

Tailoring the Hydroxyl Density of Glass Surface for Anionic Ring-Opening Polymerization of Polyamide 6 to Manufacture Thermoplastic Composites
 , , , and
, , , and
Abstract
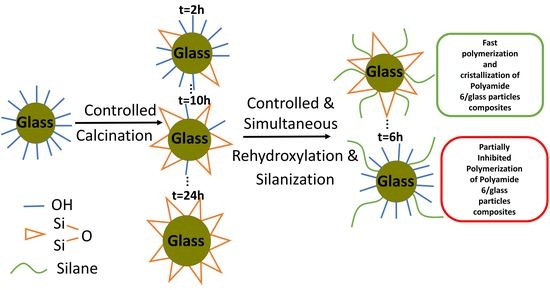
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Particle Surface Modification
2.2.1. Control of the Surface Density of Hydroxyl Groups
2.2.2. Optimization of the Treatment Time
2.2.3. Surface Modification by Silanization
2.3. Particle Surface Characterization
2.3.1. Specific Surface Area Measurement
2.3.2. Thermogravimetric Analysis (TGA)
2.3.3. Ft-Ir Spectroscopic Measurements
2.4. Differential Scanning Calorimetry (DSC) Characterization of PA6 Polymerization and Crystallization
2.4.1. Sample Preparation
2.4.2. DSC
2.5. Composite Mechanical Properties
3. Results
3.1. Characterizing and Controlling the Hydroxyl Groups Surface Density on Glass Particles
3.1.1. Effect of the Initial Calcination Time on the -OH Surface Density
3.1.2. Impact of Dehydroxylation on PA6 Polymerization
3.2. Controlling the -OH Quantity Regenerated during the Silanization Treatment
3.3. Polymerization and Crystallization Kinetics after Silane Grafting
3.3.1. Silane Surface Modification and Its Influence on Polymerization and Crystallization
3.3.2. Relevance of the Developed Protocol
3.4. Mechanical Properties
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ji, X.; Xu, Y.; Zhang, W.; Cui, L.; Liu, J. Review of functionalization, structure and properties of graphene/polymer composite fibers. Compos. Part Appl. Sci. Manuf. 2016, 87, 29–45. [Google Scholar] [CrossRef]
- Pratyush Behera, R.; Rawat, P.; Kumar Tiwari, S.; Kumar Singh, K. A brief review on the mechanical properties of Carbon nanotube reinforced polymer composites. Mater. Today Proc. 2020, 22, 2109–2117. [Google Scholar] [CrossRef]
- Altin Karataş, M.; Gökkaya, H. A review on machinability of carbon fiber reinforced polymer (CFRP) and glass fiber reinforced polymer (GFRP) composite materials. Def. Technol. 2018, 14, 318–326. [Google Scholar] [CrossRef]
- Vaidya, U.K.; Chawla, K.K. Processing of fibre reinforced thermoplastic composites. Int. Mater. Rev. 2008, 53, 185–218. [Google Scholar] [CrossRef]
- Petersen, H.; Kusano, Y.; Brøndsted, P.; Almdal, K. Preliminary characterization of glass fiber sizing. In Proceedings of the Risø International Symposium on Materials Science; Department of Wind Energy, Risø Campus, Technical University of Denmark: Lyngby, Denmark, 2013; Volumn 34, pp. 333–340. [Google Scholar]
- Thomason, J. Glass fibre sizing: A review. Compos. Part Appl. Sci. Manuf. 2019, 127, 105619. [Google Scholar] [CrossRef]
- Matisons, J.G. Silanes and Siloxanes as Coupling Agents to Glass: A Perspective. In Silicone Surface Science; Advances in Silicon, Science; Owen, M.J., Dvornic, P.R., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volumn 4, pp. 281–298. [Google Scholar]
- Plonka, R.; Mäder, E.; Gao, S.; Bellmann, C.; Dutschk, V.; Zhandarov, S. Adhesion of epoxy/glass fibre composites influenced by aging effects on sizings. Compos. Part Appl. Sci. Manuf. 2004, 35, 1207–1216. [Google Scholar] [CrossRef]
- Plueddemann, E.P. Silane Coupling Agents; Springer: Boston, MA, USA, 1991. [Google Scholar]
- Allen, K. Silanes as the interphase in adhesive bonds. J. Adhes. Sci. Technol. 1992, 6, 23–32. [Google Scholar] [CrossRef]
- Li, D.; Liu, Q.; Yu, L.; Li, X.; Zhang, Z. Correlation between interfacial interactions and mechanical properties of PA-6 doped with surface-capped nano-silica. Appl. Surf. Sci. 2009, 255, 7871–7877. [Google Scholar] [CrossRef]
- Li, Z.; Xiao, T.; Zhao, S. Effects of surface treatments on Mechanical properties of Continuous basalt fibre cords and their adhesion with rubber matrix. Fibers Polym. 2016, 17, 910–916. [Google Scholar] [CrossRef]
- van Rijswijk, K.; Teuwen, J.; Bersee, H.; Beukers, A. Textile fiber-reinforced anionic polyamide-6 composites. Part I: The vacuum infusion process. Compos. Part Appl. Sci. Manuf. 2009, 40, 1–10. [Google Scholar] [CrossRef]
- Senani, S.D.M. Interaction Organosilanes/Silice de précipitation du Milieu Hydro-Alcoolique au Milieu Aqueux. Ph.D. Thesis, Université Pierre et Marie Curie-Paris VI, Paris, France, 2004. [Google Scholar]
- Zhuravlev, L. Surface characterization of amorphous silica-a review of work from the former USSR. Colloids Surf. Physicochem. Eng. Asp. 1993, 74, 71–90. [Google Scholar] [CrossRef]
- Gonzalez-Benito, J.; Baselga, J.; Aznar, A.J. Microstructural and wettability study of surface pretreated glass fibres. J. Mater. Process. Technol. 1999, 92, 129–134. [Google Scholar] [CrossRef]
- Young, G. Interaction of water vapor with silica surfaces. J. Colloid Sci. 1958, 13, 67–85. [Google Scholar] [CrossRef]
- Zhuravlev, L. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf. Physicochem. Eng. Asp. 2000, 173, 1–38. [Google Scholar] [CrossRef]
- Erkelens, J.; Linsen, B. Quantitative determination of hydroxyl groups and water for silica. J. Colloid Interface Sci. 1969, 29, 464–468. [Google Scholar] [CrossRef]
- Christy, A.A.; Egeberg, P.K. Quantitative determination of surface silanol groups in silicagel by deuterium exchange combined with infrared spectroscopy and chemometrics. Analyst 2005, 130, 738–744. [Google Scholar] [CrossRef]
- Bermudez, V.M. Proton nuclear magnetic resonance technique for determining the surface hydroxyl content of hydrated silica gel. J. Phys. Chem. 1970, 74, 4160–4161. [Google Scholar] [CrossRef]
- Kellum, G.E.; Smith, R.C. Determination of water, silanol, and strained siloxane on silica surfaces. Anal. Chem. 1967, 39, 341–345. [Google Scholar] [CrossRef]
- Mueller, R.; Kammler, H.K.; Wegner, K.; Pratsinis, S.E. OH Surface Density of SiO2 and TiO2 by Thermogravimetric Analysis. Langmuir 2003, 19, 160–165. [Google Scholar] [CrossRef]
- Vansant, E.F.; Van Der Voort, P.; Vrancken, K.C. Characterization and Chemical Modification of the Silica Surface; Elsevier: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Brochier Salon, M.C.; Belgacem, M.N. Hydrolysis-condensation kinetics of different silane coupling agents. Phosphorus, Sulfur, Silicon Relat. Elem. 2011, 186, 240–254. [Google Scholar] [CrossRef]
- Thomason, J.L.; Nagel, U.; Yang, L.; Bryce, D. A study of the thermal degradation of glass fibre sizings at composite processing temperatures. Compos. Part Appl. Sci. Manuf. 2019, 121, 56–63. [Google Scholar] [CrossRef]
- Vicard, C.; De Almeida, O.; Cantarel, A.; Bernhart, G. Experimental study of polymerization and crystallization kinetics of polyamide 6 obtained by anionic ring opening polymerization of ϵ-caprolactam. Polymer 2017, 132, 88–97. [Google Scholar] [CrossRef]
- Labrosse, A.; Burneau, A. Characterization of porosity of ammonia catalysed alkoxysilane silica. J. -Non-Cryst. Solids 1997, 221, 107–124. [Google Scholar] [CrossRef]
- Legrand, A.; Hommel, H.; Tuel, A.; Vidal, A.; Balard, H.; Papirer, E.; Levitz, P.; Czernichowski, M.; Erre, R.; Van Damme, H.; et al. Hydroxyls of silica powders. Adv. Colloid Interface Sci. 1990, 33, 91–330. [Google Scholar] [CrossRef]
- Perro, A. Synthèse et Valorisation de Particules Colloidales de Morphologie et de Fonctionnalité de Surface Controlées. Ph.D. Thesis, Université Bordeaux 1, Talence, France, 2006. [Google Scholar]
- Szekeres, M.; Dékány, I.; De Keizer, A. Adsorption of dodecyl pyridinium chloride on monodisperse porous silica. Colloids Surf. Physicochem. Eng. Asp. 1998, 141, 327–336. [Google Scholar] [CrossRef]
- Szekeres, M.; Tóth, J.; Dékány, I. Specific surface area of Stoeber silica determined by various experimental methods. Langmuir 2002, 18, 2678–2685. [Google Scholar] [CrossRef]
- Wells, J.; Koopal, L.; de Keizer, A. Monodisperse, nonporous, spherical silica particles. Colloids Surfaces Physicochem. Eng. Asp. 2000, 166, 171–176. [Google Scholar] [CrossRef]
- de Keizer, A.; van der Ent, E.; Koopal, L. Surface and volume charge densities of monodisperse porous silicas. Colloids Surf. Physicochem. Eng. Asp. 1998, 142, 303–313. [Google Scholar] [CrossRef]
- Park, J.; Subramanian, R. Interfacial shear strength and durability improvement by monomeric and polymeric silanes in basalt fiber/epoxy single-filament composite specimens. J. Adhes. Sci. Technol. 1991, 5, 459–477. [Google Scholar] [CrossRef]
- Sever, K.; Seki, Y.; Tavman, I.H.; Erkan, G.; Cecen, V. The structure of γ-glycidoxypropyltrimethoxysilane on glass fiber surfaces: Characterization by FTIR, SEM, and contact angle measurements. Polym. Compos. 2009, 30, 550–558. [Google Scholar] [CrossRef]
- Rathor, N.; Panda, S. Aminosilane densities on nanotextured silicon. Mater. Sci. Eng. 2009, 29, 2340–2345. [Google Scholar] [CrossRef]
- Zhao, J.; Li, Y.; Guo, H.; Gao, L. Relative surface density and stability of the amines on the biochip. Chin. J. Anal. Chem. 2006, 34, 1235–1238. [Google Scholar] [CrossRef]
- Acres, R.G.; Ellis, A.V.; Alvino, J.; Lenahan, C.E.; Khodakov, D.A.; Metha, G.F.; Andersson, G.G. Molecular structure of 3-aminopropyltriethoxysilane layers formed on silanol-terminated silicon surfaces. J. Phys. Chem. 2012, 116, 6289–6297. [Google Scholar] [CrossRef]
- Wang, X.Y.; Mertz, D.; Blanco-Andujar, C.; Bora, A.; Ménard, M.; Meyer, F.; Giraudeau, C.; Bégin-Colin, S. Optimizing the silanization of thermally-decomposed iron oxide nanoparticles for efficient aqueous phase transfer and MRI applications. RSC Adv. 2016, 6, 93784–93793. [Google Scholar] [CrossRef]
- Hair, M.L. Hydroxyl groups on silica surface. J. -Non-Cryst. Solids 1975, 19, 299–309. [Google Scholar] [CrossRef]
- van Rijswijk, K.; van Geenen, A.; Bersee, H. Textile fiber-reinforced anionic polyamide-6 composites. Part II: Investigation on interfacial bond formation by short beam shear test. Compos. Part Appl. Sci. Manuf. 2009, 40, 1033–1043. [Google Scholar] [CrossRef]
- Bessell, T.; Shortall, J.B. The crystallization and interfacial bond strength of nylon 6 at carbon and glass fibre surfaces. J. Mater. Sci. 1975, 10, 2035–2043. [Google Scholar] [CrossRef]
- Gonzalez-Benito, J.; Cabanelas, J.C.; Aznar, A.J.; Vigil, M.R.; Bravo, J.; Baselga, J. Surface characterization of silanized glass fibers by labeling with environmentally sensitive fluorophores. J. Appl. Polym. Sci. 1996, 62, 375–384. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Time in Silanization Solution (without Silane) | TGA Weight Loss 1 (±0.01%) | Hydroxyls Mass Concentration COH (±0.01 mmol OH·g−1) |
|---|---|---|
| 0 h (calcinated) | 0.14 | 0.16 |
| 2 h | 0.19 | 0.21 |
| 3 h | 0.24 | 0.27 |
| 6 h | 0.31 | 0.34 |
| Particle Type | Total Weight Loss (± 0.01%) | Weight Loss Related to Hydroxyls (±0.01%) | Weight Loss Related to Silane (±0.01%) | Silane Mass Concentration (±mmol·g−1) |
|---|---|---|---|---|
| Calcinated | 0.14 | 0.14 | None | None |
| Calcinated + silanized for 2 h | 0.71 | 0.05 | 0.66 | 0.05 |
| Isothermal Synthesis | 1st Heating | Cooling | 2nd Heating | |||
|---|---|---|---|---|---|---|
| Particle Type | Peak of Crystallization (±0.4 min) | Melting Temperature | Melting Enthalpy ΔHm | Crystallization Temperature | Melting Temperature | Melting Nnthalpy ΔHm |
| Tm (±0.2 °C) | (±0.6 J.g−1) | Tc (±0.3 °C) | Tm (±0.2 °C) | (±0.6 J.g−1) | ||
| Calcinated | 9.16 | 213.1 | 67.4 | 156.1 | 201.7 | 53.0 |
| Calcinated + silanizedfor 2 h | 7.83 | 215.6 | 71.2 | 159.4 | 206.7 | 54.7 |
| Particle Type | Maximum Stress (MPa) | Strain at Break (%) | Tensile Modulus (MPa) |
|---|---|---|---|
| Calcinated | 46 (±4.91) | 1.8 (±0.22) | 4390 (±84) |
| Calcinated + silanized for 2 h | 63 (±4.09) | 3.8 (±0.31) | 3281 (±230) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belkhiri, A.; Virgilio, N.; Nassiet, V.; Welemane, H.; Chabert, F.; De Almeida, O. Tailoring the Hydroxyl Density of Glass Surface for Anionic Ring-Opening Polymerization of Polyamide 6 to Manufacture Thermoplastic Composites. Polymers 2022, 14, 3663. https://doi.org/10.3390/polym14173663
Belkhiri A, Virgilio N, Nassiet V, Welemane H, Chabert F, De Almeida O. Tailoring the Hydroxyl Density of Glass Surface for Anionic Ring-Opening Polymerization of Polyamide 6 to Manufacture Thermoplastic Composites. Polymers. 2022; 14(17):3663. https://doi.org/10.3390/polym14173663
Chicago/Turabian StyleBelkhiri, Achraf, Nick Virgilio, Valérie Nassiet, Hélène Welemane, France Chabert, and Olivier De Almeida. 2022. "Tailoring the Hydroxyl Density of Glass Surface for Anionic Ring-Opening Polymerization of Polyamide 6 to Manufacture Thermoplastic Composites" Polymers 14, no. 17: 3663. https://doi.org/10.3390/polym14173663