Preparation of a Series of Photoresponsive Polymersomes Bearing Photocleavable a 2-nitrobenzyl Group at the Hydrophobic/Hydrophilic Interfaces and Their Payload Releasing Behaviors


Abstract
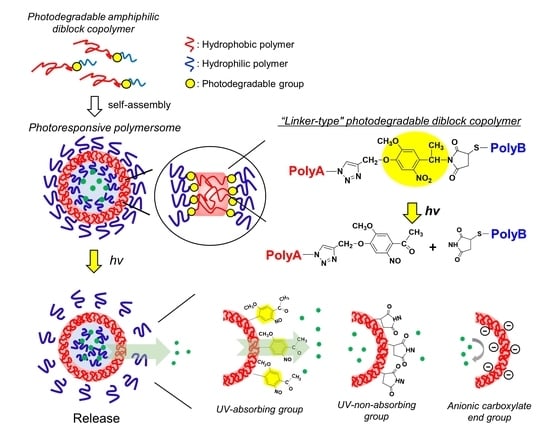
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of 1-(4-Methoxy-2-nitro-4-prop-2-ynyloxyphenyl)ethyl Bromide
2.3. Synthesis of the Heterobifunctional Linker NBEL
2.4. Synthesis of 4-Methyl-ε-caprolactone (MCL)
2.5. Synthesis of Poly(4-methyl-ε-caprolactone) (PMCL)
2.6. Synthesis of α-Carboxyl-ω-tert-butyldimethylsilyl Group-Terminated Poly(α-methyl-ε-caprolactone) (HOOC-PMCL-OTBDMS)
2.7. Synthesis of α-2-(2′,4′-Dinitrophenylthio)ethyl-ω-tert-butyldimethylsilyl-terminated Poly(α-methyl-ε-caprolactone) (DNP-PMCL-TBDMS)
2.8. Synthesis of α-Thiol-ω-tert-butyldimethylsilyl-terminated Poly(α-methyl-ε-caprolactone) (HS-PMCL-TBDMS)
2.9. Synthesis of PEG750-b-NBEL
2.10. Synthesis of PEG750-b-NBEL-Maleimide by Furan Deprotection
2.11. Synthesis of PEG750-b-NBEL-b-PMCL6600 5
2.12. Preparation of Photoresponsive Polymersomes
2.13. Preparation of Photoresponsive Polymersomes Encapsulating Fluorescein
2.14. Characterization of Photoresponsive Polymersomes
2.15. Photoirradiation
2.16. Release of Fluorescein from the Polymersome
3. Results and Discussion
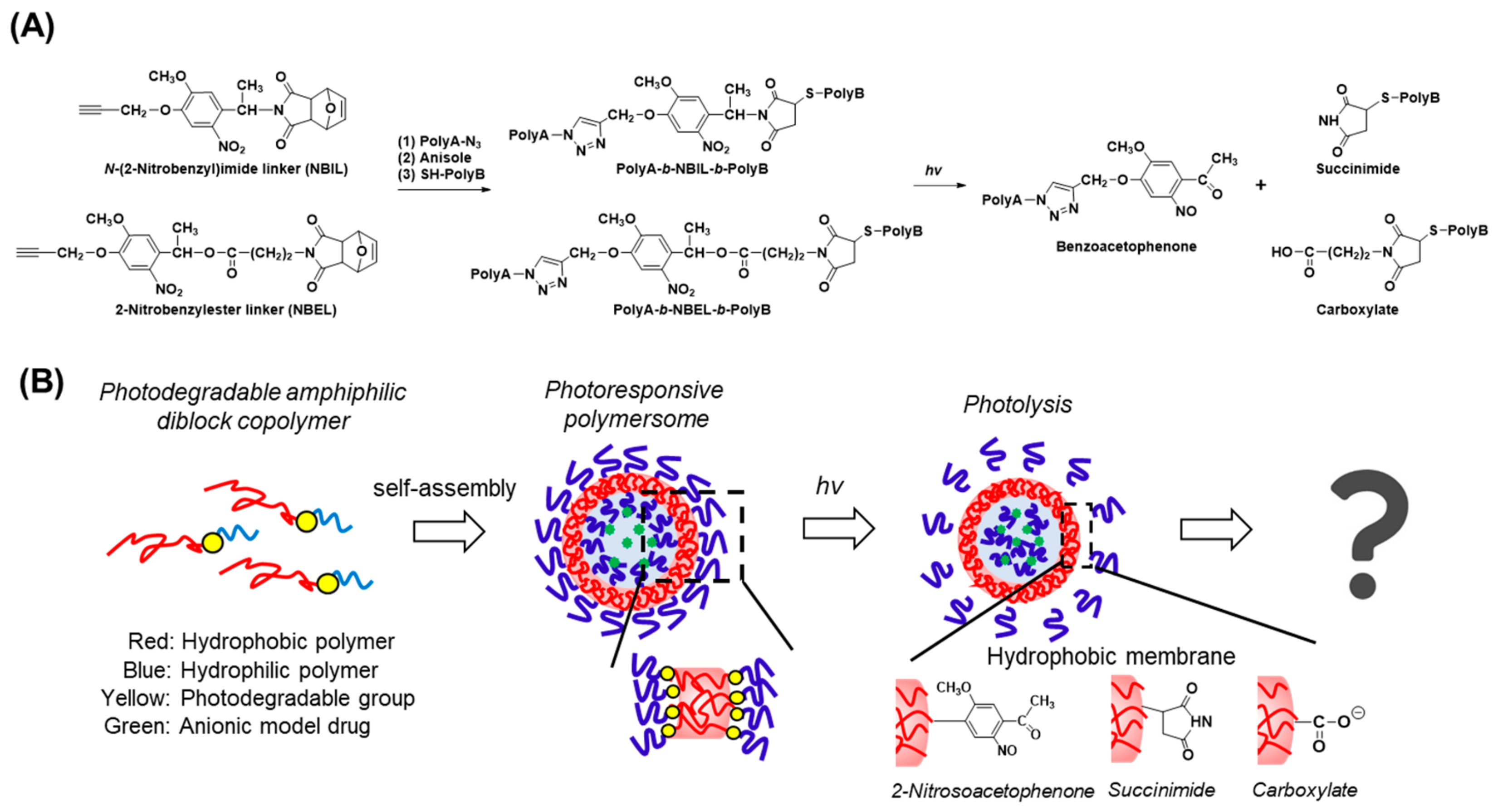
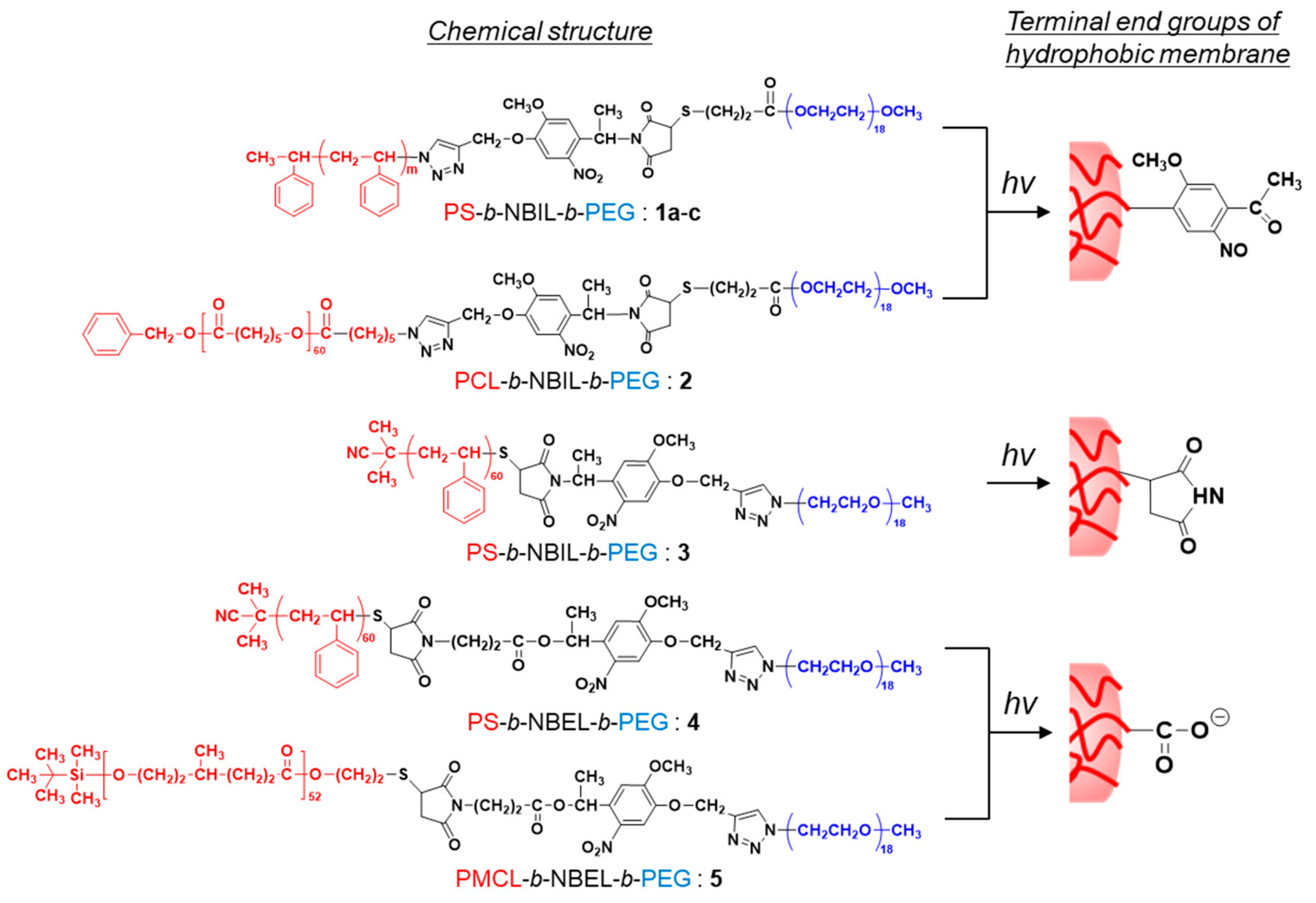
3.1. Design of Diblock Copolymers and Characterization
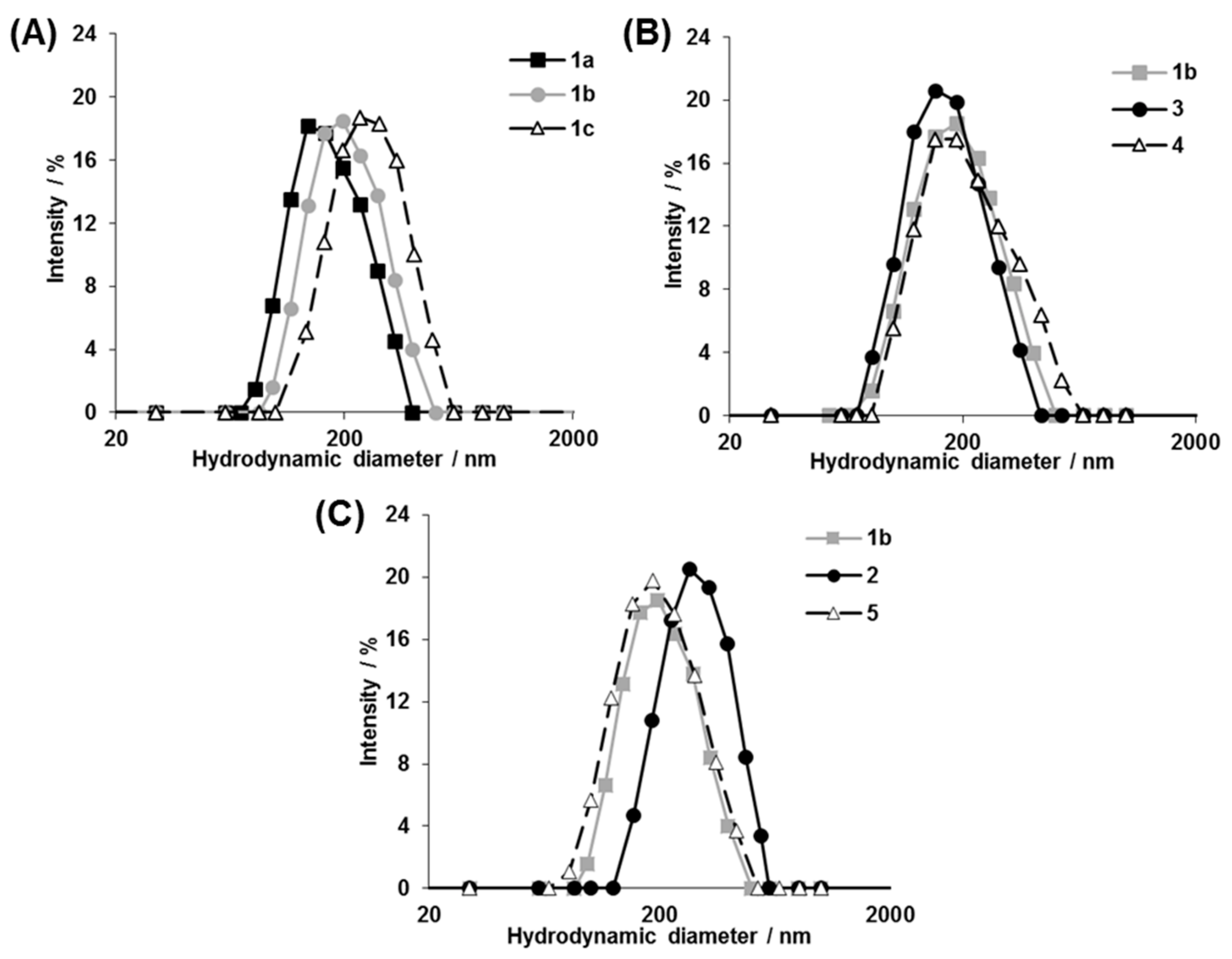
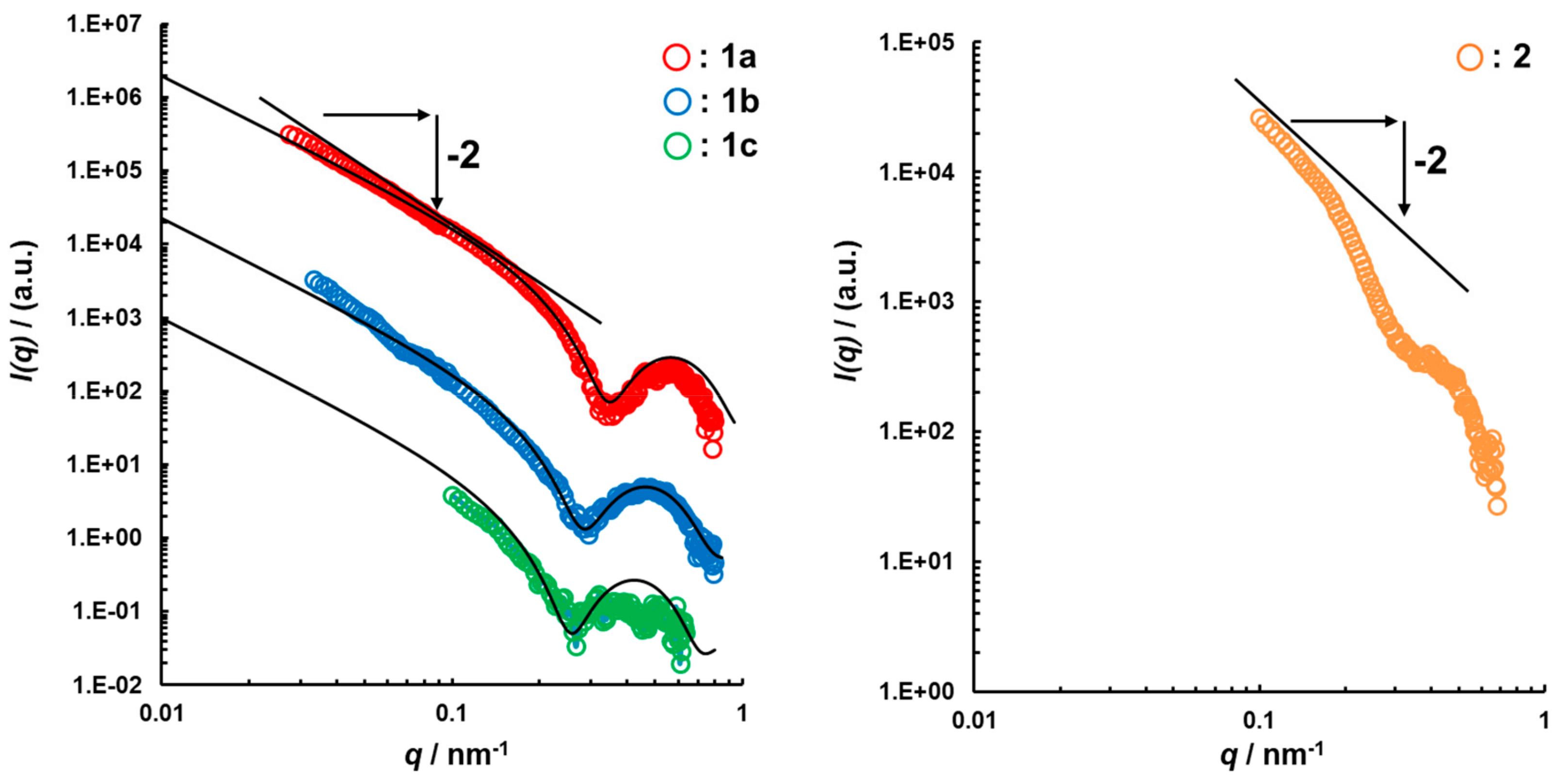
3.2. Structural Characterization of Photoresponsive Polymersomes
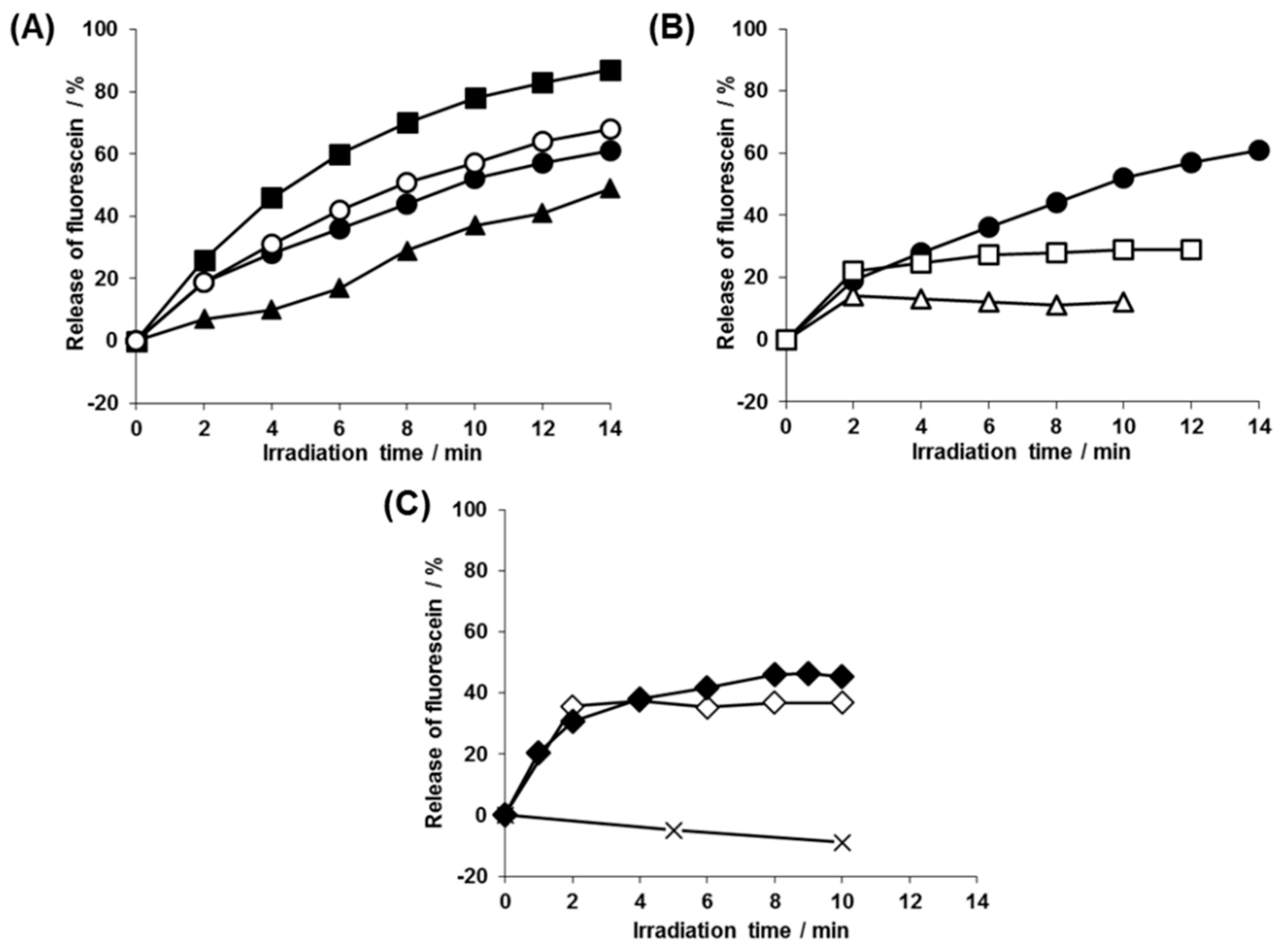
3.3. Kinetic analysis of the Photolysis Reaction of 2-nitrobenzyl Groups and Photorelease of Entrapped Payload in the Polymersomes
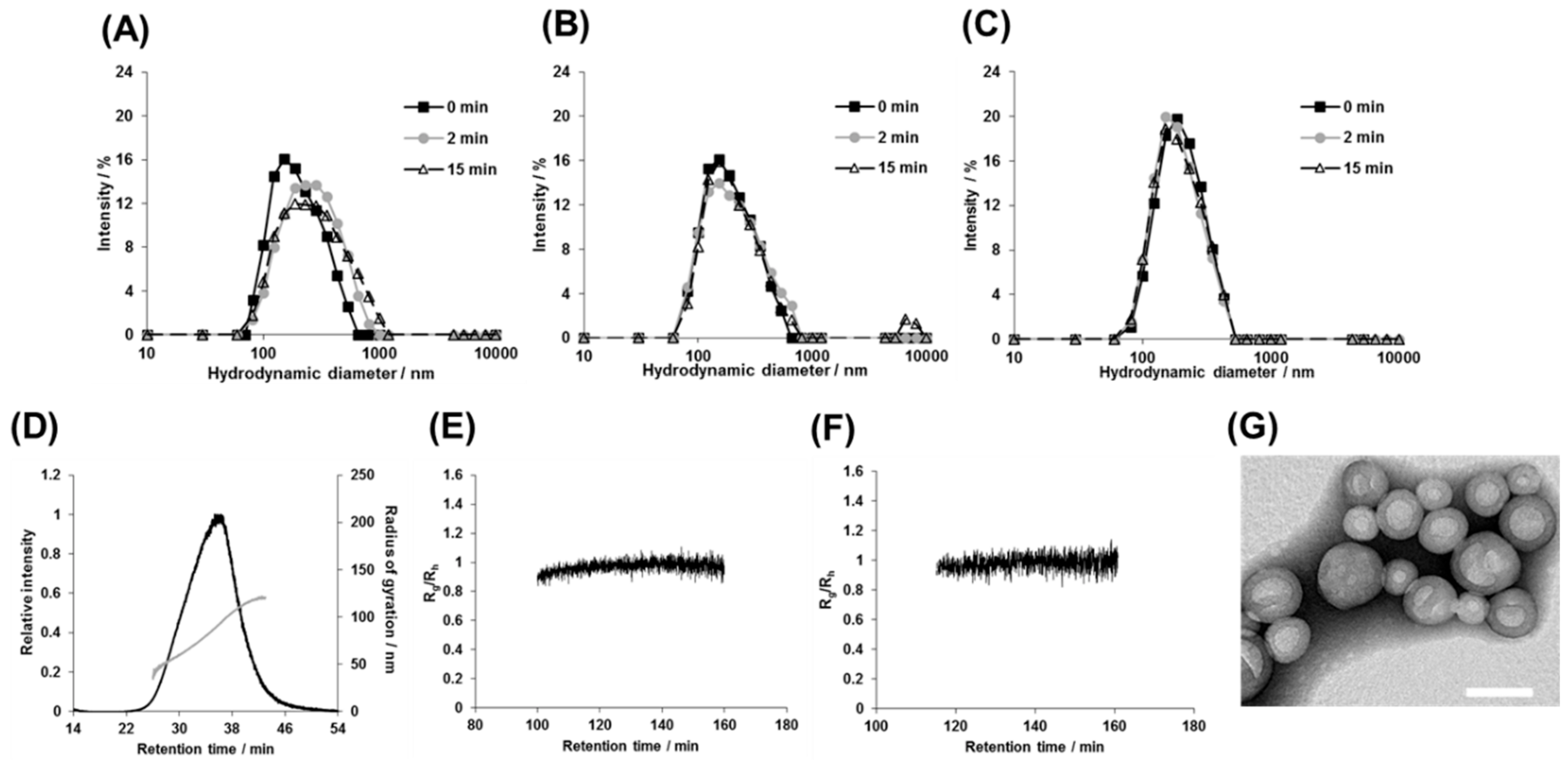
3.4. Structural Analysis of the Photoresponsive Polymersomes after Photoirradiation
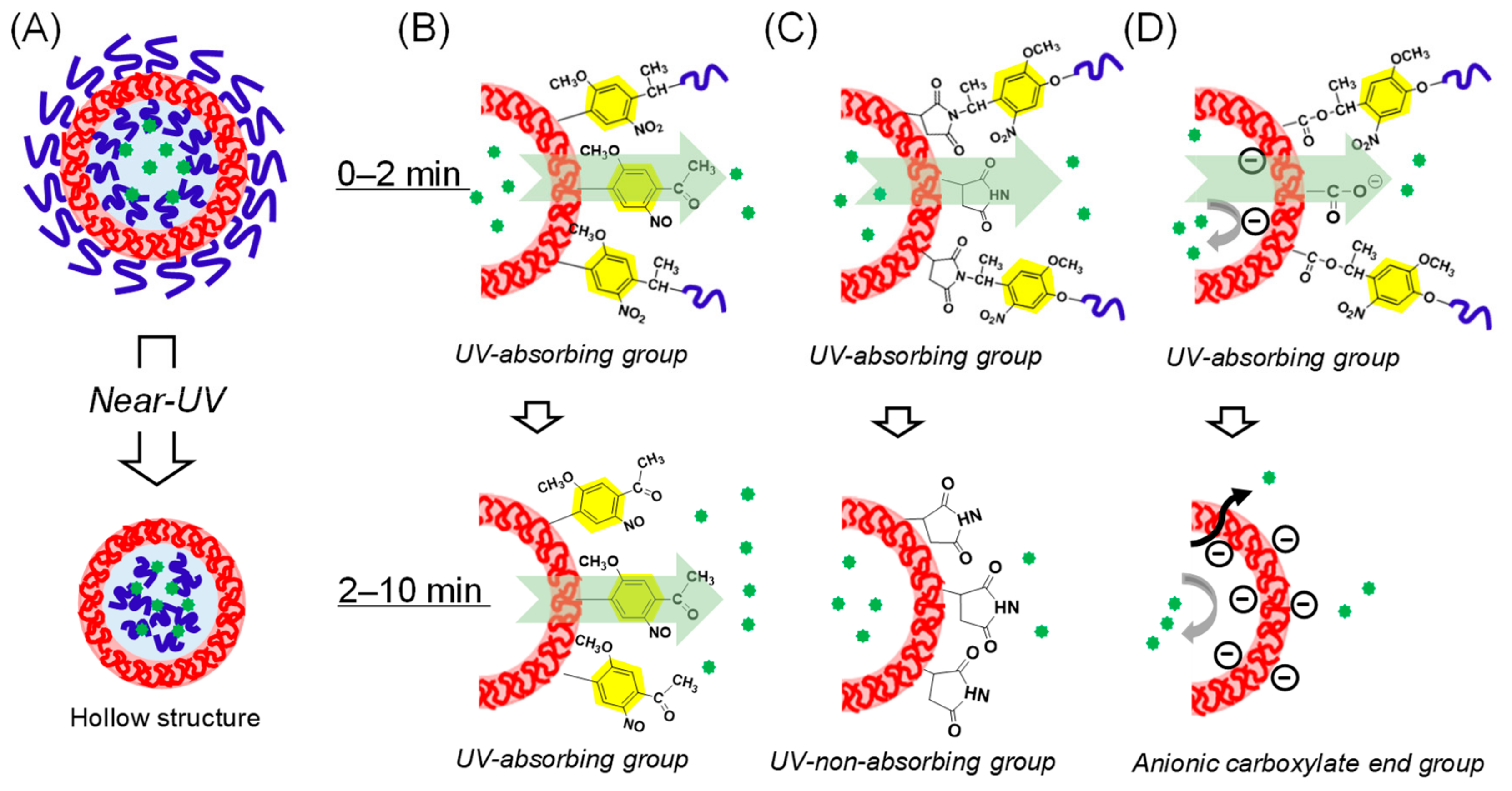
3.5. Expected Mechanism of Fluorescein Photorelease from Polymersomes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bae, Y.; Kataoka, K. Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers. Adv. Drug Deliv. Rev. 2009, 61, 768–784. [Google Scholar] [CrossRef] [PubMed]
- Discher, B.M.; Won, Y.-Y.; Ege, D.S.; Lee, J.C.-M.; Bates, F.S.; Discher, D.E.; Hammer, D.A. Polymersomes: Tough Vesicles Made from Diblock Copolymers. Science 1999, 284, 1143–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Yang, B.; Chen, S.; Du, J. Polymer vesicles: Mechanism, preparation, application, and responsive behavior. Prog. Polym. Sci. 2017, 64, 1–22. [Google Scholar] [CrossRef]
- Tang, Z.; He, C.; Tian, H.; Ding, J.; Hsiao, B.S.; Chu, B.; Chen, X. Polymeric nanostructured materials for biomedical applications. Prog. Polym. Sci. 2016, 60, 86–128. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Feijen, J. Polymersomes for drug delivery: Design, formation and characterization. J. Control. Release 2012, 161, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bonduelle, C.; Thévenot, J.; Lecommandoux, S.; Heise, A. Biologically Active Polymersomes from Amphiphilic Glycopeptides. J. Am. Chem. Soc. 2012, 134, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Hiemstra, C.; Engbers, G.H.M.; Feijen, J. Biodegradable Polymersomes. Macromolecules 2003, 36, 3004–3006. [Google Scholar] [CrossRef]
- Ghoroghchian, P.P.; Li, G.; Levine, D.H.; Davis, K.P.; Bates, F.S.; Hammer, D.A.; Therien, M.J. Bioresorbable Vesicles Formed through Spontaneous Self-Assembly of Amphiphilic Poly(ethylene oxide)-block-polycaprolactone. Macromolecules 2006, 39, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Eisenberg, A. Proton Diffusion across Membranes of Vesicles of Poly(styrene-b-acrylic Acid) Diblock Copolymers. J. Am. Chem. Soc. 2006, 128, 2880–2884. [Google Scholar] [CrossRef]
- Chiang, W.-H.; Huang, W.-C.; Chang, C.-W.; Shen, M.-Y.; Shih, Z.-F.; Huang, Y.-F.; Lin, S.-C.; Chiu, H.-C. Functionalized polymersomes with outlayered polyelectrolyte gels for potential tumor-targeted delivery of multimodal therapies and MR imaging. J. Control. Release 2013, 168, 280–288. [Google Scholar] [CrossRef]
- Lomas, H.; Canton, I.; MacNeil, S.; Du, J.; Armes, S.P.; Ryan, A.J.; Lewis, A.L.; Battaglia, G. Biomimetic pH Sensitive Polymersomes for Efficient DNA Encapsulation and Delivery. Adv. Mater. 2007, 19, 4238–4243. [Google Scholar] [CrossRef]
- Iatrou, H.; Frielinghaus, H.; Hanski, S.; Ferderigos, N.; Ruokolainen, J.; Ikkala, O.; Richter, D.; Mays, J.; Hadjichristidis, N. Architecturally Induced Multiresponsive Vesicles from Well-Defined Polypeptides. Formation of Gene Vehicles. Biomacromolecules 2007, 8, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Vader, P.; Schiffelers, R.M. Extracellular vesicles for nucleic acid delivery: Progress and prospects for safe RNA-based gene therapy. Gene Ther. 2017, 24, 157. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Shen, J.; Zhang, Z.; Yu, H.; Li, Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv. Drug Deliv. Rev. 2013, 65, 1699–1715. [Google Scholar] [CrossRef] [PubMed]
- Fleige, E.; Quadir, M.A.; Haag, R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: Concepts and applications. Adv. Drug Deliv. Rev. 2012, 64, 866–884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rutjes, F.P.J.T.; van Hest, J.C.M. pH responsive polymersome Pickering emulsion for simple and efficient Janus polymersome fabrication. Chem. Commun. 2014, 50, 14550–14553. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, G.; Hu, J.; Zhang, G.; Liu, S. Concurrent Block Copolymer Polymersome Stabilization and Bilayer Permeabilization by Stimuli-Regulated “Traceless” Crosslinking. Angew. Chem. Int. Ed. 2014, 53, 3138–3142. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-L.; Chang, H.-Y.; Sheng, Y.-J.; Tsao, H.-K. Photoresponsive Polymersomes Formed by Amphiphilic Linear–Dendritic Block Copolymers: Generation-Dependent Aggregation Behavior. Macromolecules 2012, 45, 7143–7156. [Google Scholar] [CrossRef]
- Liu, F.; Kozlovskaya, V.; Medipelli, S.; Xue, B.; Ahmad, F.; Saeed, M.; Cropek, D.; Kharlampieva, E. Temperature-Sensitive Polymersomes for Controlled Delivery of Anticancer Drugs. Chem. Mater. 2015, 27, 7945–7956. [Google Scholar] [CrossRef]
- Song, Z.; Kim, H.; Ba, X.; Baumgartner, R.; Lee, J.S.; Tang, H.; Leal, C.; Cheng, J. Polypeptide vesicles with densely packed multilayer membranes. Soft Matter 2015, 11, 4091–4098. [Google Scholar] [CrossRef]
- Katz, J.S.; Zhong, S.; Ricart, B.G.; Pochan, D.J.; Hammer, D.A.; Burdick, J.A. Modular Synthesis of Biodegradable Diblock Copolymers for Designing Functional Polymersomes. J. Am. Chem. Soc. 2010, 132, 3654–3655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabane, E.; Malinova, V.; Meier, W. Synthesis of Photocleavable Amphiphilic Block Copolymers: Toward the Design of Photosensitive Nanocarriers. Macromol. Chem. Phys. 2010, 211, 1847–1856. [Google Scholar] [CrossRef]
- Cabane, E.; Malinova, V.; Menon, S.; Palivan, C.G.; Meier, W. Photoresponsive polymersomes as smart, triggerable nanocarriers. Soft Matter 2011, 7, 9167–9176. [Google Scholar] [CrossRef] [Green Version]
- Peters, F.B.; Brock, A.; Wang, J.; Schultz, P.G. Photocleavage of the Polypeptide Backbone by 2-Nitrophenylalanine. Chem. Biol. 2009, 16, 148–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Nakahama, S.; Yamaguchi, K. A Heterobifunctional Linker Bearing Azide-reactive Alkyne and Thiol-reactive Maleimide Connected with N-(2-Nitrobenzyl)imide to Synthesize Photocleavable Diblock Copolymers. Chem. Lett. 2013, 42, 791–793. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tochigi, H.; Yamazaki, S.; Nakahama, S.; Yamaguchi, K. Synthesis of Amphiphilic Diblock Copolymer Using Heterobifunctional Linkers, Connected by a Photodegradable N-(2-Nitrobenzyl)imide Structure and Available for Two Different Click Chemistries. Bull. Chem. Soc. Jpn. 2016, 89, 481–489. [Google Scholar] [CrossRef]
- Bleul, R.; Thiermann, R.; Maskos, M. Techniques To Control Polymersome Size. Macromolecules 2015, 48, 7396–7409. [Google Scholar] [CrossRef]
- Yildiz, M.E.; Prud’homme, R.K.; Robb, I.; Adamson, D.H. Formation and characterization of polymersomes made by a solvent injection method. Polym. Adv. Technol. 2007, 18, 427–432. [Google Scholar] [CrossRef]
- Schumers, J.-M.; Fustin, C.-A.; Can, A.; Hoogenboom, R.; Schubert, U.S.; Gohy, J.-F. Are o-nitrobenzyl (meth)acrylate monomers polymerizable by controlled-radical polymerization? J. Polym. Sci. A Polym. Chem. 2009, 47, 6504–6513. [Google Scholar] [CrossRef]
- Giddings, J. Field-flow fractionation: analysis of macromolecular, colloidal, and particulate materials. Science 1993, 260, 1456–1465. [Google Scholar] [CrossRef]
- Williams, S.K.R.; Runyon, J.R.; Ashames, A.A. Field-Flow Fractionation: Addressing the Nano Challenge. Anal. Chem. 2011, 83, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Sanada, Y.; Akiba, I.; Hashida, S.; Sakurai, K.; Shiraishi, K.; Yokoyama, M.; Yagi, N.; Shinohara, Y.; Amemiya, Y. Composition Dependence of the Micellar Architecture Made from Poly(ethylene glycol)-block-Poly(partially benzyl-esterified aspartic acid). J. Phys. Chem. B 2012, 116, 8241–8250. [Google Scholar] [CrossRef]
- Schmidt, B.; Loeschner, K.; Hadrup, N.; Mortensen, A.; Sloth, J.J.; Bender Koch, C.; Larsen, E.H. Quantitative Characterization of Gold Nanoparticles by Field-Flow Fractionation Coupled Online with Light Scattering Detection and Inductively Coupled Plasma Mass Spectrometry. Anal. Chem. 2011, 83, 2461–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vezočnik, V.; Rebolj, K.; Sitar, S.; Ota, K.; Tušek-Žnidarič, M.; Štrus, J.; Sepčić, K.; Pahovnik, D.; Maček, P.; Žagar, E. Size fractionation and size characterization of nanoemulsions of lipid droplets and large unilamellar lipid vesicles by asymmetric-flow field-flow fractionation/multi-angle light scattering and dynamic light scattering. J. Chromat. A 2015, 1418, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Trollsås, M.; Kelly, M.A.; Claesson, H.; Siemens, R.; Hedrick, J.L. Highly Branched Block Copolymers: Design, Synthesis, and Morphology. Macromolecules 1999, 32, 4917–4924. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn a | Mw/Mn b | Hydrophobic Polymer (Mn) a | PEG(wt %) a | |
|---|---|---|---|---|
| 1a | 5700 | 1.05 | 4100 | 13.2 |
| 1b | 7600 | 1.07 | 6200 | 9.9 |
| 1c | 9300 | 1.05 | 8000 | 8.0 |
| 2 | 7700 | 1.05 | 6300 | 9.7 |
| 3 | 8000 | 1.05 | 6500 | 9.4 |
| 4 | 7600 | 1.03 | 6200 | 9.9 |
| 5 | 8000 | 1.08 | 6600 | 9.4 |
| % of Fluorescein Released/Second | |||||||
|---|---|---|---|---|---|---|---|
| 1a | 1b | 1c | 2 | 3 | 4 | 5 | |
| 0–2 min | 0.22 | 0.16 | 0.059 | 0.16 | 0.11 | < 0 | 0.26 |
| 2–10 min | 0.11 | 0.070 | 0.062 | 0.080 | < 0 | < 0 | 0.030 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, S.; Yamada, T.; Kubo, G.; Sakurai, K.; Yamaguchi, K.; Nakanishi, J. Preparation of a Series of Photoresponsive Polymersomes Bearing Photocleavable a 2-nitrobenzyl Group at the Hydrophobic/Hydrophilic Interfaces and Their Payload Releasing Behaviors. Polymers 2019, 11, 1254. https://doi.org/10.3390/polym11081254
Yamamoto S, Yamada T, Kubo G, Sakurai K, Yamaguchi K, Nakanishi J. Preparation of a Series of Photoresponsive Polymersomes Bearing Photocleavable a 2-nitrobenzyl Group at the Hydrophobic/Hydrophilic Interfaces and Their Payload Releasing Behaviors. Polymers. 2019; 11(8):1254. https://doi.org/10.3390/polym11081254
Chicago/Turabian StyleYamamoto, Shota, Takafumi Yamada, Genki Kubo, Kazuo Sakurai, Kazuo Yamaguchi, and Jun Nakanishi. 2019. "Preparation of a Series of Photoresponsive Polymersomes Bearing Photocleavable a 2-nitrobenzyl Group at the Hydrophobic/Hydrophilic Interfaces and Their Payload Releasing Behaviors" Polymers 11, no. 8: 1254. https://doi.org/10.3390/polym11081254