
The Relationship between Free Volume and Cooperative Rearrangement: From the Temperature-Dependent Neutron Total Scattering Experiment of Polystyrene

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Sample Preparing and Neutron Total Scattering
2.3. Molecular Dynamics Simulations
2.4. Fourier Transforms of MD Simulations
2.5. Define Cooperative Rearrangement Regions
3. Results
3.1. From Neutron Total Scattering to 3D Most Probable All-Atom Structure of PS
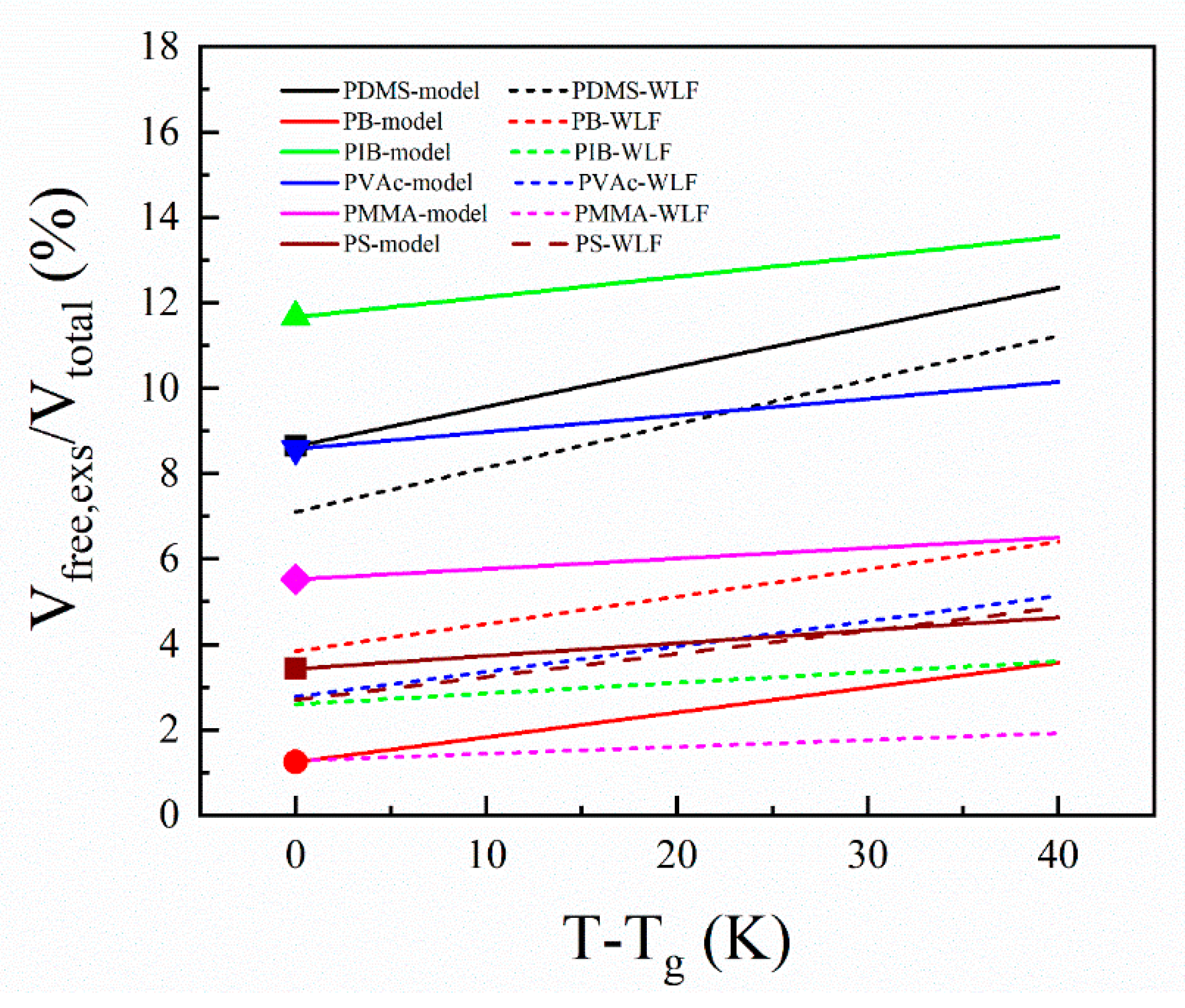
3.2. From a 3D Most Probable All-Atom Structure of PS to a Generalized Equation of Excess Free Volume
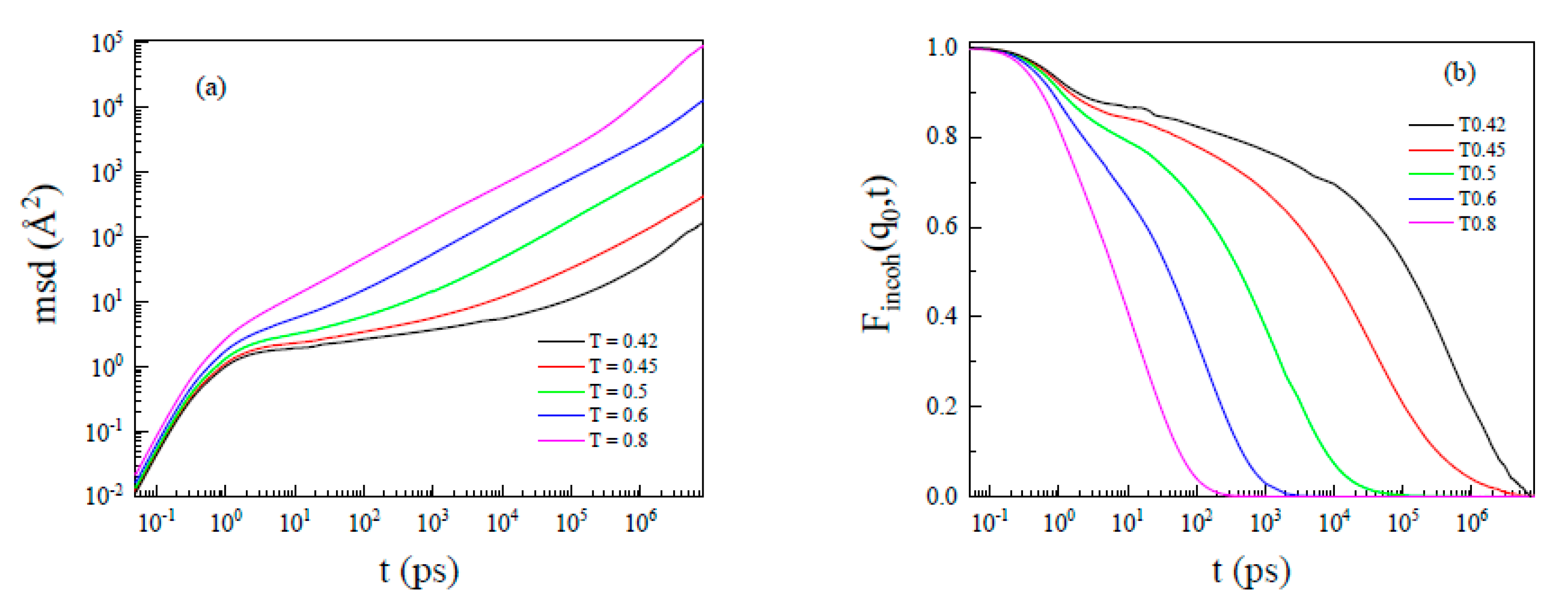
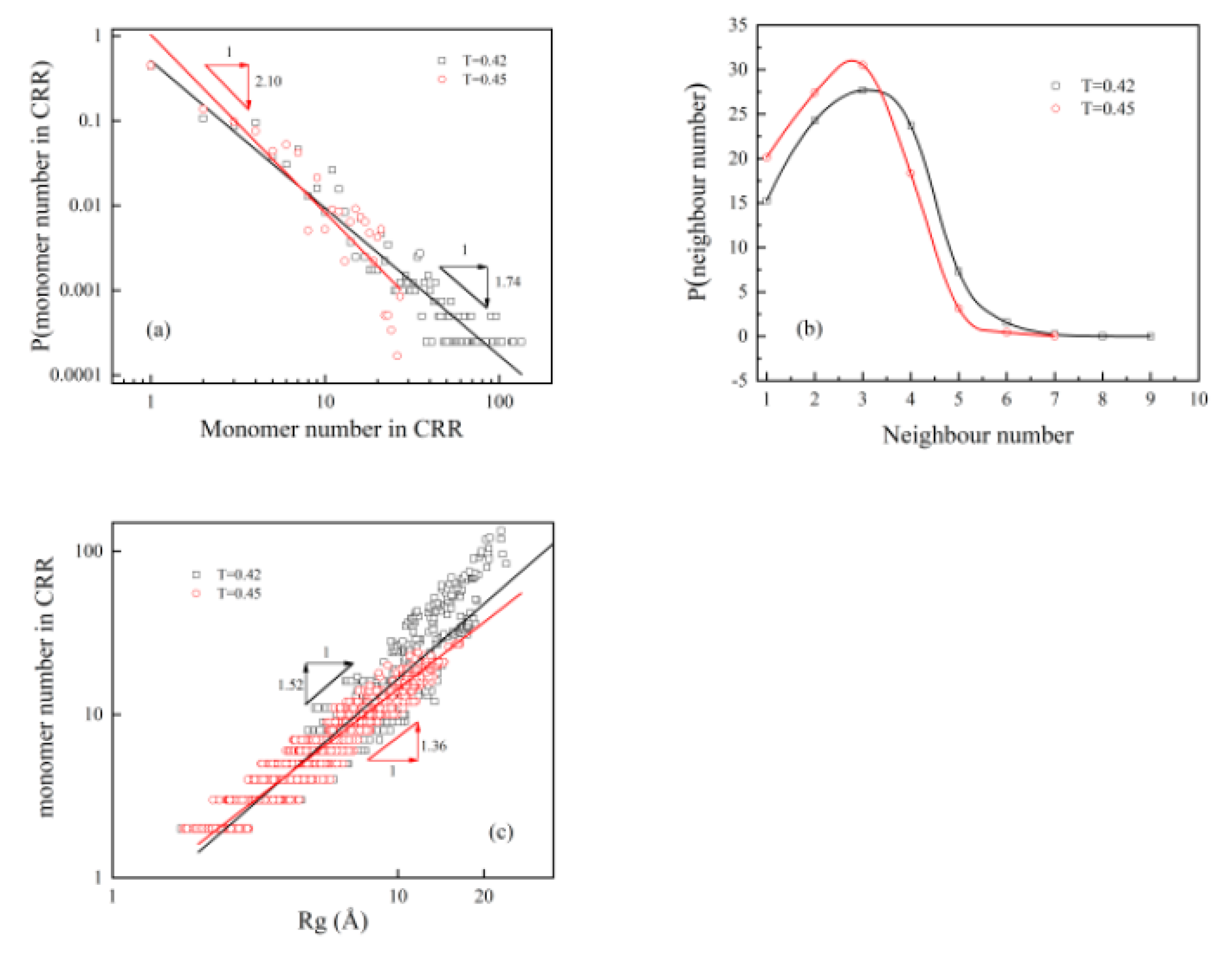
3.3. From the Slow Down Dynamics in the Cage to Heterogeneity
3.4. Relationship between “Static Cage” and “Dynamic Cage”
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vogel, H. The Temperature Dependence Law of the Viscosity of Fluids. Physikalische Zeitschrift 1921, 22, 645–646. [Google Scholar]
- Fox, T.G.; Flory, P.J. Further Studies on the Melt Viscosity of Polyisobutylene. J. Phys. Chem. 1951, 55, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, A.K.; Doolittle, D.B. Studies in Newtonian Flow. V. Further Verification of the Free Space Viscosity Equation. J. Appl. Phys. 1959, 28, 901–905. [Google Scholar] [CrossRef]
- Cohen, M.H.; Turnbull, D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 1959, 31, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Simha, R.; Boyer, R.F. On a General Relation Involving Glass Temperature and Coefficients of Expansion of Polymers. J. Chem. Phys. 1962, 37, 1003–1007. [Google Scholar] [CrossRef]
- White, R.P.; Lipson, J.E.G. Polymer Free Volume and Its Connection to the Glass Transition. Macromolecules 2016, 49, 3987–4007. [Google Scholar] [CrossRef]
- Horn, N.R. A critical review of free volume and occupied volume calculation methods. J. Membr. Sci. 2016, 518, 289–294. [Google Scholar] [CrossRef]
- Binder, K.; Kob, W. Glassy Materials and Disordered Solids: An Introduction to Their Statistical Mechanics; World Scientific: Singapore, 2006. [Google Scholar]
- Benigni, P. Thermodynamic analysis of the classical lattice-hole model of liquids. J. Non-Cryst. Solids 2020, 534, 119942. [Google Scholar] [CrossRef]
- Benigni, P. CALPHAD modeling of the glass trasition for a pure substance, coupling thermodynamics and relaxation kinetics. Calphad 2021, 72, 102238. [Google Scholar] [CrossRef]
- Tournier F., R. Thermodynamic Origin of the Vitreous Transition. Materials 2011, 4, 869–892. [Google Scholar] [CrossRef] [Green Version]
- Sanditov, D.S.; Mashanov, A.A.; Darmaev, M.V. Energy of Atomic Delocalization and Glass Temperature of Amorphous Substances. Polym. Sci. Ser. A 2020, 62, 79–84. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1970. [Google Scholar]
- Yuan, G.C.; Cheng, H.; Han, C.C. The Glass Formation of a Repulsive System with also a Short Range Attractive Potential: A Re-Interpretation of the Free Volume Theory. Polymer 2017, 131, 272–286. [Google Scholar] [CrossRef]
- Adam, G.; Gibbs, J.H. On the Temperature Dependence of Cooperative Relaxation Properties in Glass-Forming Liquids. J. Chem. Phys. 1965, 43, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.X.; Yunker, P.J.; Habdas, P.; Yodh, A.G. Cooperative Rearrangement Regions and Dynamical Heterogeneities in Colloidal Glasses with Attractive Versus Repulsive Interactions. Phys. Rev. Lett. 2011, 107, 208303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weeks, E.R.; Crocker, J.C.; Levitt, A.C.; Schofield, A.; Weitz, D.A. Imaging of Structural Relaxation Near the Colloidal Glass Transition. Science 2000, 287, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Ojovan, M.I. Glass Formation. In Encyclopedia of Glass Science, Technology, History, and Culture; Wiley: Hoboken, NJ, USA, 2021; pp. 249–259. [Google Scholar]
- Ojovan, M.I.; Louzguine-Luzgin, D.V. Revealing Structural Changes at Glass Transition via Radial Distribution Functions. J. Phys. Chem. B 2020, 124, 3186–3194. [Google Scholar] [CrossRef]
- Hoshino, T.; Fujinami, S.; Nakatani, T.; Kohmura, Y. Dynamical Heterogeneity near Glass Transition Temperature under Shear Conditions. Phys. Rev. Lett. 2020, 124, 118004. [Google Scholar] [CrossRef]
- Cotton, J.P.; Decker, D.; Benoit, H.; Farnoux, B.; Higgins, J.; Jannink, G.; Ober, R.; Picot, C.; des Cloizeaux, J. Conformation of Polymer Chain in the Bulk. Macromolecules 1974, 7, 863–872. [Google Scholar] [CrossRef]
- Mondello, M.; Yang, H.-J.; Furuya, H.; Roe, R.-J. Molecular Dynamics Simulation of Atactic Polystyrene. 1. Comparison with X-ray Scattering Data. Macromolecules 1994, 27, 3566–3574. [Google Scholar] [CrossRef]
- Furuya, H.; Mondello, M.; Yang, H.-J.; Roe, R.-J.; Erwin, R.W.; Han, C.C.; Smith, S.D. Molecular dynamics simulation of atactic polystyrene. 2. Comparison with Neutron Scattering Data. Macromolecules 1994, 27, 5674–5680. [Google Scholar] [CrossRef]
- Jiao, G.S.; Zuo, T.S.; Ma, C.L.; Han, Z.H.; Zhang, J.R.; Chen, Y.; Zhao, J.P.; Cheng, H.; Han, C.C. The 3d most-probable all-atom structure of atactic polystyrene during glass formation: A neutron total scattering study. Macromolecules 2020, 53, 5140–5146. [Google Scholar] [CrossRef]
- ISIS Neutron and Muon Source. Available online: http://www.isis.stfc.ac.uk (accessed on 7 September 2021).
- Bowron, D.T.; Soper, A.K.; Jones, K.; Ansell, S.; Birch, S.; Norris, J.; Perrott, L.; Riedel, D.; Rhodes, N.J.; Wakefield, S.R.; et al. NIMROD:The Near and InterMediate Range Order Diffractometer of the ISIS second target station. Rev. Sci. Instrum. 2010, 81, 033905. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhang, Y.; Montazerian, M.; Gulbiten, O.; Mauro, J.C.; Zanotto, E.D.; Yue, Y. Understanding Glass through Differential Scanning Calorimetry. Chem. Rev. 2019, 119, 7848–7939. [Google Scholar] [CrossRef]
- Gundrun–Rountines for Reducing Total Scattering Data. Available online: https://www.isis.stfc.ac.uk/Pages/Gudrun.aspx (accessed on 7 September 2021).
- Material Studio; Accelrys Software Inc.: San Diego, CA. Available online: http://accelrys.com/products/materials-studio/ (accessed on 7 September 2021).
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Severance, D.L. Aromatic-aromatic interactions: Free energy profiles for the benzene dimer in water, chloroform, and liquid benzene. J. Am. Chem. Soc. 1990, 112, 4768–4774. [Google Scholar] [CrossRef]
- Glaser, J.; Nguyen, T.D.; Anderson, J.A.; Lui, P.; Spiga, F.; Millan, J.A.; Morse, D.C.; Glotzer, S.C. Strong scaling of general-purpose molecular dynamics simulations on GPUs. Comput. Phys. Commun. 2015, 192, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.A.; Lorenz, C.D.; Travesset, A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J. Comput. Phys. 2008, 227, 5342–5359. [Google Scholar] [CrossRef]
- Bedrov, D.; Smith, G.; Douglas, J.F. Structural and dynamic heterogeneity in a telechelic polymer solution. Polymer 2004, 45, 3961–3966. [Google Scholar] [CrossRef]
- Bennemann, C.; Baschnagel, J.; Paul, W.; Binder, K. Molecular-dynamic simulation of a glassy polymer melt: Rouse model and cage effect. Comput. Theor. Polym. Sci. 2001, 11, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Sanditov, D.S.; Ojovan, M.I. Relaxation aspects of the liquid–glass transition. Physics-Uspekhi 2019, 62, 111–130. [Google Scholar] [CrossRef]
- Orwoll, R.A. Physical Properties of Polymers Handbook; Springer: Cham, Switzerland, 2007; pp. 93–101. [Google Scholar]
- Frick, B.; Alba-Simionesco, C.; Andersen, K.H.; Willner, L. Influence of density and temperature on the microscopic structure and the segmenatl relaxation of polybutadiene. Phys. Rev. E 2003, 67, 051801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, R.H.; Pant, P.V.K. Molecular packing and diffusion in polyisobutylene. Macromolecules 1991, 24, 6325. [Google Scholar] [CrossRef]
- Zhao, J.; Simon, S.L.; McKenna, G.B. Using 20-million-year-old amber to test the super-Arrhenius behaviour of glass-forming systems. Nat. Commun. 2013, 4, 1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larini, L.; Ottochian, A.; Michele, C.D.; Leporini, D. Universal scaling between structural relaxation and vibrational dynamics in glss-forming liquids and polymers. Nat. Phys. 2008, 4, 42–45. [Google Scholar] [CrossRef]
- Ottochian, A.; Michele, C.D.; Leporini, D. Universal divergenceless scaling between structural relaxation and caged dynamics in glass-forming systems. J. Chem. Phys. 2009, 131, 224517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betancourt, B.A.P.; Starr, F.W.; Douglas, J.F. String-like collective motion in the α- and β-relaxation of a coarse-grained polymer melt. J. Chem. Phys. 2018, 148, 104508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puosi, F.; Tripodo, A.; Leporini, D. Fast Vibrational Modes and Slow Heterogeneous Dynamics in Polymers and Viscous Liquids. Int. J. Mol. Sci. 2019, 20, 5708. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-H.; Mallamace, F.; Mou, C.-Y.; Broccio, M.; Corsaro, C.; Faraone, A.; Liu, L. The violation of the Stokes–Einstein relation in supercooled water. Proc. Natl. Acad. Sci. USA 2006, 103, 12974. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Jose, P.P. Violation of Stokes–Einstein and Stokes–Einstein–Debye relations in polymers at the gas-supercooled liquid coexistence. J. Phys. Condens. Matter 2020, 33, 055401. [Google Scholar]
- Kawasaki, T.; Kim, K. Spurious violation of the Stokes–Einstein–Debye relation in supercooled water. Sci. Rep. 2019, 9, 8118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcasu, A.Z.; Benmouna, M.; Han, C.C. Interpretation of dynamic scattering from polymer solutions. Polymer 1980, 21, 866–890. [Google Scholar] [CrossRef] [Green Version]
- Berthier, L. Dynamic heterogeneity in amorphous materials. Physics 2011, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Salez, T.; Salez, J.; Dolnoki-Veress, K.; Raphael, E.; Forrest, J.A. Cooperative strings and glassy interfaces. Proc. Natl. Acad. Sci. USA 2015, 112, 8227–8231. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.N.; Cheng, X.; Rivers, M.L.; Jaeger, H.M.; Nagel, S. the Packing of granular polymer chains. Science 2009, 326, 408–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, C.; Glotzer, S.C.; Poole, P.H.; Kob, W.; Plimpton, S.J. Spatial correlations of mobility and immobility in a glass-forming Lennard-Jones liquid. Phys. Rev. E 1999, 60, 3107–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Tg (K) | Calculation Results According to Equations (6)–(8) | WLF Results from Literature a | ||
|---|---|---|---|---|---|
| Fractional Excess Free volume at Tg (%) | αL-αG (×10−4 K−1) | Fractional Excess Free volume at Tg (%) | Slope (×10−4) | ||
| PDMS | 150 | 8.6 | 9.3 b | 7.1 | 10.3 |
| PB | 172 | 1.2 | 5.8 c | 3.8 | 6.4 |
| PIB | 205 | 11.7 | 4.7 d | 2.6 | 2.5 |
| PVAc | 305 | 8.6 | 3.9 b | 2.8 | 5.9 |
| PMMA | 381 | 5.5 | 2.5 b | 1.3 | 1.6 |
| PS-h8 | 373 | 1.5 | 3.0 | 2.7 | 5.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Z.; Jiao, G.; Ma, C.; Zuo, T.; Han, C.C.; Cheng, H. The Relationship between Free Volume and Cooperative Rearrangement: From the Temperature-Dependent Neutron Total Scattering Experiment of Polystyrene. Polymers 2021, 13, 3042. https://doi.org/10.3390/polym13183042
Han Z, Jiao G, Ma C, Zuo T, Han CC, Cheng H. The Relationship between Free Volume and Cooperative Rearrangement: From the Temperature-Dependent Neutron Total Scattering Experiment of Polystyrene. Polymers. 2021; 13(18):3042. https://doi.org/10.3390/polym13183042
Chicago/Turabian StyleHan, Zehua, Guisheng Jiao, Changli Ma, Taisen Zuo, Charles C. Han, and He Cheng. 2021. "The Relationship between Free Volume and Cooperative Rearrangement: From the Temperature-Dependent Neutron Total Scattering Experiment of Polystyrene" Polymers 13, no. 18: 3042. https://doi.org/10.3390/polym13183042