GSK3787-Loaded Poly(Ester Amide) Particles for Intra-Articular Drug Delivery

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Materials
2.2. General Methods
2.3. Preparation of GSK3787-Loaded Particles (PBSe-GSK3787)
2.4. Preparation of Non-Drug-Loaded Particles (PBSe-NDL)
2.5. Preparation of Dye-Labeld Particles
2.6. Scanning Electron Microscopy
2.7. Measurement of Drug Loading and Encapsulation Efficiency
2.8. Atomic Force Microscopy
2.9. In Vitro Release of GSK3787
2.10. Primary Chondrocyte Harvest and Culture
2.11. Cytotoxicity of GSK3787 to IMAC Cells
2.12. Cytotoxicity of PBSe-GSK3787 and PBSe-NDL Particles to IMAC Cells
2.13. Brightfield Imaging of IMAC Cells Treated with PBSe-GSK3787 Particles
2.14. Confocal Microscopy of IMAC Cells Treated with PBSe-GSK3787 Particles
2.15. Ex Vivo Intra-Articular Injection of PBSe-GSK3787 Particles
3. Results and Discussion
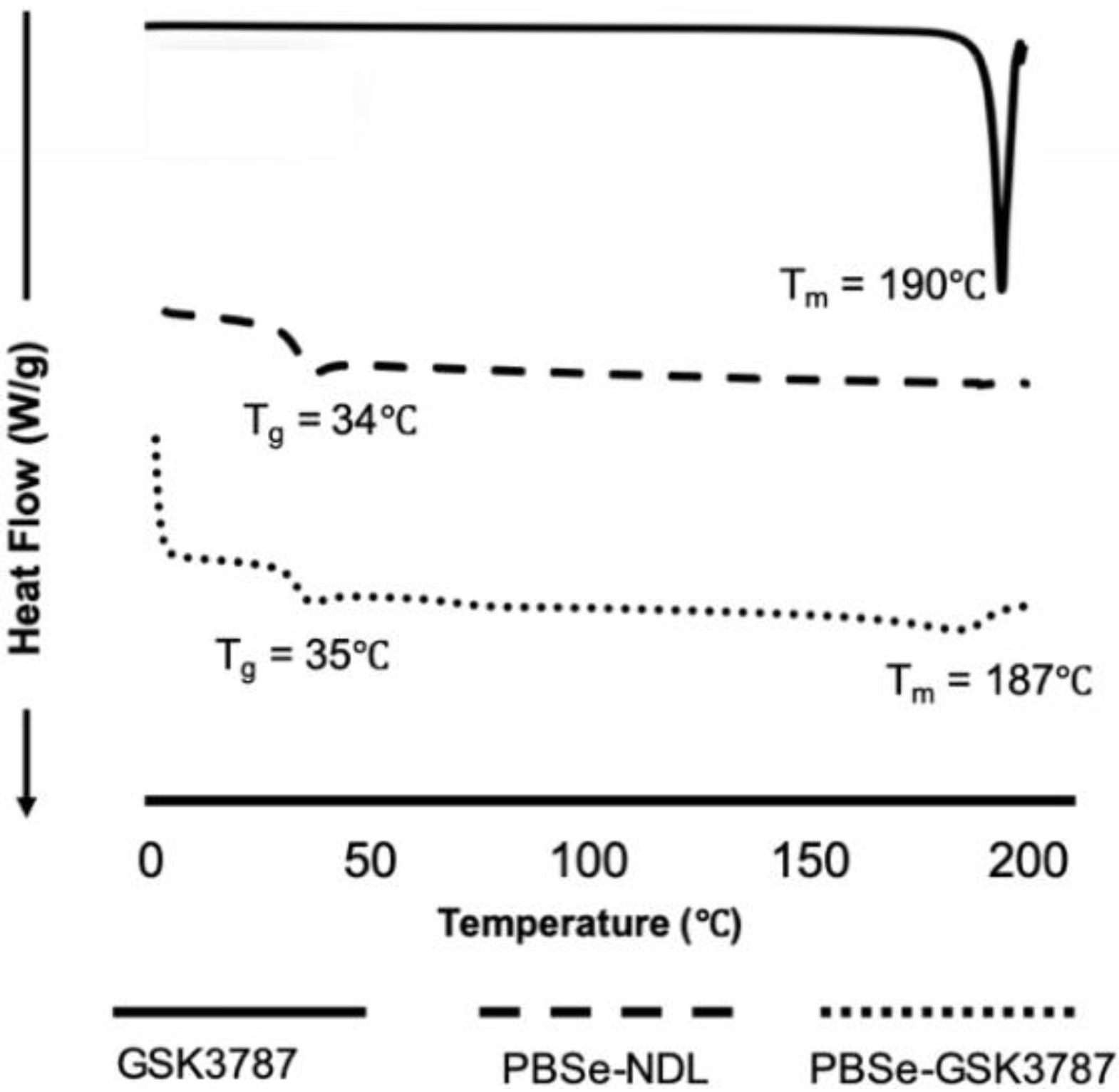
3.1. Preparation and Characterization of PBSe Particles
3.2. In Vitro Release of GSK3787
3.3. Cytotoxicity of GSK3787, PBSe-GSK3787, and PBSe-NDL to Primary Cell Cultures
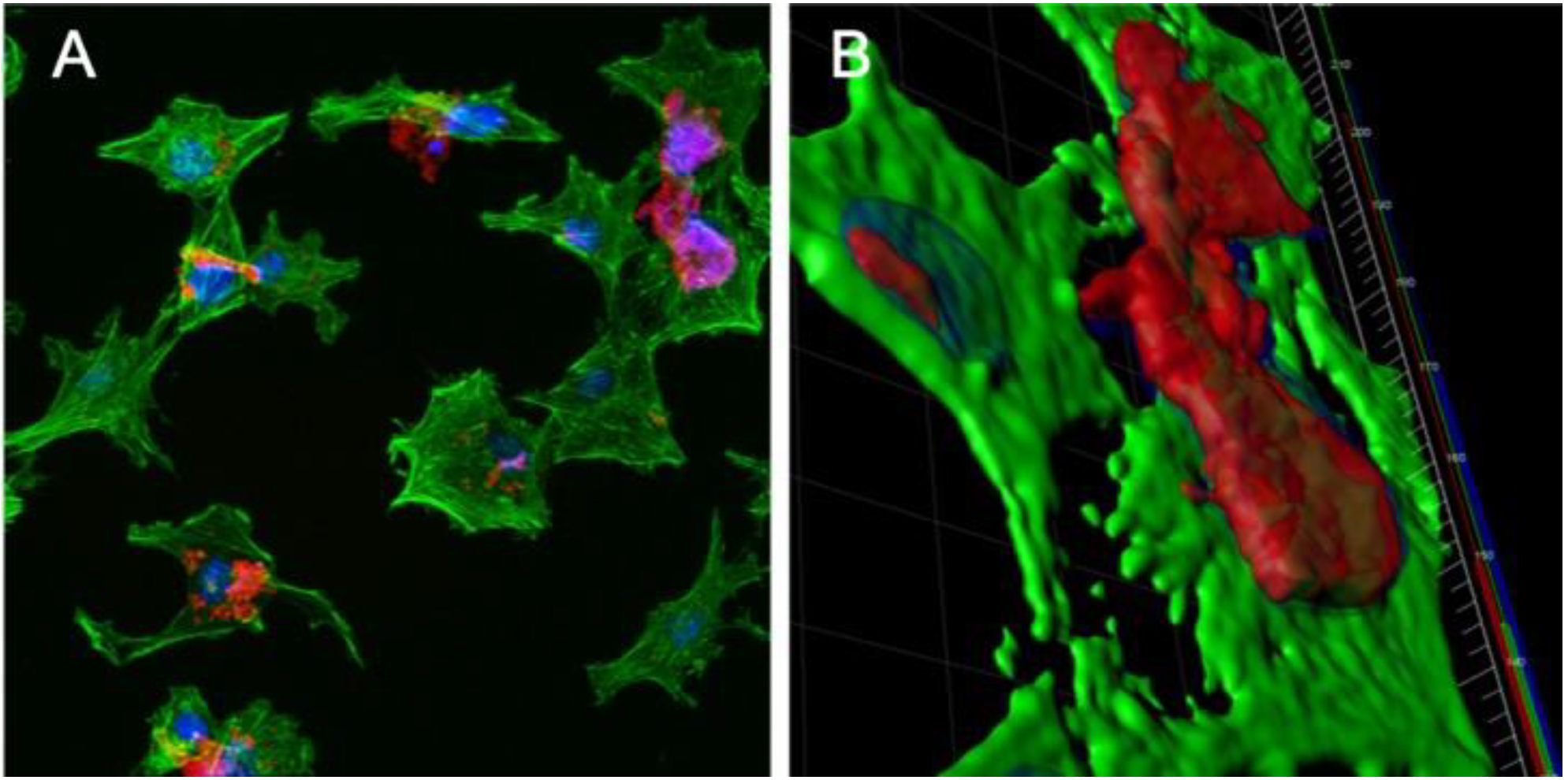
3.4. Confocal Microscopy of IMAC Cells Treated with PBSe-GSK3787-NR
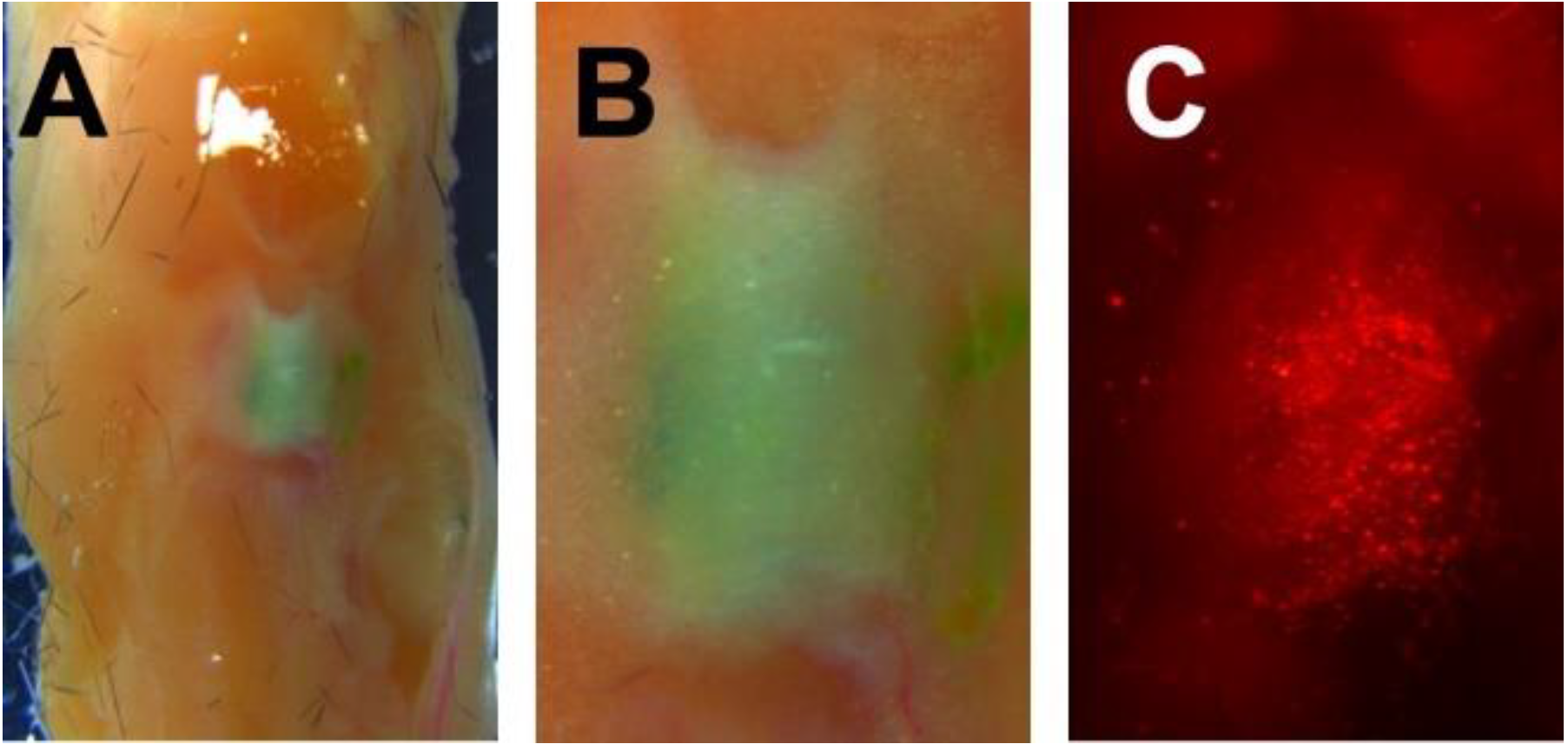
3.5. Ex Vivo Intra-Articular Injections
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis: Literature update. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Suri, P.; Morgenroth, D.C.; Hunter, D.J. Epidemiology of osteoarthritis and associated comorbidities. PM R 2012, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Neogi, T. The epidemiology and impact of pain in osteoarthritis. Osteoarthr. Cartil. 2013, 21, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Hurley, M.V.; Walsh, N.E.; Mitchell, H.L.; Pimm, T.J.; Patel, A.; Williamson, E.; Jones, R.H.; Dieppe, P.A.; Reeves, B.C. Clinical effectiveness of a rehabilitation program integrating exercise, self-management, and active coping strategies for chronic knee pain: A cluster randomized trial. Arthritis Rheum. 2007, 57, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Brakke, R.; Singh, J.; Sullivan, W. Physical therapy in persons with osteoarthritis. PM R 2012, 4, 53–58. [Google Scholar] [CrossRef]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J. Clin. Investig. 2006, 116, 4–15. [Google Scholar] [CrossRef]
- Mahmud, T.; Scott, D.L.; Bjarnason, I. A unifying hypothesis for the mechanism of NSAID related gastrointestinal toxicity. Ann. Rheum. Dis. 1996, 55, 211–213. [Google Scholar] [CrossRef] [Green Version]
- Douglas, R.J. Corticosteroid injection into the osteoarthritic knee: Drug selection, dose, and injection frequency. Int. J. Clin. Pract. 2012, 66, 699–704. [Google Scholar] [CrossRef]
- Hunter, D.J. Osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2011, 25, 801–814. [Google Scholar] [CrossRef]
- Grayson, C.W.; Decker, R.C. Total joint arthroplasty for persons with osteoarthritis. PM R 2012, 4, 97–103. [Google Scholar] [CrossRef]
- Losina, E.; Paltiel, A.D.; Weinstein, A.M.; Yelin, E.; Hunter, D.J.; Chen, S.P.; Klara, K.; Suter, L.G.; Solomon, D.H.; Burbine, S.A.; et al. Lifetime medical costs of knee osteoarthritis management in the United States: Impact of extending indications for total knee arthroplasty. Arthritis Care Res. 2015, 67, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Bryan, R.; Terkeltaub, R. Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 2015, 11, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grothe, K.; Flechsenhar, K.; Paehler, T.; Ritzeler, O.; Beninga, J.; Saas, J.; Herrmann, M.; Rudolphi, K. IkappaB kinase inhibition as a potential treatment of osteoarthritis - results of a clinical proof-of-concept study. Osteoarthr. Cartil. 2017, 25, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigoglou, S.; Papavassiliou, A.G. The NF-kappaB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef]
- Kelly, S.; Chapman, R.J.; Woodhams, S.; Sagar, D.R.; Turner, J.; Burston, J.J.; Bullock, C.; Paton, K.; Huang, J.; Wong, A.; et al. Increased function of pronociceptive TRPV1 at the level of the joint in a rat model of osteoarthritis pain. Ann. Rheum. Dis. 2015, 74, 252–259. [Google Scholar] [CrossRef]
- Malfait, A.M.; Miller, R.J. Emerging Targets for the Management of Osteoarthritis Pain. Curr. Osteoporos. Rep. 2016, 14, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Ratneswaran, A.; LeBlanc, E.A.; Walser, E.; Welch, I.; Mort, J.S.; Borradaile, N.; Beier, F. Peroxisome proliferator-activated receptor delta promotes the progression of posttraumatic osteoarthritis in a mouse model. Arthr. Rheumatol. 2015, 67, 454–464. [Google Scholar] [CrossRef]
- Shearer, B.G.; Wiethe, R.W.; Ashe, A.; Billin, A.N.; Way, J.M.; Stanley, T.B.; Wagner, C.D.; Xu, R.X.; Leesnitzer, L.M.; Merrihew, R.V.; et al. Identification and characterization of 4-chloro-N-(2-{[5-trifluoromethyl)-2-pyridyl]sulfonyl}ethyl)benzamide (GSK3787), a selective and irreversible peroxisome proliferator-activated receptor delta (PPARdelta) antagonist. J. Med. Chem. 2010, 53, 1857–1861. [Google Scholar] [CrossRef]
- Lee, C.H.; Olson, P.; Hevener, A.; Mehl, I.; Chong, L.W.; Olefsky, J.M.; Gonzalez, F.J.; Ham, J.; Kang, H.; Peters, J.M.; et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2006, 103, 3444–3449. [Google Scholar] [CrossRef] [Green Version]
- Schmuth, M.; Haqq, C.M.; Cairns, W.J.; Holder, J.C.; Dorsam, S.; Chang, S.; Lau, P.; Fowler, A.J.; Chuang, G.; Moser, A.H.; et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J. Investig. Dermatol. 2004, 122, 971–983. [Google Scholar] [CrossRef] [Green Version]
- Tan, N.S.; Icre, G.; Montagner, A.; Bordier-ten-Heggeler, B.; Wahli, W.; Michalik, L. The nuclear hormone receptor peroxisome proliferator-activated receptor beta/delta potentiates cell chemotactism, polarization, and migration. Mol. Cell. Biol. 2007, 27, 7161–7175. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y. Intra-articular drug delivery systems for arthritis treatment. Rheumatol. Curr. Res. 2012, 2, 1000e106. [Google Scholar] [CrossRef] [Green Version]
- Bajpayee, A.G.; Wong, C.R.; Bawendi, M.G.; Frank, E.H.; Grodzinsky, A.J. Avidin as a model for charge driven transport into cartilage and drug delivery for treating early stage post-traumatic osteoarthritis. Biomaterials 2014, 35, 538–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maudens, P.; Jordan, O.; Allemann, E. Recent advances in intra-articular drug delivery systems for osteoarthritis therapy. Drug. Discov. Today 2018, 23, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jiang, D.; Wang, Z.; Wu, G.; Miao, L.; Huang, L. Intra-articular delivery of liposomal celecoxib-hyaluronate combination for the treatment of osteoarthritis in rabbit model. Int. J. Pharm. 2013, 441, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, D.; Wang, J.; Wu, L.; Li, W.; Chen, J.; Cai, B.C.; Cheng, H. Development of nanoparticles-in-microparticles system for improved local retention after intra-articular injection. Drug Deliv. 2014, 21, 342–350. [Google Scholar] [CrossRef]
- Janssen, M.; Timur, U.T.; Woike, N.; Welting, T.J.; Draaisma, G.; Gijbels, M.; van Rhijn, L.W.; Mihov, G.; Thies, J.; Emans, P.J. Celecoxib-loaded PEA microspheres as an auto regulatory drug-delivery system after intra-articular injection. J. Control. Release 2016, 244, 30–40. [Google Scholar] [CrossRef]
- Villamagna, I.J.; Gordon, T.N.; Hurtig, M.B.; Beier, F.; Gillies, E.R. Poly(ester amide) particles for controlled delivery of celecoxib. J. Biomed. Mater. Res. Part A 2019, 107A, 1235–1243. [Google Scholar] [CrossRef]
- Li, Y.; Cao, J.; Han, S.; Liang, Y.; Zhang, T.; Zhao, H.; Wang, L.; Sun, Y. ECM based injectable thermo-sensitive hydrogel on the recovery of injured cartilage induced by osteoarthritis. Artif. Cells Nanomed. Biotechnol. 2018, 46, 152–160. [Google Scholar] [CrossRef]
- Petit, A.; Sandker, M.; Muller, B.; Meyboom, R.; van Midwoud, P.; Bruin, P.; Redout, E.M.; Versluijs-Helder, M.; van der Lest, C.H.; Buwalda, S.J.; et al. Release behavior and intra-articular biocompatibility of celecoxib-loaded acetyl-capped PCLA-PEG-PCLA thermogels. Biomaterials 2014, 35, 7919–7928. [Google Scholar] [CrossRef]
- Prince, D.A.; Villamagna, I.J.; Borecki, A.; Beier, F.; de Bruyn, J.R.; Hurtig, M.; Gillies, E.R. Thermoresponsive and covalently cross-linkable hydrogels for intra-articular drug delivery. ACS Appl. Biol. Mater. 2019, 2, 3498–3507. [Google Scholar] [CrossRef]
- Geiger, B.C.; Wang, S.; Padera, R.F.; Grodzinsky, A.J.; Hammond, P.T. Cartilage-penetrating nanocarriers improve delivery and efficacy of growth factor treatment of osteoarthritis. Sci. Transl. Med. 2018, 10, eaat8800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Bendele, A.M.; Blanks, R.C.; Bodick, N. Sustained efficacy of a single intra-articular dose of FX006 in a rat model of repeated localized knee arthritis. Osteoarthr. Cartil. 2015, 23, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soleimani, A.; Drappel, S.; Carlini, R.; Goredema, A.; Gillies, E.R. Structure–Property relationships for a series of poly(ester amide)s containing amino acids. Ind. Eng. Chem. Res. 2014, 53, 1452–1460. [Google Scholar] [CrossRef]
- Place, E.S.; George, J.H.; Williams, C.K.; Stevens, M.M. Synthetic polymer scaffolds for tissue engineering. Chem. Soc. Rev. 2009, 38, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Gil, M.H.; Simões, P.N. Biodegradable poly(ester amide)s—A remarkable opportunity for the biomedical area: Review on the synthesis, characterization and applications. Prog. Polym. Sci. 2014, 39, 1291–1311. [Google Scholar] [CrossRef]
- Knight, D.K.; Gillies, E.R.; Mequanint, K. Strategies in functional poly(ester amide) syntheses to study human coronary artery smooth muscle cell interactions. Biomacromolecules 2011, 12, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Rizkalla, A.S.; Mequanint, K. Versatile biodegradable poly(ester amide)s derived from α-Amino acids for vascular tissue engineering. Materials 2010, 3, 2346–2368. [Google Scholar] [CrossRef] [Green Version]
- Tsitlanadze, G.; Machaidze, M.; Kviria, T.; Djavakhishvili, N.; Chu, C.C.; Katsarava, R. Biodegradation of amino-acid-based poly(ester amide)s: In vitro weight loss and preliminary in vivo studies. J. Biomater. Sci. Polym. Ed. 2004, 15, 1–24. [Google Scholar] [CrossRef]
- Knight, D.K.; Gillies, E.R.; Mequanint, K. Biomimetic L-aspartic acid-derived functional poly(ester amide)s for vascular tissue engineering. Acta Biomater. 2014, 10, 3484–3496. [Google Scholar] [CrossRef]
- Cao, K.; Flegg, D.S.; Lin, S.; Lagugné-Labarthet, F.; Mequanint, K.; Gillies, E.R. Fabrication and in situ cross-linking of carboxylic-acid-functionalized poly(ester amide) scaffolds for tissue engineering. ACS Appl. Polym. Mater. 2019, 1, 2360–2369. [Google Scholar] [CrossRef]
- Murase, S.K.; del Valle, L.J.; Kobauri, S.; Katsarava, R.; Puiggalí, J. Electrospun fibrous mats from a l-phenylalanine based poly(ester amide): Drug delivery and accelerated degradation by loading enzymes. Polym. Degrad. Stab. 2015, 119, 275–287. [Google Scholar] [CrossRef]
- Said, S.S.; Pickering, J.G.; Mequanint, K. Controlled delivery of fibroblast growth factor-9 from biodegradable poly(ester amide) fibers for building functional neovasculature. Pharm. Res. 2014, 31, 3335–3347. [Google Scholar] [CrossRef] [PubMed]
- Rudnik-Jansen, I.; Colen, S.; Berard, J.; Plomp, S.; Que, I.; van Rijen, M.; Woike, N.; Egas, A.; van Osch, G.; van Maarseveen, E.; et al. Prolonged inhibition of inflammation in osteoarthritis by triamcinolone acetonide released from a polyester amide microsphere platform. J. Control. Release 2017, 253, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Mullin, N.; Hobbs, J.K. A non-contact, thermal noise based method for the calibration of lateral deflection sensitivity in atomic force microscopy. Rev. Sci. Instrum. 2014, 85, 113703. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.R.; Benedetti, L.R.; Bradley, D.K.; Hilsabeck, T.J.; Izumi, N.; Khan, S.; Kyrala, G.A.; Ma, T.; Pak, A. Comparison of implosion core metrics: A 10 ps dilation X-ray imager vs a 100 ps gated microchannel plate. Rev. Sci. Instrum. 2016, 87, 11E311. [Google Scholar] [CrossRef]
- Gosset, M.; Berenbaum, F.; Thirion, S.; Jacques, C. Primary culture and phenotyping of murine chondrocytes. Nat. Protoc. 2008, 3, 1253–1260. [Google Scholar] [CrossRef]
- Jelvehgari, M.; Montazam, S. Comparison of microencapsulation by emulsion-solvent extraction/ evaporation technique using derivatives cellulose and acrylate-methacrylate copolymer as carriers. Jundishapur J. Nat. Pharm. Prod. 2012, 7, 144–152. [Google Scholar]
- Rosca, I.D.; Watari, F.; Uo, M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J. Control. Release 2004, 99, 271–280. [Google Scholar] [CrossRef]
- Aranberri, I.; Beverley, K.J.; Binks, B.P.; Clint, J.H.; Fletcher, P.D.I. How do emulsions evaporate. Langmuir 2002, 18, 3471–3475. [Google Scholar] [CrossRef]
- Dilling, W.L.; Tefertiller, N.B.; Kallos, G.J. Evaporation rates and reactivities of methylene chloride, chloroform, 1,1,1-trichloroethane, trichloroethylene, tetrachloroethylene, and other chlorinated compounds in dilute aqueous solutions. Environ. Sci. Technol. 1975, 9, 833–838. [Google Scholar] [CrossRef]
- Edwards, S.H.R. Intra-articular drug delivery: The challenges to extend drug residence time in the joint. Vet. J. 2011, 11, 1479–1484. [Google Scholar] [CrossRef] [PubMed]
- Kontomaris, S.-V. The Hertz model in AFM nanoindentation experiments: Applications in biological samples and biomaterials. Micro. Nanosyst. 2018, 10, 11–22. [Google Scholar] [CrossRef]
- Athanasiou, K.A.; Rosenwasser, M.P.; Buckwalter, J.A.; Malinin, T.I.; Mow, V.C. Interspecies comparisons of in situ intrinsic mechanical properties of distal femoral cartilage. J. Orthop. Res. 1991, 9, 330–340. [Google Scholar] [CrossRef]
- Schinagl, R.M.; Gurskis, D.; Chen, A.C.; Sah, R.L. Depth-dependent confined compression modulus of full-thickness bovine articular cartilage. J. Orthop. Res. 1997, 15, 499–506. [Google Scholar] [CrossRef]
- Malone, M.A.; Kaushik, N.; Waheed, A. Intra-articular steroids: How soon and how often after the first injection? SM J. Commun. Med. 2016, 2, 1014. [Google Scholar]
- Guo, K.; Chu, C.C. Biodegradable and injectable paclitaxel-loaded poly(ester amide) microspheres: Fabrication and characterization. J. Biomed. Mater. Res. Part B 2008, 89B, 491–500. [Google Scholar] [CrossRef]
- Lamplot, J.D.; Liu, B.; Yin, L.; Zhang, W.; Wang, Z.; Luther, G.; Wagner, E.; Li, R.; Nan, G.; Shui, W.; et al. Reversibly immortalized mouse articular chondrocytes acquire long-term proliferative capability while retaining chondrogenic phenotype. Cell Transplant. 2015, 24, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.I.; Argyle, D.J.; Clements, D.N. In vitro models for the study of osteoarthritis. Vet. J. 2016, 209, 40–49. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Composition | Z-Average Diameter (DLS) (nm) | Particle Diameter (SEM) (nm) | GSK3787 Loading (wt.%) | GSK3787 Encapsulation Efficiency (%) | Young’s Modulus (MPa) | Tg (°C) | Tm (°C) |
|---|---|---|---|---|---|---|---|
| PBSe-NDL | 790 ± 64 | 870 ± 74 | - | - | 7.0 ± 1.4 | 34 | - |
| PBSe-GSK3787 | 530 ± 54 | 580 ± 290 | 8.1 ± 0.4 | 94.0 ± 4.8 | 2.8 ± 1.0 | 35 | 187 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villamagna, I.J.; McRae, D.M.; Borecki, A.; Mei, X.; Lagugné-Labarthet, F.; Beier, F.; Gillies, E.R. GSK3787-Loaded Poly(Ester Amide) Particles for Intra-Articular Drug Delivery. Polymers 2020, 12, 736. https://doi.org/10.3390/polym12040736
Villamagna IJ, McRae DM, Borecki A, Mei X, Lagugné-Labarthet F, Beier F, Gillies ER. GSK3787-Loaded Poly(Ester Amide) Particles for Intra-Articular Drug Delivery. Polymers. 2020; 12(4):736. https://doi.org/10.3390/polym12040736
Chicago/Turabian StyleVillamagna, Ian J., Danielle M. McRae, Aneta Borecki, Xueli Mei, François Lagugné-Labarthet, Frank Beier, and Elizabeth R. Gillies. 2020. "GSK3787-Loaded Poly(Ester Amide) Particles for Intra-Articular Drug Delivery" Polymers 12, no. 4: 736. https://doi.org/10.3390/polym12040736