
Understanding the Thermal Degradation Mechanism of High-Temperature-Resistant Phthalonitrile Foam at Macroscopic and Molecular Levels

 ,
, 
Abstract
:1. Introduction
2. Experimental
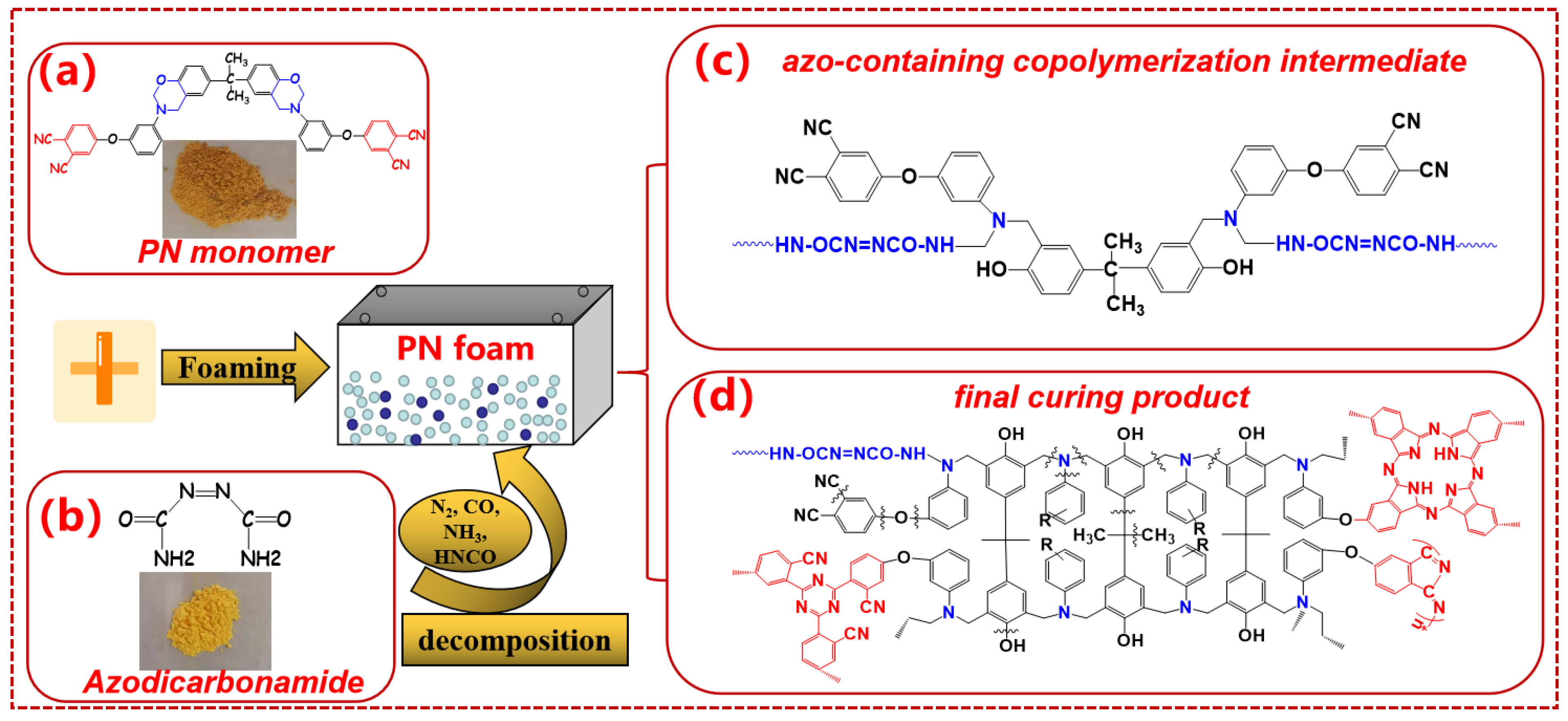
2.1. Materials
2.2. Preparation
2.3. Characterization
3. Results and Discussions
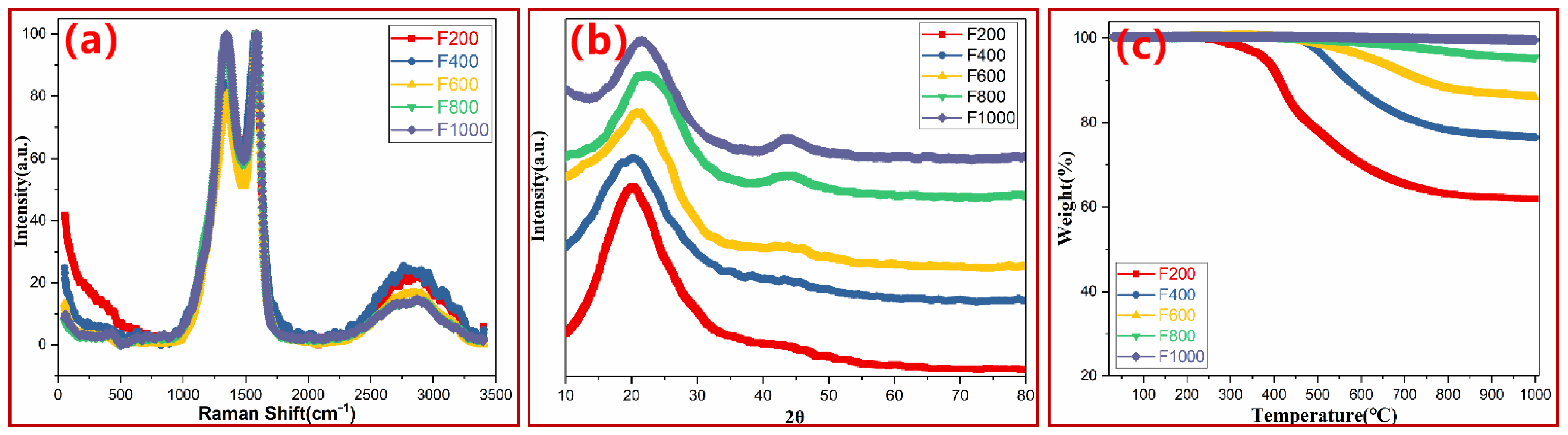
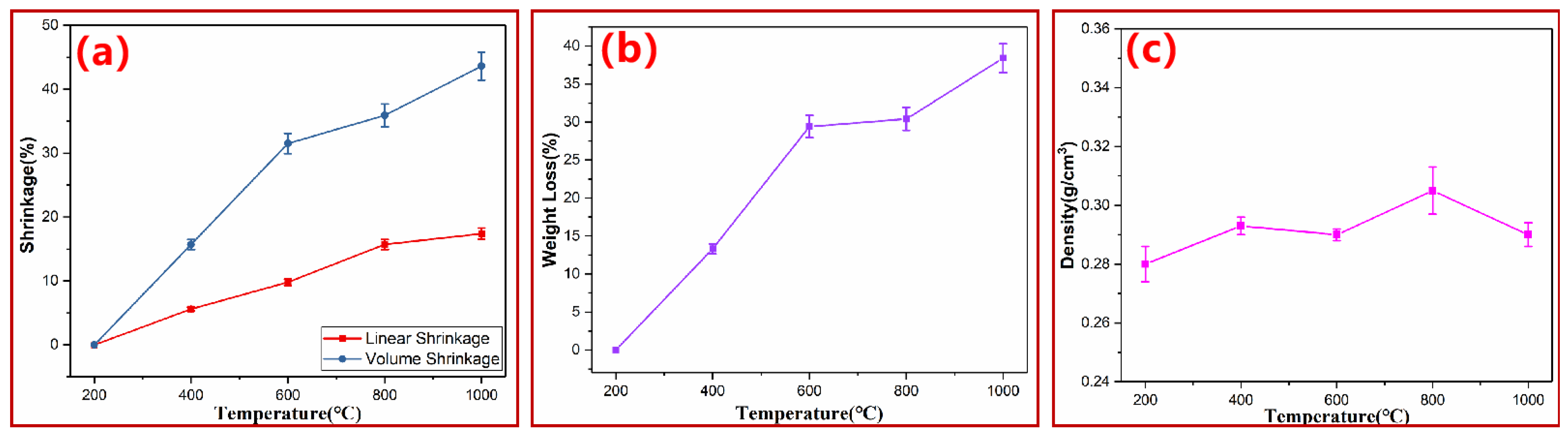
3.1. Macroscopic Thermal Degradation Behavior of PN Foam
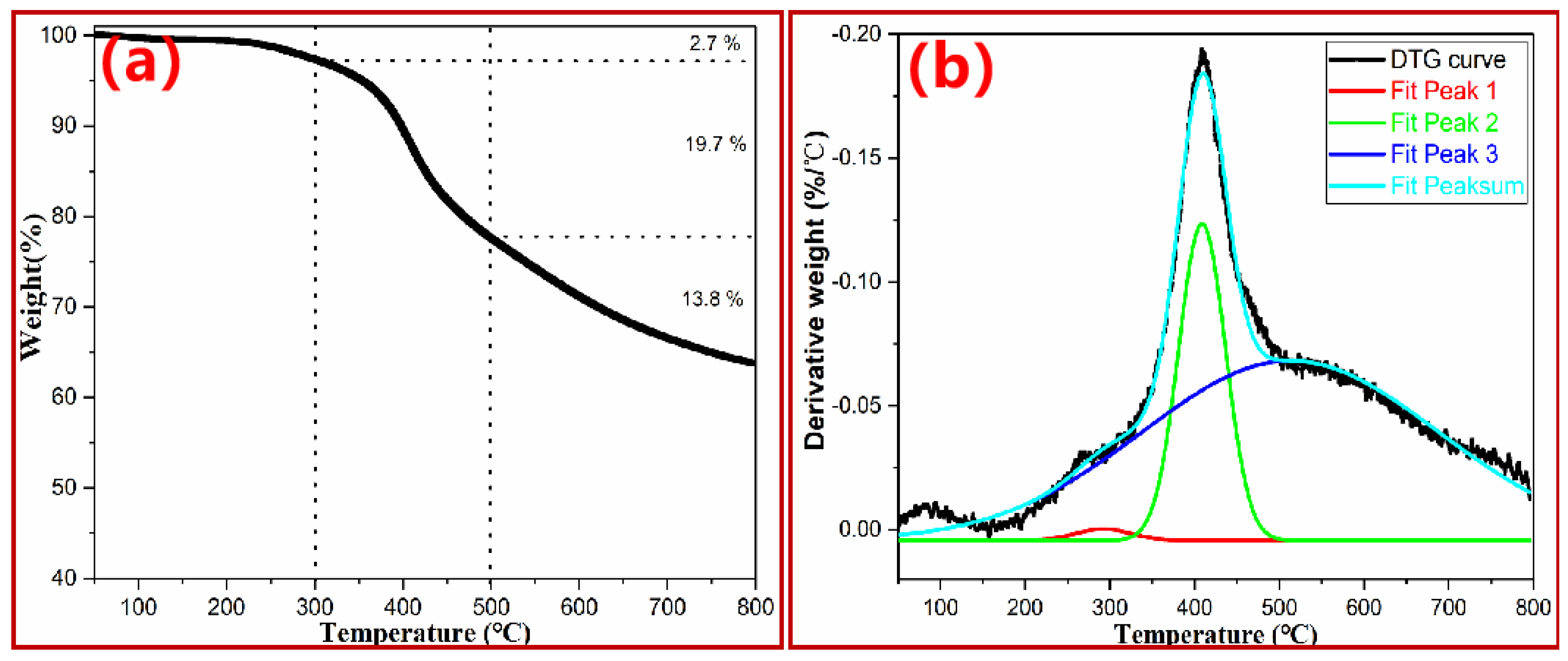
3.2. General Thermal Degradation Behavior of PN Foam by TGA–DTG
3.3. Monitoring Thermal Degradation of PN Foam by TGA–FTIR
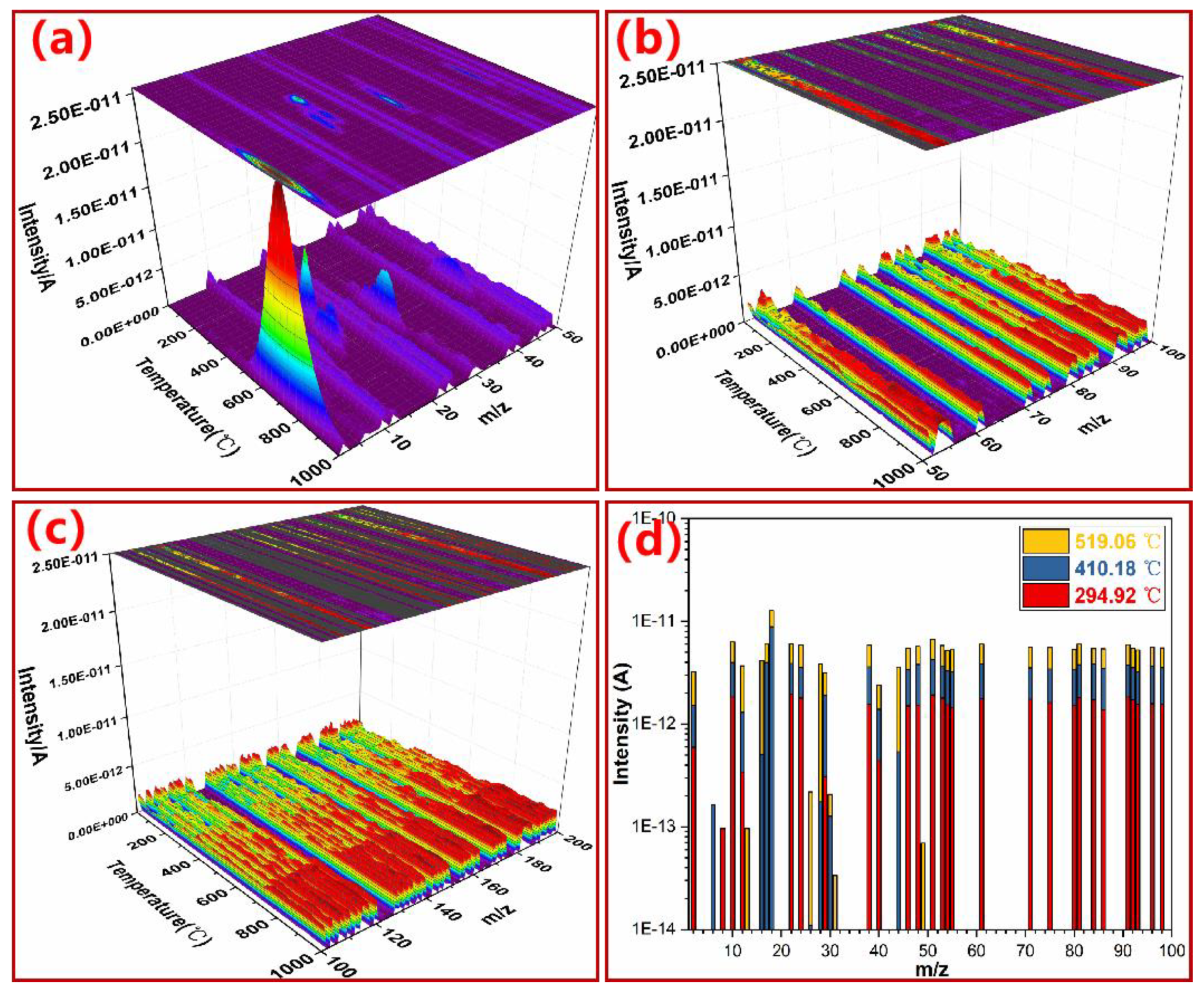
3.4. Monitoring Thermal Degradation of PN Foam by TGA–MS
3.5. Possible Thermal Degradation Mechanisms of PN Foam
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Derradji, M.; Wang, J.; Liu, W. Phthalonitrile Resins and Composites; William Andrew Publishing: New York, NY, USA, 2018; pp. 107–174. [Google Scholar]
- Ye, J.; Zhang, S.; Wu, M.; Liu, X.; Liu, X. Thermal, mechanical and dielectric property enhancement of benzoxazine-containing phthalonitrile resin: The effect of functional oligomeric polyphenyl ether. Polymer 2023, 280, 126040. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, L.; Lin, J.; Du, L. A theoretical insight into the curing mechanism of phthalonitrile resins promoted by aromatic amines. Phys. Chem. Chem. Phys. 2021, 23, 17300–17309. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lei, W.; Liu, Q.; Li, Y.; Li, K.; Wang, P.; Feng, W. A tailor-made method to recycle slow-curing thermoset of phthalonitrile by constructing self-composite with the improved properties. Compos. Commun. 2021, 26, 100791. [Google Scholar] [CrossRef]
- Xu, M.; Ren, D.; Chen, L.; Li, K.; Liu, X. Understanding of the polymerization mechanism of the phthalonitrile-based resins containing benzoxazine and their thermal stability. Polymer 2018, 143, 28–39. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, X.; Wei, J.; Xu, M.; Tong, L.; Zhao, R.; Liu, X. Morphological, electrical, thermal and mechanical properties of phthalocyanine/multi-wall carbon nanotubes nanocomposites prepared by masterbatch dilution. J. Polym. Res. 2012, 19, 9969. [Google Scholar] [CrossRef]
- Fu, T.; Zhao, X.; Chen, L.; Fu, T.; Zhao, X.; Chen, L.; Wu, W.; Zhao, Q.; Wang, X.; Guo, D.; et al. Bioinspired color changing molecular sensor toward early fire detection based on transformation of phthalonitrile to phthalocyanine. Adv. Funct. Mater. 2019, 29, 1806586. [Google Scholar] [CrossRef]
- Xu, X.; Xu, M.; Liu, T.; Ren, D.; Liu, X. Understanding the curing behaviors and properties of phthalonitrile containing benzoxazine with a new type of aniline curing agent. Polym. Test. 2022, 107, 107487. [Google Scholar] [CrossRef]
- Bai, S.; Sun, X.; Wu, M.; Shi, X.; Chen, X.; Yu, X.; Zhang, Q. Effects of pure and intercalated halloysites on thermal properties of phthalonitrile resin nanocomposites. Polym. Degrad. Stab. 2020, 177, 109192. [Google Scholar] [CrossRef]
- Chen, X.; Cai, Y.; Qu, X.; Chen, J.; Zheng, D. Preparation of a self-catalyzed amino-epoxy phthalonitrile resin with a large processing window. J. Mater. Sci. 2022, 57, 1545–1553. [Google Scholar] [CrossRef]
- Kolesnikov, T.I.; Orlova, A.M.; Tsegelskaya, A.Y.; Cherkaev, G.V.; Kechekyan, A.S.; Buzin, A.I.; Dmitryakov, P.V.; Belousov, S.I.; Abramov, I.G.; Serushkina, O.V. Dual-curing propargyl-phthalonitrile imide-based thermoset: Synthesis, characterization and curing behavior. Eur. Polym. J. 2021, 161, 110865. [Google Scholar] [CrossRef]
- Yakovlev, M.V.; Morozov, O.S.; Afanaseva, E.S.; Babkin, A.V.; Kepman, A.V. Tri-functional phthalonitrile monomer as stiffness increasing additive for easy processable high performance resins. React. Funct. Polym. 2020, 146, 104409. [Google Scholar] [CrossRef]
- Chaussoy, N.; Brandt, D.; Gérard, J.F. Phthalonitrile functionalized resoles—Use of 2,3-dicyanohydroquinone as a versatile monomer for resins with very high thermal stability. Polym. Degrad. Stab. 2023, 110420. [Google Scholar] [CrossRef]
- Nechausov, S.; Aleksanova, A.; Morozov, O.; Babkin, A.; Kepman, A.; Avdeev, V.; Bulgakov, B. Heat-Resistant Phthalonitrile-Based Resins for 3D Printing via Vat Photopolymerization. ACS Appl. Polym. Mater. 2022, 4, 6958–6968. [Google Scholar] [CrossRef]
- Bulgakov, B.A.; Sulimov, A.V.; Babkin, A.V.; Timoshkin, I.A.; Solopchenko, A.V.; Kepman, A.V.; Avdeev, V.V. Phthalonitrile-carbon fiber composites produced by vacuum infusion process. J. Compos. Mater. 2017, 51, 4157–4164. [Google Scholar] [CrossRef]
- Yang, X.; Li, K.; Xu, M.; Jia, K.; Liu, X. Designing a low-temperature curable phenolic/benzoxazine-functionalized phthalonitrile copolymers for high performance composite laminates. J. Polym. Res. 2017, 24, 195. [Google Scholar] [CrossRef]
- Bulgakov, B.A.; Morozov, O.S.; Timoshkin, I.A.; Babkin, A.V.; Kepman, A.V. Bisphthalonitrile-based thermosets as heat-resistant matrices for fiber reinforced plastics. Polym. Sci. Ser. C 2021, 63, 64–101. [Google Scholar] [CrossRef]
- Derradji, M.; Wang, J.; Liu, W. High performance ceramic-based phthalonitrile micro and nanocomposites. Mater. Lett. 2016, 182, 380–385. [Google Scholar] [CrossRef]
- Liu, C.; Qiao, Y.; Jia, H.; Li, N.; Chen, Y.; Jian, X. Improved mechanical properties of basalt fiber/phthalonitrile composites modified by thermoplastic poly (phthalazinone ether nitrile)s. Polymer 2021, 228, 123947. [Google Scholar] [CrossRef]
- Yang, X.; Li, K.; Xu, M.; Liu, X. Significant improvement of thermal oxidative mechanical properties in phthalonitrile GFRP composites by introducing microsilica as complementary reinforcement. Compos. Part B Eng. 2018, 155, 425–430. [Google Scholar] [CrossRef]
- Sun, B.G.; Shi, H.Q.; Yang, K.X.; Lei, Q.; Li, Y.Q.; Fu, Y.Q.; Hu, N.; Guo, Y.; Zhou, H.; Fu, S.Y. Effects of 3-aminophenylacetylene on mechanical properties at elevated temperatures of carbon fiber/phthalonitrile composites. Compos. Commun. 2020, 18, 55–61. [Google Scholar] [CrossRef]
- Sun, B.G.; Lei, Q.; Guo, Y.; Shi, H.Q.; Sun, J.B.; Yang, K.X.; Zhou, H.; Li, Y.Q.; Hu, N.; Wang, H. Enhanced mechanical properties at 400 °C of carbon fabric reinforced phthalonitrile composites by high temperature postcure. Compos. Part B Eng. 2019, 166, 681–687. [Google Scholar] [CrossRef]
- Derradji, M.; Ramdani, N.; Zhang, T.; Wang, J.; Gong, L.; Xu, X.; Lin, Z.; Henniche, A.; Rahoma, H.; Liu, W. Effect of silane surface modified titania nanoparticles on the thermal, mechanical, and corrosion protective properties of a bisphenol-A based phthalonitrile resin. Prog. Org. Coat. 2016, 90, 34–43. [Google Scholar] [CrossRef]
- Laskoski, M.; Keller, T.M.; Qadri, S.B. Direct conversion of highly aromatic phthalonitrile thermosetting resins into carbon nanotube containing solids. Polymer 2007, 48, 7484–7489. [Google Scholar] [CrossRef]
- Lei, Y.; Zhao, R.; Xu, M.; Zhao, X.; Yang, X.; Guo, H.; Zhong, J.; Liu, X. Production of empty and iron-filled multiwalled carbon nanotubes from iron–phthalocyanine polymer and their electromagnetic properties. J. Mater. Sci. Mater. Electron. 2012, 23, 921–927. [Google Scholar] [CrossRef]
- Weng, Z.; Zhang, K.; Qi, Y.; Zhang, T.; Xia, M.; Hu, F.; Zhang, S.; Liu, C.; Wang, J.; Jian, X. Scalable fabrication of heteroatom-doped versatile hierarchical porous carbons with an all-in-one phthalonitrile precursor and their applications. Carbon 2020, 159, 495–503. [Google Scholar] [CrossRef]
- Zeng, J.; Xie, W.; Zhou, H.; Zhao, T.; Xu, B.; Jiang, Q.; Algadi, H.; Zhou, Z.; Gu, H. Nitrogen-doped graphite-like carbon derived from phthalonitrile resin with controllable negative magnetoresistance and negative permittivity. Adv. Compos. Hybrid Mater. 2023, 6, 64. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.; Roy, S.; Chu, E.; See, K.; Hu, X. Phthalonitrile-based carbon foam with high specific mechanical strength and superior electromagnetic interference shielding performance. ACS Appl. Mater. Interfaces 2016, 8, 7422–7430. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Wang, D.; Li, Y.; Li, K.; Liu, Q.; Wang, P.; Feng, W.; Liu, Q.; Yang, X. High temperature resistant polymer foam based on bi-functional benzoxazine-phthalonitrile resin. Polym. Degrad. Stab. 2022, 201, 110003. [Google Scholar] [CrossRef]
- Lei, W.; Wang, D.; Liu, Q.; Li, K.; Li, Y.; Zhong, F.; Liu, Q.; Wang, P.; Feng, W.; Yang, X. Large-scale preparation of uniform millet bread-like durable benzoxazine-phthalonitrile foam with outstanding mechanical and thermal properties. Polymers 2022, 14, 5410. [Google Scholar] [CrossRef]
- Tay, Y.S.; Liu, M.; Lim, J.S.K.; Chen, H.; Hu, X. Phthalonitrile prepolymer and PAN blends: New strategy for precursor stabilization and pyrolytic char yield enhancement. Polym. Degrad. Stab. 2020, 172, 109056. [Google Scholar] [CrossRef]
- Mortezaeikia, V.; Tavakoli, O.; Khodaparasti, M.S. A review on kinetic study approach for pyrolysis of plastic wastes using thermogravimetric analysis. J. Anal. Appl. Pyrolysis 2021, 160, 105340. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Lei, W.; Liu, X.; Zeng, Q.; Liu, Q.; Feng, W.; Li, K.; Wang, P. Thermal degradation behaviors of poly (arylene ether nitrile) bearing pendant carboxyl groups. Polym. Degrad. Stab. 2021, 191, 109668. [Google Scholar] [CrossRef]
- Liang, B.; Hu, J.; Yuan, P.; Li, C.; Li, R.; Liu, Y.; Zeng, K.; Yang, G. Kinetics of the pyrolysis process of phthalonitrile resin. Thermochim. Acta 2019, 672, 133–141. [Google Scholar] [CrossRef]
- Liang, B.; Wang, J.; Hu, J.; Li, C.; Li, R.; Liu, Y.; Zeng, K.; Yang, G. TG-MS-FTIR study on pyrolysis behavior of phthalonitrile resin. Polym. Degrad. Stab. 2019, 169, 108954. [Google Scholar] [CrossRef]
- Zhou, T.; Xiao, H.; Peng, W.; Liang, B.; Liu, Y.; Lv, J.; Hu, J.; Zeng, K.; Yang, G. Study on pyrolysis behaviors of L-tyrosine-based phthalonitrile resin. Polym. Test. 2020, 86, 106506. [Google Scholar] [CrossRef]
- Guo, X.; Liang, B.; Chen, M.; He, X.; Xiao, H.; Zeng, K.; Zhou, T.; Hu, J.; Yang, G. Study on pyrolysis behavior of bio-based adenine containing phthalonitrile resin obtained by powder metallurgy-like process. Polym. Degrad. Stab. 2021, 188, 109569. [Google Scholar] [CrossRef]
- Lobanova, M.S.; Aleshkevich, V.V.; Yablokova, M.Y.; Morozov, O.S.; Babkin, A.V.; Kepman, A.V.; Avdeev, V.V.; Bulgakov, B.A. Kinetics of the oxidative aging of phthalonitrile resins and their effects on the mechanical properties of thermosets. Thermochim. Acta 2023, 724, 179492. [Google Scholar] [CrossRef]
- Jin, F.L.; Zhao, M.; Park, M.; Park, S. Recent trends of foaming in polymer processing: A review. Polymers 2019, 11, 953. [Google Scholar] [CrossRef]
- Fraleoni-Morgera, A.; Chhikara, M. Polymer-based nano-composites for thermal insulation. Adv. Eng. Mater. 2019, 21, 1801162. [Google Scholar] [CrossRef]
- Członka, S.; Strąkowska, A.; Kairytė, A. Effect of walnut shells and silanized walnut shells on the mechanical and thermal properties of rigid polyurethane foams. Polym. Test. 2020, 87, 106534. [Google Scholar] [CrossRef]
- Jiao, L.; Xiao, H.; Wang, Q.; Sun, J. Thermal degradation characteristics of rigid polyurethane foam and the volatile products analysis with TG-FTIR-MS. Polym. Degrad. Stab. 2013, 98, 2687–2696. [Google Scholar] [CrossRef]
- ISO 845:2006; Cellular Plastics and Rubbers—Determination of Apparent Density. International Organization of Standards: Geneva, Switzerland, 2006.
- Han, M.; Yin, X.; Ren, S.; Duan, W.; Zhang, L.; Cheng, L. Core/shell structured C/ZnO nanoparticles composites for effective electromagnetic wave absorption. RSC Adv. 2016, 6, 6467–6474. [Google Scholar] [CrossRef]
- Naskar, A.; Paul, S. Non-destructive measurement of grinding-induced deformation-depth using grazing incidence X-ray diffraction technique. NDT E Int. 2022, 126, 102592. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Kleinsorge, B.; Morrison, N.A.; Hart, A.; Stolojan, V.; Robertson, J. Stress reduction and bond stability during thermal annealing of tetrahedral amorphous carbon. J. Appl. Phys. 1999, 85, 7191–7197. [Google Scholar] [CrossRef]
- Zhou, D.W.; Liang, L.Y.; Lu, M.G. Dimeric liquid crystalline thermosets from azo-containing diglycidyl ether cured by anhydride. Polym. Bull. 2011, 66, 1111–1123. [Google Scholar] [CrossRef]
- Ayer, M.A.; Simon, Y.C.; Weder, C. Azo-containing polymers with degradation on-demand feature. Macromolecules 2016, 49, 2917–2927. [Google Scholar] [CrossRef]
- Lisa, G.; Păiuş, C.; Raicu-Luca, A.; Hurduc, N. Azo-polysiloxanes thermal stability study: Thermal stability of azo-polysiloxanes with biological applications. High Perform. Polym. 2012, 24, 530–537. [Google Scholar] [CrossRef]
- Kumazawa, H.; Inoue, M.; Kasuya, T. Photocatalytic degradation of volatile and nonvolatile organic compounds on titanium dioxide particles using fluidized beds. Ind. Eng. Chem. Res. 2003, 42, 3237–3244. [Google Scholar] [CrossRef]
- Benhammada, A.; Trache, D. Thermal decomposition of energetic materials using TG-FTIR and TG-MS: A state-of-the-art review. Appl. Spectrosc. Rev. 2020, 55, 724–777. [Google Scholar] [CrossRef]
- Bai, J.; Yu, C.; Li, L.; Wu, P.; Luo, Z.; Ni, M. Experimental study on the NO and N2O formation characteristics during biomass combustion. Energy Fuels 2013, 27, 515–522. [Google Scholar] [CrossRef]
- Yang, S.; Zhu, X.; Wang, J.; Jin, X.; Liu, Y.; Qian, F.; Zhang, S.; Chen, J. Combustion of hazardous biological waste derived from the fermentation of antibiotics using TG–FTIR and Py–GC/MS techniques. Bioresour. Technol. 2015, 193, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R. Chemical Bonds and Bonds Energy; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gaseous Volatile | m/z | Tr of Gaseous Product (°C) | Trm of Benzoxazine-Containing PN Foam (°C) | Trm of Resorcinol-Based PN Resin (°C) [35] | Trm of l-Tyrosine-Based PN Resin (°C) [36] | Trm of Adenine-Containing PN Resin (°C) [37] |
|---|---|---|---|---|---|---|
| H2O | 18 | 340–620 | 406, 540 | 560 | 550 | Not given |
| NH3 | 17 | 350–810 | 403, 501 | 580 | 566 | varied |
| CO2 | 44 | 360–840 | 407, 595, 776 | 550 | 437 | Not given |
| CH4 | 16 | 400–670 | 403, 539, 601 | 590 | 444 | 590, 890 |
| CO | 28 | 400–720 | 483, 590 | 590, 720 | 439 | Not given |
| H2 | 2 | 500–920 | 744 | Not given | 766 | Not given |
| HCN/NO? | 27/30 | 380–1000 | 403, 561, 766 | HCN, 600 | HCN, 576 | HCN, 600 |
| Ar-R | >78 | varied | varied | C6H6, 590 | Not given | 500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Li, Y.; Lei, W.; Bai, Z.; Zhan, Y.; Li, Y.; Li, K.; Wang, P.; Feng, W.; Liu, Q. Understanding the Thermal Degradation Mechanism of High-Temperature-Resistant Phthalonitrile Foam at Macroscopic and Molecular Levels. Polymers 2023, 15, 3947. https://doi.org/10.3390/polym15193947
Yang X, Li Y, Lei W, Bai Z, Zhan Y, Li Y, Li K, Wang P, Feng W, Liu Q. Understanding the Thermal Degradation Mechanism of High-Temperature-Resistant Phthalonitrile Foam at Macroscopic and Molecular Levels. Polymers. 2023; 15(19):3947. https://doi.org/10.3390/polym15193947
Chicago/Turabian StyleYang, Xulin, Yi Li, Wenwu Lei, Zhongxiang Bai, Yingqing Zhan, Ying Li, Kui Li, Pan Wang, Wei Feng, and Qi Liu. 2023. "Understanding the Thermal Degradation Mechanism of High-Temperature-Resistant Phthalonitrile Foam at Macroscopic and Molecular Levels" Polymers 15, no. 19: 3947. https://doi.org/10.3390/polym15193947