined through visual observations of thermally equilibrated samples. Block copolymer gels formed from ordered packed micelle phases of cubically-packed spherical micelles are isotropic at length scales comparable to the wavelength of light. Block copolymer gels formed from ordered packed micelle phases of hexagonally packed cylindrical micelles display optical anisotropy at this length scale and this anisotropy is qualitatively probed with birefringence. Samples are placed in a temperature bath for at least one hour to thermally equilibrate and then probed through crossed polarizers. The static phase diagram for this block copolymer in water is presented in Figure 2. In this figure, the “disordered” region is an optically isotropic, clear, viscous liquid. The “multiphase” region is a turbid, white solution. The “cubic” and “cylindrical” regions labeled in Figure 2 are both clear gels, but the cubic region is optically isotropic while the cylindrical region shows observable birefringence under crossed polarizers. The hashed gray lines in Figure 2 indicate regions where the samples showed inhomogeneous regions of birefringence instead of a homogeneous birefringence after an equilibration time of one hour, possibly indicating mixed or coexisting phases.
A similar phase behavior to that shown in Figure 2 has been reported previously [6,27-29]. Different structures have been assigned to the 33–40 wt% region in the literature. One report describes the 35 wt% sample as being a clear gel that transitions from “FCC” at 25 °C to “FCC?” at 45 °C [27]. Another report describes the 35 wt% sample as transitioning from a cubic phase at 25 °C to a hexagonal phase at 45 °C [6]. Our previous work has shown agreement with the existence of an OOT between a cubically-packed spherical micelle phase and a hexagonally-packed cylindrical micelle phase between the concentrations of 33–40 wt% [18]. In Figure 2, a 35 wt% block copolymer at 25 °C exists as a stiff clear isotropic gel, while the same sample at 35 °C is a stiff clear gel that displays birefringence. Some of the ordered micelle crystal phases, including those at 35 wt%, determined from visual observations and birefringence measurements are confirmed by SANS, as will be discussed in a following section, and they are indicated on Figure 2 by boxed symbols.
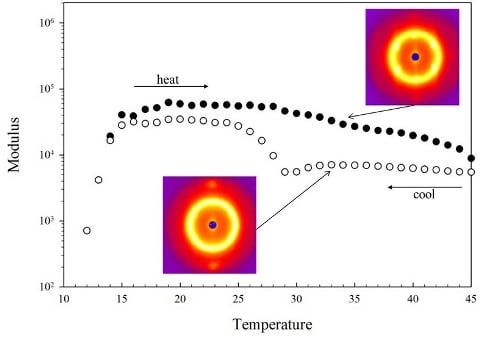
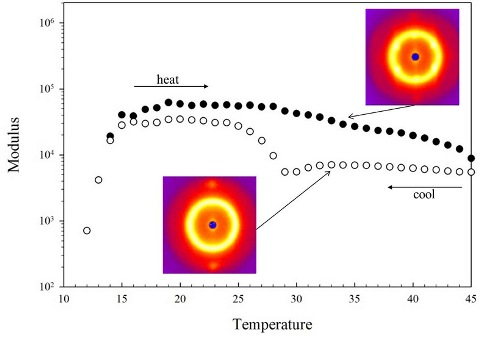
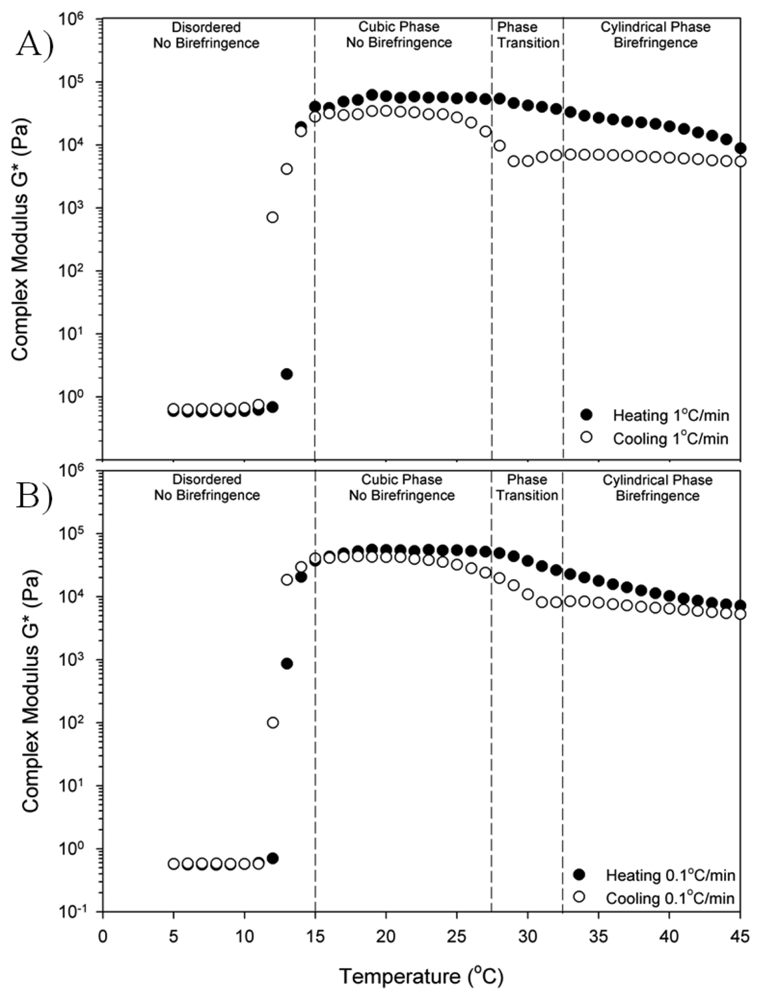
The thermorevsibility of this block copolymer in the temperature range between 5–45 °C is confirmed by measuring the complex modulus as shown in Figure 3. The complex modulus is measured at a fixed frequency of 1 rad/s in the linear regime (σo = 10 Pa for disordered phase, σo = 100 Pa for ordered phase) as the temperature increases (closed) and decreases (open) at a rate of 1 °C/min in Figure 3(A) and at a rate of 0.1 °C/min in Figure 3(B). The linear regime was chosen to allow the moduli to be a probe of structure; higher stresses would align and deform the structure, complicating interpretation of the results. In the linear regime, we did not observe these complications. For both heating ramp rates, the samples were heated to 50 °C and then the temperature ramp reversed. This upper temperature was chosen to avoid sampling the “multiphase” region for excessive time as the impact of that transition was not the focus of this work. There is an increase in the modulus at the liquid to gel transition as the micelles form and begin to pack into an ordered phase. The gelation temperature of the 35 wt% block copolymer is 13 °C and is defined, for convenience, to be the point where the storage modulus G′ equals the loss modulus G″. This gelation temperature marking the disorder-order transition is similar to other reported values.
The different regions of phase behavior from Figure 2 are indicated by dashed lines and labels in Figure 3(A) and Figure 3(B). The OOT region from the crystal phase of cubically-packed spherical micelles (cubic) to a hexagonally-packed cylindrical micelle phase (cylindrical) is the same in both Figure 3(A) and Figure 3(B). The modulus is lower in the cylindrical phase than the cubic, but the transition is anisotropic around the OOT. The moduli in the cubic phase and the cylindrical phase show a hysteresis between the increasing and decreasing temperature ramps with the lower values occurring as the temperature ramp is decreasing. The hysteresis is observed at both temperature ramp rates, but the hysteresis is greater for the faster rate of 1 °C/min (Figure 3(A)). The hysteresis did not disappear at even the slowest rates probed. In both cases, the transition appears sharper on cooling (cubic phase to cylindrical phase) than on heating (cylindrical phase to cubic phase). The ODT exhibits weak dependence on the temperature ramp rate, but this is small compared to the impact on the OOT. Other block copolymers, which exhibit only the cubic phase, do not show this dependence of mechanical properties on thermal history [14].
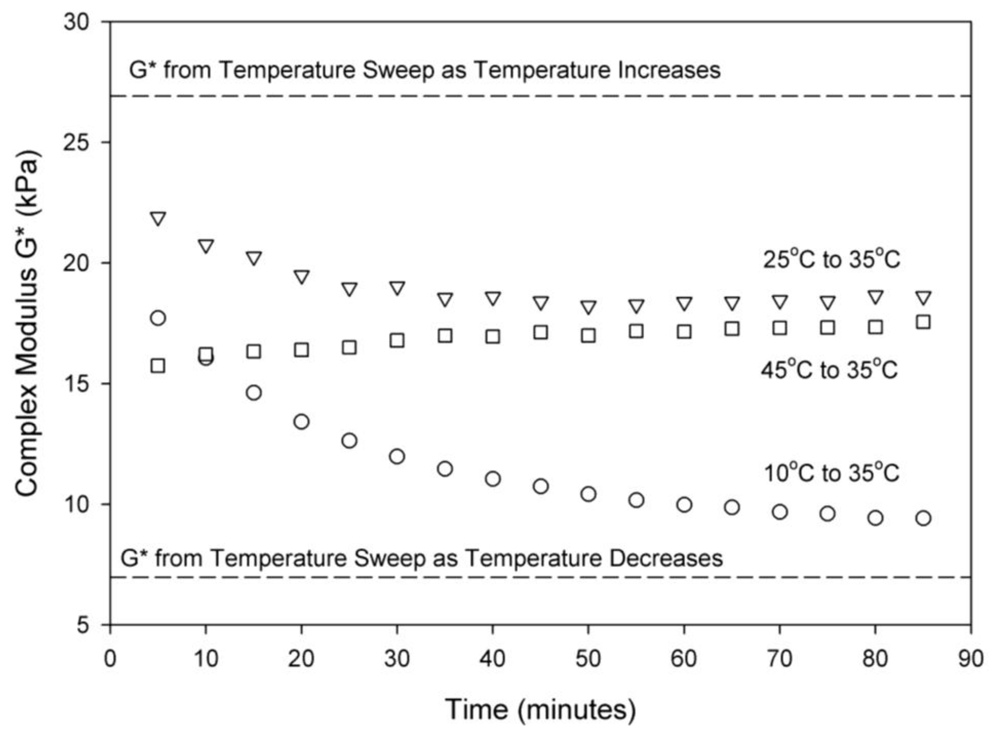
The effects of three different thermal histories on the hexagonally-packed cylindrical micelle crystal phase are studied for a 35 wt% block copolymer at 35 °C and are presented in Figure 4. Two of the experiments represent a temperature step increase, from the cubic structure to the cylindrical (25 °C → 35 °C) and from one temperature in the cylindrical structure region to another (45 °C → 35 °C). Figure 4 shows both of these experiments reached a similar steady-state complex modulus at 35 °C. A third experiment was conducted with the solution first equilibrated at 10 °C for thirty minutes and then quenched directly to 35 °C. Going directly from 10 °C, where the block copolymer is in a disordered state, to a cylindrical gel resulted in a different steady state complex modulus. The complex modulus values at 35 °C that were observed from the hysteresis in Figure 3(A) also depends on the thermal history and are shown as the dashed lines in Figure 4. It is clear that the thermal history does have an effect on the mechanical properties of the cylindrical structure. To avoid the potential persistence of one phase into another, all samples are cooled to a disordered state at 10 °C on the rheometer to “melt” any residual structure prior to heating to the test temperature. Figure 2 shows that a disordered liquid is observed for 35 wt% block copolymer samples at 10 °C. At 10 °C, SANS shows a disordered micelle peak and the rheology at this temperature is liquid-like; no shear-induced structure is seen at this low temperature and mechanical measurements (rheology) show similar transients from temperatures below 10 °C. Based on these observations, we conclude that cooling to 10 °C is able to “melt” any crystalline order and return the system to a reproducible state [34]. This procedure allows us to go from the disordered phase to a cylindrical phase and thus minimize any residual thermal history from the cubic phase. It is interesting to note the long timescales involved with formation and organization of this phase. For all three thermal histories, times on the order of an hour are required for the mechanical measurements to reach a stable value.
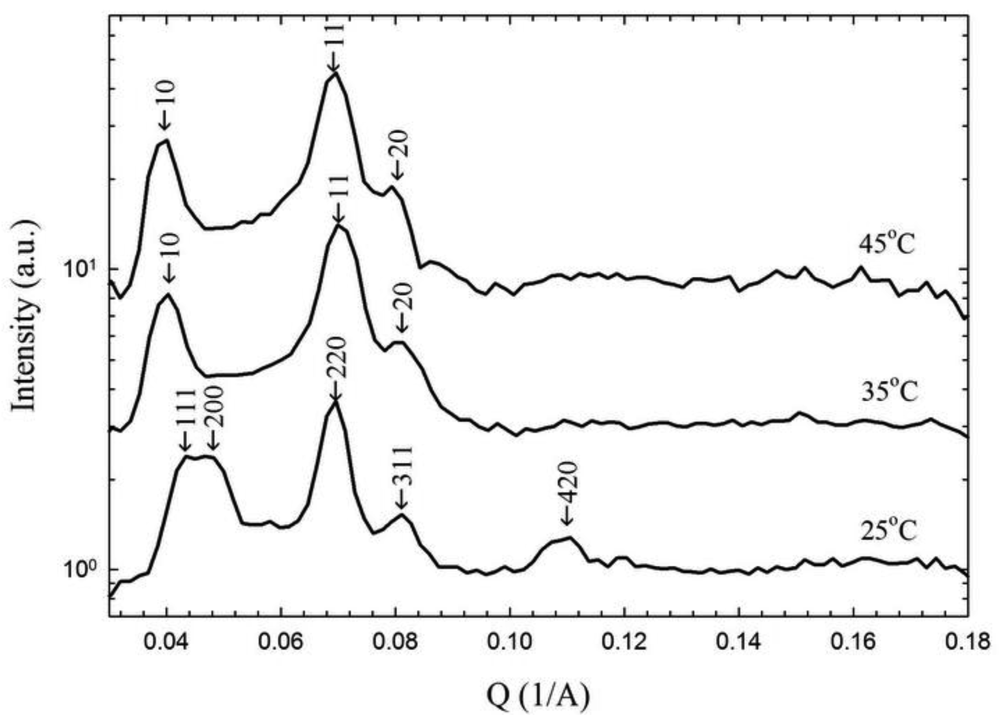
The phases determined from the birefringence and rheology experiments are confirmed with small angle scattering. Small angle X-ray scattering (SAXS) provides peak resolution, but we are not able to align the samples with shear so these results represent scattering from a multicrystalline or powder structure. SAXS data for a 35 wt% block copolymer in water sample at three different temperatures is shown in Figure 5. In all three cases, the sample was equilibrated for hours at the test temperature after heating from the “melt” at cold temperatures. In Figure 5, the intensity versus scattering angle shows peaks indicative of the structure. The peak positions of a cubic lattice are observed at 25 °C , while the 35 and 45 °C data is consistent with hexagonally-packed cylinders . At 25 °C, we have indexed the peaks assuming an FCC crystal and the peak positions are consistent with this type of packing. However, it is not possible to negate other crystal structures observed in packed spheres. A mixture of FCC and hexagonally close packed layers (HCP) was observed for this Pluronic system at 30 wt% and 23 °C; which would also be consistent with our SAXS data [28]. The crystal structure clearly changes dramatically on an increase in temperature from 25 °C. The differences between the two higher temperatures are not as significant, and the peaks in Figure 5 are indexed assuming hexagonally packed cylinders.
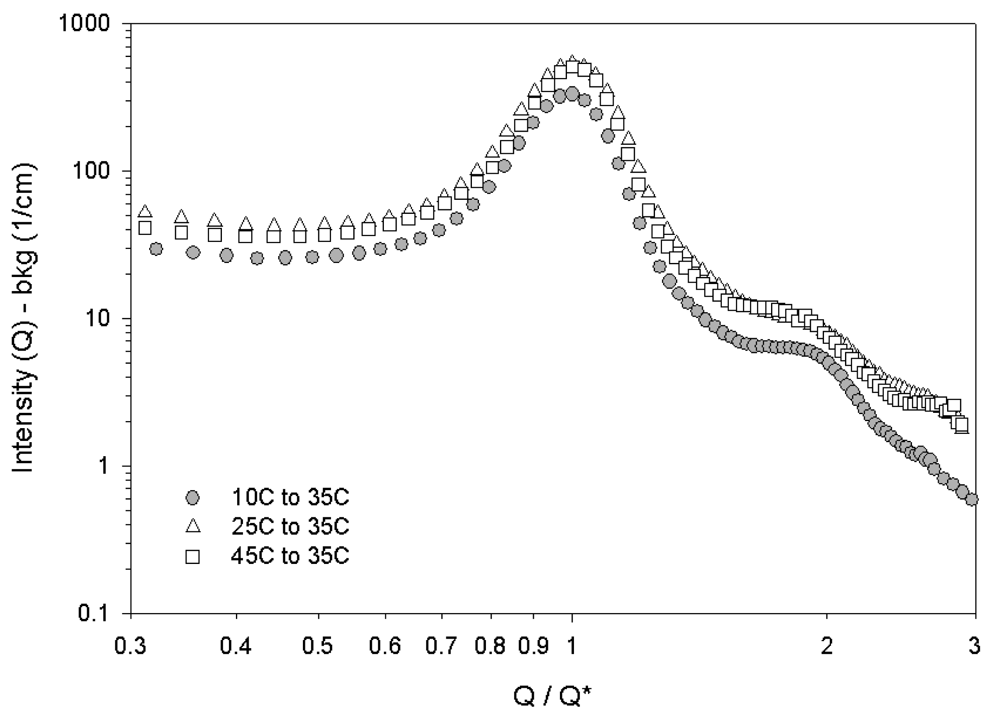
The structure of 35 wt% solution at 35 °C has already been identified as hexagonally-packed cylinders in Figure 2. We are able to apply different thermal histories in our SANS experiments. Figure 6 shows the SANS data of scattered intensity as a function of Q/Q* for a 35 wt% block copolymer sample after different thermal histories where Q* is the position of the first peak. The horizontal axis is normalized in this way simply to highlight the relative peak positions. These peak positions are consistent with the hexagonally packed-cylindrical micelle phase . [18,35]. The peak at is not observed in SANS due to the wavelength smearing and resolution issues that are inherent with neutron scattering experiments, but this was visible in the SAXS shown in Figure 5. The SANS scattering for the sample quenched from disordered to 35 °C is shown in gray. The peak positions do not change based on the thermal history but the SANS second peak in Figure 6 is more resolved for the quenched sample. We also observe a slight decrease in the overall scattering intensity, which is on an absolute scale for SANS. It is possible that the impact of thermal history is on the organization of the crystallites, quenching may result in a different textural lengthscale for the multicrystalline structure. It is also possible that small regions of a different crystal structure are seen in the system and SANS is not sensitive enough to resolve the details of the structure. For this reason, we will align the crystal with shear to better resolve the structure.
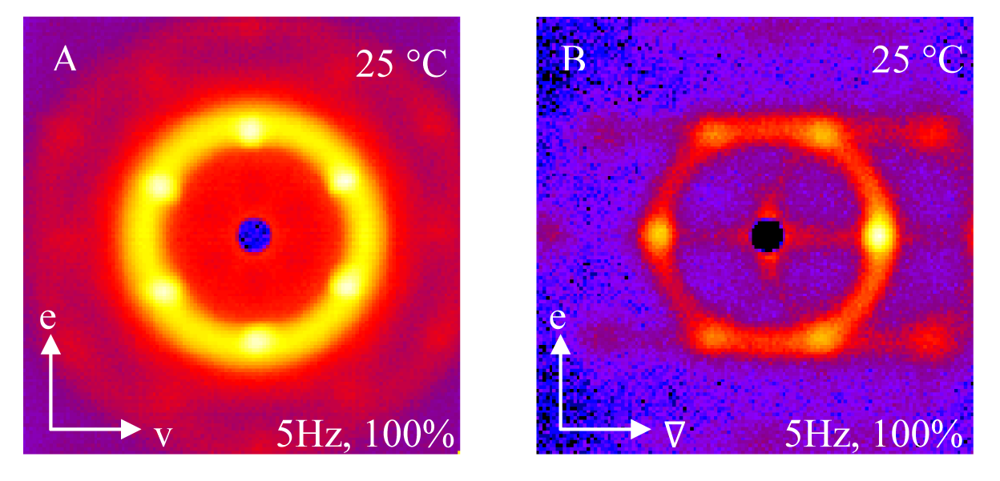
Small angle neutron scattering (SANS) using a Couette shear cell is described previously and shown in Figure 1; the use of the shear cell allows us to align the crystals as these are soft materials. Previous work has shown that these structures align easily under shear and maintain that alignment after cessation of the applied shear [18]. Figure 7 shows the radial and tangential scattering patterns from a 35 wt% block copolymer sample under oscillation (5 Hz and 100% strain) at 25 ° C. The samples are quenched from the disordered state (10 °C) to 25 ° C and allowed to equilibrate. The radial scattering in Figure 7(A) shows a six-fold symmetry that is indicative of the scattering arising from a three-fold or six-fold planar system as in the alignment of spherical close-packed colloidal crystal systems [36]. These planes of close-packed cubic crystals will form a face-centered-cubic lattice (FCC), a hexagonal close-packed lattice (HCP) or a homogenous mixture of FCC and HCP known as a random close packed lattice (RCP). A mixed phase of HCP and FCC has already been reported for this block copolymer at similar concentrations and temperatures. The scattering in Figure 7(A) shows three hexagonal rings that correspond to the planes of the FCC and HCP crystals as predicted by diffraction models [37,38]. Differentiation between FCC, HCP and RCP is possible with a second projection of the structure, so we consider the tangential scattering. The lower resolution of the tangential scattering in Figure 7(B) makes this difficult but the tangential scattering with six peaks including the smearing of the top and bottom peaks resembles that expected for RCP. The radial and tangential scattering patterns in Figure 7(A) and Figure 7(B) for 35 wt% block copolymer at 25 ° C are characteristic of a RCP crystal of spherical micelles and will be referred to as such throughout this paper [16,36].
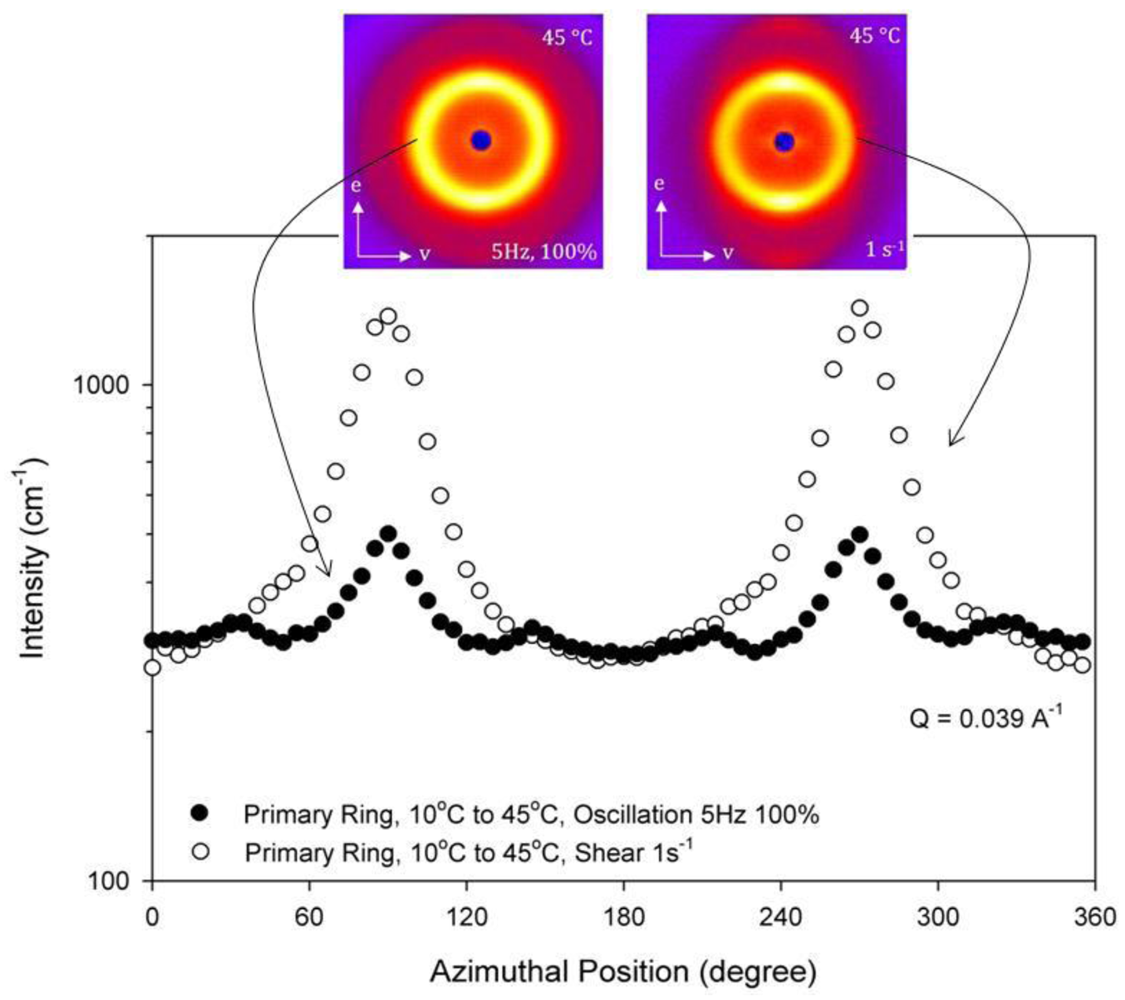
At 45 °C, the 35 wt% block copolymer solution is hexagonally-packed cylinders, but the structure appears to be sensitive to details of applied shear fields. The radial scattering patterns for a 35 wt% block copolymer at 45 °C are shown in Figure 8 again quenched from a disordered state to 45 °C. These scattering patterns are characteristic of the radial scattering of the hexagonally-packed cylindrical phase as indicated by the two bright peaks along the vorticity axis as shown in Figure 8 [18,39]. The radial scattering pattern shown to the left in Figure 8 is measured under the oscillatory shear (5 Hz and 100% strain) and the radial scattering pattern shown to the right in Figure 8 under simple shear (γ˙ = 1 s−1. Figure 8 shows the intensity in the primary ring (Q = 0.039 Å−1) as a function of azimuthal angle with 0° defined as the shear direction. Figure 8 shows peaks in the vorticity direction (90° and 270°, where 0° is defined to be in the shear (v) direction) for both types of shear, but the oscillatory shear data also shows four smaller peaks at 30°, 150°, 210° and 330°. These peak positions correspond to the six-fold peaks observed in the crystal phase of cubically-packed spherical micelles shown in Figure 7(A). This mixed pattern has not been reported previously in sheared samples. Given that the samples are birefringent at 45 °C and the key features of a phase of aligned cylinders exist, we will identify the 45 °C sample as a hexagonally-packed cylindrical phase (HEX). The sensitivity to the nature of the shear field, coupled with transients and hysteresis demonstrate the sensitivity of this transition. It is also important to note that both of these samples are contained in the same shear cell, so the hysteresis cannot be attributed to a temperature gradient in the shear cell; if that were the case, the mixed structure would be observed in all cases, independent of the nature of the shear applied.
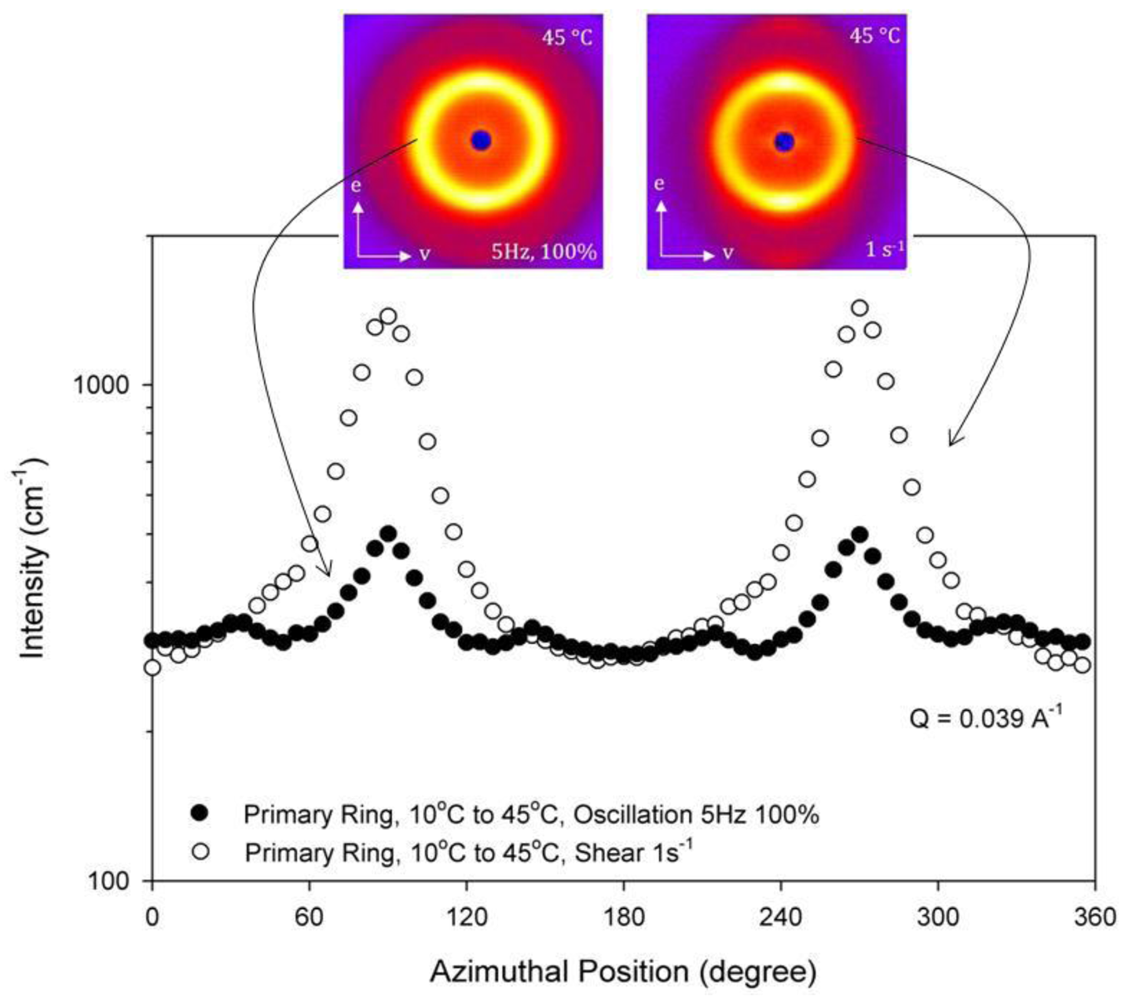
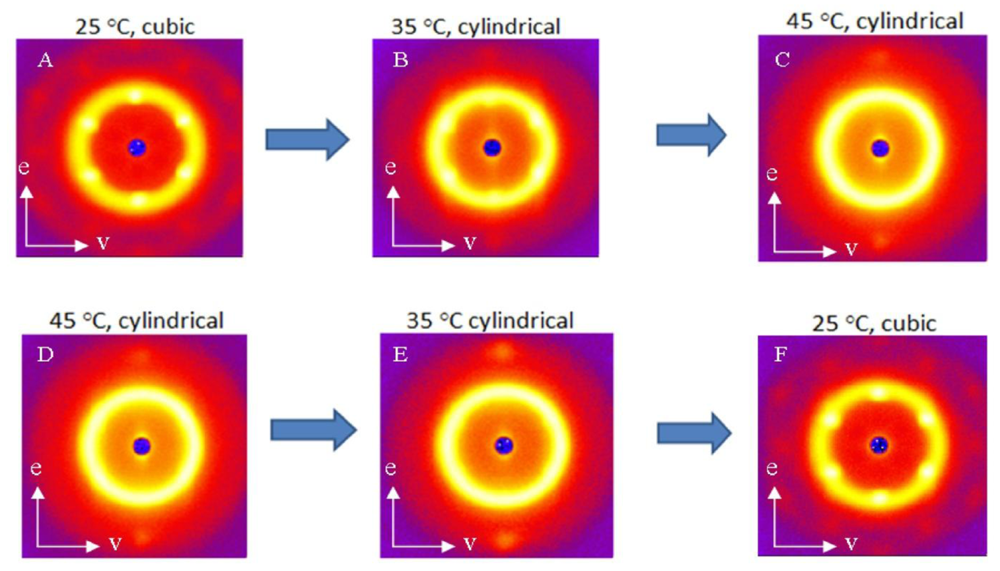
The SANS scattering profiles of the block copolymer near the OOT are shown in Figure 9. In this figure, different thermal histories have been applied and the impact on the nanoscale structure shown.The block copolymer gel displays an ordered RCP spherical micelle crystal phase at 25 °C in Figure 9(A) as discussed previously with Figure 7. At 45 °C in Figure 9(D), two bright diffraction spots are observed in the vorticity direction as seen in Figure 8 for the crystal of hexagonally-packed cylindrical micelles (HEX). The 35 °C ordered micelle gel structure depends on the thermal history of the sample as seen in the differences between the scattering patterns of Figure 9(B) and Figure 9(E). In Figure 9(A) and Figure 9(D), the sample was quenched from the disordered state to the temperature of interest and allowed to equilibrate for 45 minutes. From these two base states, the sample is stepped to higher or lower temperature and equilibrated for 45 minutes prior to collecting structural data. Figure 9(B) shows a six-fold symmetry that is indicative of a cubically-packed spherical micelle phase instead of a hexagonally-packed cylindrical micelle phase. In Figure 9(E), the sample was cooled from a hexagonally-packed cylindrical to 35 ° C and instead shows the two bright diffraction spots in the vorticity direction that are characteristic of a HEX phase. The anisotropic OOT between the cubic and cylindrical micelle phases is thus also observed using SANS in addition to the rheology measurements in Figures 3 and 4. Figures 9(C) and Figures 9(F) indicate that the hysteresis persists only over a small temperature range as the RCP and HEX structures are restored at the appropriate temperatures. It is worth noting that the RCP phase observed in Figure 9(F) is extremely aligned as higher order rings of peaks are visible, providing an optimal temperature profile to use to achieve highly aligned block copolymer micelle crystals and nanocomposites. These results provide an indication that specific thermal histories will allow for more organized crystals, which is key to templating processes.
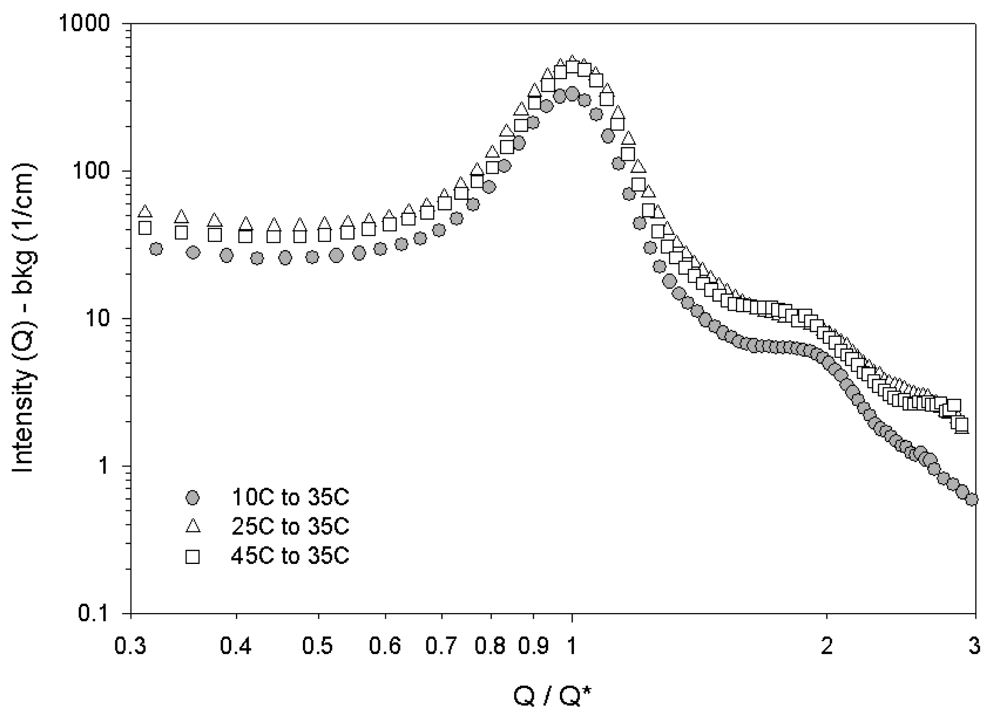
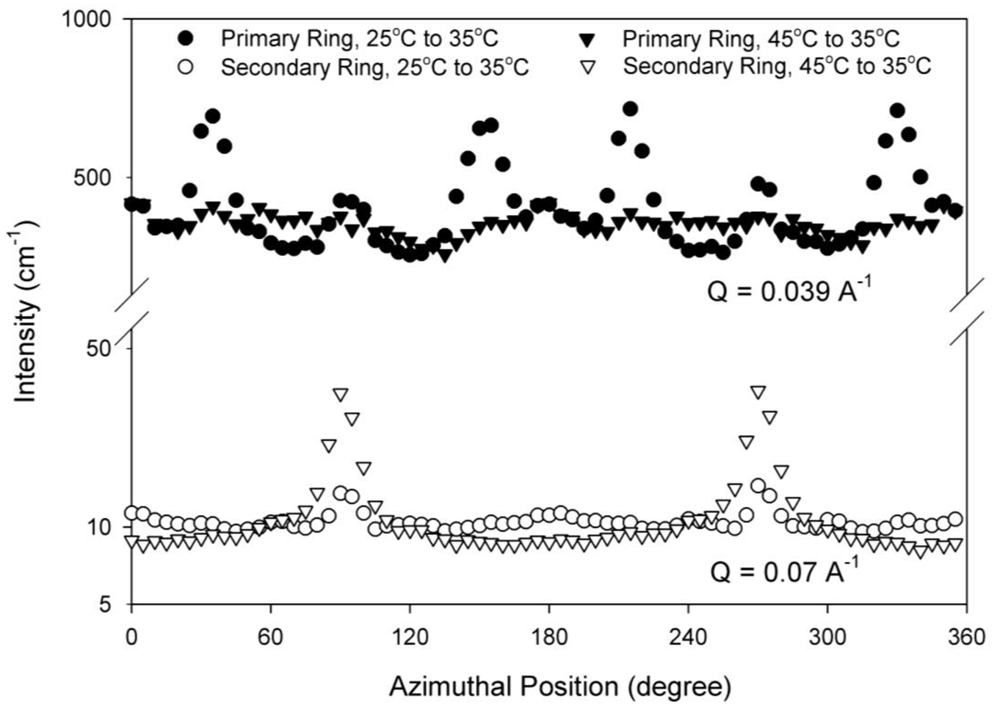
These differences in the SANS scattering profiles between Figures 9(B) and Figures 9(E) are more clearly observed by comparing the intensities in the first ring as a function of azimuthal angle in Figure 10. The primary ring at a scattering vector of 0.039 Å−1 in Figure 10 is the innermost ring of peaks and the secondary ring at a scattering vector of 0.07 Å−1 is the next ring of peaks. The cubic order persists when heated from 25 °C to 35 °C as seen in Figure 10 by the presence of the six peaks in the primary ring. These six peaks are not seen in the samples cooled from 45 °C to 35 °C. The persistence of the six peaks into the cylindrical micelle phase on heating but not on cooling shows that thermal history is an important factor in the resulting nanoscale structure of the HEX phase. The azimuthal intensity dependence in the secondary ring shows the two bright diffraction spots in the vorticity direction that are characteristic of a HEX phase for both thermal histories but are more distinct for the sample cooled within the HEX phase. Figures 9(A) and Figures 9(F) show scattering from samples at 25 °C with very different thermal histories (one quenched from the disordered phase and the other cooled from the HEX phase). The azimuthal intensities of these two-dimensional SANS scattering profiles for these two thermal histories are seen to be identical (data not shown). Thus thermal history effects do not influence the nanoscale structure of the RCP phase.
4. Discussion
The anisotropic OOT between the RCP phase and HEX phase in Pluronic® P123 exists at both the macroscale and the nanoscale as observed through rheology and SANS experiments. At the nanoscale the RCP phase at 25 °C persists into the HEX phase at 35 °C indicating that the OOT from spherical micelles to cylindrical micelles is more hindered than cylindrical micelle breakup into spheres since no indication of the cubic system is shown in a transition from 35 °C to 25 ° C. The persistence of the six peaks from the RCP phase into the HEX phase at the nanoscale in Figure 10 indicates that the same hysteresis due to thermal history effects in Figure 3 in the macroscale rheology measurements can also be observed with SANS for the HEX phase.
Anisotropic order-order transitions in other block copolymers have been previously documented in the melt and in solution [40,41]. Similar behavior to that observed here was seen in a triblock copolymer melt of poly(styrene-b-isoprene-b-styrene) where it was reported that fluctuations with cubic symmetry were evident in the stable hexagonal phase even after shear alignment [40,41]. An OOT between a cubic phase and cylindrical phase has been shown previously in block copolymer solutions to have a hysteresis that affects the phase transition properties [40-42]. In a system of symmetric styrene-isoprene diblock copolymer, the existence of supercooled cylinders are found to persist into the cubic phase. The stability of these supercooled cylinders depends on the concentration of the block copolymer. In some cases this metastable phase persists in the system after a month of annealing when the block copolymer concentrations are high [41].
This anisotropy in both the literature and our system is likely due to the difference between the spherical micelles forming cylindrical micelles and the cylindrical micelles breaking up into spherical micelles during the OOT. This difficulty of the system to transition from spherical to cylindrical micelles could be due to the need for the cubically-packed spherical micelle phase to reorient itself to form and pack the cylindrical micelles into a hexagonally-packed cylindrical micelle phase. For the reverse OOT, the cylindrical micelles will begin to break up into spherical micelles which will more easily repack into a cubically-packed spherical micelle phase. It is hypothesized here that it is the hindrance of rearranging the spherical micelles into cylindrical micelles and then packing of the cylindrical micelles into a hexagonally-packed cylindrical micelle phase that gives rise to this anisotropic OOT. It is also hypothesized that the cylindrical micelles break apart easily into spherical micelles and repack into a cubically-packed spherical micelle phase quickly allowing the block copolymer to go from the hexagonally-packed cylindrical micelle phase to the cubically-packed spherical micelle phase more easily than the reverse phase transition.
We show that the block copolymer demonstrates a thermal history between the RCP phase and HEX phase and will approach different steady state moduli values. This thermal history has also been shown at the nanoscale in the HEX phase for block copolymers but not the RCP phase. Allowing the micelle crystal to equilibrate at 10 °C for half an hour minimizes these thermal history effects. This suggests that more care must be given when studying block copolymer systems with more than one ordered phase to ensure that a stronger micelle gel such as the cubically-packed spherical micelle phase is not remaining dominant in a weaker micelle gel such as the hexagonally-packed cylindrical micelle phase.
5. Conclusions
The Pluronic® P123 block copolymer system forms a cubically-packed spherical micelle phase (RCP) and a hexagonally-packed cylindrical micelle phase (HEX). The hexagonally-packed cylindrical micelle phase forms a weaker gel that is more sensitive to thermal history and different shear fields than the cubically-packed spherical micelle phase. A hysteresis exists in the order-order transition between the RCP phase and the HEX phase. These thermal history effects are observed at both the macroscale (rheology) and nanoscale (SANS) for the hexagonally-packed cylindrical micelle phase. We propose that the cubically-packed spherical micelle phase persists into the hexagonally-packed cylindrical micelle phase due to the hindrance of rearranging the spherical micelles into cylindrical micelles and then packing of the cylindrical micelles into a hexagonally-packed cylindrical micelle phase and that this mechanism gives rise to the observed anisotropic order-order transition. One ramification of this work is the guidance offered to nanocomposite preparation in the hexagonally-packed cylinder phase; to avoid mixed phases, the thermal history should be chosen such that the hexagonally-packed phase is reached directly from the isotropic phase and that simple shear be applied rather than oscillatory shear.





Acknowledgments
This work utilized facilities supported in part by the National Science Foundation under Agreement No. DMR-0454672. We acknowledge the support of the National Institute of Standards and Technology, U.S. Department of Commerce, in providing the neutron research facilities used in this work. We acknowledge the PPG foundation and the National Science Foundation (CTS-9871110 and CTS-0521079) for equipment. T.A.L. acknowledges the National Science Foundation Graduate Research Fellowship Program for providing research funding.
References
- Förster, S.; Antonietti, M. Amphiphilic Block Copolymers in Structure-Controlled Nanomaterial Hybrids. Adv. Mater. 1998, 10, 195–217. [Google Scholar]
- Hashimoto, T. Phase Transition and Self-Assembly in Block Copolymers. Macromol. Symp. 2001, 174, 69–84. [Google Scholar]
- Malmsten, M.; Lindman, B. Self-Assembly in Aqueous Block Copolymer Solutions. Macromolecules 1992, 25, 5440–5445. [Google Scholar]
- Mortensen, K.; Brown, W. Poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) Triblock Copolymers in Aqueous Solution. The Influence of Relative Block Size. Macromolecules 1993, 26, 4128–4135. [Google Scholar]
- Mortensen, K.; Pedersen, J.S. Structural Study on the Micelle Formation of Poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) Triblock Copolymer in Aqueous Solution. Macromolecules 1993, 26, 805–812. [Google Scholar]
- Wanka, G.; Hoffmann, H.; Ulbricht, W. Phase Diagrams and Aggregation Behavior of Poly(oxyethylene)-Poly(oxypropylene)-Poly(oxyethylene) Triblock Copolymers in Aqueous Solutions. Macromolecules 1994, 27, 4145–4159. [Google Scholar]
- Alexandridis, P.; Holzwarth, J.F.; Hatton, T.A. Micellization of Poly(ethylene oxide)-Poly(propylene oxide)-Poly(ethylene oxide) Triblock Copolymers in Aqueous Solutions: Thermodynamics of Copolymer Association. Macromolecules 1994, 27, 2414–2425. [Google Scholar]
- Alexandridis, P.; Alan Hatton, T. Poly(ethylene oxide)-Poly(propylene oxide)-Poly(ethylene oxide) Block Copolymer Surfactants in Aqueous Solutions and at Interfaces: Thermodynamics, Structure, Dynamics, and Modeling. Colloids Surf. A Physicochem. Eng. Asp. 1995, 96, 1–46. [Google Scholar]
- Alexandridis, P.; Nivaggioli, T.; Hatton, T.A. Temperature Effects on Structural Properties of Pluronic P104 and F108 PEO-PPO-PEO Block Copolymer Solutions. Langmuir 1995, 11, 1468–1476. [Google Scholar]
- Alexandridis, P.; Zhou, D.; Khan, A. Lyotropic Liquid Crystallinity in Amphiphilic Block Copolymers: Temperature Effects on Phase Behavior and Structure for Poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) Copolymers of Different Composition. Langmuir 1996, 12, 2690–2700. [Google Scholar]
- Mortensen, K. Structural Studies of Aqueous Solutions of PEO - PPO - PEO Triblock Copolymers, Their Micellar Aggregates and Mesophases; A Small-Angle Neutron Scattering Study. J. Phys. Conden. Matter 1996, 8, A103–A124. [Google Scholar]
- Mortensen, K.; Batsberg, W.; Hvidt, S. Effects of PEO-PPO Diblock Impurities on the Cubic Structure of Aqueous PEO-PPO-PEO Pluronics Micelles: fcc and bcc Ordered Structures in F127. Macromolecules 2008, 41, 1720–1727. [Google Scholar]
- Cheng, J.Y.; Mayes, A.M.; Ross, C.A. Nanostructure Engineering by Templated Self-Assembly of Block Copolymers. Nat. Mater 2004, 3, 823–828. [Google Scholar]
- Pozzo, D.C.; Hollabaugh, K.R.; Walker, L.M. Rheology and Phase Behavior of Copolymer-Templated Nanocomposite Materials. J. Rheol. 2005, 49, 759–782. [Google Scholar]
- Pozzo, D.C.; Walker, L.M. Small-Angle neutron Scattering of Silica Nanoparticles Templated in PEO-PPO-PEO Cubic Crystals. Colloid. Surf. A Physicochem. Eng. Asp. 2007, 294, 117–129. [Google Scholar]
- Pozzo, D.C.; Walker, L.M. Shear Orientation of Nanoparticle Arrays Templated in a Thermoreversible Block Copolymer Micellar Crystal. Macromolecules 2007, 40, 5801–5811. [Google Scholar]
- Domach, M.M.; Walker, L.M. Stabilizing Biomacromolecules in Nontoxic Nano-Structured Materials. J. Assoc. Lab. Autom. 2010, 15, 136–144. [Google Scholar]
- Pozzo, D.C.; Walker, L.M. Macroscopic Alignment of Nanoparticle Arrays in Soft Crystals of Cubic and Cylindrical Polymer Micelles. Eur. Phys. J. E Soft Matter Biolog. Phys. 2008, 26, 183–189. [Google Scholar]
- Walz, M.; Wolff, M.; Voss, N.; Zabel, H.; Magerl, A. Micellar Crystallization with a Hysteresis in Temperature. Langmuir 2010, 26, 14391–14394. [Google Scholar]
- Hamley, I.W.; Pople, J.A.; Diat, O. A thermally Induced Transition from a Body-Centred to a Face-Centred Cubic Lattice in a Diblock Copolymer Gel. Colloid Polym. Sci. 1998, 276, 446–450. [Google Scholar]
- Hamley, I.W.; Daniel, C.; Mingvanish, W.; Mai, S.-M.; Booth, C.; Messe, L.; Ryan, A.J. From Hard Spheres to Soft Spheres: The Effect of Copolymer Composition on the Structure of Micellar Cubic Phases Formed by Diblock Copolymers in Aqueous Solution. Langmuir 2000, 16, 2508–2514. [Google Scholar]
- McConnell, G.A.; Gast, A.P.; Huang, J.S.; Smith, S.D. Disorder-Order Transitions in Soft Sphere Polymer Micelles. Phys. Rev. Lett. 1993, 71, 2102. [Google Scholar]
- McConnell, G.A.; Gast, A.P. Melting of Ordered Arrays and Shape Transitions in Highly Concentrated Diblock Copolymer Solutions. Macromolecules 1997, 30, 435–444. [Google Scholar]
- Pople, J.A.; Hamley, I.W.; Fairclough, J.P.A.; Ryan, A.J.; Komanschek, B.U.; Gleeson, A.J.; Yu, G.E.; Booth, C. Ordered Phases in Aqueous Solutions of Diblock Oxyethylene/Oxybutylene Copolymers Investigated by Simultaneous Small-Angle X-ray Scattering and Rheology. Macromolecules 1997, 30, 5721–5728. [Google Scholar]
- Bang, J.; Lodge, T.P.; Wang, X.; Brinker, K.L.; Burghardt, W.R. Thermoreversible, Epitaxial fcc <–> bcc Transitions in Block Copolymer Solutions. Phys. Rev. Lett. 2002, 89, 215505. [Google Scholar]
- Lodge, T.P.; Bang, J.; Park, M.J.; Char, K. Origin of the Thermoreversible fcc-bcc Transition in Block Copolymer Solutions. Phys. Rev. Lett. 2004, 92, 145501–145504. [Google Scholar]
- Newby, G.E.; Hamley, I.W.; King, S.M.; Martin, C.M.; Terrill, N.J. Structure, Rheology and Shear Alignment of Pluronic Block Copolymer Mixtures. J. Colloid Interf. Sci. 2009, 329, 54–61. [Google Scholar]
- Soni, S.S.; Brotons, G.; Bellour, M.; Narayanan, T.; Gibaud, A. Quantitative SAXS Analysis of the P123/Water/Ethanol Ternary Phase Diagram. J. Physi. Chem. B 2006, 110, 15157–15165. [Google Scholar]
- Holmqvist, P.; Alexandridis, P.; Lindman, B. Modification of the Microstructure in Block Copolymer-Water Systems by Varying the Copolymer Composition and the Type: Small-Angle X-ray Scattering and Deuterium-NMR Investigation. J. Phys. Chem. B 1998, 102, 1149–1158. [Google Scholar]
- Pozzo, D.C. Templating Nanoparticles Using Thermo-reversible Soft Crystals. Ph.D. Thesis, Carnegie Mellon University, Pittsburgh, PA, USA, 2006. [Google Scholar]
- Glinka, C.J.; Barker, J.G.; Hammouda, B.; Krueger, S.; Moyer, J.J.; Orts, W.J. The 30 m Small-Angle Neutron Scattering Instruments at the National Institute of Standards and Technology. J. Appl. Crystallogr. 1998, 31, 430–445. [Google Scholar]
- Straty, G.C.; Hanley, H.J.M.; Glinka, C.J. Shearing Apparatus for Neutron Scattering Studies on Fluids: Preliminary Results for Colloidal Suspensions. J. Stat. Phys. 1991, 62, 1015–1023. [Google Scholar]
- Kline, S.R. Reduction and Analysis of SANS and USANS Data Using Igor Pro. J. Appl. Crystallogr. 2006, 39, 895–900. [Google Scholar]
- LaFollette, T.A. Block Copolymer Robustness and Stability of Templated Protein Nanoparticles. Ph.D. Thesis, Carnegie Mellon University, Pittsburgh, PA, USA, 2010. [Google Scholar]
- Förster, S.; Timmann, A.; Konrad, M.; Schellbach, C.; Meyer, A.; Funari, S.S.; Mulvaney, P.; Knott, R. Scattering Curves of Ordered Mesoscopic Materials. J. Phys. Chem. B 2005, 109, 1347–1360. [Google Scholar]
- Zhang, J.; Alsayed, A.; Lin, K.H.; Sanyal, S.; Zhang, F.; Pao, W.J.; Balagurusamy, V.S.K.; Heiney, P.A.; Yodh, A.G. Template-Directed Convective Assembly of Three-Dimensional Face-Centered-Cubic Colloidal Crystals. Appl. Phys. Lett. 2002, 81, 3176–3178. [Google Scholar]
- Loose, W.; Ackerson, B.J. Model Calculations for the Analysis of Scattering Data from Layered Structures. J. Chem. Phys. 1994, 101, 7211–7220. [Google Scholar]
- McConnell, G.A.; Lin, M.Y.; Gast, A.P. Long Range Order in Polymeric Micelles under Steady Shear. Macromolecules 1995, 28, 6754–6764. [Google Scholar]
- Alberda van Ekenstein, G.; Polushkin, E.; Nijland, H.; Ikkala, O.; ten Brinke, G. Shear Alignment at Two Length Scales: Comb-Shaped Supramolecules Self-Organized as Cylinders-within-Lamellar Hierarchy. Macromolecules 2003, 36, 3684–3688. [Google Scholar]
- Ryu, C.Y.; Lodge, T.P. Thermodynamic Stability and Anisotropic Fluctuations in the Cylinder-to-Sphere Transition of a Block Copolymer. Macromolecules 1999, 32, 7190–7201. [Google Scholar]
- Park, M.J.; Char, K.; Bang, J.; Lodge, T.P. Interplay between Cubic and Hexagonal Phases in Block Copolymer Solutions. Langmuir 2005, 21, 1403–1411. [Google Scholar]
- Ryu, C.Y.; Vigild, M.E.; Lodge, T.P. Fluctuations with Cubic Symmetry in a Hexagonal Copolymer Microstructure. Phys. Rev. Lett. 1998, 81, 5354–5357. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
LaFollette, T.A.; Walker, L.M. Structural and Mechanical Hysteresis at the Order-Order Transition of Block Copolymer Micellar Crystals. Polymers 2011, 3, 281-298. https://doi.org/10.3390/polym3010281
LaFollette TA, Walker LM. Structural and Mechanical Hysteresis at the Order-Order Transition of Block Copolymer Micellar Crystals. Polymers. 2011; 3(1):281-298. https://doi.org/10.3390/polym3010281
Chicago/Turabian StyleLaFollette, Theresa A., and Lynn M. Walker. 2011. "Structural and Mechanical Hysteresis at the Order-Order Transition of Block Copolymer Micellar Crystals" Polymers 3, no. 1: 281-298. https://doi.org/10.3390/polym3010281