Synthesis and Self-Assembly of Poly(N-Vinylcaprolactam)-b-Poly(ε-Caprolactone) Block Copolymers via the Combination of RAFT/MADIX and Ring-Opening Polymerizations

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
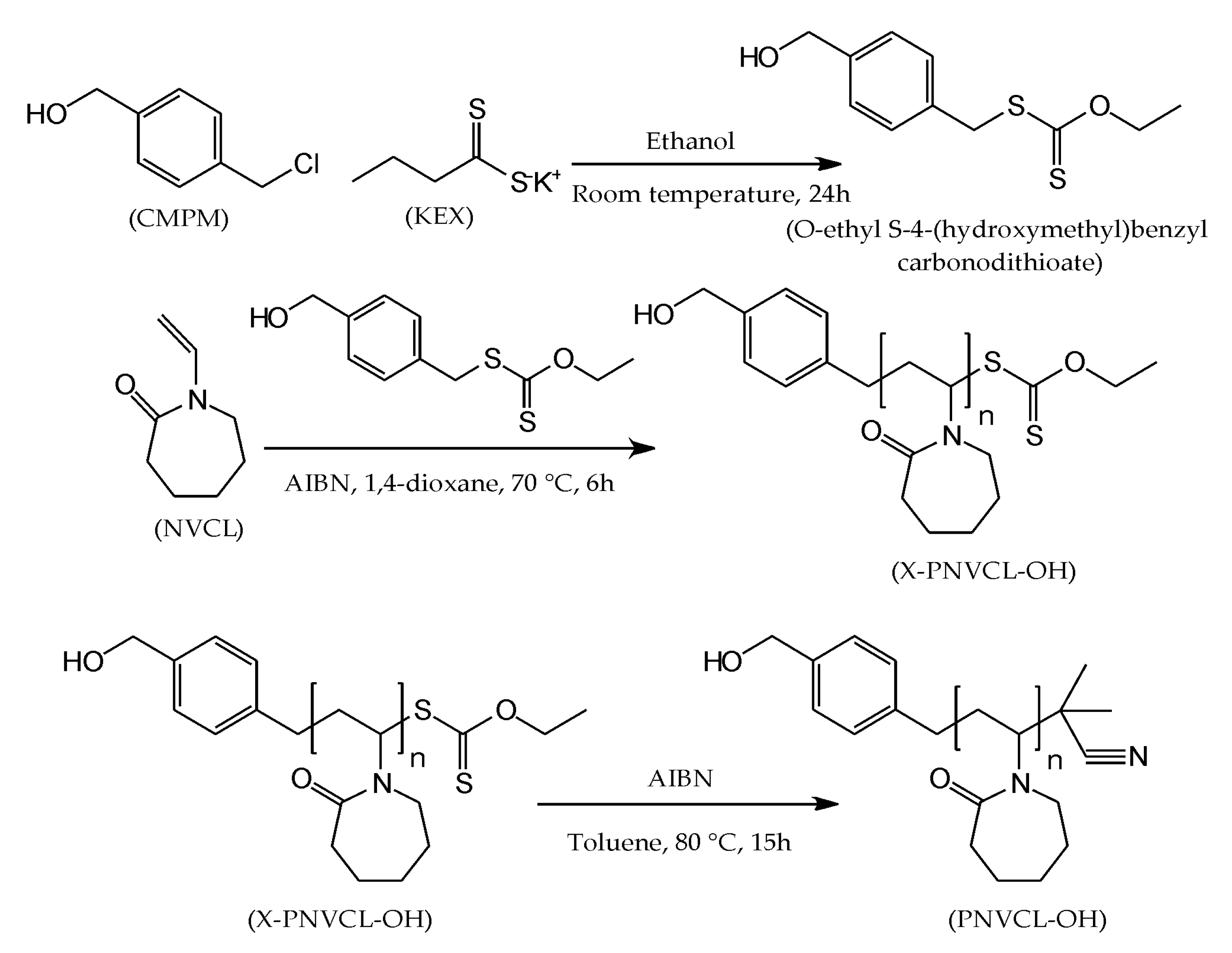
2.2.1. Synthesis of Hydroxyl-Terminated Poly(N-Vinylcaprolactam) (PNVCL–OH)
Synthesis of the O-Ethyl S-4-(Hydroxymethyl) Benzyl Carbonodithioate Chain Transfer Agent
RAFT/MADIX Polymerization of NVCL
Xanthate End Group Removal from PNVCL Homopolymer by a Radical-Induced Process
2.2.2. Ɛ-CL Ring-Opening Polymerization Initiated by PNVCL–OH Homopolymer
2.2.3. Micelles Preparation
2.3. Characterizations
3. Results
3.1. Synthesis of Hydroxyl-Terminated PNVCL Homopolymers (PNVCL–OH)
3.2. ε-CL Ring-Opening Polymerization Initiated by PNVCL–OH Homopolymer
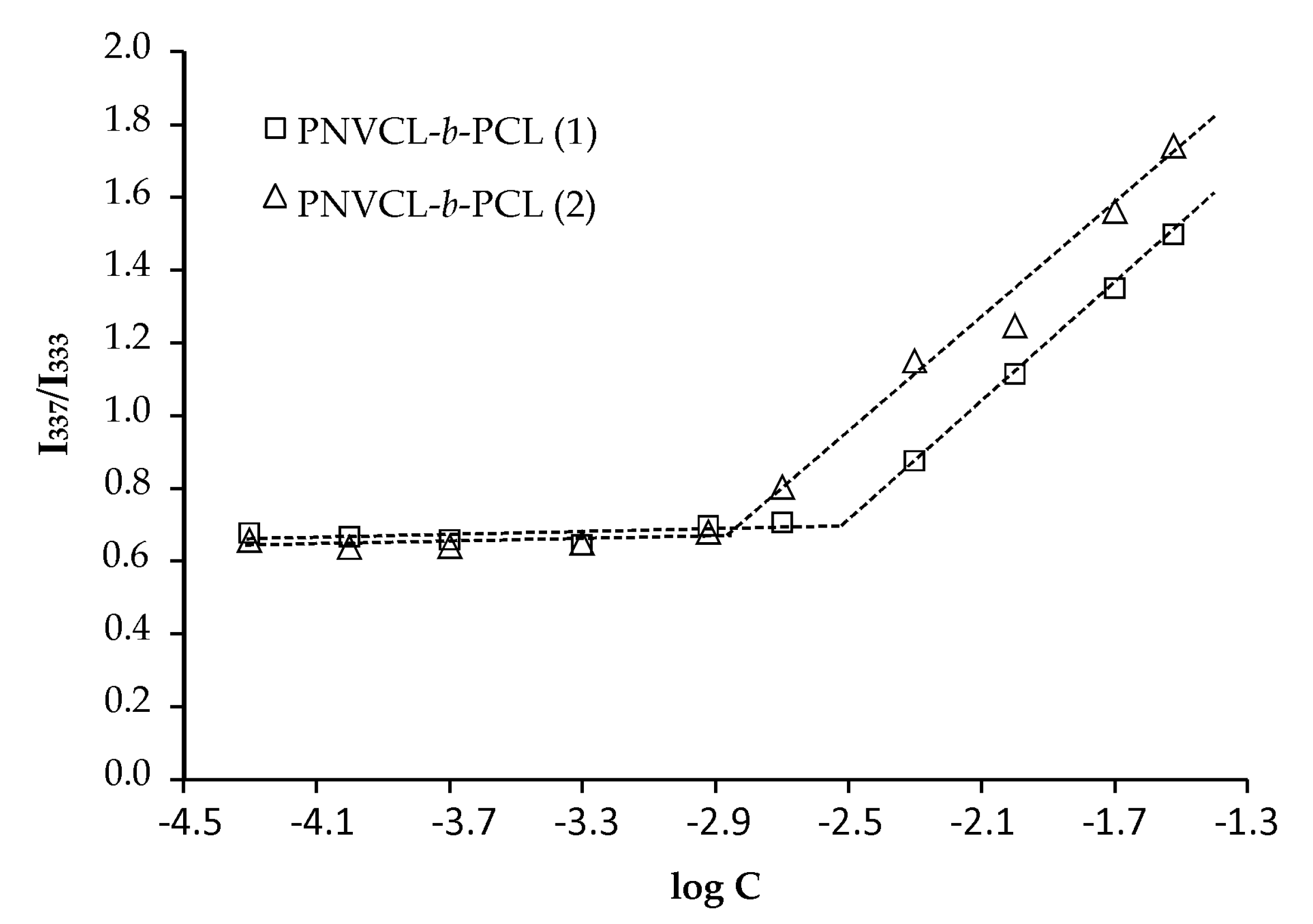
3.3. Determination of the CMC of the Block Copolymers
3.4. Micelles Morphology and Effect of the Hydrophobic Block Length on the Hydrodynamic Diameter of the Polymeric Micelles
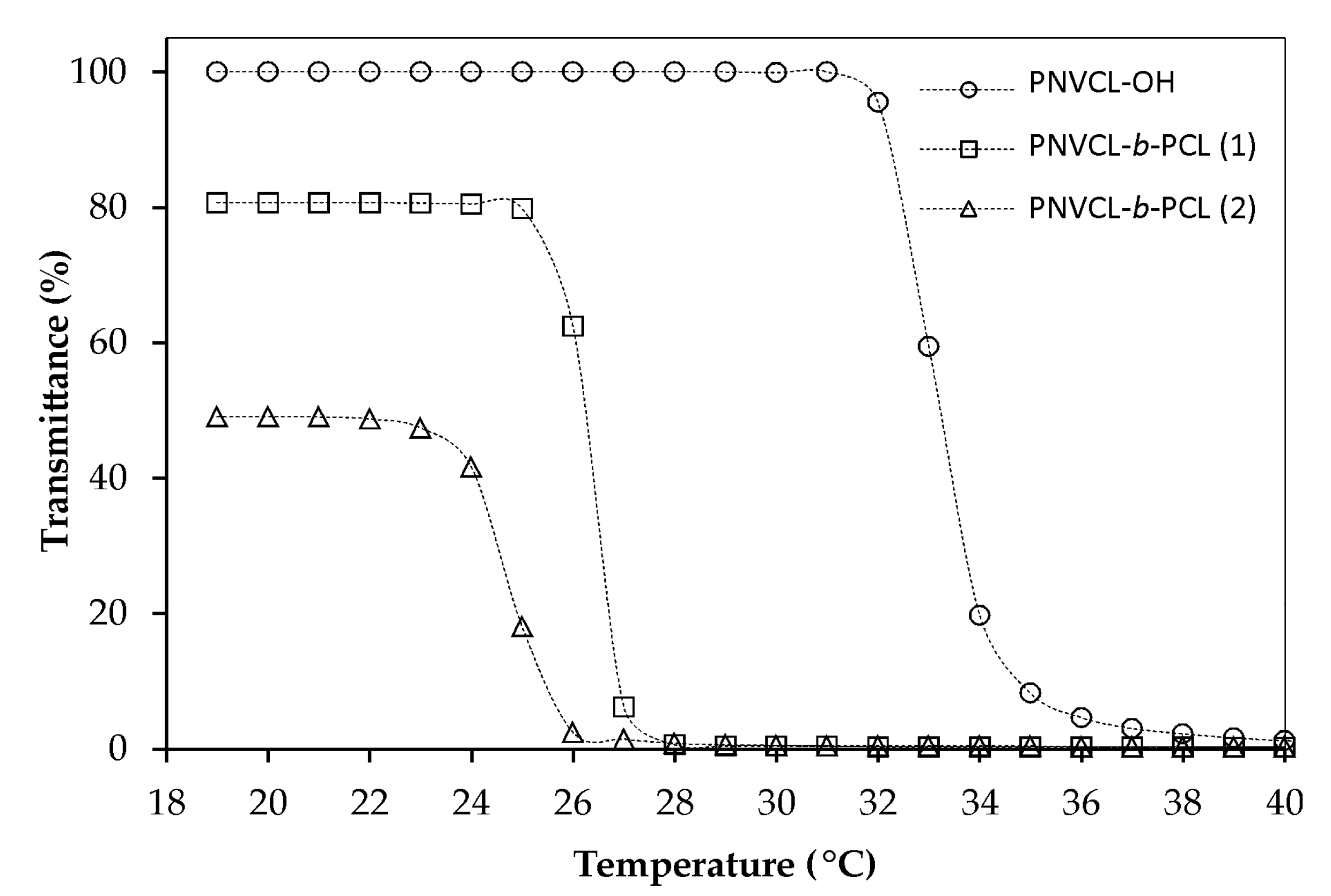
3.5. Effect of the PNVCL-b-PCL Block Copolymer Concentration and Hydrophobic Block Length on the LCST of the Polymeric Micelles
3.6. Effect of Temperature on the Hydrodynamic Diameter of the Polymeric Micelles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chrysostomou, V.; Pispas, S. Stimuli-responsive amphiphilic PDMAEMA-b-PLMA copolymers and their cationic zwitterionic analogs. J. Polym. Sci. Part A Polym. Chem. 2018, 56, 598–610. [Google Scholar] [CrossRef]
- Muller, J.; Marchandeau, F.; Prelot, B.; Zajac, J.; Robin, J.-J.; Monge, S. Self-organization in water of well-defined amphiphilic poly(vinyl acetate)-b-poly(vinyl alcohol) diblock copolymers. Polym. Chem. 2015, 6, 3063–3073. [Google Scholar] [CrossRef]
- Imran, M.; Shah, M.R.; Shafiullah, M. Chapter 10—Amphiphilic block copolymers-based micelles for drug delivery. In Design and Development of New Nanocarriers; Grumezescu, A.M., Ed.; William Andrew: Norwich, NY, USA, 2018; pp. 365–400. [Google Scholar]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yi, J.; Yin, Z.; Wang, S.; Yang, Q.; Wu, S.; Song, X.; Zhang, G. Synthesis and self-assembly of new amphiphilic thermosensitive poly(N-vinylcaprolactam)/poly(ε-caprolactone) block copolymers via the combination of ring-opening polymerization and click chemistry. J. Polym. Res. 2013, 20, 262–270. [Google Scholar] [CrossRef]
- Topuzogullari, M.; Bulmus, V.; Dalgakiran, E.; Dincer, S. pH-and temperature-responsive amphiphilic diblock copolymers of oligoethyleneglycol methacrylate synthesized by RAFT polymerization. Polymer 2014, 55, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Sponchioni, M.; Palmiero, U.C.; Moscatelli, D. Thermo-responsive polymers: Applications of smart materials in drug delivery and tissue engineering. Mater. Sci. Eng. C 2019, 102, 589–605. [Google Scholar] [CrossRef]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef]
- Liu, Q.-S.; Wu, W.-H.; Zhu, M.-F.; Qin, Z. Reducing the formation of six-membered ring ester during thermal degradation of biodegradable PHBV to enhance its thermal stability. Polym. Degrad. Stab. 2009, 94, 18–24. [Google Scholar] [CrossRef]
- Gandhi, A.; Paul, A.; Sen, S.O.; Sen, K.K. Studies on thermoresponsive polymers: Phase behavior, drug delivery and biomedical applications. Asian J. Pharm. 2015, 10, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Zarrintay, P.; Jouyandeh, M.; Ganjali, M.R.; Hadavand, B.S.; Mozafari, M.; Sheiko, S.S.; Vantankhan-Varnoosfaderani, M.; Gutiérrez, T.; Saeb, M.R. Thermo-sensitive polymers in medical: A review. Eur. Polym. J. 2019, 117, 402–423. [Google Scholar] [CrossRef]
- Lemanowicz, M.; Gierczycki, A.; Kuźnik, W.; Milczyńska, J.; Bulanda, P. Application of thermosensitive polymers in stabilization colloids. Adv. Powder Technol. 2016, 27, 471–480. [Google Scholar] [CrossRef]
- Cao, P.-F.; Mangadlao, J.D.; Advincula, R.C. Stimuli-responsive polymers and their potential applications in oil-gas industry. Polym. Rev. 2015, 55, 706–733. [Google Scholar] [CrossRef]
- Reese, C.E.; Mikhonin, A.V.; Kamenjicki, M.; Tikhonov, A.; Asher, S.A. Nanogel nanosecond photonic crystal optical switching. J. Am. Chem. Soc. 2004, 126, 1493–1496. [Google Scholar] [CrossRef] [PubMed]
- Jańczewski, D.; Tomczak, N.; Han, M.Y.; Vancso, G.J. Stimulus responsive PNIPAM/QD hybrid microspheres by copolymerization with surface engineered QDs. Macromolecules 2009, 42, 1801–1804. [Google Scholar] [CrossRef]
- Medeiros, S.F.; Santos, A.M.; Fessi, H.; Elaissari, A. Stimuli-responsive magnetic particles for biomedical applications. Int. J. Pharm. 2011, 403, 139–161. [Google Scholar] [CrossRef]
- Cortez-Lemus, N.A.; Licea-Claverie, A. Poly(N-vinylcaprolactam), a comprehensive review on a thermoresponsive polymer becoming popular. Prog. Polym. Sci. 2016, 53, 1–51. [Google Scholar] [CrossRef]
- Kumar, A.; Deepak; Sharma, S.; Afgan, S.; Kumar, R.; Keshari, A.K.; Srivastava, R. Development of graft copolymer of carboxymethylcellulose and N-vinylcaprolactam towards strong antioxidant and antibacterial polymeric materials. Int. J. Biol. Macromol. 2018, 112, 780–787. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, L.; Fu, X.; Song, X.; Yang, Q.; Zhang, G. Synthesis and self-assembly of a new amphiphilic thermosensitive poly(N-vinylcaprolactam)/poly(ε-caprolactone) block copolymer. Polym. Bull. 2014, 71, 1–18. [Google Scholar] [CrossRef]
- Napoli, A.; Sebok, D.; Senti, A.; Meier, W. Block Copolymers in Nanoscience; Lazzari, M., Liu, G., Lecommandoux, S., Eds.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 39–71. [Google Scholar]
- Glaied, O.; Delaite, C.; Riess, G. Synthesis of PCL-b-PVAc block copolymers by combination of click chemistry, ROP, and RAFT polymerization. Polym. Bull. 2012, 68, 607–621. [Google Scholar] [CrossRef]
- Mishra, A.K.; Patel, V.K.; Viswakarma, N.K.; Biswas, C.S.; Raula, M.; Mishra, A.; Mandal, T.K.; Ray, B. Synthesis of well-defined amphiphilic poly(ε-caprolactone)-b-poly(N-vinylpyrrolidone) block copolymers via the combination of ROP and xanthate-mediated raft polymerization. Macromolecules 2011, 44, 2465–2473. [Google Scholar] [CrossRef]
- Yu, Y.C.; Kang, H.U.; Youk, J.H. Synthesis and micellar characterization of thermosensitive amphiphilic poly(ε-caprolactone)-b-poly(N-vinylcaprolactam) block copolymers. Colloid Polym. Sci. 2012, 290, 1107–1113. [Google Scholar] [CrossRef]
- Ramesh, K.; Mishra, A.K.; Patel, V.K.; Vishwakarma, N.K.; Biswas, C.S.; Paira, T.K.; Mandal, T.K.; Maiti, P.; Misra, N. Synthesis of well-defined amphiphilic (D,L-lactide)-b-poly(N-vinylpyrrolidone) block copolymers using ROP and xanthate-mediated RAFT polymerization. Polymer 2012, 53, 5743–5753. [Google Scholar] [CrossRef]
- Kang, H.U.; Yu, Y.C.; Shin, S.J.; Kim, J.; Youk, J.H. One-pot synthesis of poly(N-vinylpyrrolidone)-b-poly(ε-caprolactone) block copolymers using a dual initiator for RAFT polymerization and ROP. Macromolecules 2013, 46, 1291–1295. [Google Scholar] [CrossRef]
- Yu, Y.C.; Li, G.; Kim, J.; Youk, J.H. One-pot synthesis of poly(N-vinylcaprolactam)-based biocompatible block copolymers using a dual initiator for ROP and RAFT polymerization. Polymer 2013, 54, 6119–6124. [Google Scholar] [CrossRef]
- Kang, H.U.; Yu, Y.C.; Shin, S.J.; Youk, J.H. One-step synthesis of block copolymers using a hydroxyl-functionalized trithiocarbonate RAFT agents as a dual initiator for RAFT polymerization and ROP. J. Polym. Sci. A Polym. Chem. 2013, 51, 774–779. [Google Scholar] [CrossRef]
- Mishara, A.K.; Vishwakarma, N.K.; Patel, V.K.; Biswas, C.S.; Paira, T.K.; Mandal, T.K.; Maiti, P.; Ray, B. Synthesis, characterization, and solution behavior of well-defined double hydrophilic linear amphiphilic poly(N-isopropulacrylamide)-b-poly(ε-caprolactone)-b-poly(N-isopropylacrylamide) triblock copolymers. Colloid Polym. Sci. 2014, 292, 1405–1418. [Google Scholar] [CrossRef]
- Cong, H.; Jingang, L.; Li, L.; Zheng, S. Thermoresponsive gelation behavior of poly(N-isopropylacrylamide)-block-poly(N-vinylpyrrolidone)-block-poly(N-isopropylacrylamide) triblock copolymers. Eur. Polym. J. 2014, 61, 23–32. [Google Scholar] [CrossRef]
- Öztürk, T.; Kiliçlioğlu, A.; Savaş, B.; Hazer, B. Synthesis and characterization of poly(ε-caprolactone-co-ethylene glycol) star-type amphiphilic copolymers by “click” chemistry and ring-opening polymerization. J. Macromol. Sci. Part A 2018, 55, 588–594. [Google Scholar] [CrossRef]
- Konishcheva, E.; Häussinger, D.; Lörcher, S.; Meier, W. Key aspects to yield low dispersity of PEO-b-PCL diblock copolymers and their mesoscale self-assembly. Eur. Polym. J. 2016, 83, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Ponjavic, M.; Nikolic, M.; Jevtic, S.; Rogan, J.; Stevanovic, S.; Djonlagic, J. Influence of a low content of PEO segment on the thermal, surface and morphological properties of triblock and diblock PCL copolymers. Macromol. Res. 2016, 24, 323–335. [Google Scholar] [CrossRef]
- Ali, R.; Farah, A.; Binkhathlan, Z. Development and characterization of methoxy poly(ethylene oxide)-block-poly(ε-caprolactone) (PEO-b-PCL) micelles as vehicles for the solubilization and delivery of tacrolimus. Saudi Pharm. J. 2017, 25, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Kheiri Manjili, H.; Ghasemi, P.; Malvandi, H.; Mousavi, M.S.; Attari, E.; Danafar, H. Pharmacokinetics and in vivo delivery of curcumin by copolymeric mPEG-PCL micelles. Eur. J. Pharm. Biopharm. 2017, 116, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.; Chae, S.Y.; Nahm, J.-W. Thermosensitive poly(N-isopropylacrylamide)-b-poly(ε-caprolactone) nanoparticles for efficient drug delivery system. Polymer 2006, 47, 4571–4580. [Google Scholar] [CrossRef]
- Wu, Q.; Yi, J.; Wang, S.; Liu, D.; Song, X.; Zhang, G. Synthesis and self-assembly of new amphiphilic thermosensitive poly(N-vinylcaprolactam)/poly(D,L-lactide) block copolymers via the combination of ring-opening polymerization and click chemistry. Polym. Bull. 2015, 72, 1449–1466. [Google Scholar] [CrossRef]
- Binauld, S.; Delafresnaye, L.; Charleux, B.; D’agosto, F.; Lansalot, M. Emulsion polymerization of vinyl acetate in the presence of different hydrophilic polymersobtained by RAFT/MADIX. Macromolecules 2014, 47, 3461–3472. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P.; Mars, C.A. Reversible addition-fragmentation chain transfer polymerization: End group modification for functionalized polymers and chain transfer agent recovery. Macromolecules 2005, 38, 2033–2036. [Google Scholar] [CrossRef]
- Wu, Q.H.; Wang, C.; Zhang, D.; Song, X.M.; Liu, D.L.; Wang, L.P.; Zhang, G.L. Synthesis and micellization of a new amphiphilic star-shaped poly(D,L-lactide)/polyphosphoester block copolymer. React. Funct. Polym. 2012, 72, 372–377. [Google Scholar] [CrossRef]
- Postma, A.; Davis, T.P.; Evans, R.A.; Li, G.; Moad, G.; O’Shea, M. Synthesis of well-defined polystyrene with primary amine end groups through the use of phthalimido-functional RAFT agents. Macromolecules 2006, 39, 5293–5306. [Google Scholar] [CrossRef]
- Boyko, B. N-Vinylcaprolactam based Bulk and Microgels: Synthesis, Structural Formation and Characterization by Dynamic Light Scattering. 173 f. Ph.D. Thesis, Dresden University of Technology, Desden, Germany, 2004. [Google Scholar]
- Kirsh, Y.E.; Yanul, N.A.; Kalninsh, K.K. Structural transformations and water associate interactions in poly-N-vinylcaprolactam-water system. Eur. Polym. J. 1999, 35, 305–316. [Google Scholar] [CrossRef]
- Lebedev, V.T.; Tőrők, G.; Cser, L.; Káli, G.; Sibilev, A.I. Molecular dynamics of poly(N-vinylcaprolactam) hydrate. Appl. Phys. A 2002, 74, 478–480. [Google Scholar] [CrossRef]
- Liu, J.; Detrembleur, C.; Pauw-Gillet, M.-C.; Mornet, S.; Duguet, E.; Jérôme, C. Gold Nanorods coated with a Thermo-responsive Poly(ethylene glycol)-b-poly(N-vinylcaprolactam) corona as Drug System for Remotely Near Infrared Triggered Release. Polym. Chem. 2014, 5, 799–813. [Google Scholar] [CrossRef] [Green Version]
- Fallon, M.; Halligan, S.; Pezzoli, R.; Geever, L.; Higginbotham, C. Synthesis and characterization of novel temperature and pH sensitive physically cross-linked poly(N-vinylcaprolactam-co-itaconic acid) hydrogels for drug delivery. Gels 2019, 5, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usanmaz, A.; Ozdemir, T.; Polat, O. Solid state polymerization of NVCL via gamma irradiation and characterization. J. Macromol. Sci. Part A 2009, 46, 597–606. [Google Scholar] [CrossRef]
- Durkut, S.; Elçin, Y.M. Synthesis and characterization of thermosensitive poly(N-vinylcaprolactam)-g-collagen. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Deng, X.; Yang, H. Biodegradable poly(ε-caprolactone)-poly(ethylene glycol) block copolymers: Characterization and their use as drug carriers for a controlled delivery systems. Biomaterials 2003, 24, 3563–3570. [Google Scholar] [CrossRef]
- Sownthari, K.; Suthanthiraraj, S.A. Synthesis and characterization of an electrolyte system based on a biodegradable polymer. Express Polym. Lett. 2013, 7, 495–504. [Google Scholar] [CrossRef]
- Wang, Q.; Tao, L.; Yang, Z.; Raj, W.; Zhang, Y.; Wang, T.; Pietrasik, J. Macroscopic and microscopic shape memory effects of block copolymers prepared via ATRP. J. Polym. Sci. 2020, 58, 20–24. [Google Scholar] [CrossRef]
- Soto, A.P.; Gilroy, J.B.; Winnik, M.A.; Manners, I. Pointed-oval-shaped micelles from crystalline-coil block copolymers by crystallization-driven living self-assembly. Angew. Chem. Int. Ed. 2010, 49, 8220–8223. [Google Scholar] [CrossRef]
- Maeda, Y.; Tsubota, M.; Ikeda, I. Temperature-responsive graft copolymers with poly(propylene glycol) side chains. Macromol. Rapid Commun. 2003, 24, 242–245. [Google Scholar] [CrossRef]
- Viholal, H.; Laukkanen, A.; Valtola, L.; Tenhu, H.; Hirvonen, J. Cytotoxicity of thermosensitive polymers poly(N-isopropylacrylamide), poly(N-vinylcaprolactam) and amphiphilically modified poly(N-vinylcaprolactam). Biomaterials 2005, 26, 3055–3064. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Conv. a (%) | Mn theo.b (g mol−1) | Mn NMRa (g mol−1) | Mn SECc (g mol−1) | Đ c |
|---|---|---|---|---|---|
| X-PNVCL–OH | 34.0 | 7342 | 6379 | 3903 | 1.44 |
| PNVCL–OH | - | - | 6480 | 4177 | 1.43 |
| Reaction | Conv. c (%) | Mn theo.d (g mol−1) | Mn NMRc (g mol−1) | Mn SECe (g mol−1) | Ð e | CMC f 10−3 (mg mL−1) | Zav g (nm) | PdI g |
|---|---|---|---|---|---|---|---|---|
| PNVCL-b-PCL (1) a | 92.4 | 10638 | 9629 | 6517 | 1.48 | 2.9 | 86 | 0.203 |
| PNVCL-b-PCL (2) b | 97.6 | 14306 | 12719 | 9007 | 1.57 | 1.4 | 117 | 0.237 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moraes, R.M.; Carvalho, L.T.; Alves, G.M.; Medeiros, S.F.; Bourgeat-Lami, E.; Santos, A.M. Synthesis and Self-Assembly of Poly(N-Vinylcaprolactam)-b-Poly(ε-Caprolactone) Block Copolymers via the Combination of RAFT/MADIX and Ring-Opening Polymerizations. Polymers 2020, 12, 1252. https://doi.org/10.3390/polym12061252
Moraes RM, Carvalho LT, Alves GM, Medeiros SF, Bourgeat-Lami E, Santos AM. Synthesis and Self-Assembly of Poly(N-Vinylcaprolactam)-b-Poly(ε-Caprolactone) Block Copolymers via the Combination of RAFT/MADIX and Ring-Opening Polymerizations. Polymers. 2020; 12(6):1252. https://doi.org/10.3390/polym12061252
Chicago/Turabian StyleMoraes, Rodolfo M., Layde T. Carvalho, Gizelda M. Alves, Simone F. Medeiros, Elodie Bourgeat-Lami, and Amilton M. Santos. 2020. "Synthesis and Self-Assembly of Poly(N-Vinylcaprolactam)-b-Poly(ε-Caprolactone) Block Copolymers via the Combination of RAFT/MADIX and Ring-Opening Polymerizations" Polymers 12, no. 6: 1252. https://doi.org/10.3390/polym12061252