Production and Characterization of Non-Isocyanate Polyurethane/SiO2 Films Through a Sol-Gel Process for Thermal Insulation Applications


Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Information
2.2. Synthesis of 5-Membered Cyclic Carbonates
2.3. Synthesis of Silica Nanoparticles
2.4. Synthesis of Non-Isocyanate Polyurethane
2.5. Synthesis of PHUs/SiO2 Materials
2.6. Names of Materials
2.7. Preparation of Samples for Analyzes
2.8. Characterization Techniques
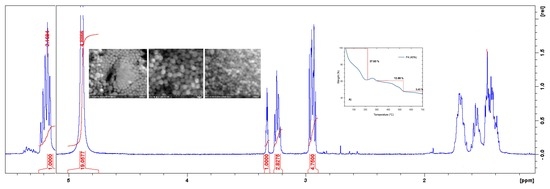
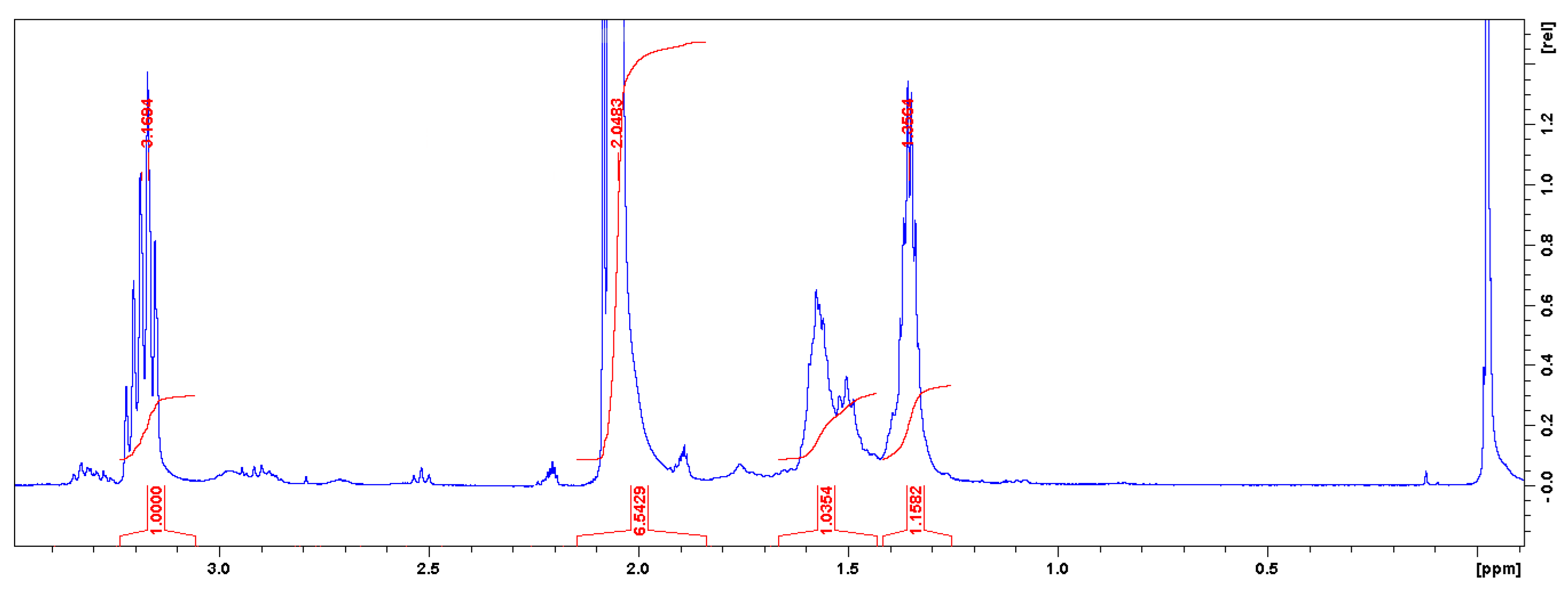
2.8.1. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.8.2. Fourier Transform Infrared (IR) Spectroscopy
2.8.3. Ultraviolet-Visible Microscopy (UV-Vis) Spectroscopy
2.8.4. Scanning Electron Microscopy (SEM)
2.8.5. Transmission Electron Microscopy (TEM)
2.8.6. Energy Dispersive Spectroscopy (EDS)
2.8.7. Thermogravimetric Analysis (TGA) and Derivate Thermogravimetric Analysis (DTG)
2.8.8. Thermal Conductivity (TC) Measurement
3. Results and Discussion
3.1. Characterizations of Cyclic Carbonates
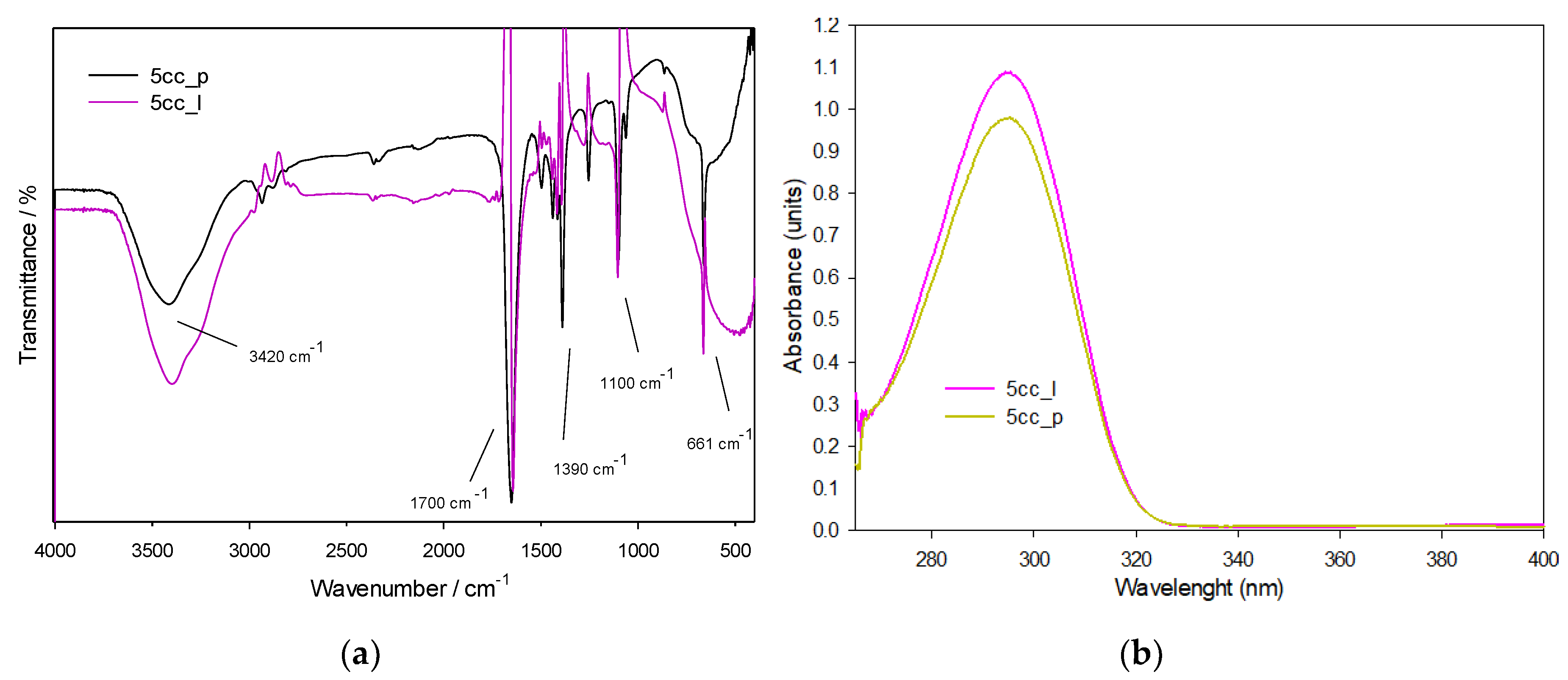
3.1.1. FTIR and UV-Vis Spectroscopies
3.1.2. Thermogravimetric Analysis and Derivate Thermogravimetric Analysis
3.2. Characterizations of SiO2 Nanoparticles
3.2.1. FTIR and UV-Vis Spectroscopies
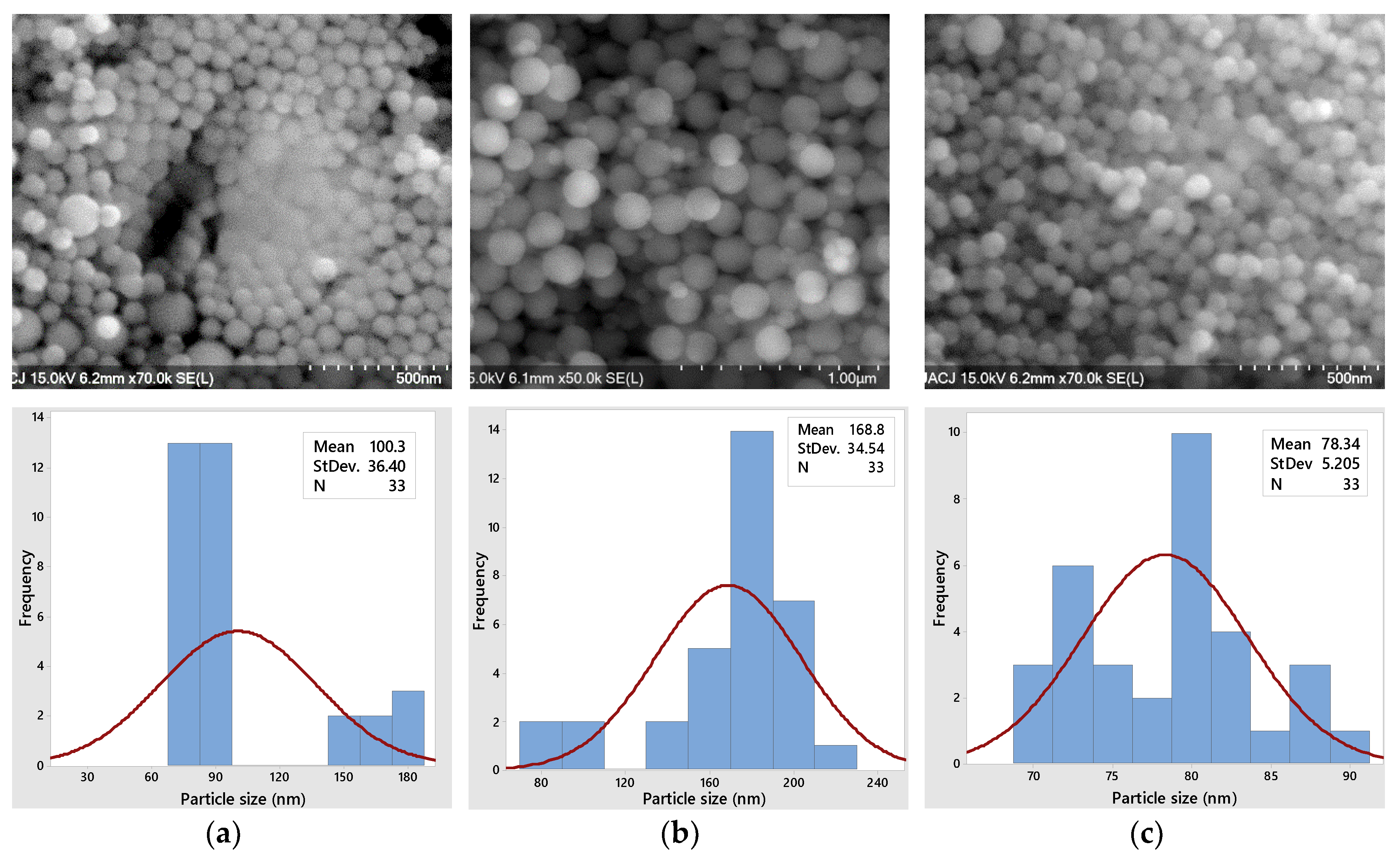
3.2.2. Scanning Electron Microscopy
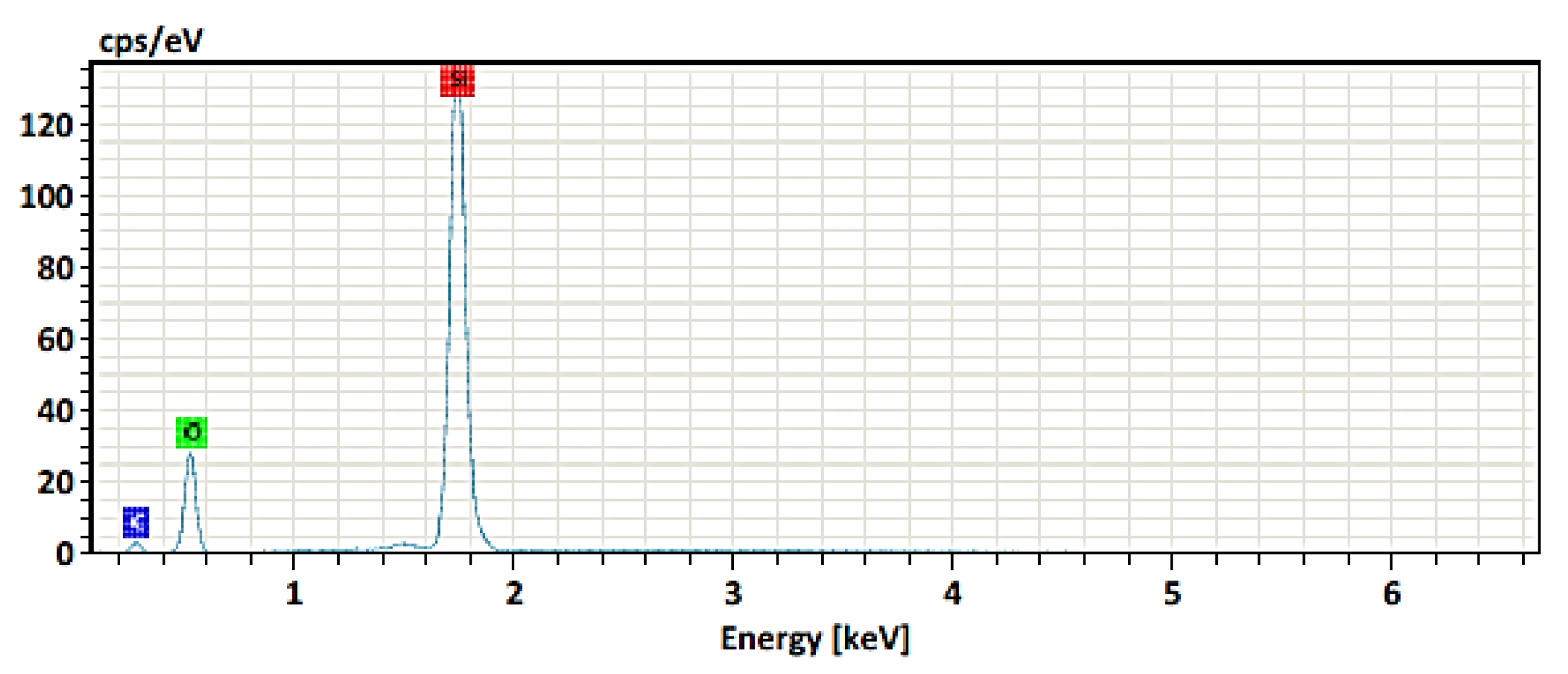
3.2.3. Electron Dispersive Spectroscopy Analysis
3.2.4. Transmission Electron Microscopy and EDS analysis
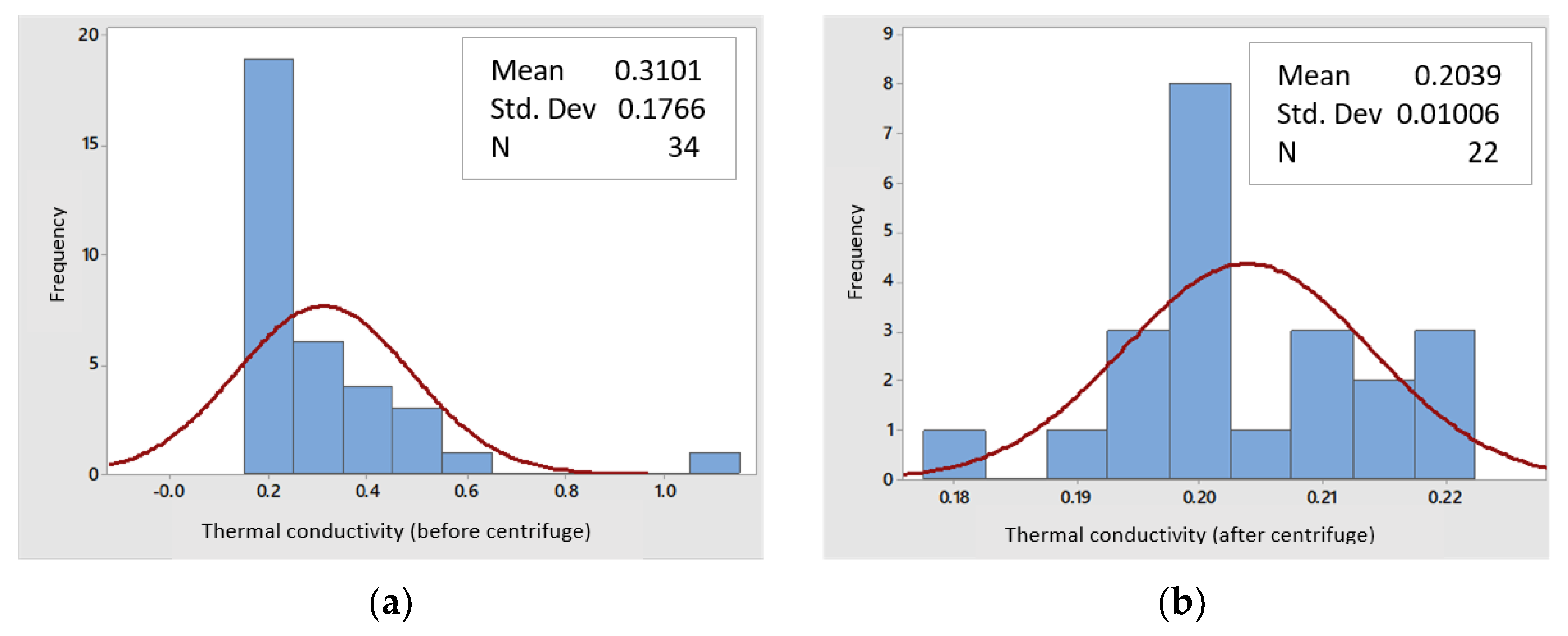
3.2.5. Thermal Conductivity
3.3. Characterizations of Non-Isocyanate Polyurethane Materials
3.3.1. FTIR and UV-Vis Spectroscopies
3.3.2. Scanning Electron Microscopy
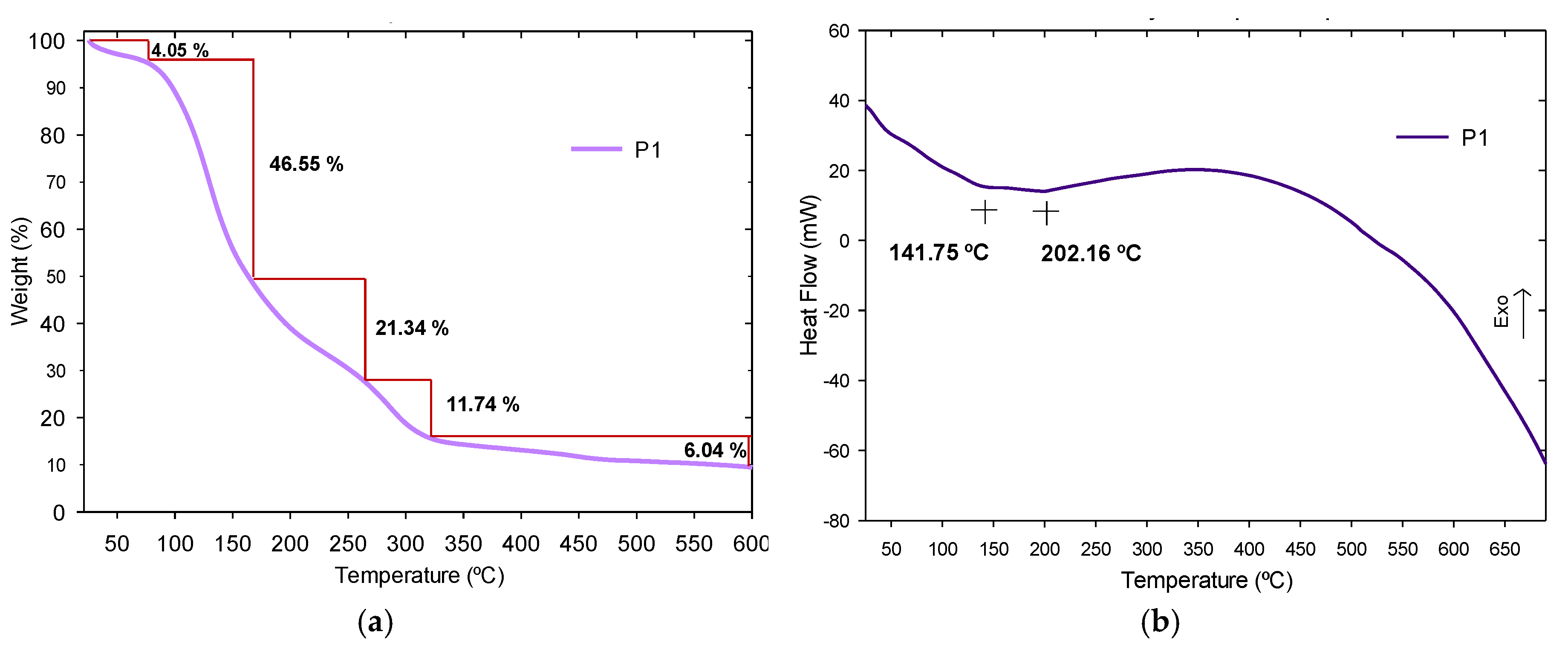
3.3.3. Thermogravimetric Analysis and Derivate Thermogravimetric Analysis
3.4. Characterizations of PHUs/SiO2 Polymeric Materials
3.4.1. FTIR and UV-Vis Spectroscopies
3.4.2. Scanning Electron Microscopy
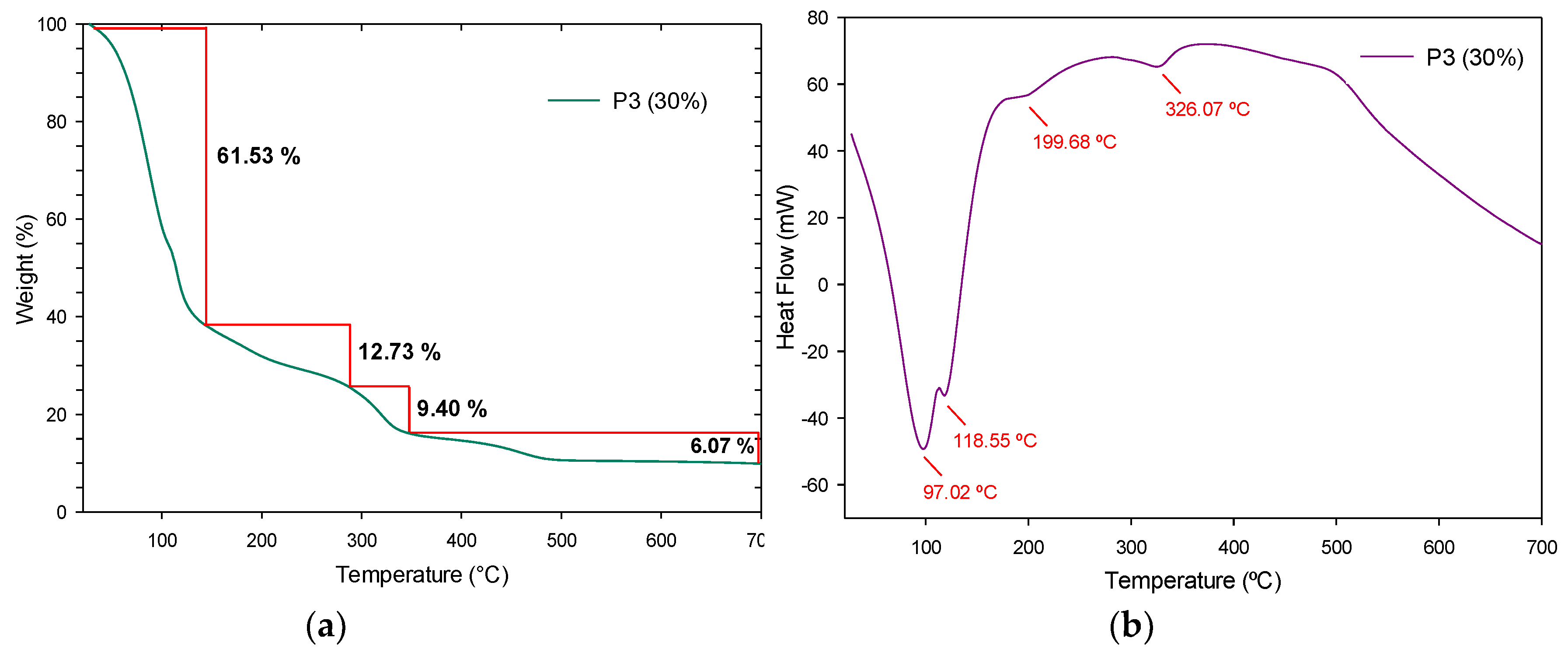
3.4.3. Thermogravimetric Analysis and Derivate Thermogravimetric Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Delebecq, E.; Pascault, J.; Boutevin, B.; Ganachaud, F. On the Versatility of Urethane/Urea Bonds: Reversibility, Blocked Isocyanate, and Non-isocyanate Polyurethane. Am. Chem. Soc. 2013, 113, 80–118. [Google Scholar] [CrossRef] [PubMed]
- Cornille, A.; Auvergne, R.; Figovsky, O.; Boutevina, B.; Caillol, S. A perspective approach to sustainable routes for non-isocyanate polyurethanes. Eur. Polym. J. 2017, 87, 535–552. [Google Scholar] [CrossRef]
- Merenyi, S. REACH: Regulation (EC) No 1907/2006, Munich, GRIN Verlag. Consolidated, version (June 2012) with an Introduction and Future Prospects Regarding the Area of Chemicals Legislation. 19 Octorber 2012. Available online: https://www.grin.com/document/203248 (accessed on 29 August 2019).
- Federation of European Rigid Polyurethane Foam Association. Report N 1, Thermal Insulation Materials Made of Rigid Polyurethane Foam (PUR/PIR), Properties-Manufacture. 2006. Available online: https://uia.org/s/or/en/1100037301 (accessed on 29 August 2019).
- Figovsky, O.; Shapovalov, L.; Leykin, A. Nonisocyanate Polyurethanes Based on Cyclic Carbonates and Nanostructured Composites. Sci. ISR Advant. 2012, 14, 95–106. [Google Scholar] [CrossRef]
- Figovsky, O.; Shapovalov, L.; Leykin, A.; Birukova, O.; Potashnikova, R. Progress in elaboration of nonisocyanate polyurethanes based on cyclic carbonates. PU Mag. 2012, 3, 1–9. [Google Scholar] [CrossRef]
- Rahman, I.A.; Padavettan, V. Synthesis of Silica nanoparticles by Sol-Gel: Size-dependent properties, surface modification, and applications in silica-polymer nanocompositesa review. J. Nanomater. 2012, 2012, 132424. [Google Scholar] [CrossRef]
- Díaz, H.; Guzmán, J.; Meneses, E.; García, R.; Martínez, R. Efficient Synthesis of Organic Carbonates to the Multigram Level from CO2 Using a New Chitin-Supported Catalyst. Catal. Lett. 2017, 147, 2260–2268. [Google Scholar] [CrossRef]
- Moreno-Wong, S.J.C.P.; Alvarado-Rivera, J.; Moreno, G.; Acosta-Enriquez, M.C.; Castillo, S.J. Chemical and structural characterization of PbS-SiO2 Core-shell structures synthesized by ultrasonic wave-assisted chemical bath deposition. Microsc. Soc. Am. 2015, 21, 1033–1034. [Google Scholar] [CrossRef]
- Ibrahim, K.; Zikry, A.; Sharaf, M. Preparation of spherical silica nanoparticles: Stober silica. J. Am. Sci. 2010, 6, 985–989. [Google Scholar] [CrossRef]
- Mota, M.L. Sintesis de oligofenilenos modificados con cadenas alquílicas y grupos CN y estudio de sus propiedades de cristal líquido y luminiscencia. Ph.D. Thesis, Facultad de Ciencias Químicas, Universidad Autónoma de Coahuila, Saltillo, México, 2014. [Google Scholar]
- Mota, M.L.; Carrillo, A.; Ambrosio, R. Synthesis and Study of Chemical, Thermal, Mesomorphic, and Optical Properties of Terphenyls Modified with Nitrile Groups. J. Chem. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Felix-Serrano, I.; Trejo-Carbajal, N.; Rodríguez-González, R.J.; Larios-López, L.; Moggio, I.; Arias, E.; Torres-Lubián, R.; Navarro-Rodríguez, D. Synthesis and liquid-crystalline properties of methacrylate monomers carrying a p-terphenyl laterally substituted with one or two cyano groups. J. Mol. Liq. 2017, 241, 347–354. [Google Scholar] [CrossRef]
- Liu, G.; Wu, G.; Chen, J.; Huo, S.; Jin, C.; Kong, Z. Synthesis and properties of POSS-containing gallic acid-based non-isocyanate polyurethanes coatings. Polym. Degrad. Stab. 2015, 121, 247–252. [Google Scholar] [CrossRef]
- Patil, A.O. Poly (Alkyl Epoxides) Process for Making, and Lubricant Compositions Having Same. U.S. Patent 0263469 A1, 27 October 2011. [Google Scholar]
- Gaikwad, M.S.; Gite, V.V.; Mahulikar, P.P.; Hundiwale, D.G.; Yemul, O.S. Eco-friendly polyurethane coatings from cottonseed and karanja oil. Prog. Org. Coat. 2015, 86, 164–172. [Google Scholar] [CrossRef]
- Hosgor, Z.; Kayaman-Apohan, N.; Karatas, S.; Menceloğlu, Y.; Güngör, A. Preparation and characterization of phosphine oxide based polyurethane/silica nanocomposite via non-isocyanate route. Prog. Org. Coat. 2010, 69, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Heinze, T.; El Seoud, O.A.; Koschella, A. Cellulose Derivatives: Synthesis, Structure, and Properties; Springer: Berlin, Germany, 2018. [Google Scholar] [CrossRef]
- National Institute of Standards and Technology. Search for Species Data by Vibrational Energy Value. 2017. Available online: https://webbook.nist.gov/chemistry (accessed on 29 August 2019).
- Cornille, A.; Michaud, G.; Simon, F.; Fouquay, S.; Auvergne, A.; Boutevin, A.; Caillol, S. Promising mechanical and adhesive properties of isocyanate-free poly(hydroxyurethane). Eur. Polym. J. 2016, 84, 404–420. [Google Scholar] [CrossRef]
- Abdelaal, H.M. Fabrication of hollow silica microspheres utilizing a hydrothermal approach. Chin. Chem. Lett. 2014, 25, 627–629. [Google Scholar] [CrossRef]
- Ryu, M.T.S. Fictive temperature measurement of amorphous SiO2 films by IR method. J. Non-Cryst. Solids 2006, 352, 3929–3935. [Google Scholar] [CrossRef]
- Nazeran, N.; Moghaddas, J. Synthesis and characterization of silica aerogel reinforced rigid polyurethane foam for thermal insulation application. J. Non-Cryst. Solids 2017, 461, 1–11. [Google Scholar] [CrossRef]
- Nikabadi, H.R.; Shahtahmasebi, N.; Rokn-Abadi, M.R.; Mohagheghi, M.M.B.; Goharshadi, E.K. Gradual growth of gold nanoseeds on silica for SiO2@gold homogeneous nano core/shell applications by the chemical reduction method. Phys. SCR 2013, 87, 2–7. [Google Scholar] [CrossRef]
- Cornejo-Monroy, D.; Sánchez-Ramírez, J.F.; Pescador-Rojas, J.A.; Herrera-Pérez, J.L.; González-Araoz, M.P.; Guarneros, C. Nanoesferas monodispersas de SiO2: Síntesis controlada y caracterización. Superf. y Vacío 2009, 22, 44–48. [Google Scholar]
- Amirthalingam, T.; Kalirajan, J.; Chockalingam, A. Use of Silica-Gold Core Shell Structured Nanoparticles for Targeted Drug Delivery System. Nanomed. Nanotechnol. 2011, 2, 2–6. [Google Scholar] [CrossRef]
- Noriega, N.E.; Hernández, J.F.; Sandoval, J.L.; de los Angeles Ramírez Ambriz, M.; Abdelkefi, A.; Cornejo, D. Fabricación y caracterización de nanopartículas de sílice huecas para uso en aislantes térmicos mediante un análisis factorial. Cultura científica y tecnológica 2016, 57. Available online: http://erevistas.uacj.mx/ojs/index.php/culcyt/article/view/839 (accessed on 29 August 2019).
- Singh, L.P. Sol-Gel processing of silica nanoparticles and their applications. Adv. Colloid Interface Sci. 2014, 214, 17–37. [Google Scholar] [CrossRef]
- Cornille, A.; Guillet, C.; Benyahya, S.; Negrell, C.; Boutevin, B.; Caillol, S. Room temperature flexible isocyanate-free polyurethane foams. Eur. Polym. J. 2016, 84, 873–888. [Google Scholar] [CrossRef]
- Matsukizono, H.; Endo, T. Synthesis of Bi- and Trifunctional Cyclic Carbonates Based on Trimethylolpropane and Their Application to Networked Polyhydroxyurethanes. J. Mater. Sci. Res. 2016, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Tasaganva, R.G.; Doddamani, R.V.; Inamdar, S.R.; Kariduraganavar, M.Y. Synthesis of thermally stable new polyurethanes containing nitro-substituted 1,3, 4-oxadiazole chromophores for second order nonlinear optical applications. Opt.-Int. J. Light Electron. Opt. 2015, 126, 4991–5000. [Google Scholar] [CrossRef]
- Beniah, G.; Fortman, D.J.; Heath, W.H.; Dichtel, W.R.; Torkelson, J.M. Non-Isocyanate Polyurethane Thermoplastic Elastomer: Amide-Based Chain Extender Yields Enhanced Nanophase Separation and Properties in Polyhydroxyurethane. Macromolecules 2017, 50, 4425–4434. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, F.C. Spectrometric Identification of Organic Compounds; John Wiley & Sons: Hoboken, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Kamaci, M.; Kaya, I. New low-band gap polyurethanes containing azomethine bonding: Photophysical, electrochemical, thermal and morphological properties. J. Taiwan Inst. Chem. Eng. 2016, 59, 536–546. [Google Scholar] [CrossRef]
- Sotelo-lerma, M.; Zingaro, R.A.; Castillo, S.J. Preparation of CdTe coatings using the chemical deposition method. J. Organomet. Chem. 2014, 623, 81–86. [Google Scholar] [CrossRef]
- Tryznowski, M.; Gołofit, T.; Świderska, A. Poly(hydroxyurethane)s with diethyl tartrate-based amide backbone by an isocyanate-free route: Use as adhesives. Polymer 2018, 144, 1–6. [Google Scholar] [CrossRef]
- Carré, C.; Zoccheddu, H.; Delalande, S.; Pichon, P.; Avérous, L. Synthesis and characterization of advanced biobased thermoplastic nonisocyanate polyurethanes, with controlled aromatic-aliphatic architectures. Eur. Polym. J. 2016, 84, 759–769. [Google Scholar] [CrossRef]
- Comí, M.; Lligadas, G.; Ronda, J.C.; Galià, M.; Cádiz, V. Adaptive bio-based polyurethane elastomers engineered by ionic hydrogen bonding interactions. Eur. Polym. J. 2017, 91, 408–419. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Identification | Sample Description |
|---|---|
| 5cc_l | Liquid phase of cyclic carbonates |
| 5cc_p | Precipitated phase of cyclic carbonates |
| 5cc | Cyclic carbonates including both liquid and precipitated phase |
| P1 | Poly(hydroxy)urethanes (PHUs) |
| P2 (20%) | 20% of NPs concentration within the polymer (PHUs/SiO2) |
| P3 (30%) | 30% of NPs concentration within the polymer (PHUs/SiO2) |
| P4 (40%) | 40% of NPs concentration within the polymer (PHUs/SiO2) |
| Interaction | Bond | cm−1 |
|---|---|---|
| A. Stretching | O–H | 3420 |
| B. Stretching | C–H | 2950 |
| C. Stretching | C=O | 1780 |
| D. Bending | C–H | 1390 |
| E. Stretching | O–C–O | 1100 |
| F. Stretching | O–C–O | 661 |
| Element | At. No. | Line s. | Netto | Mass [%] | Mass Norm. [%] | Atom [%] | abs.error [%] (1 sigma) | abs.error [%] (2 sigma) | abs.error [%] (3 sigma) |
|---|---|---|---|---|---|---|---|---|---|
| Silicon | 14 | K-Serie | 376313 | 45.63 | 51.88 | 36.10 | 1.92 | 3.84 | 5.76 |
| Oxygen | 8 | K-Serie | 53245 | 31.24 | 35.51 | 43.38 | 3.69 | 7.38 | 11.07 |
| Carbon | 6 | K-Serie | 5076 | 11.09 | 12.61 | 20.51 | 1.76 | 3.51 | 5.27 |
| Sum 87.96 | 100.00 | 100.00 |
| El | AN | Series | unn. C [wt.%] | Norm. C [wt.%] | C Atom. C [at. %] | Error | (1 Sigma) [wt. %] |
|---|---|---|---|---|---|---|---|
| O | 8 | K-Series | 19.52 | 54.61 | 67.87 | - | 3.62 |
| Si | 14 | K-Series | 16.22 | 45.39 | 32.13 | - | 1.01 |
| C | 6 | K-Series | 0.00 | 0.00 | 0.00 | - | 0.00 |
| Cu | 29 | K-Series | 0.00 | 0.00 | 0.00 | - | 0.00 |
| Total: | 35.75 | 100.00 | 100.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noriega, N.E.; Carrillo, A.; Castillo, S.J.; Mota, M.L. Production and Characterization of Non-Isocyanate Polyurethane/SiO2 Films Through a Sol-Gel Process for Thermal Insulation Applications. Polymers 2019, 11, 1596. https://doi.org/10.3390/polym11101596
Noriega NE, Carrillo A, Castillo SJ, Mota ML. Production and Characterization of Non-Isocyanate Polyurethane/SiO2 Films Through a Sol-Gel Process for Thermal Insulation Applications. Polymers. 2019; 11(10):1596. https://doi.org/10.3390/polym11101596
Chicago/Turabian StyleNoriega, Natalia E., Amanda Carrillo, Santos J. Castillo, and María L. Mota. 2019. "Production and Characterization of Non-Isocyanate Polyurethane/SiO2 Films Through a Sol-Gel Process for Thermal Insulation Applications" Polymers 11, no. 10: 1596. https://doi.org/10.3390/polym11101596
APA StyleNoriega, N. E., Carrillo, A., Castillo, S. J., & Mota, M. L. (2019). Production and Characterization of Non-Isocyanate Polyurethane/SiO2 Films Through a Sol-Gel Process for Thermal Insulation Applications. Polymers, 11(10), 1596. https://doi.org/10.3390/polym11101596