Mg(II) Coordination Polymers Based on Flexible Isomeric Tetracarboxylate Ligands: Syntheses, Structures, Structural Transformation and Luminescent Properties


Abstract
:
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Materials
2.3. Preparations
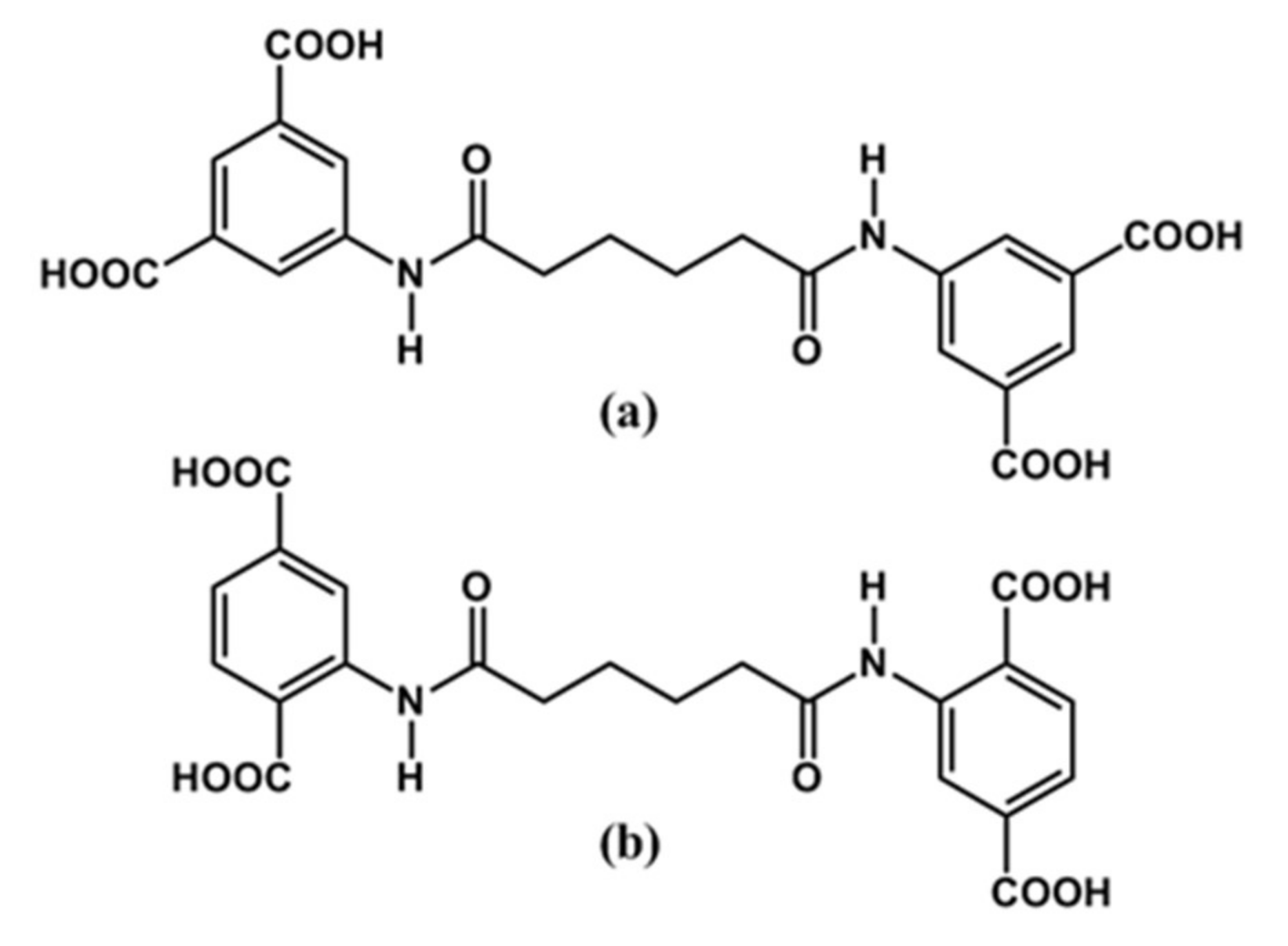
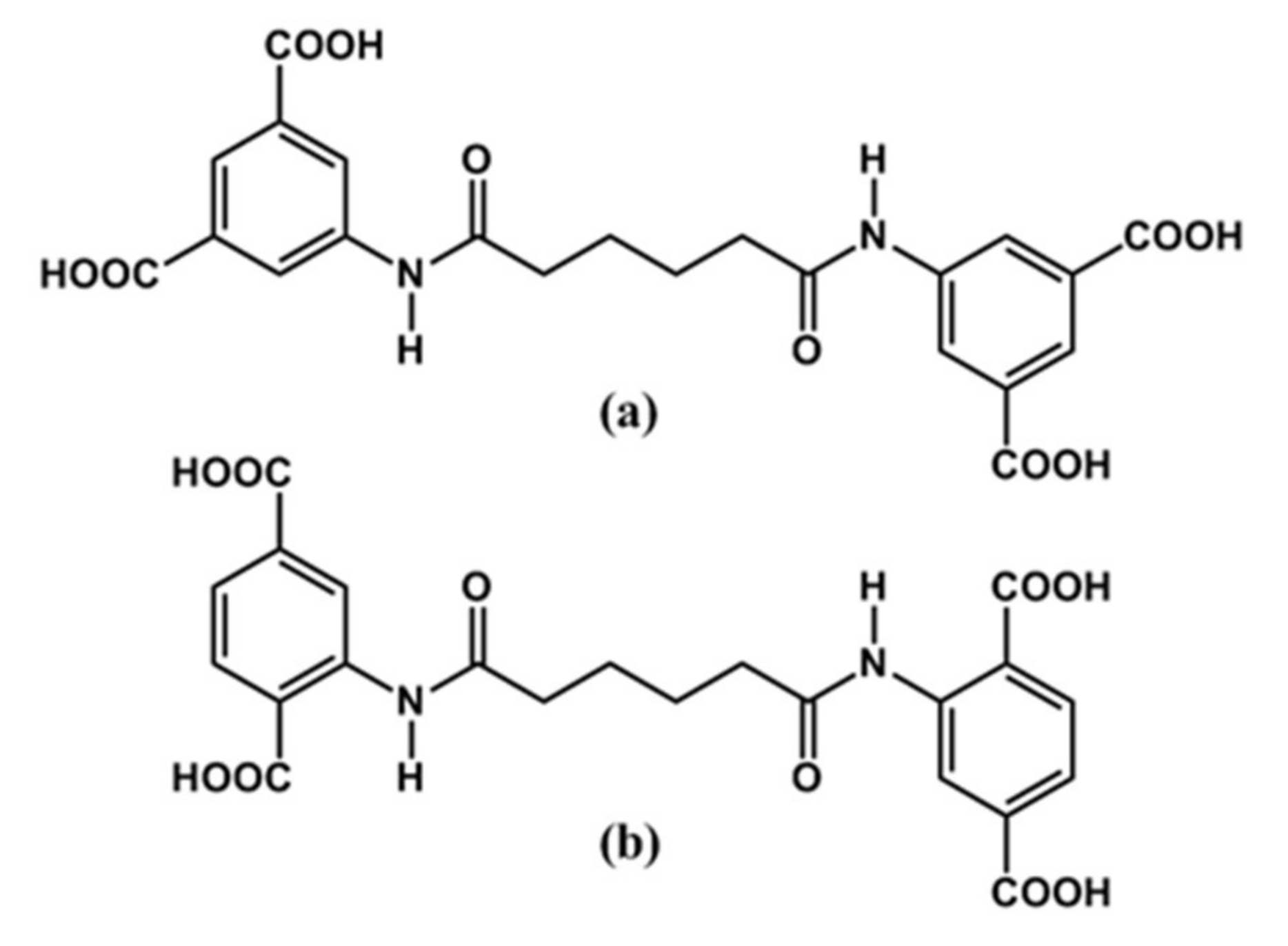
2.3.1. Bis(3,5-dicarboxyphenyl)adipoamide, H4L1
2.3.2. Bis(2,5-dicarboxyphenyl)adipoamide, H4L2
2.3.3. {[Mg2(L1)(H2O)2]·2EtOH.3H2O}n, 1
2.3.4. [Mg2(L1)(H2O)8]n, 2
2.3.5. {[Mg2(L2)(H2O)6]·H2O}n, 3
2.4. X-ray Crystallography
3. Results and Discussion
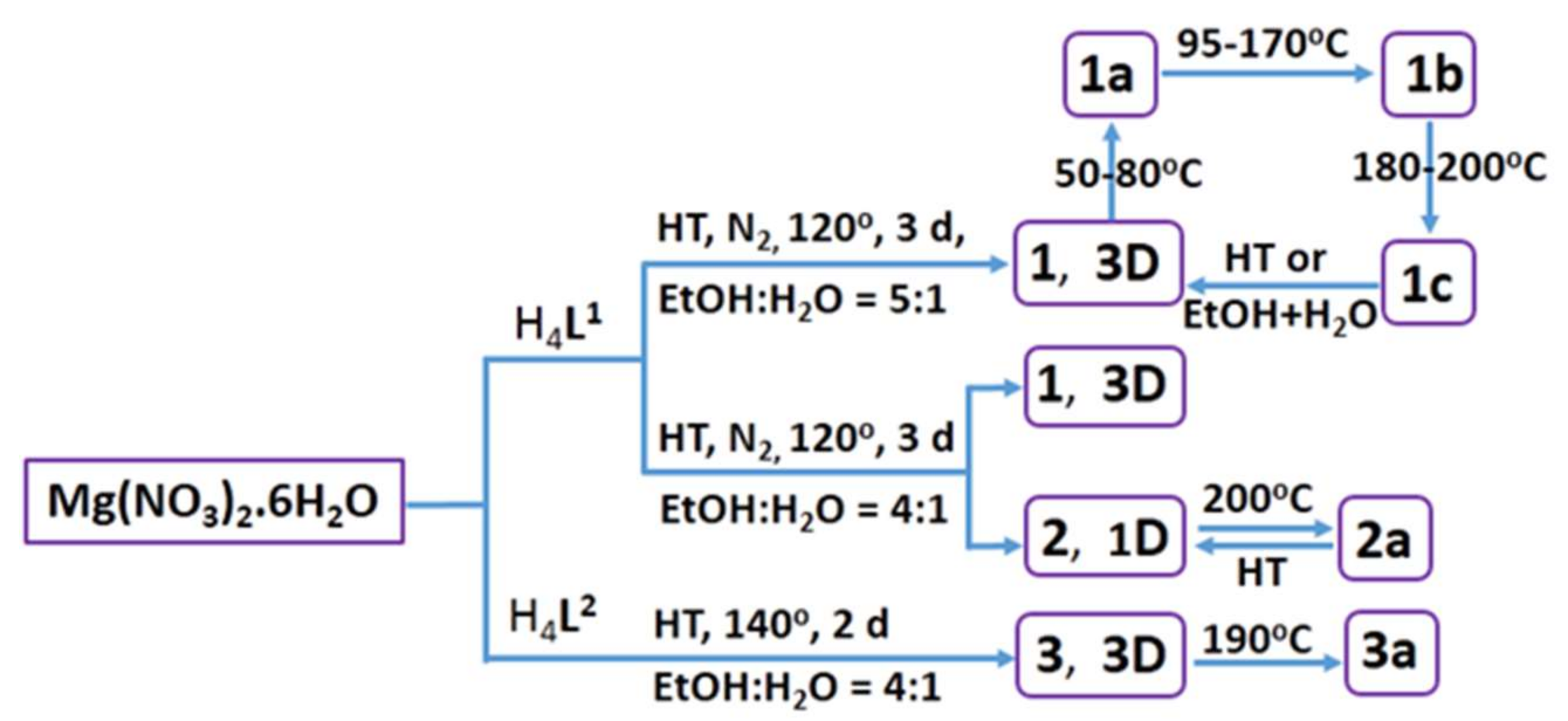
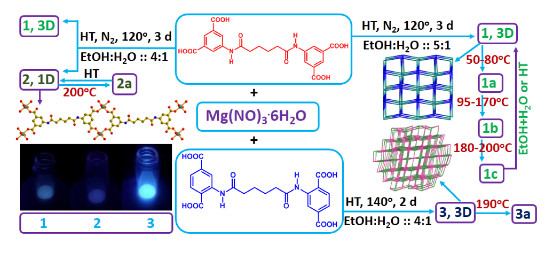
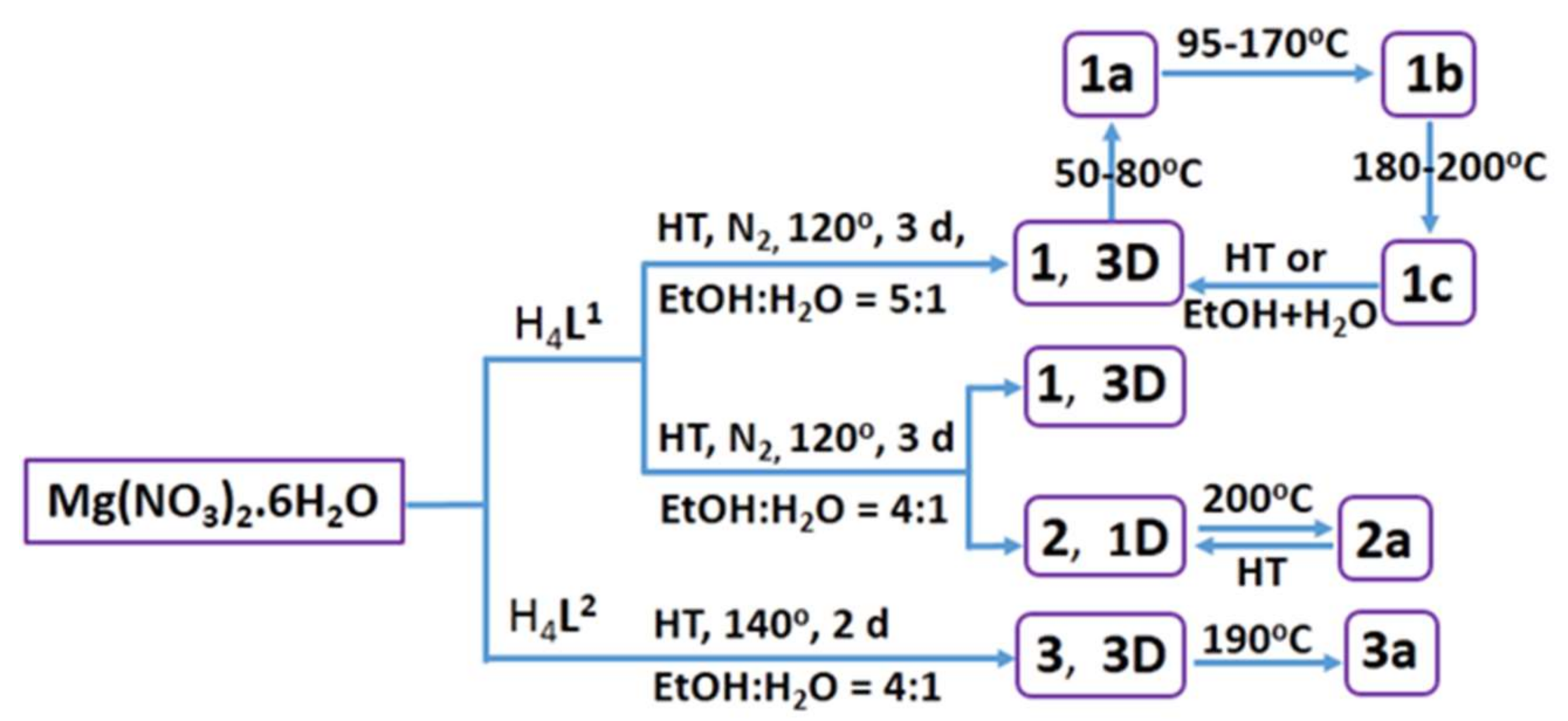
3.1. Synthesis
3.2. Structrual Descriptions
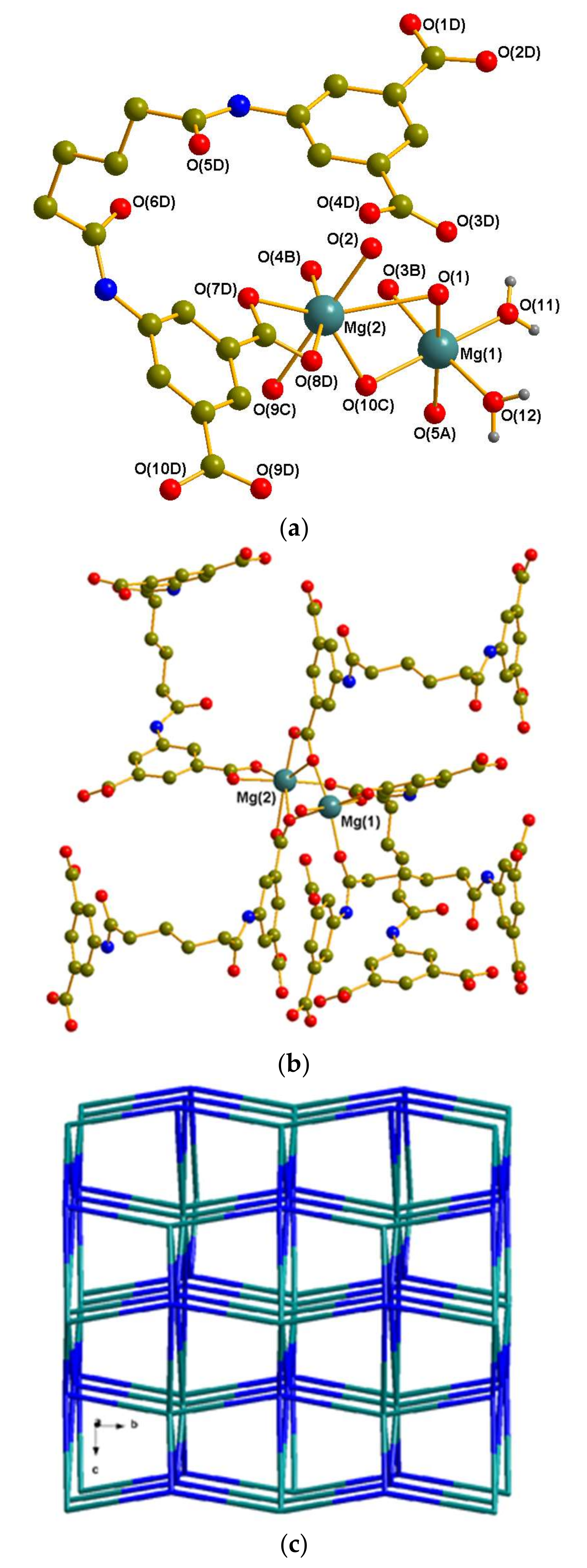
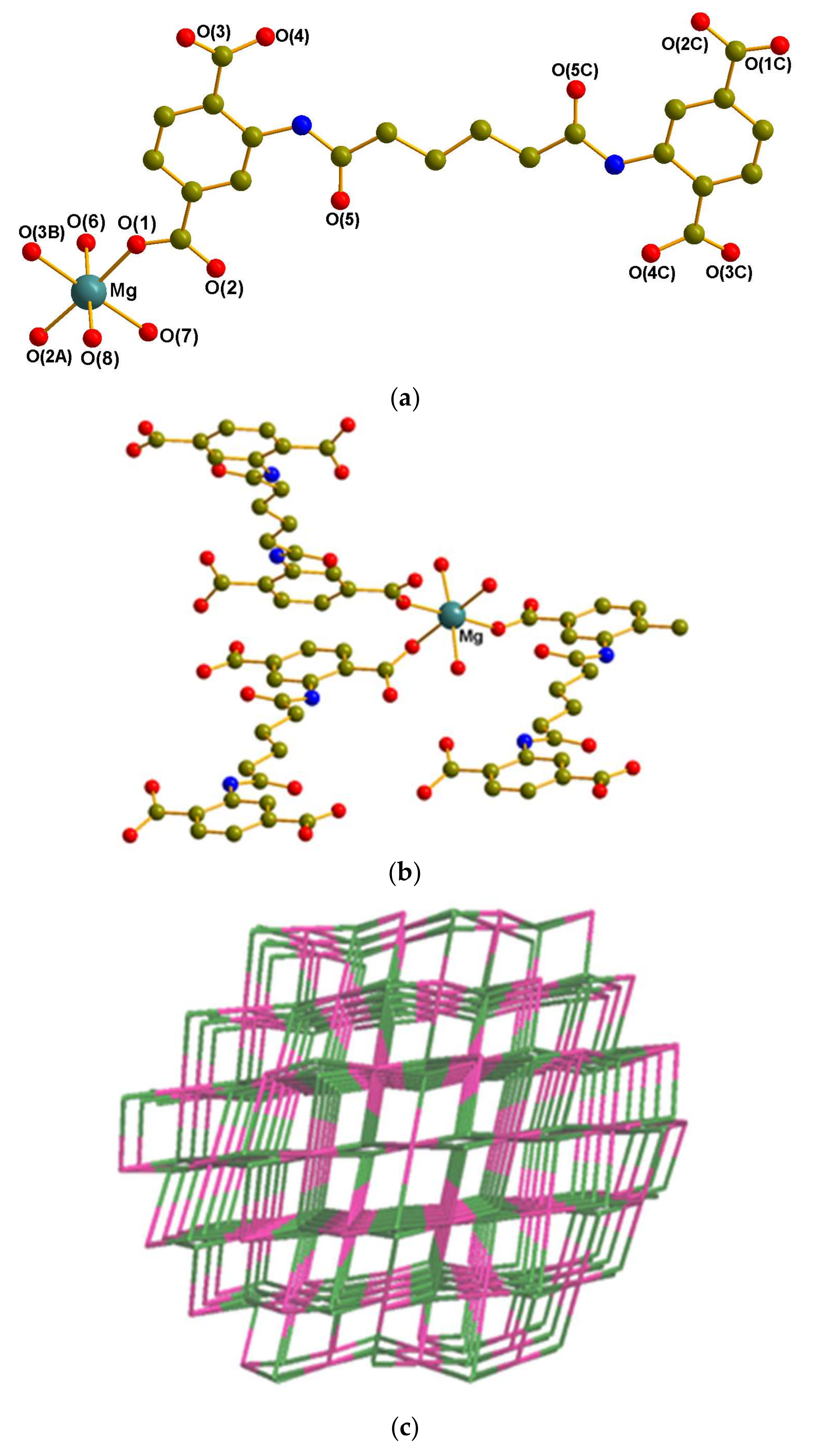
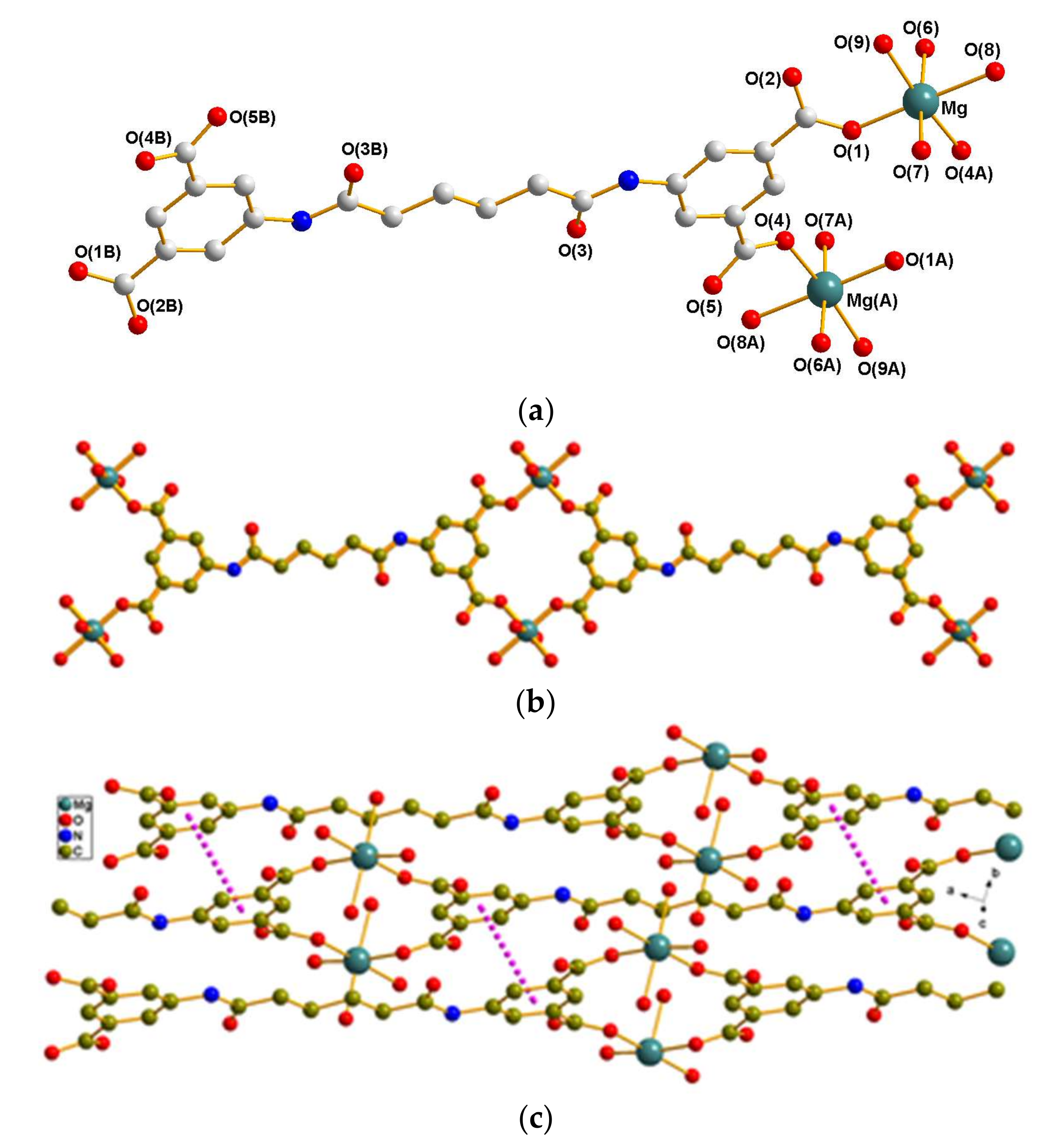
3.2.1. Crystal Structure of 1
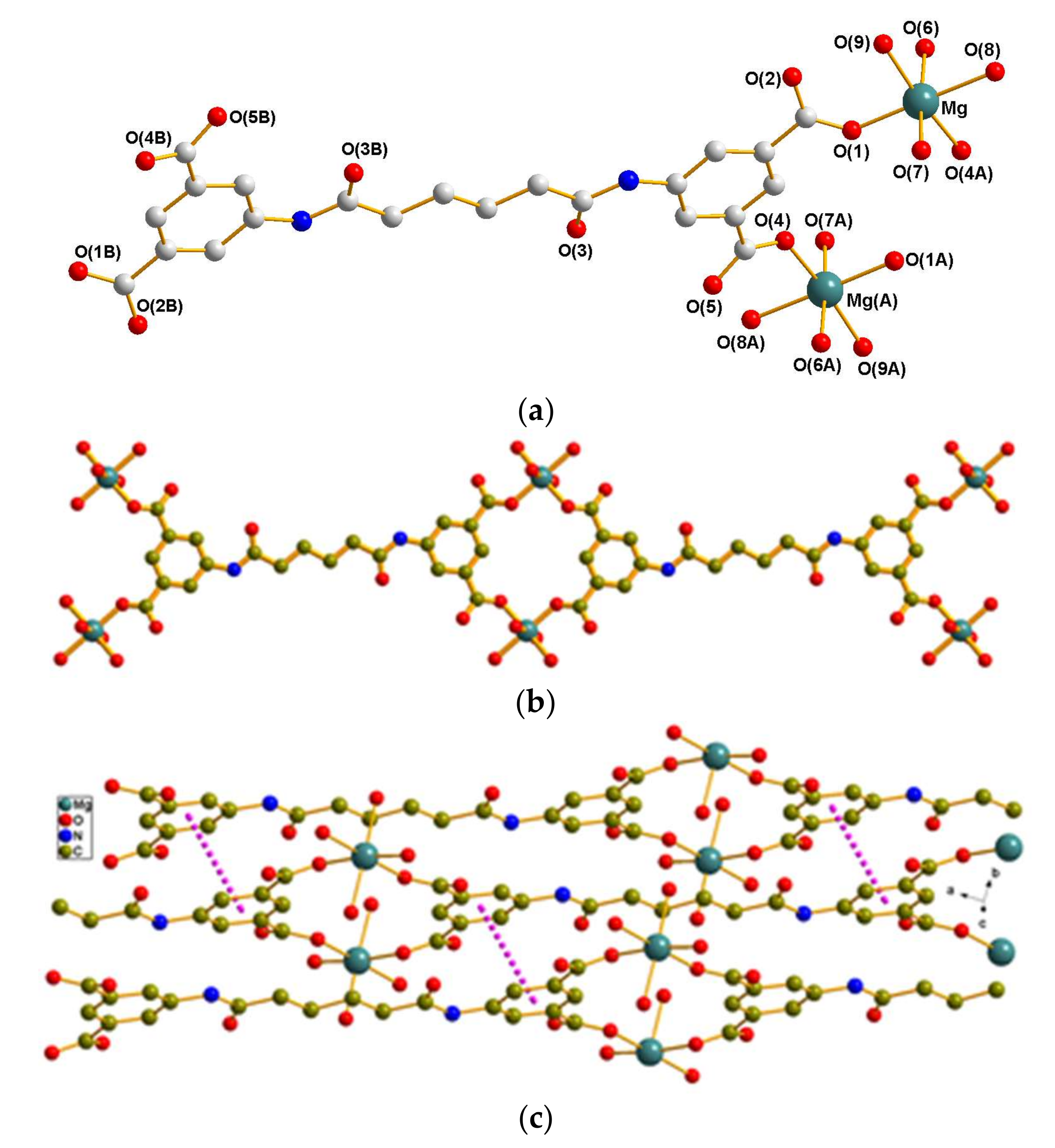
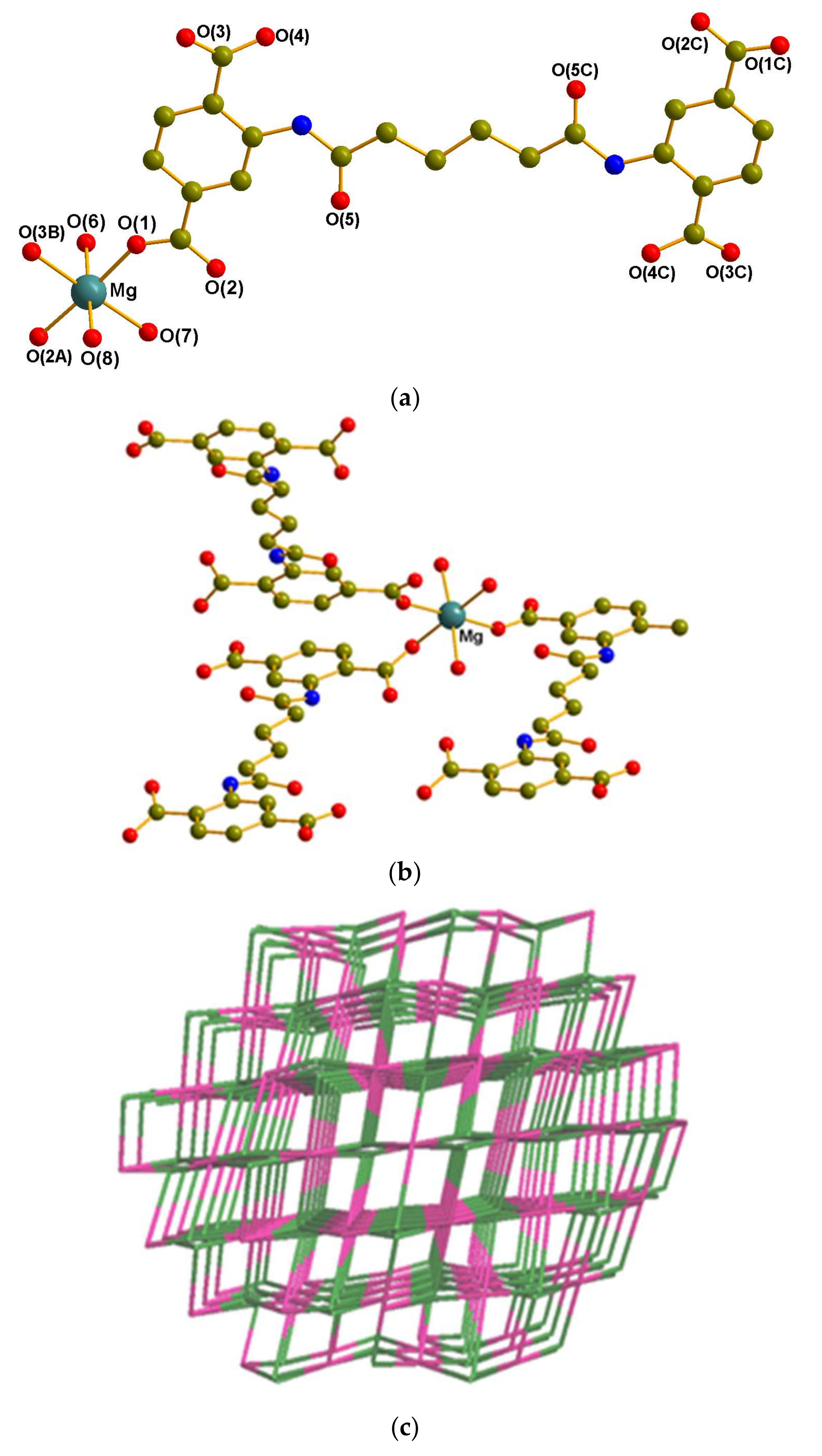
3.2.2. Crystal Structure of 2
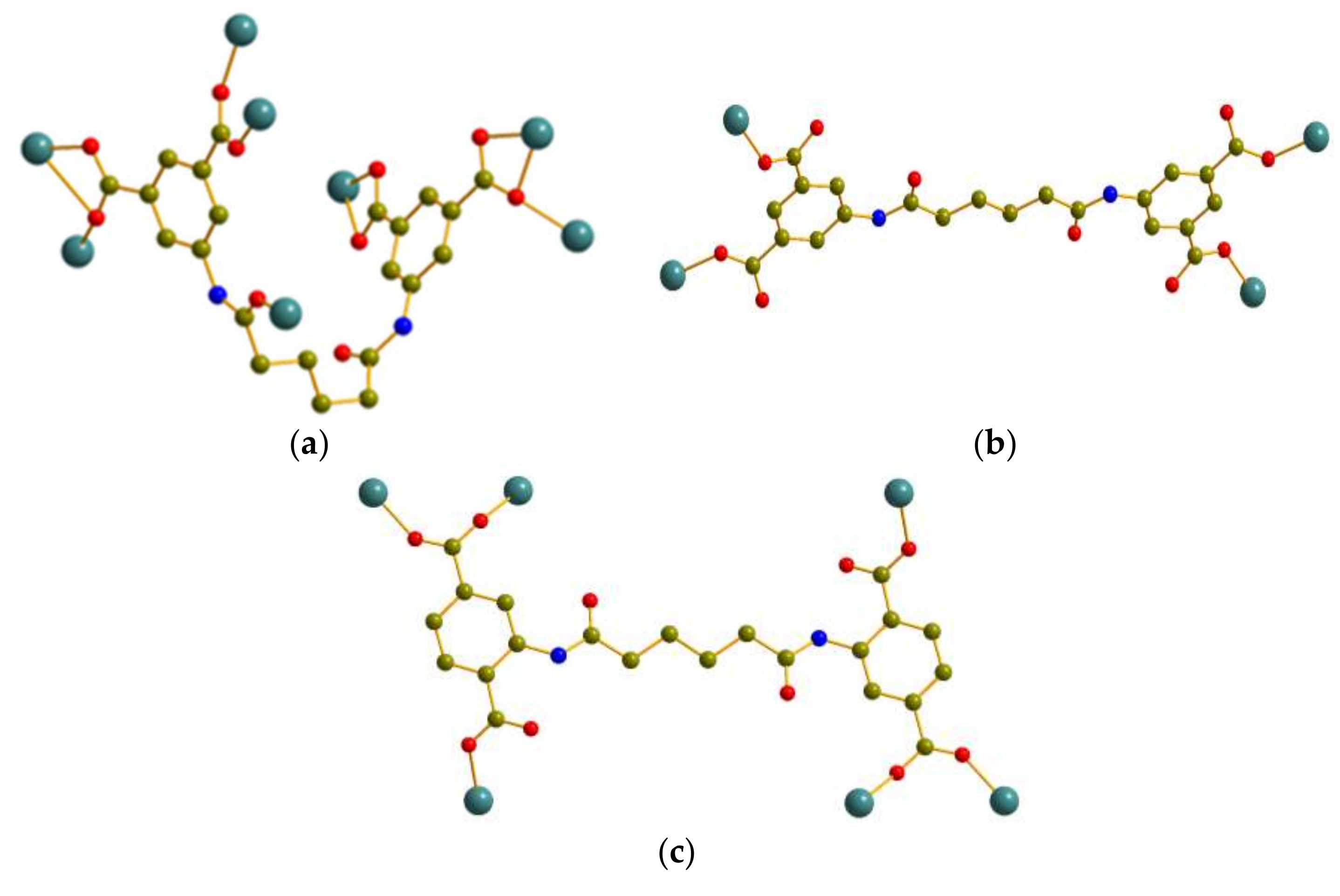
3.2.3. Crystal Structure of 3
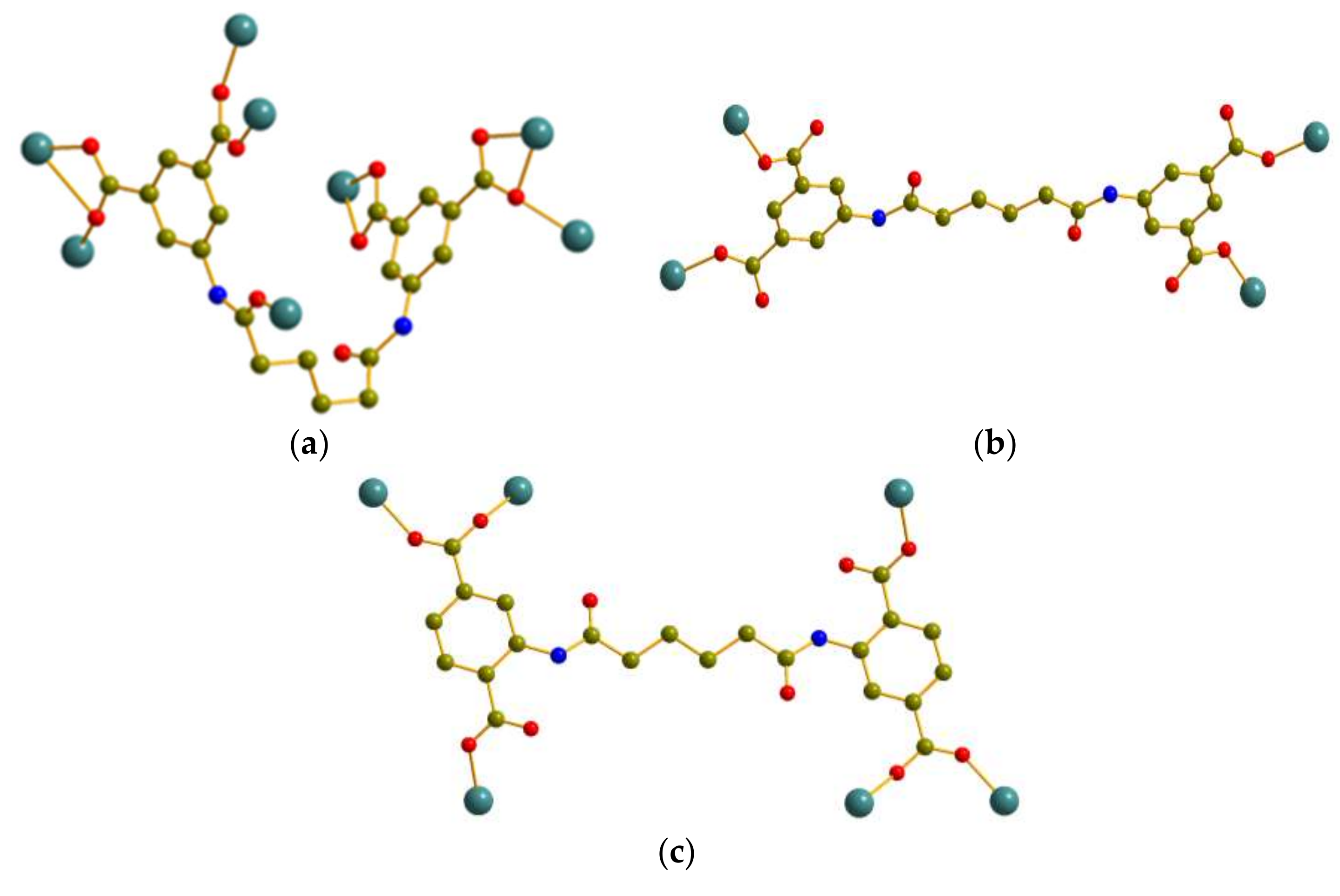
3.3. Coordination Modes and Ligand Conformation
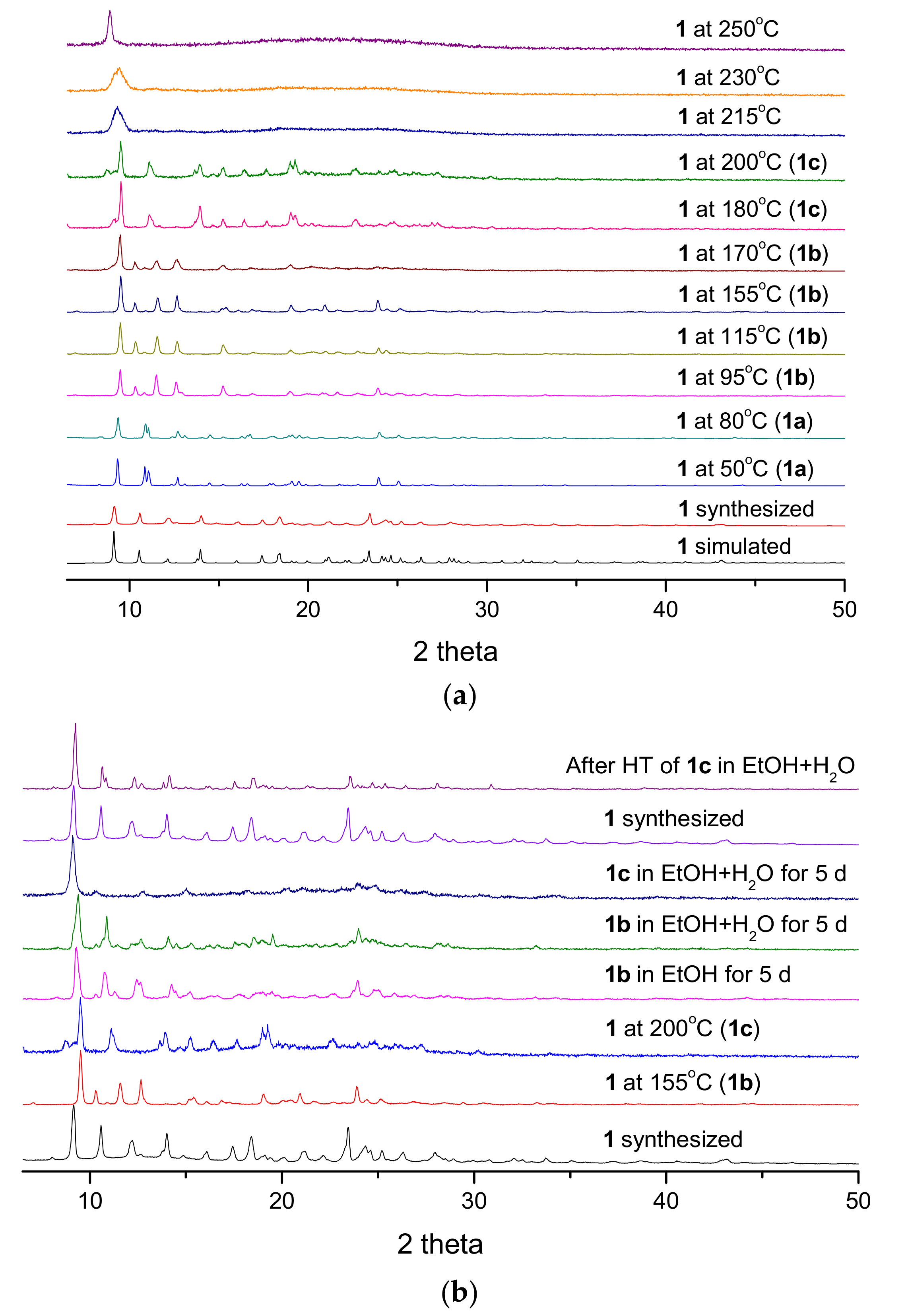
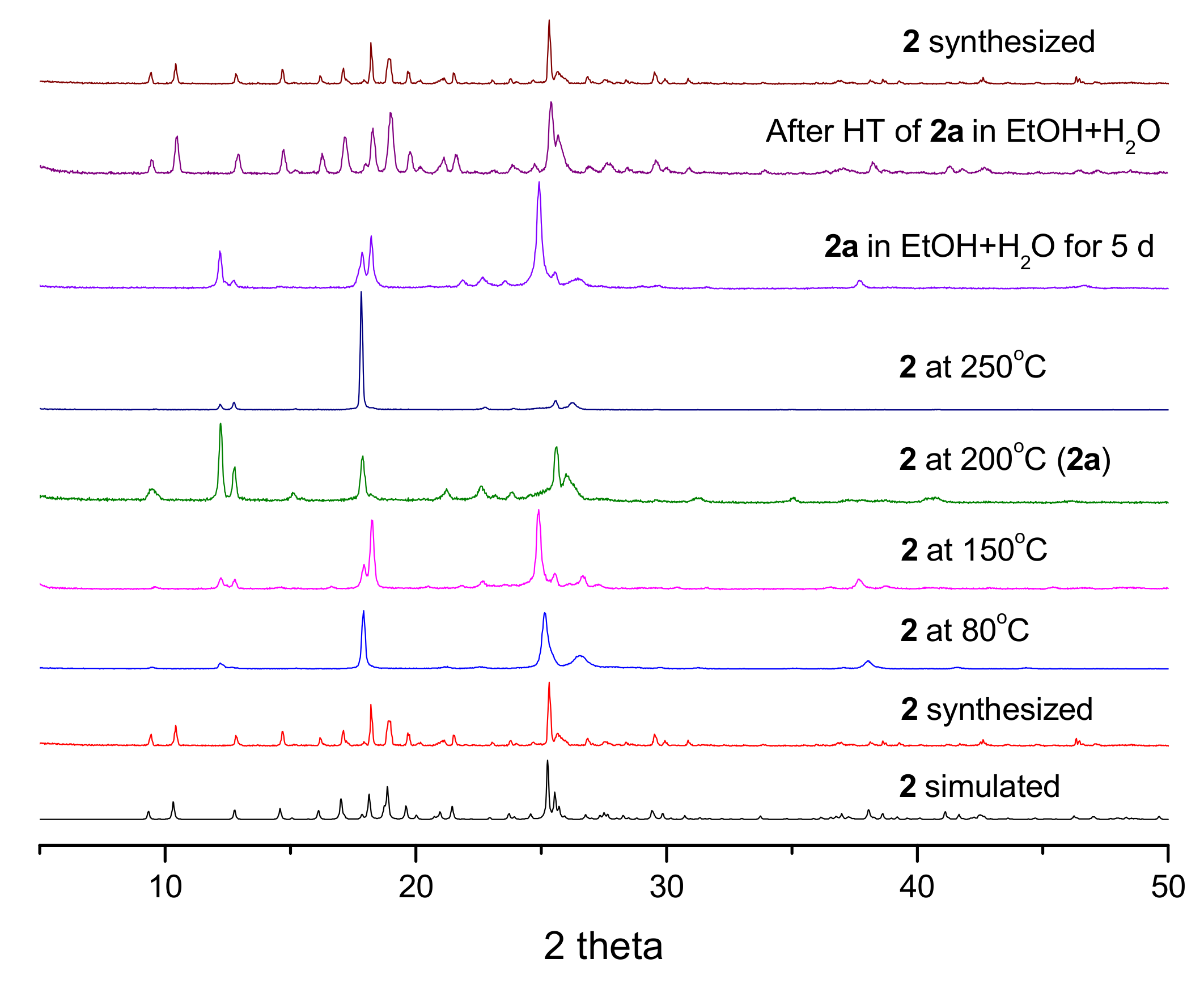
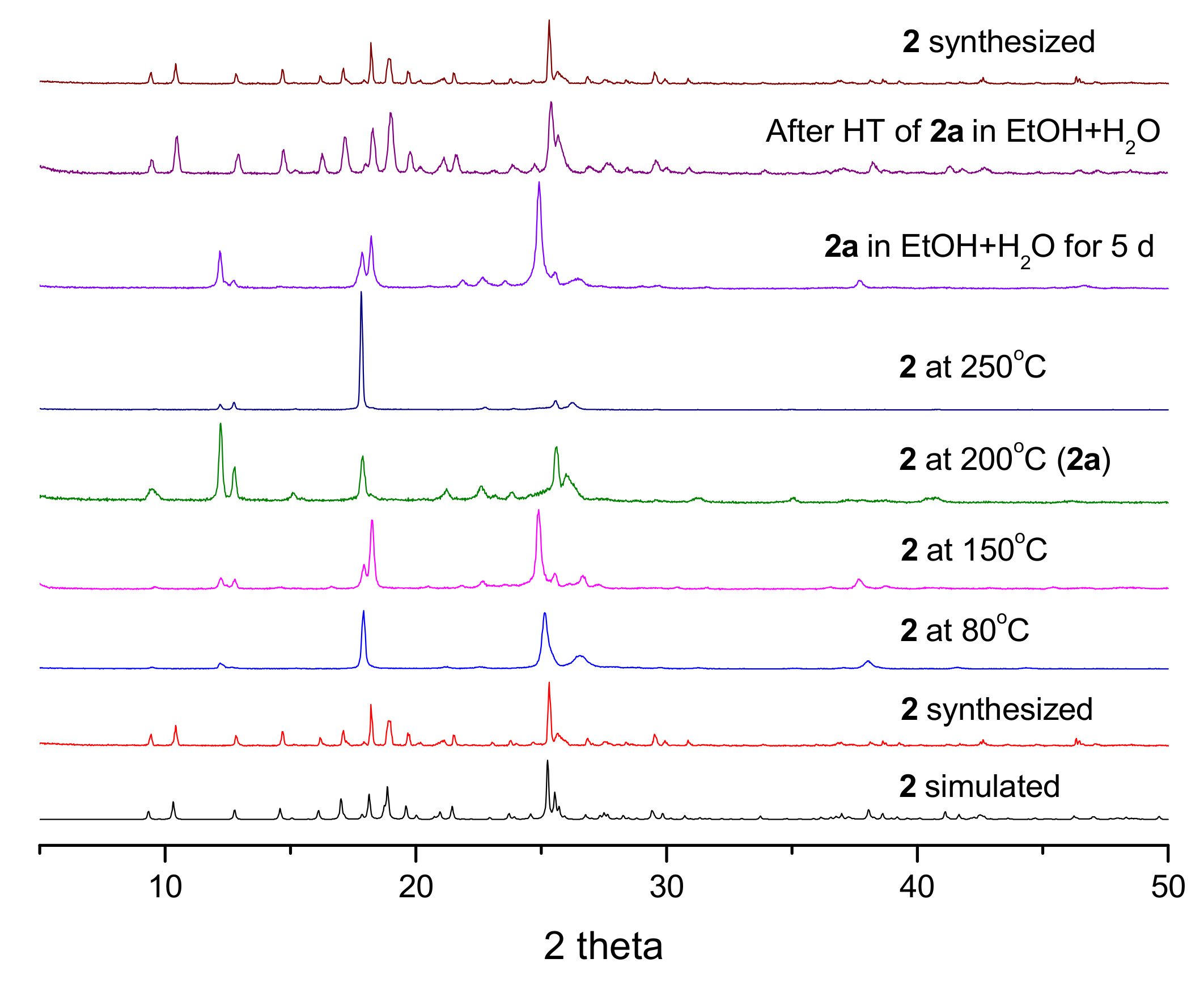
3.4. PXRD Patterns and Thermogravimetric Analysis
3.5. Structural Transformation
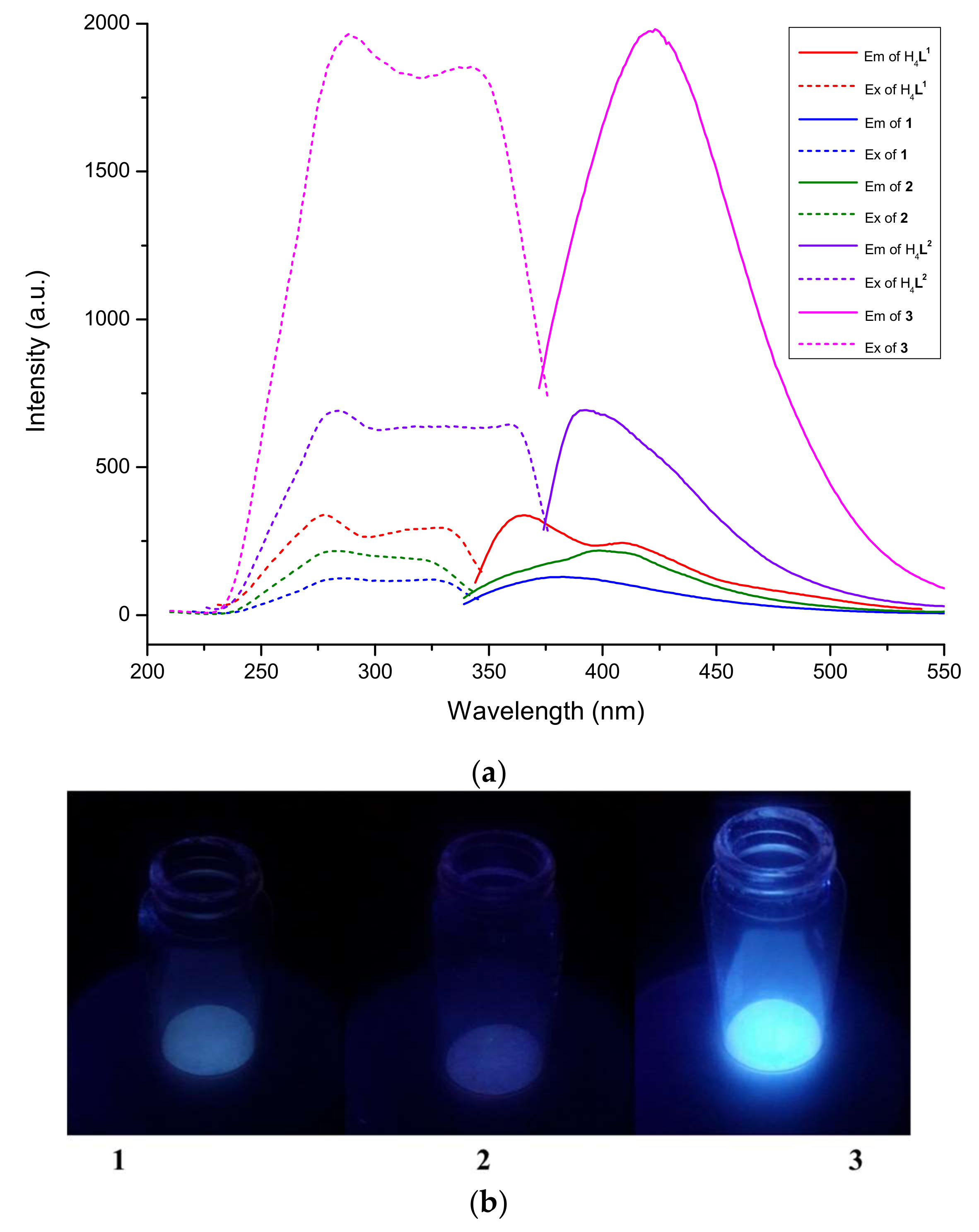
3.6. Absorption and Luminescence Properties
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tiekink, E.R.; Vittal, J. Frontiers in Crystal Engineering; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Batten, S.R.; Neville, S.M.; Turner, D.R. Coordination Polymers: Design, Analysis and Application; Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar]
- MacGillivray, L.R. Metal-Organic Frameworks: Design and Application; John Wiley & Sons: New York, NY, USA, 2010. [Google Scholar]
- Schröder, M. Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis; Springer: New York, NY, USA, 2010. [Google Scholar]
- Getman, R.B.; Bae, Y.S.; Wilmer, C.E.; Snurr, R.Q. Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal-organic frameworks. Chem. Rev. 2012, 112, 703–723. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Hupp, J.T. Rational design, synthesis, purification, and activation of metal-organic framework materials. Acc. Chem. Res. 2010, 43, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Thapa, K.B.; Chen, J.-D. Crystal engineering of coordination polymers containing flexible bis-pyridyl-bis-amide ligands. CrystEngComm 2015, 17, 4611–4626. [Google Scholar] [CrossRef]
- Blake, A.J.; Champness, N.R.; Cooke, P.A.; Nicolson, J.E.B.; Wilson, C. Multi-modal bridging ligands; effects of ligand functionality, anion and crystallisation solvent in silver(I) co-ordination polymers . J. Chem. Soc. Dalton Trans. 2000, 3811–3819. [Google Scholar] [CrossRef]
- Vittal, J.J. Supramolecular structural transformations involving coordination polymers in the solid state. Coord. Chem. Rev. 2007, 251, 1781–1795. [Google Scholar] [CrossRef]
- Kole, G.K.; Vittal, J.J. Solid-state reactivity and structural transformations involving coordination polymers. Chem. Soc. Rev. 2013, 42, 1755–1775. [Google Scholar] [CrossRef] [PubMed]
- Horike, S.; Matsuda, R.; Tanaka, D.; Mizuno, M.; Endo, K.; Kitagawa, S. Immobilization of sodium ions on the pore surface of a porous coordination polymer. J. Am. Chem. Soc. 2006, 128, 4222–4223. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guo; Liu, B.; Chen, W.-T.; Huang, J.-S. A novel 2-d honeycomb-like lithium coordination polymer containing 42-membered rings. Cryst. Growth Des. 2005, 5, 841–843. [Google Scholar] [CrossRef]
- Senthil Raja, D.; Lin, P.C.; Liu, W.R.; Zhan, J.X.; Fu, X.Y.; Lin, C.H. Multidimensional (0D to 3D) alkaline-earth metal diphosphonates: Synthesis, structural diversity, and luminescence properties. Inorg. Chem. 2015, 54, 4268–4278. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.A.; Boissonnault, J.A.; Cirera, J.; Gulland, R.; Paesani, F.; Cohen, S.M. Chemically crosslinked isoreticular metal-organic frameworks. Chem. Commun. 2013, 49, 3200–3202. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.-Y.; Si, C.-D.; Fan, Y.; Hu, D.-C.; Yao, X.-Q.; Yang, Y.-X.; Liu, J.-C. Effect of n-donor ligands and metal ions on the coordination polymers based on a semirigid carboxylic acid ligand: Structures analysis, magnetic properties, and photoluminescence. Cryst. Growth Des. 2016, 16, 2062–2073. [Google Scholar] [CrossRef]
- Gao, C.-Y.; Wang, D.; Li, J.-P.; Wang, Y.; Yang, W. Assemblies of metal–organic frameworks based on a tetrapodal linker for luminescence sensing of tetrahydrofuran. CrystEngComm 2016, 18, 2857–2863. [Google Scholar] [CrossRef]
- Zang, S.Q.; Liang, R.; Fan, Y.J.; Hou, H.W.; Mak, T.C. Divalent zinc and cadmium coordination polymers of a new flexible tetracarboxylate ligand: Syntheses, crystal structures and properties. Dalton Trans. 2010, 39, 8022–8032. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Yu, C.; Li, Y.; Lah, M.S. A 3-dimensional coordination polymer with a rare lonsdaleite topology constructed from a tetrahedral ligand. CrystEngComm 2012, 14, 7174–7177. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xing, F.; Bai, Y.-L.; Zhao, Y.; Li, M.-X.; Zhu, S. Synthesis and structure of semirigid tetracarboxylate copper(II) porous coordination polymers and their versatile high-efficiency catalytic dye degradation in neutral aqueous solution. Cryst. Growth Des. 2016, 16, 2277–2288. [Google Scholar] [CrossRef]
- Gai, Y.L.; Jiang, F.L.; Chen, L.; Bu, Y.; Su, K.Z.; Al-Thabaiti, S.A.; Hong, M.C. Photophysical studies of europium coordination polymers based on a tetracarboxylate ligand. Inorg. Chem. 2013, 52, 7658–7665. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Huang, C.; Xue, X.; Li, M.; Hou, H.; Fan, Y. Ni(II) coordination polymers constructed from the flexible tetracarboxylic acid and different n-donor ligands: Structural diversity and catalytic activity. Cryst. Growth Des. 2015, 15, 4507–4517. [Google Scholar] [CrossRef]
- Li, X.; Jiang, F.; Chen, L.; Wu, M.; Lu, S.; Pang, J.; Zhou, K.; Chen, X.; Hong, M. Two microporous metal–organic frameworks constructed from trinuclear cobalt(II) and cadmium(II) cluster subunits. CrystEngComm 2016, 18, 2239–2243. [Google Scholar] [CrossRef]
- Zhang, X.; Hou, L.; Liu, B.; Cui, L.; Wang, Y.-Y.; Wu, B. Syntheses, structures, and luminescent properties of six new zinc(II) coordination polymers constructed by flexible tetracarboxylate and various pyridine ligands. Cryst. Growth Des. 2013, 13, 3177–3187. [Google Scholar] [CrossRef]
- Li, Q.-Q.; Zhang, W.-Q.; Ren, C.-Y.; Fan, Y.-P.; Li, J.-L.; Liu, P.; Wang, Y.-Y. Reaction-determined assemblies of 0D to 3D complexes: Structural diversities and luminescence properties. CrystEngComm 2016, 18, 3358–3371. [Google Scholar] [CrossRef]
- Kirillov, A.M.; Karabach, Y.Y.; Kirillova, M.V.; Haukka, M.; Pombeiro, A.J.L. Topologically unique 2D heterometallic CuII/Mg coordination polymer: Synthesis, structural features, and catalytic use in alkane hydrocarboxylation. Cryst. Growth Des. 2012, 12, 1069–1074. [Google Scholar] [CrossRef]
- Lin, Z.-J.; Han, L.-W.; Wu, D.-S.; Huang, Y.-B.; Cao, R. Structure versatility of coordination polymers constructed from a semirigid tetracarboxylate ligand: Syntheses, structures, and photoluminescent properties. Cryst. Growth Des. 2013, 13, 255–263. [Google Scholar] [CrossRef]
- Fromm, K.M. Coordination polymer networks with s-block metal ions. Coord. Chem. Rev. 2008, 252, 856–885. [Google Scholar] [CrossRef]
- Zhai, Q.-G.; Bu, X.; Zhao, X.; Mao, C.; Bu, F.; Chen, X.; Feng, P. Advancing magnesium–organic porous materials through new magnesium cluster chemistry. Cryst. Growth Des. 2016, 16, 1261–1267. [Google Scholar] [CrossRef]
- Banerjee, D.; Parise, J.B. Recent advances in s-block metal carboxylate networks. Cryst. Growth Des. 2011, 11, 4704–4720. [Google Scholar] [CrossRef]
- Zhang, Q.; Hao, H.; Zhang, H.; Wang, S.; Jin, J.; Sun, D. Two enantiomorphic MgII/carboxylate coordination polymers produced from spontaneous resolution with an achiral bipyridyl dicarboxylate ligand. Eur. J. Inorg. Chem. 2013, 2013, 1123–1126. [Google Scholar] [CrossRef]
- Chen, R.-Y.; Tian, D.; Hu, T.-L.; Chang, Z. Two Mg(II) coordination polymers based on the flexible carboxylic ligands: Synthesis, crystal structures, luminescent and adsorption properties. Inorg. Chem. Commun. 2014, 49, 131–135. [Google Scholar] [CrossRef]
- XSCANS, Release 2.21; Siemens Energy and Automation Inc.: Madison, WI, USA, 1995.
- SMART/SAINT/ASTRO, Release 4.03; Siemens Energy & Automation, Inc.: Madison, WI, USA, 1995.
- SHELXTL 5.10; Bruker Analytical X-ray Instruments Inc.: Karlsruche, Germany, 1997.
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. [Google Scholar] [CrossRef]
- Spek, A.L. Single-crystal structure validation with the programplaton. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Cheng, P.-C.; Kuo, P.-T.; Liao, Y.-H.; Xie, M.-Y.; Hsu, W.; Chen, J.-D. Ligand-isomerism controlled structural diversity of Zn(II) and Cd(II) coordination polymers from mixed dipyridyladipoamide and benzenedicarboxylate ligands. Cryst. Growth Des. 2012, 13, 623–632. [Google Scholar] [CrossRef]
- Cheng, P.-C.; Kuo, P.-T.; Xie, M.-Y.; Hsu, W.; Chen, J.-D. Structure-directing roles of auxiliary polycarboxylate ligands in the formation of Zn(II) and Cd(II) coordination polymers based on a flexible N,N′-di(3-pyridyl)dodecanediamide. CrystEngComm 2013, 15, 6264–6272. [Google Scholar] [CrossRef]
- Song, Y.; Feng, M.-L.; Wu, Z.-F.; Huang, X.-Y. Solvent-assisted construction of diverse Mg-TDC coordination polymers. CrystEngComm 2015, 17, 1348–1357. [Google Scholar] [CrossRef]
- Leong, W.L.; Batabyal, S.K.; Kasapis, S.; Vittal, J.J. Influence of chiral ligands on the gel formation of a Mg(II) coordination polymer. CrystEngComm 2015, 17, 8011–8014. [Google Scholar] [CrossRef]
- Turro, N.J. Modern Molecular Photochemistry; University Science Books: Sausalito, CA, USA, 1991. [Google Scholar]
- Cui, Y.; Yue, Y.; Qian, G.; Chen, B. Luminescent functional metal-organic frameworks. Chem. Rev. 2012, 112, 1126–1162. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Li, G.; Hua, J.; Shi, Z.; Feng, S. Syntheses, topological structures and properties of six metal–organic frameworks constructed from a flexible tetracarboxylate ligand. CrystEngComm 2015, 17, 3162–3170. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Formula | C26H38Mg2N2O17 | C22H32Mg2N2O18 | C22H30Mg2N2O17 |
| Fw | 699.20 | 661.11 | 643.10 |
| crystal system | Orthorhombic | Monoclinic | Monoclinic |
| space group | Pna21 | P21/n | P21/c |
| a, Å | 16.7546 (2) | 10.4098 (2) | 10.1719 (2) |
| b, Å | 14.7326 (2) | 7.4955 (1) | 9.5855 (2) |
| c, Å | 12.8347 (2) | 18.2183 (3) | 15.4358 (3) |
| α,° | 90 | 90 | 90 |
| β,° | 90 | 96.671 (1) | 105.778 (1) |
| γ,° | 90 | 90 | 90 |
| V, Å3 | 3168.10 (8) | 1411.89 (4) | 1448.33 (5) |
| Z | 4 | 2 | 2 |
| Dcalc, g/cm3 | 1.466 | 1.555 | 1.475 |
| μ(Mo Kα), mm−1 | 0.157 | 0.174 | 0.165 |
| Range (2θ) for data collection, deg | 1.841 ≤ 2θ ≤ 28.314 | 2.152 ≤ 2θ ≤ 28.294 | 2.742 ≤ 2θ ≤ 26.00 |
| independent reflections | 7742 [R (int) = 0.0433] | 3490 [R (int) = 0.0162] | 2845 [R (int) = 0.0314] |
| data/restraints/parameters | 7742/9/436 | 3490/0/223 | 2845/0/198 |
| quality-of-fit indicator c | 1.057 | 1.059 | 1.047 |
| final R indices [I > 2σ(I)] a, b | R1 = 0.0486, wR2 = 0.1242, | R1 = 0.0399, wR2 = 0.1125 | R1 = 0.0520, wR2 = 0.1456 |
| R indices (all data) | R1 = 0.0671, wR2 = 0.1366 | R1 = 0.0425, wR2 = 0.1148 | R1 = 0.0670, wR2 = 0.1586 |
| Compound | Excitation λ(ex) nm | Emission λ(em) nm |
|---|---|---|
| H4L1 | 278, 316, 328 | 365, 408 |
| 1 | 287, 324 | 384 |
| 2 | 283 | 399 |
| H4L2 | 284, 331, 369 | 391 |
| 3 | 289, 342 | 423 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thapa, K.B.; Yang, X.-K.; Chen, J.-D. Mg(II) Coordination Polymers Based on Flexible Isomeric Tetracarboxylate Ligands: Syntheses, Structures, Structural Transformation and Luminescent Properties. Polymers 2018, 10, 371. https://doi.org/10.3390/polym10040371
Thapa KB, Yang X-K, Chen J-D. Mg(II) Coordination Polymers Based on Flexible Isomeric Tetracarboxylate Ligands: Syntheses, Structures, Structural Transformation and Luminescent Properties. Polymers. 2018; 10(4):371. https://doi.org/10.3390/polym10040371
Chicago/Turabian StyleThapa, Kedar Bahadur, Xiang-Kai Yang, and Jhy-Der Chen. 2018. "Mg(II) Coordination Polymers Based on Flexible Isomeric Tetracarboxylate Ligands: Syntheses, Structures, Structural Transformation and Luminescent Properties" Polymers 10, no. 4: 371. https://doi.org/10.3390/polym10040371