Effect of Bidispersity on Dynamics of Confined Polymer Films


1
Department of Fire Command, China People’s Police University, Langfang 065000, China
2
Xinjiang Laboratory of Phase Transitions and Microstructures in Condensed Matter Physics, College of Physical Science and Technology, Yili Normal University, Yining 835000, China
3
State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Science, Changchun 130022, China
4
School of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, China
*
Authors to whom correspondence should be addressed.
Polymers 2018, 10(12), 1327; https://doi.org/10.3390/polym10121327
Submission received: 6 November 2018
/
Revised: 26 November 2018
/
Accepted: 28 November 2018
/
Published: 30 November 2018
(This article belongs to the Special Issue Theory and Simulations of Entangled Polymers)
Abstract
:Using Monte Carlo simulations, we studied the effect of bidispersity on the dynamics of polymer films capped between two neutral walls, where we chose three representative compositions for bidispersed polymer films. Our results demonstrate that the characteristic entanglement length is an important parameter to clarify the effect of the bidispersity on the dynamics of polymer films. For the short chains, shorter than the characteristic entanglement length, the average number of near-neighboring particles increases with the decrease of the film thickness and limits the diffusivity of the short chains, which is independent of the film compositions. However, the dynamics of the long chains, of which is above the characteristic entanglement length, is determined by the film’s composition. In our previous paper, we inferred from the structures and entanglements of the bidisperse system with short and long chains that the constraint release contributes significantly to the relaxation mechanism of long chains. By calculating the self-diffusion coefficient of long chains, we confirmed this prediction that, with a lower weight fraction of long chains, the self-diffusion coefficient of long chains decreases slowly with the decrease of the film thickness, which is similar to that of short chains. With a higher weight fraction of long chains, the competition between the disentanglement and the increased in the local degree of confinement which resulted in the self-diffusion coefficient of long chains varying non-monotonically with the film thickness. Furthermore, for the bidisperse system with long and long chains, the diffusivity of long chains was not affected by the constraint release, which varied nonmonotonically with the decrease of the film thickness due to the competition between the disentanglement and the enhanced confinement. Herein, compared with the previous work, we completely clarified the relationship between the structures and dynamics for three representative compositions of bidisperse polymer films, which contains all possible cases for bidisperse systems. Our work not only establishes a unified understanding of the dependency of dynamics on the bidispersity of polymer films, but also helps to understand the case of polydispersity, which can provide computational supports for various applications for polymer films.
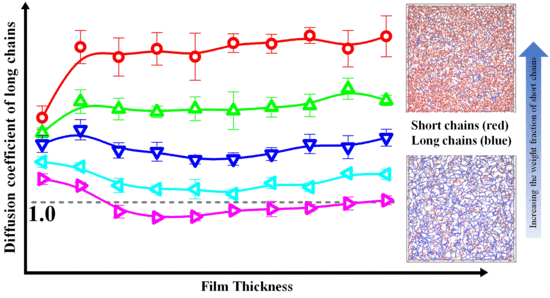
1. Introduction
The dynamic properties of thin polymer films are the key physical factors for their widespread technological applications such as surface coatings, adhesives, lubricants, composite materials, etc. [1,2,3,4,5,6], and therefore have received intense interests in the past decades. However, much of this attention has focused on elucidating the changes in the monodisperse polymer films under different types of confinement [7,8,9,10,11,12,13,14,15,16,17]. The fundamental role that the inevitable existence of polydispersity in polymer films plays in their dynamic properties has not yet been well-understood.
Polydispersity is known to affect the structural and thermodynamic properties [18,19] and even the processing of polymer melts [20,21]. The tube model has given a specific description of the effects of polydispersity on the dynamics of bulk systems [22]. For monodisperse polymer melts, the relaxation time of the surrounding chains determines the lifetime of an entanglement that confines a chain inside a tube, which means that chains crawl out the tube like a slithering snake, known as “reptation” [22]. In contrast, for polydisperse polymer melts, due to the fast relaxation of short chains, the entanglements formed by short and long chains can quickly dissolve, which results in the lifetime dependence of the entanglements on the involved chain length [23]. As a consequence, because of the possible motion in a direction perpendicular to its contour line [24], the long chains relax more quickly than that of a pure reptation process [23].
However, a unified understanding of the dependency of the dynamic properties of polymer films on polydispersity has not been achieved. The experiments investigating the effects of bidispersity and tridispersity on a wall slip of polymer films have indicated that short chains can enhance the slip of entangled chains on non-wetting surfaces, which may come from the effects of chain end groups or short chain enrichment on the effective friction coefficient [25,26,27]. In fact, simulation studies have also revealed a significant slip for short chains [28], which indicates that the simulations provide an excellent tool to obtain a molecular scale understanding. The dynamic Monte Carlo study has found that polydispersity can reduce the confinement effects on the orientation and relaxation time of polymer chains, where the polydispersity can provide more degrees of freedom for chain configurations [29], In our previous work, we have shown that short chains can significantly affect the local situations of entanglements, and predicted the relaxation mechanisms of long chains based on constraint release dynamics [30], Actually, the entanglement plays a key role in the dynamics properties of polymers. Nevertheless, a link between the changes of entanglements and the changes of dynamics of polymer films induced by polydispersity has hardly been explicitly established in previous studies.
In this work, using Monte Carlo simulations, we constructed the simplest case of polydispersity, a bidisperse mixture, to investigate the effect of polydispersity on the dynamics of confined polymer films, which can represent the dynamics of polymer chains in bulk melts as well as in confined cases [17,31,32,33]. We considered three model systems in our simulations: The first model system consisted of two short chains of which the molecular lengths were both below the characteristic entanglement length; the second one consisted of short chains of which the molecular length was below the characteristic entanglement length, and long chains of which the molecular length was above the characteristic entanglement length; the third one consisted of two long chains of which the molecular lengths were both above the characteristic entanglement length. In our previous work, we only calculated the structures and entanglements of the second model system [30]. However, in this work, we chose three representative compositions of bidisperse polymer films (containing all possible cases for bidisperse systems), thus we could completely clarify the relationship between the structures and dynamics for bidisperse polymer films. The degree of entanglement of polymers was extracted by a geometric analysis method (Z1) [34,35,36,37], which has been proven to be suitable for analyzing anisotropic samples [13,14,38], The remainder of this article is organized as follows: In Simulation Methods, we described the simulation methods and the corresponding simulation details. In Results and Discussion, we systematically investigated the effect of bidispersity on the dynamics of short and long polymers. Our simulations revealed the molecular mechanisms of the effect of bidispersity on the dynamics of the polymers in these confined systems and among which differences depend on the film composition. In Conclusions, we briefly summarized our results and provided some concluding remarks.
2. Model and Simulation Method
We constructed a coarse-grained computational model for polymer melts capped between two hard walls and performed the simulations using the lattice-based Monte Carlo model introduced by Shaffer (S-BFM); interested readers could read our previous papers and other references for more details [17,30,31,32,33]. In our model, each monomer on the polymer chains occupied a single site on the primary cubic lattice with a lattice constant ap = 1, and the bond lengths were allowed to fluctuate with the values of 1, 21/2, and 31/2. Polymer configurations could evolve through local displacements of one single monomer by forbidding double occupancy of the primary lattice sites and maintaining chain connectivity.
In this work, lengths are given in the unit of primary lattice spacing (ap), and times are given in the unit of attempted Monte Carlo steps (MCs) per monomer. We have demonstrated that the critical entanglement length from the unentangled to the entangled region in the bulk is Ne ≈ 33 by the Z1 algorithm in our previous study [17], which is agreement with previous works [31,32,39]. Hence, we chose the length of Ns = 10 and Nl = 20 for the first model system, Ns = 20 and Nl = 150 for the second one, and Ns = 150 and Nl = 300 for the third one, to cover both the unentangled and entangled regions for chains. We defined the weight fraction of the longer chains in the system as φl = Nlnl/(Nsns + Nlnl), where ns and nl are the numbers of the shorter and longer chains, respectively. We set φl as 16.7%, 33.3%, 50.0%, 66.7% and 83.3% (see Table 1 for details).
We put both the shorter and longer chains into a rectangular simulation box with dimensions of (Lx, Ly, Lz), where periodic boundary conditions were applied in the x and y directions. Moreover, we fixed two flat and impenetrable walls at z = 0 and Lz + 1 in the z direction, which were constructed of stationary particles (each particle occupies a single lattice site). Thus, we could consider the neutral walls by the excluded volume interaction between the monomers and the wall particles. In all cases, we set the fraction of occupied lattice sites as one-half, which has been shown to represent polymer melts in this model [39]. We adjusted the film thickness from 2 to 54 in our simulations (see Table 2 for details).
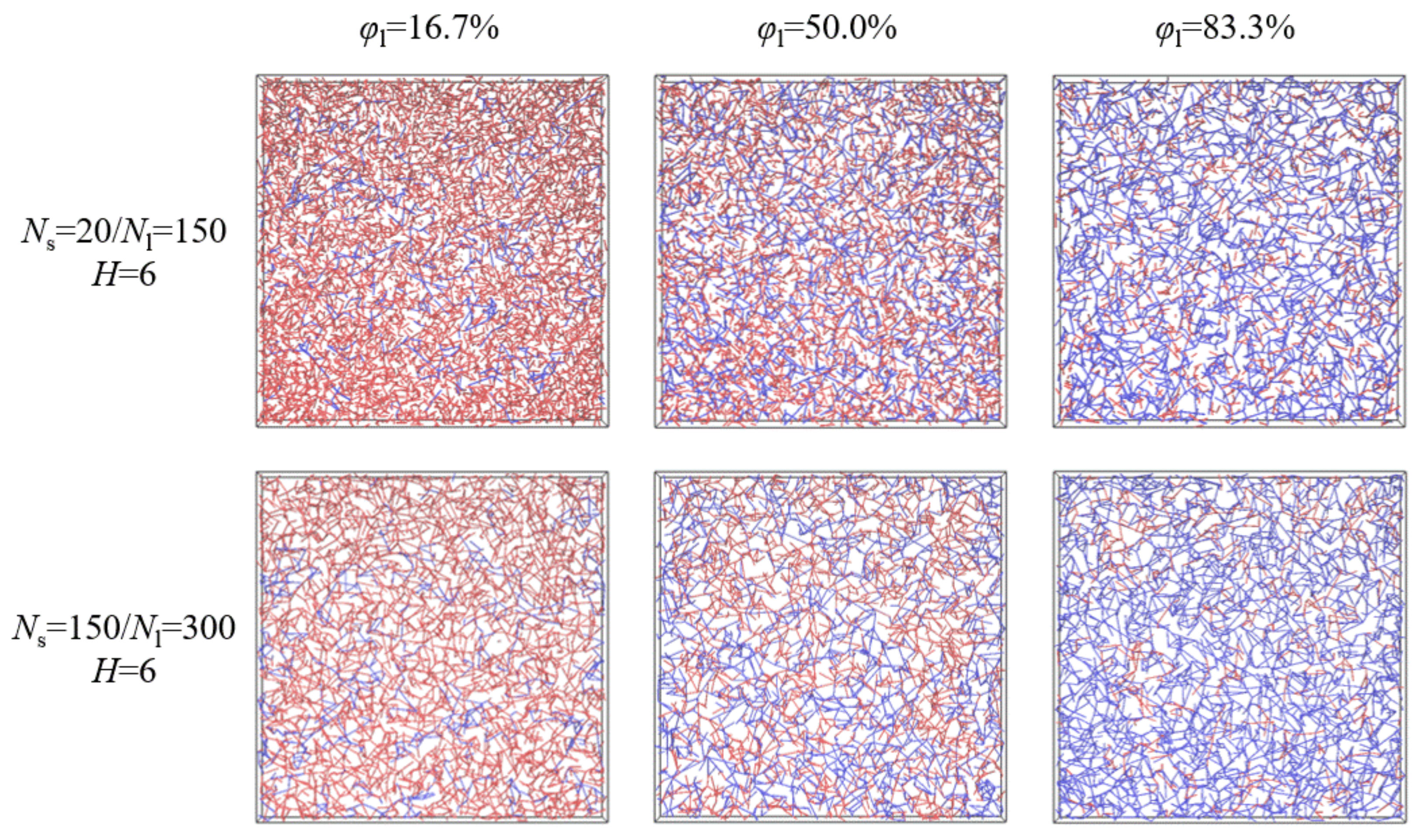
First, we put both the shorter and longer chains on the lattice and run 106–108 MCs to equilibrate the system, where the chain crossing is allowed. Then, 106–108 MCs are run to gradually remove those crossings. Finally, we further ran 107–108 MCs to obtain the equilibrium configurations. For each composition of the model system, 10 parallel simulations with different random seeds were performed to average the results. The Z1 algorithm uses geometrical moves to monotonically reduce the chain contour lengths to the limit of infinitely thin primitive path (PP) thickness, which is suitable to analyze chain entanglements in the equilibrium configurations [34,35,36,37]. As shown in Figure 1, for polymer films of Ns = 20/Nl = 150, the density of entanglements formed among long chains played a key role in the confinement of the long-chain mobility, which evidently increases with the increase of the weight fraction of the longer chains for the same film thickness. In contrast, for polymer films of Ns = 150/Nl = 300, since the molecular length of the shorter chains is above the characteristic entanglement length, entanglements formed among shorter and longer chains all had a great effect on the chain mobility, and the total density of entanglements hardly varied with the weight fraction for the same film thickness. As discussed in our previous paper, we could obtain the diffusion coefficient by extrapolation in the limit of large simulation time [17]. Although this apparent diffusion coefficient is different from a true diffusion constant obtained for a sufficiently long time, we could use this apparent diffusion coefficient as a measure of dynamic difference, which was valid for both Rouse and reptation regions [30].
In order to compare the un-constrained monodisperse polymer melts, we also simulated the cases in the absence of walls (periodic boundary conditions are applied in all three directions). The simulation details and corresponding results are shown in Table 3.
3. Results and Discussion
3.1. Model System of Ns = 10/Nl = 20
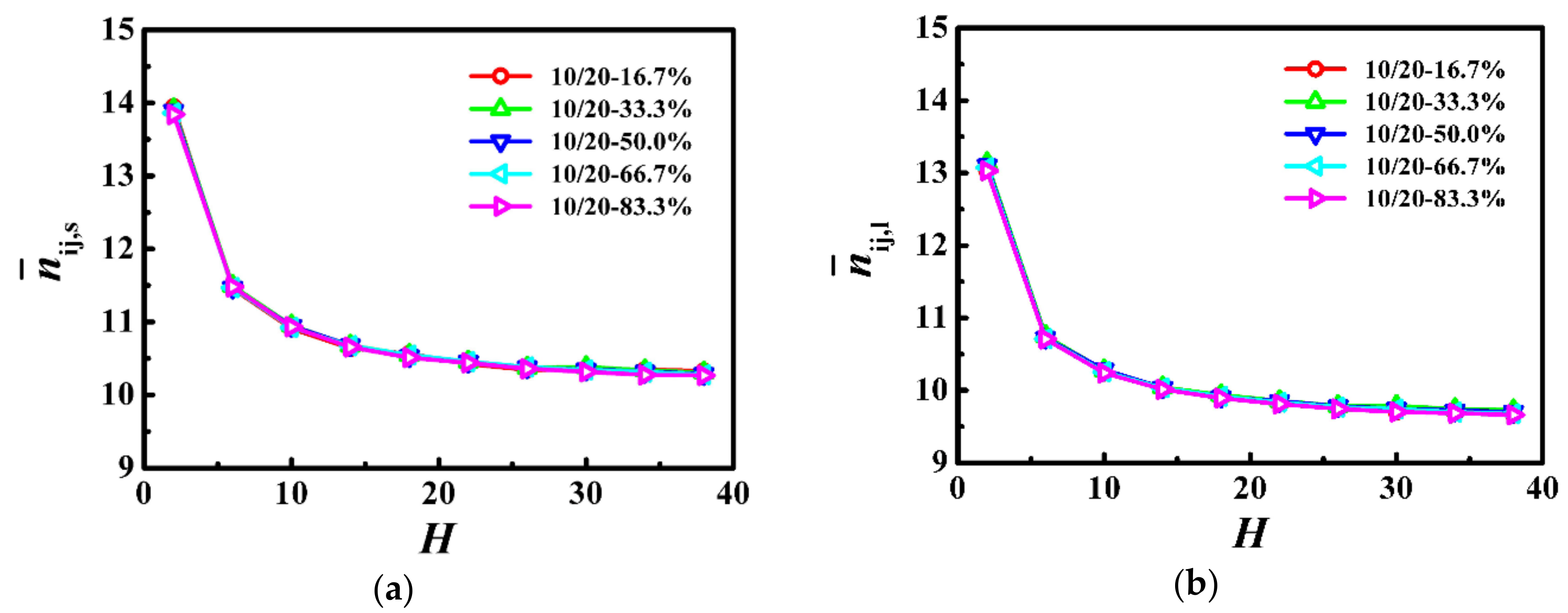
Firstly, we used the parameter of the number of near-neighboring “particles” per monomer () to evaluate the local degree of confinement, where i is the monomer of a given chain and j includes monomers from other chains as well as wall particles within the distance of 31/2 on the primary cubic lattice [17,30]. Figure 2 shows for the shorter and longer chains as a function of the film thickness, respectively. shows the similar trend, that is it is the largest for the thinnest film, which decreased rapidly as the film thickness was increased, and finally became a slowly decreasing function of the film thickness, which indicates that the local degree of confinement is independent of the weight fraction in the bidisperse polymer films.
To further investigate the effect of bidispersity on the dynamics of polymer films, in Figure 3, we respectively calculated the self-diffusion coefficient of the center-of-mass for the shorter (Dcm,s) and longer (Dcm,l) chains as a function of the film thickness, and normalized the diffusion coefficient by that in the unconstrained monodisperse melts (see Dcm,b in Table 3). For the shorter and longer chains, the diffusion coefficient is a slowly decreasing function with the decrease of the film thickness in thicker films, whereas it becomes a rapidly decreasing function with the decrease of the film thickness in thinner films. Moreover, our results indicate that the self-diffusion coefficient of the shorter and longer chains increased with the decrease of the weight fraction of the longer chains for the same film thickness. Besides, in bidisperse systems, the longer chains reside away from the walls and the shorter chains are close to the walls [30]. This spatial inhomogeneity may cause the diffusion coefficients of both components are always smaller than those of bulk systems. Thus, due to the fast relaxation of the shorter chains, the diffusion coefficient of the longer chains increases with the decrease of the weight fraction of the longer chains.
As Ns = 10 and Nl = 20 were both below the characteristic entanglement length (Ne ≈ 33), they formed few entanglements which hardly had any influence on their dynamics. With the slow increase of the average number of near-neighboring particles (see Figure 2), the self-diffusion coefficients of the shorter and longer chains decreased slowly with the decrease of the film thickness (see Figure 3). For the thinnest film, the average number of near-neighboring particles exhibited a rapid increase (see Figure 2), which resulted in the lowest diffusivity of the shorter and longer polymers (see Figure 3). Our results indicate that the local degree of confinement regulates the dynamics of polymers in bidisperse polymer films, where the shorter and longer chains are both in the unentangled region.
According to the Rouse theory [22,40], the self-diffusion coefficient of polymers depends on chain length as Dcm = kBT/(ζN) for monodisperse melts, where ζ represents the monomeric friction coefficient. However, the chain ends have a greater mobility than the central section of the chain, and the friction coefficient is proportional to chain length until the chains become sufficiently long that the enhanced mobility of the ends only plays a minor role in the overall diffusion [41,42,43]. Since the friction coefficient is a mean-field measure, for bidisperse polymer melts, the average friction coefficient can be generalized through a simple linear additivity of the overall friction coefficient and becomes ζB = φsζs + φlζl, where φi is the weight fraction of polymer Ni [44]. The diffusion coefficient of the Ni polymer is Dcm,i =kBT/(ζBNi). The value of Dcm calculated from this model agreed well with that measured from simulations [44]. In this model system, for the same film thickness, as the weight fraction of the longer chains was decreased, the weight fraction of the shorter chains was increased, and consequently the average friction coefficient of monomers exhibited a decrease which enhances the dynamics of the shorter and longer chains (see Figure 3), even though the local degrees of confinement remained rather unchanged (see Figure 2).
3.2. Model System of Ns = 20/Nl = 150
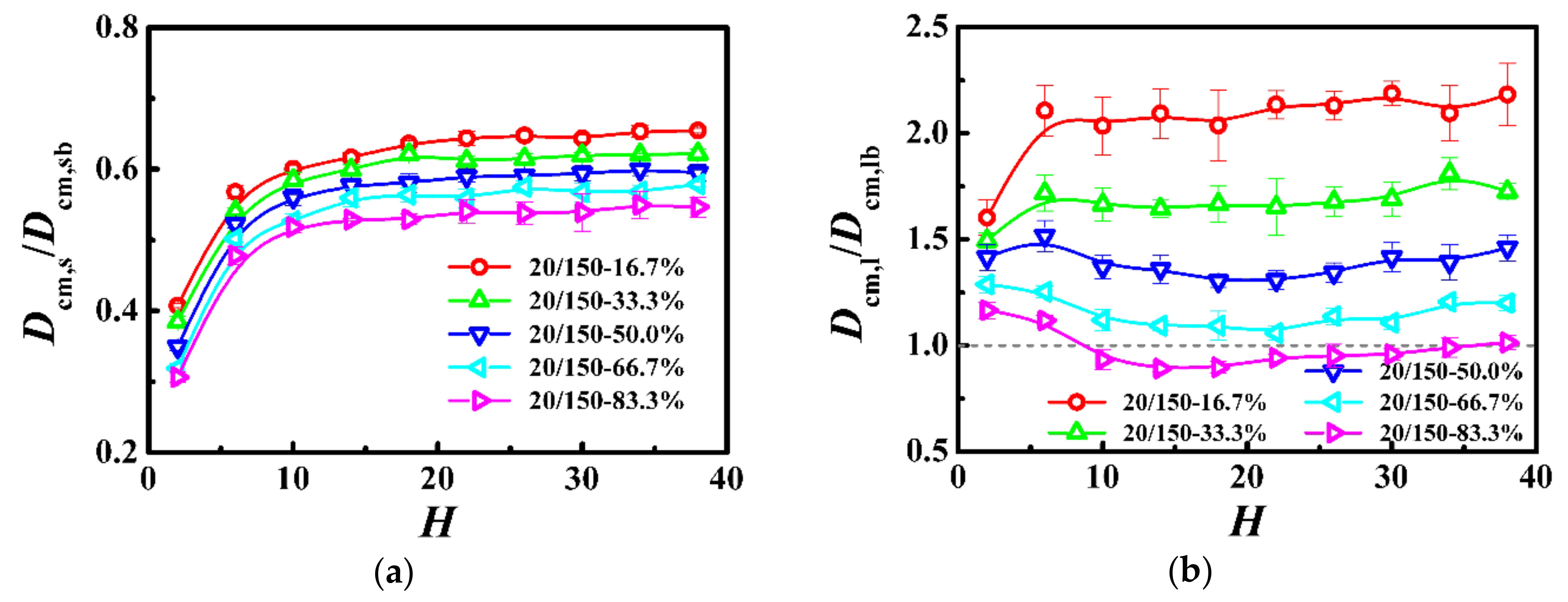
We have investigated the local degree of confinement for the model system Ns = 20 and Nl = 150 in our previous work [30], where the variations of the average number of near-neighboring particles were similar to those of the model system of Ns = 10 and Nl = 20 (see Figure 2). Although the disentanglement of the longer chains was in agreement with the monodisperse systems [13,14,45], we found that the number of short−long entanglements exhibited a dramatic increase whereas that of long−long entanglements exhibited a slight decrease with the decrease of the weight fraction of the longer chains.
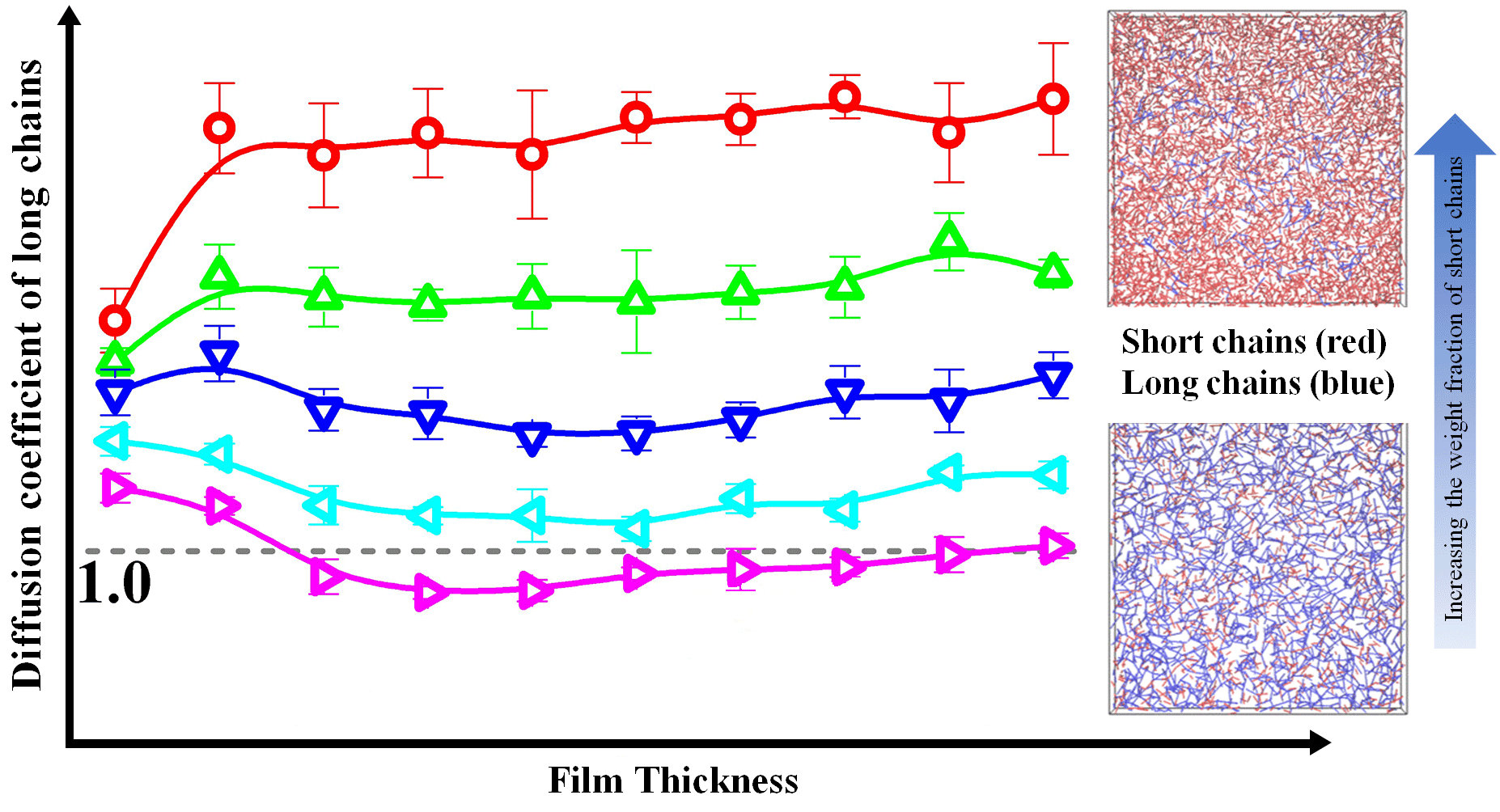
To further investigate the effect of bidispersity on the dynamics of this model, we calculated the self-diffusion coefficient of the shorter (Dcm,s) and longer (Dcm,l) chains as a function of the film thickness in Figure 4, where the diffusion coefficient was normalized with respect to that in the unconstrained monodisperse melts (see Dcm,b in Table 3). For the shorter chains, the diffusion coefficient slowly decreased in thicker films and then rapidly decreased in thinner films with the decrease of the film thickness. In contrast, the variations of dynamics of the longer chains with the film thickness depend on the weight fraction. When the weight fraction of the longer chains is lower than one-half, the diffusion coefficient is a slowly decreasing function with the decrease of the film thickness in thicker films and becomes a rapidly decreasing function with the decrease of the film thickness in ultrathin films, which is similar to the variations of the dynamics of the shorter chains. However, if the weight fraction of the longer chains is not lower than one-half, the diffusion coefficient is a slowly decreasing function with the decrease of the film thickness, when the film thickness is greater than the bulk chain dimension (H > 2Rgl,b), and an increasing function with the decrease of the film thickness when the film thickness is smaller than the bulk chain dimension (H < 2Rgl,b). Moreover, when the weight fraction of the longer chains is equal to one-half, the diffusion coefficient in ultrathin films fluctuates slightly. Besides, the previous study has demonstrated that the diffusion coefficient of long chains is smaller than that of the bulk samples [46]. However, in this model system, compared to the long-long entanglements, the short-long entanglements dissolved quickly due to the fast relaxation of the shorter chains, which resulted in the diffusion coefficient of the longer chains greater than that of the bulk samples, especially for the lower weight fraction of the longer chains.
Because the molecular length of the longer chains was above the characteristic entanglement length (Nl = 150 > Ne ≈ 33), the shorter chains could form entanglements with them. However, those entanglements dissolved quickly due to the fast relaxation of the shorter chains and have an insignificant influence on their dynamics. As the film thickness was decreased, the self-diffusion coefficient of the shorter chains decreased slowly (see Figure 4) with a slow increase of the average number of near-neighboring particles (see Reference [30]). For the thinnest film, the rapid increase of the average number of near-neighboring particles (see Reference [30]) resulted in the lowest diffusivity of the shorter polymers (see Figure 4). Our results indicate that, the dynamics of the shorter chains in this model system was regulated by the local degree of confinement, which is similar to the dynamics of polymers in the unentangled bidisperse polymer films.
The Struglinski-Graessley parameter is defined as Gr = NlNe2/Ns3, where Nl and Ns are respectively the lengths of the longer and shorter chains and Ne is the entanglement length. It can be interpreted as the ratio between the relaxation time of the longer chains due to the pure reptation and the relaxation time of the tube caused by constraint release [22]. For Gr larger than 1: The relaxation process is governed by the constraint release mechanisms [23,24,47]. The tube dilation model also predicted that, for higher weight fraction of the longer chains (φl > 1/Gr), the number of long-long entanglements was high enough to restrict the longer chains within a tube even after the relaxation of the shorter chains. The decline of the entanglements causes the dilated tube diameter for the longer chains to increase to a certain extent, which is called “restricted tube dilation”. For the lower weight fraction of the longer chains (φl < 1/Gr), however, the number of remaining long-long entanglements is not sufficient, so that the tube diameter can grow without limits and thus the process is called “free dilation” [23,24]. Previous molecular dynamics simulations have proved that the constraint release is the dominant relaxation mechanism in the melts of long and short chains [48,49]. In addition, a dissipative particle dynamics simulation has also provided strong evidence for the tube dilation of the long chains in bidisperse polymer melts [50], which supports the validity of the tube dilation model [23].
In this model system, the Struglinski-Graessley parameter was Gr = 20.4 > 1, which indicates that the constraint release mechanism plays a key role in the relaxation process of the longer chains. For the lower weight fraction of the longer chains, our results show that, as the average number of near-neighboring particles increased slowly (see Reference [30]), the self-diffusion coefficient of the longer chains decreased slowly with the decrease of the film thickness (see Figure 4); for the thinnest film, the average number of near-neighboring particles rapidly increased (see Reference [30]), which significantly limited the diffusivity of the longer polymers (see Figure 4). The change agrees with the prediction in our previous work [30], which is similar to that of the dynamics of the short chains. For the higher weight fraction of the longer chains, as the film thickness was decreased (H > 2Rgl,b), the number of long-long entanglements remained unchanged (see Reference [30]), but the average number of near-neighboring particles slowly increased (see Reference [30]), which resulted in a slight reduction of the diffusivity of the longer polymers (see Figure 4). With a further decrease in the film thickness (H < 2Rgl,b), the number of long-long entanglements rapidly decreased (see Reference [30]), which enhanced the dynamics of the longer polymers (see Figure 4). When the weight fraction of the longer chains was equal to one-half, in ultrathin films, the competition between the disentanglement and the increase in the local degree of confinement resulted in a slight fluctuation of the diffusion coefficient (see Figure 4).
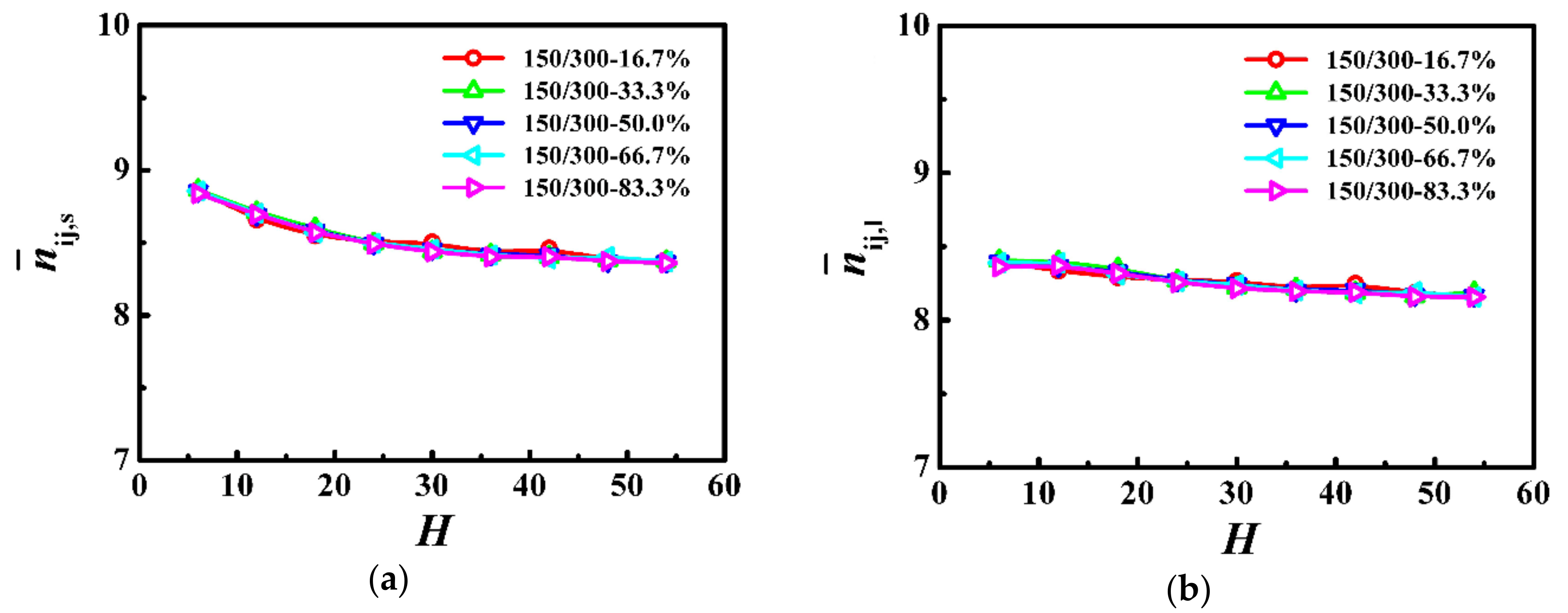
3.3. Model System of Ns = 150/Nl = 300
In Figure 5, we calculated the average number of near-neighboring particles per monomer as a function of the film thickness, which is a slowly decreasing function with the increase of the film thickness for both the shorter and longer chains. Moreover, the results from the average number of near-neighboring particles per monomer represent the local degrees of confinement for the shorter and longer chains, which are independent of the weight fraction in the polymer films.
In Figure 6, we respectively calculated the average number of entanglements for each shorter () and longer () chains in the PP network as a function of the film thickness. For the shorter and longer chains, the average number of entanglements was approximately independent of the film thickness in thicker films, however, it rapidly decreased with the decrease of the film thickness when the thickness was smaller than the coil size of the polymer (H < 2Rgs,b or H < 2Rgl,b). In addition, our results indicate that the weight fraction in this model system had an insignificant influence on the degree of chain entanglements.
We further respectively calculated the self-diffusion coefficient of the center-of-mass for the shorter (Dcm,s) and longer (Dcm,l) chains as a function of the film thickness in Figure 7, and normalized the diffusion coefficient by that in the unconstrained monodisperse melts (see Dcm,b in Table 3). For the shorter and longer chains, the diffusion coefficient was a slowly decreasing function with the decrease of the film thickness when the thickness was greater than the bulk chain dimension (H > 2Rgs,b or H > 2Rgl,b) and became a rapidly increasing function with the decrease of the film thickness when it was smaller than the bulk chain dimension (H < 2Rgs,b or H < 2Rgl,b). Moreover, our results indicate that polymers exhibited higher diffusivity for the lower weight fraction of the longer chains in the polymer films.
In this model system, we fixed the lengths of the shorter and longer chains as Ns = 150 and Nl = 300, and the Struglinski-Graessley parameter was Gr = 0.1 < 1. The tube dilation model and the tube reptation model draw the same conclusions for polymer melts, with Gr smaller than 1, and agree that the chains reptated inside an undilated tube, and therefore, the relaxation of the longer chain component is not affected by constraint release [23,24,46]. The entanglements evidently impose a crucial effect on the dynamics of polymers in spite of the weight fraction in the polymer films. For the shorter and longer chains, as the film thickness was decreased (H > 2Rgs,b or H > 2Rgl,b), the number of entanglements remained almost unchanged (see Figure 6), but the average number of near-neighboring particles slowly increased (see Figure 5), which results in a slight reduction of the diffusivity of the polymers (see Figure 7). With a further decrease in the film thickness (H < 2Rgs,b or H < 2Rgl,b), the number of entanglements rapidly decreased (see Figure 6), which enhanced the dynamics of the polymers (see Figure 7).
4. Conclusions
In this work, we considered three model systems: First, when the molecular lengths of the shorter and longer chains were both below the characteristic entanglement length; second, when the molecular length of the shorter chains was below the characteristic entanglement length whereas that of the long chains was above it; third, when the molecular lengths of the shorter and longer chains were both above the characteristic entanglement length.
Our results demonstrate that, for short chains of which the molecular length is below the characteristic entanglement length, their dynamics is regulated by the local degree of confinement so that the diffusion coefficient decreases slowly in thicker films and then rapidly in thinner films with the decrease of the film thickness, which is independent of the composition of bidisperse polymer films. In contrast, the dynamics of long chains of which the molecular length was above the characteristic entanglement length depended on the film composition, which can be interpreted in terms of reptation and constraint release dynamics. For bidisperse film compositions with two long chains (Struglinsky-Graessley parameter Gr < 1.0), the dynamics of long chains was not affected by constraint release. Due to the competition between the decrease of the number of entanglements and the increase of the average number of near-neighboring particles, the diffusion coefficient varied nonmonotonically with the film thickness; the diffusion coefficient slowly decreased when the film thickness was greater than the bulk chain dimension (H > 2Rg,lb) and rapidly increased when the film thickness was smaller (H < 2Rg,lb) with the decrease of the film thickness. For bidisperse film compositions with short and long chains (Gr > 1.0), constraint release contributed significantly to the relaxation mechanism of the long chains. For lower weight fraction of long chains, the fast relaxation of short chains dilated the tube diameter of long chains, and consequently the local degree of confinement instead of the entanglements had a marked effect on the dynamics of long chains. As the film thickness was decreased, the increase of the average number of near-neighboring particles slowed down the diffusivity of long chains. However, for the higher weight fraction of long chains, after the relaxation of short chains, the long-long entanglements were in sufficient quantities to restrict the long chains within a tube, which implies that the dynamics of long chains is hardly affected by constraint release. The diffusion coefficient varied nonmonotonically with the decrease of the film thickness due to the competition between the disentanglement and the enhanced confinement. Compared with the previous work, we completely clarified the relationship between the structures and dynamics for all possible cases for bidisperse systems, which not only establishes a unified understanding of the dependency of dynamics on the bidispersity of polymer films, but also helps to understand cases of polydispersity.
Author Contributions
S.L., Q.C., M.D. and T.S. conceived and designed the simulations; S.L. and Q.C. performed the simulations; S.L., Q.C. and M.D. analyzed the data; S.L., Q C., M.D. and T.S. interpreted the results and wrote the paper.
Funding
This research was funded by the National Natural Science Foundation of China (Nos. 51473168, 21604086 and 21647114), the Key Research Program of Frontier Sciences, CAS, Grant QYZDY-SSW-SLH027, the Research Project of Science and Technology of Higher Education Institutions in Hebei Province, China, Grant No. Z2018213, and the Cultivation Fund of National Natural Science Foundation of China Supported by China People’s Police University, Grant No. ZKJJPY201710.
Acknowledgments
We are grateful to Martin Kroöger (ETH Zuürich) for help with Z1 code.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Park, M.; Harrison, C.; Chaikin, P.M.; Register, R.A.; Adamson, D.H. Block copolymer lithography: Periodic arrays of ~1011 holes in 1 square centimeter. Science 1997, 276, 1401–1404. [Google Scholar] [CrossRef]
- Feller, L.M.; Cerritelli, S.; Textor, M.; Hubbell, J.A.; Tosatti, S.G. Influence of poly (propylene sulfide-block-ethylene glycol) di-and triblock copolymer architecture on the formation of molecular adlayers on gold surfaces and their effect on protein resistance: A candidate for surface modification in biosensor research. Macromolecules 2005, 38, 10503–10510. [Google Scholar] [CrossRef]
- Olson, D.C.; Piris, J.; Collins, R.T.; Shaheen, S.E.; Ginley, D.S. Hybrid photovoltaic devices of polymer and ZnO nanofiber composites. Thin Solid Films 2006, 496, 26–29. [Google Scholar] [CrossRef]
- Stockelhuber, K.W.; Svistkov, A.S.; Pelevin, A.G.; Heinrich, G. Impact of filler surface modification on large scale mechanics of styrene butadiene/silica rubber composites. Macromolecules 2011, 44, 4366–4381. [Google Scholar] [CrossRef]
- Saeed, F.; Ansarifar, A.; Ellis, R.J.; Haile-Meskel, Y.; Irfan, M.S. Two advanced styrene-butadiene/polybutadiene rubber blends filled with a silanized silica nanofiller for potential use in passenger car tire tread compound. J. Appl. Polym. Sci. 2012, 123, 1518–1529. [Google Scholar] [CrossRef]
- Wu, S. Polymer Interface and Adhesion; Marcel Dekker: New York, NY, USA, 1982. [Google Scholar]
- Batistakis, C.; Michels, M.A.J.; Lyulin, A.V. Confinement-induced stiffening of thin elastomer films: Linear and nonlinear mechanics vs. local dynamics. Macromolecules 2014, 47, 4690–4703. [Google Scholar] [CrossRef]
- Batistakis, C.; Michels, M.A.J.; Lyulin, A.V. Glassy boundary layers vs enhanced mobility in capped polymer films. J. Chem. Phys. 2013, 139, 024906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batistakis, C.; Lyulin, A.V.; Michels, M.A.J. Slowing down versus acceleration in the dynamics of confined polymer films. Macromolecules 2012, 45, 7282–7292. [Google Scholar] [CrossRef]
- Bäumchen, O.; Fetzer, R.; Jacobs, K. Reduced interfacial entanglement density affects the boundary conditions of polymer flow. Phys. Rev. Lett. 2009, 103, 247801. [Google Scholar] [CrossRef] [PubMed]
- Bodiguel, H.; Fretigny, C. Reduced viscosity in thin polymer films. Phys. Rev. Lett. 2006, 97, 266105. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Massa, M.V.; Dalnoki-Veress, K.; Brown, H.R.; Jones, R.A. Chain entanglement in thin freestanding polymer films. Phys. Rev. Lett. 2005, 94, 127801. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.M. Spatial distribution of entanglements in thin free-standing films. Phys. Rev. E 2016, 94, 012503. [Google Scholar] [CrossRef] [PubMed]
- Sussman, D.M.; Tung, W.-S.; Winey, K.I.; Schweizer, K.S.; Riggleman, R.A. Entanglement reduction and anisotropic chain and primitive path conformations in polymer melts under thin film and cylindrical confinement. Macromolecules 2014, 47, 6462–6472. [Google Scholar] [CrossRef]
- Sarabadani, J.; Milchev, A.; Vilgis, T.A. Structure and dynamics of polymer melt confined between two solid surfaces: A molecular dynamics study. J. Chem. Phys. 2014, 141, 044907. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, D.; Han, C.C.; Liao, Q. Dynamics of polymer melts confined by smooth walls: Crossover from nonentangled region to entangled region. J. Chem. Phys. 2007, 126, 204907. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Ding, M.; Shi, T. Effects of polymer–wall interactions on entanglements and dynamics of confined polymer films. J. Phys. Chem. B 2017, 121, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, J.; Paul, W.; Tries, V.; Binder, K. Statics and dynamics of bidisperse polymer melts: A Monte Carlo study of the bond-fluctuation model. Macromolecules 1998, 31, 3856–3867. [Google Scholar] [CrossRef]
- Thompson, P.A.; Grest, G.S.; Robbins, M.O. Phase transitions and universal dynamics in confined films. Phys. Rev. Lett. 1992, 68, 3448–3451. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, W.; White, J.L.; Spruiell, J.E. Experimental investigation of the influence of molecular weight distribution on the rheological properties of polypropylene melts. Polym. Eng. Sci. 1980, 20, 1166–1176. [Google Scholar] [CrossRef]
- Minoshima, W.; White, J.L.; Spruiell, J.E. Experimental investigations of the influence of molecular weight distribution on melt spinning and extrudate swell characteristics of polypropylene. J. Appl. Polym. Sci. 1980, 25, 287–306. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1988; Volume 73. [Google Scholar]
- Doi, M.; Graessley, W.W.; Helfand, E.; Pearson, D.S. Dynamics of polymers in polydisperse melts. Macromolecules 1987, 20, 1900–1906. [Google Scholar] [CrossRef]
- Marrucci, G. Relaxation by reptation and tube enlargement: A model for polydisperse polymers. J. Polym. Sci. Part B Polym. Phys. 1985, 23, 159–177. [Google Scholar] [CrossRef]
- Sabzevari, S.M.; Cohen, I.; Wood-Adams, P.M. Wall slip of bidisperse linear polymer melts. Macromolecules 2014, 47, 3154–3160. [Google Scholar] [CrossRef]
- Sabzevari, S.M.; Cohen, I.; Wood-Adams, P.M. Wall slip of tridisperse polymer melts and the effect of unentangled versus weakly entangled chains. Macromolecules 2014, 47, 8033–8040. [Google Scholar] [CrossRef]
- Sabzevari, S.M.; McGraw, J.D.; Wood-Adams, P.M. Short chains enhance slip of highly entangled polystyrenes during thin film dewetting. RSC Adv. 2016, 6, 91163–91170. [Google Scholar] [CrossRef] [Green Version]
- Eslami, H.; Muüller-Plathe, F. Viscosity of nanoconfined polyamide-6,6 oligomers: Atomistic reverse nonequilibrium molecular dynamics simulation. J. Phys. Chem. B 2010, 114, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Rorrer, N.A.; Dorgan, J.R. Effects of polydispersity on confined homopolymer melts: A Monte Carlo study. J. Chem. Phys. 2014, 141, 214905. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Ding, M.; Shi, T. Effect of bidispersity on structure and entanglement of confined polymer films. J. Phys. Chem. B 2017, 121, 7502–7507. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.S. Effects of chain topology on polymer dynamics: Bulk melts. J. Chem. Phys. 1994, 101, 4205–4213. [Google Scholar] [CrossRef]
- Shaffer, J.S. Effects of chain topology on polymer dynamics: Configurational relaxation in polymer melts. J. Chem. Phys. 1995, 103, 761–772. [Google Scholar] [CrossRef]
- Termonia, Y. Two-dimensional nanometric confinement of entangled polymer melts. Polymer 2011, 52, 5193–5196. [Google Scholar] [CrossRef]
- Kröger, M. Shortest multiple disconnected path for the analysis of entanglements in two-and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Shanbhag, S.; Kröger, M. Primitive path networks generated by annealing and geometrical methods: Insights into differences. Macromolecules 2007, 40, 2897–2903. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Kröger, M. Combined molecular algorithms for the generation, equilibration and topological analysis of entangled polymers: Methodology and performance. Int. J. Mol. Sci. 2009, 10, 5054–5089. [Google Scholar] [CrossRef] [PubMed]
- Hoy, R.S.; Foteinopoulou, K.; Kröger, M. Topological analysis of polymeric melts: Chain-length effects and fast-converging estimators for entanglement length. Phys. Rev. E 2009, 80, 031803. [Google Scholar] [CrossRef] [PubMed]
- Baig, C.; Mavrantzas, V.G.; Kroöger, M. Flow effects on melt structure and entanglement network of linear polymers: Results from a nonequilibrium molecular dynamics simulation study of a polyethylene melt in steady shear. Macromolecules 2010, 43, 6886–6902. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Heermann, D.W.; Kremer, K. Dynamics of polymer solutions and melts. Reptation predictions and scaling of relaxation times. J. Chem. Phys. 1991, 95, 7726–7740. [Google Scholar] [CrossRef]
- Rouse, P.E. A theory of the linear viscoelastic properties of dilute solutions of coiling polymers. J. Chem. Phys. 1953, 21, 1272–1280. [Google Scholar] [CrossRef]
- Kremer, K.; Grest, G.S. Dynamics of entangled linear polymer melts: A molecular-dynamics simulation. J. Chem. Phys. 1990, 92, 5057–5086. [Google Scholar] [CrossRef]
- Harmandaris, V.A.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic molecular dynamics simulation of polydisperse linear polyethylene melts. Macromolecules 1998, 31, 7934–7943. [Google Scholar] [CrossRef]
- Smith, S.W.; Hall, C.K.; Freeman, B.D. Molecular dynamics study of transport coefficients for hard-chain fluids. J. Chem. Phys. 1995, 102, 1057–1073. [Google Scholar] [CrossRef]
- Barsky, S. Molecular dynamics study of diffusion in bidisperse polymer melts. J. Chem. Phys. 2000, 112, 3450–3456. [Google Scholar] [CrossRef]
- Vladkov, M.; Barrat, J.-L. Local dynamics and primitive path analysis for a model polymer melt near a surface. Macromolecules 2007, 40, 3797–3804. [Google Scholar] [CrossRef]
- Eslami, H.; Muüller-Plathe, F. How thick is the interphase in an ultrathin polymer film? Coarse-grained molecular dynamics simulations of polyamide-6,6 on graphene. J. Phys. Chem. C 2013, 117, 5249–5257. [Google Scholar] [CrossRef]
- Viovy, J.L.; Rubinstein, M.; Colby, R.H. Constraint release in polymer melts: Tube reorganization versus tube dilation. Macromolecules 1991, 24, 3587–3596. [Google Scholar] [CrossRef]
- Baig, C.; Stephanou, P.S.; Tsolou, G.; Mavrantzas, V.G.; Kröger, M. Understanding dynamics in binary mixtures of entangled cis-1,4-polybutadiene melts at the level of primitive path segments by mapping atomistic simulation data onto the tube model. Macromolecules 2010, 43, 8239–8250. [Google Scholar] [CrossRef]
- Stephanou, P.S.; Mavrantzas, V.G. Accurate prediction of the linear viscoelastic properties of highly entangled mono and bidisperse polymer melts. J. Chem. Phys. 2014, 140, 214903. [Google Scholar] [CrossRef] [PubMed]
- Langeloth, M.; Masubuchi, Y.; Böhm, M.C.; Müller-Plathe, F. Reptation and constraint release dynamics in bidisperse polymer melts. J. Chem. Phys. 2014, 141, 194904. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative primitive path networks with the shorter (red) and longer (blue) chains for polymer films of Ns = 20/Nl = 150 and Ns = 150/Nl = 300, where the film thickness is fixed as H = 6 and the weight fraction of the longer chains is changed from φl = 16.7% to φl = 83.3%.
Figure 1.
Representative primitive path networks with the shorter (red) and longer (blue) chains for polymer films of Ns = 20/Nl = 150 and Ns = 150/Nl = 300, where the film thickness is fixed as H = 6 and the weight fraction of the longer chains is changed from φl = 16.7% to φl = 83.3%.

Figure 2.
Average number of near-neighboring particles per monomer for (a) shorter (
) and (b) longer () chains as a function of the film thickness (H) in the model system of Ns = 10 and Nl = 20.
Figure 2.
Average number of near-neighboring particles per monomer for (a) shorter (
) and (b) longer () chains as a function of the film thickness (H) in the model system of Ns = 10 and Nl = 20.

Figure 3.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of the film thickness (H) in the model system of Ns = 10 and Nl = 20.
Figure 3.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of the film thickness (H) in the model system of Ns = 10 and Nl = 20.
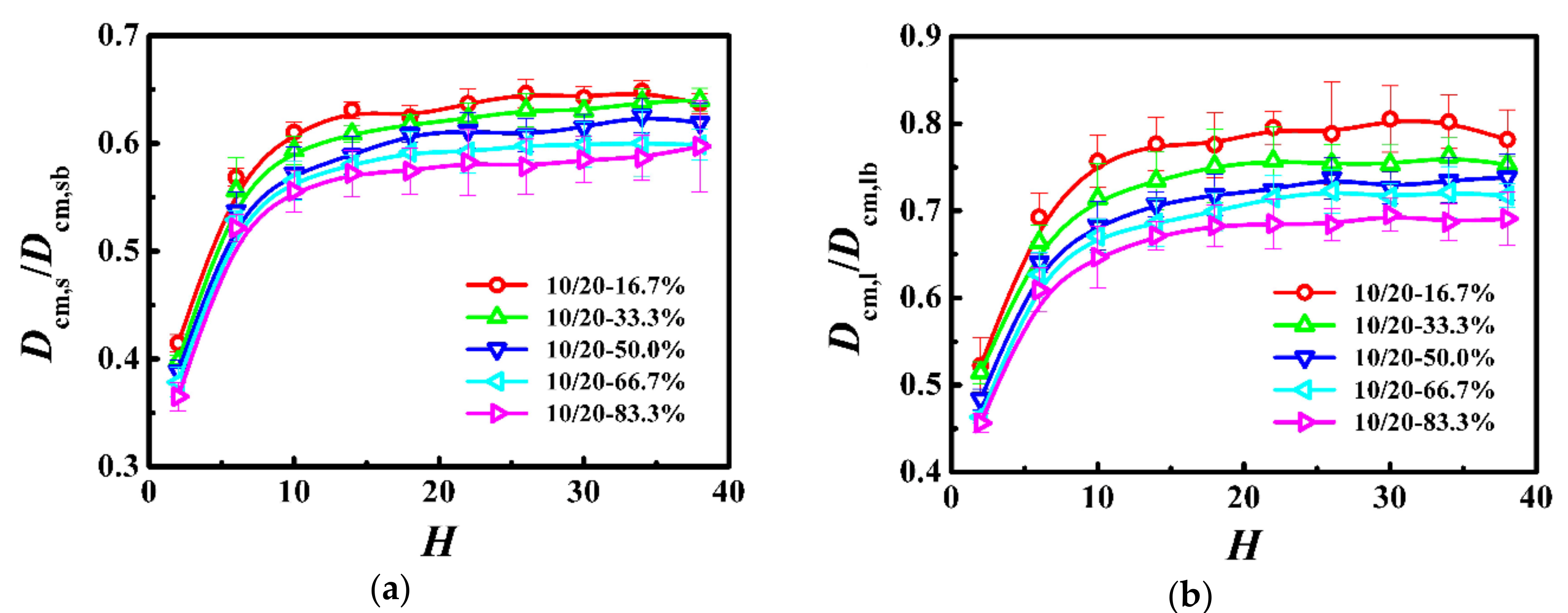
Figure 4.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of film thickness (H) in the model system of Ns = 20 and Nl = 150.
Figure 4.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of film thickness (H) in the model system of Ns = 20 and Nl = 150.

Figure 5.
Average number of near-neighboring particles per monomer for (a) shorter () and (b) longer () chains as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.
Figure 5.
Average number of near-neighboring particles per monomer for (a) shorter () and (b) longer () chains as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.
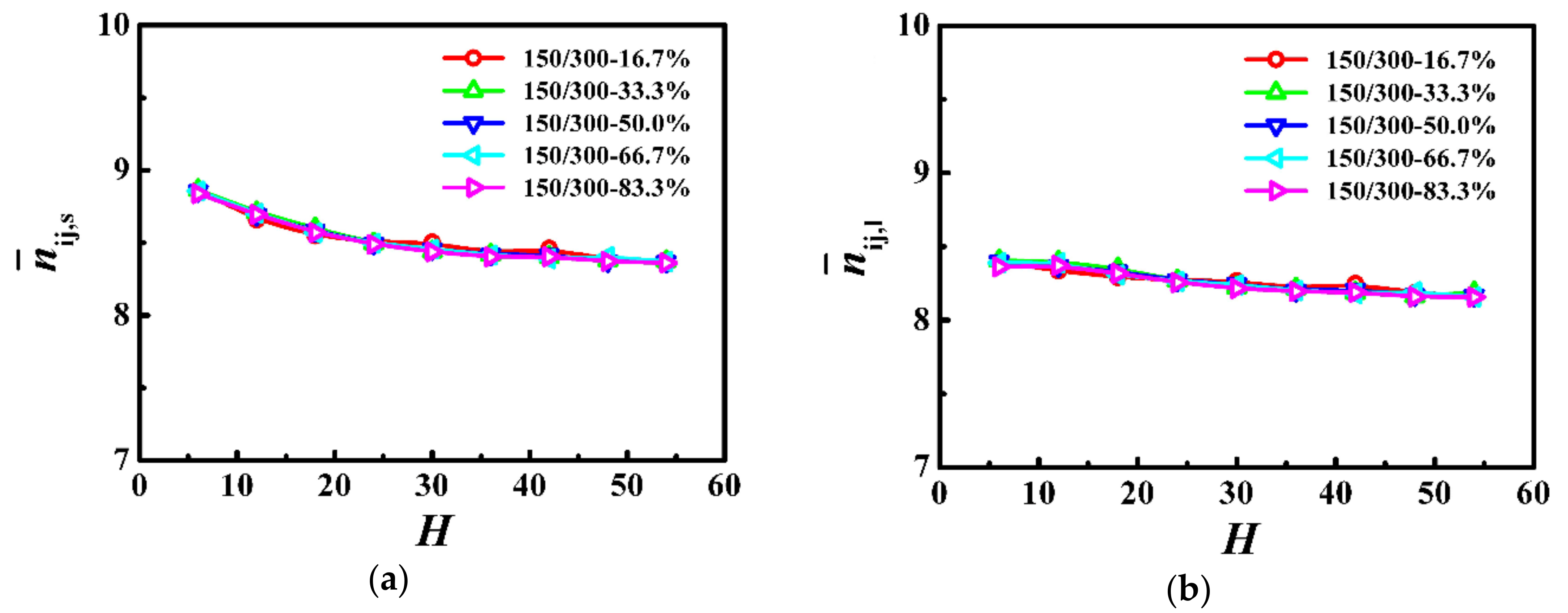
Figure 6.
Average number of entanglements for each (a) shorter () and (b) longer () chain as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.
Figure 6.
Average number of entanglements for each (a) shorter () and (b) longer () chain as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.
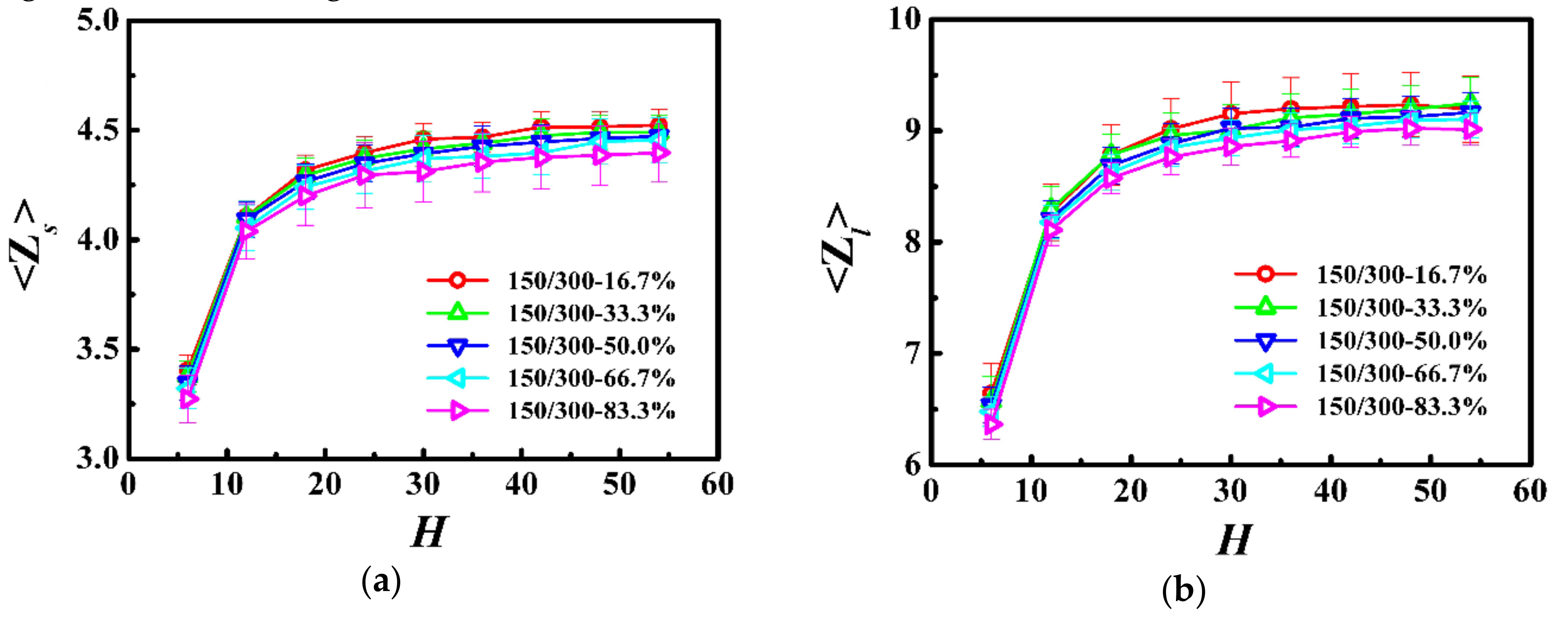
Figure 7.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.
Figure 7.
Normalized self-diffusion coefficient of the center-of-mass for (a) shorter (Dcm,s/Dcm,sb) and (b) longer (Dcm,l/Dcm,lb) chains as a function of the film thickness (H) in the model system of Ns = 150 and Nl = 300.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Compositions of bidisperse polymer melts 1.
| System (Ns/Nl-φl) | ns | nl |
|---|---|---|
| 10/20-16.7% | 1120 | 112 |
| 10/20-33.3% | 900 | 225 |
| 10/20-50.0% | 674 | 337 |
| 10/20-66.7% | 450 | 450 |
| 10/20-83.3% | 224 | 562 |
| 20/150-16.7% | 4500 | 120 |
| 20/150-33.3% | 3600 | 240 |
| 20/150-50.0% | 2700 | 360 |
| 20/150-66.7% | 1800 | 480 |
| 20/150-83.3% | 900 | 600 |
| 150/300-16.7% | 960 | 96 |
| 150/300-33.3% | 768 | 192 |
| 150/300-50.0% | 576 | 288 |
| 150/300-66.7% | 386 | 386 |
| 150/300-83.3% | 192 | 480 |
1ns and nl are the numbers of the shorter and longer chains with the lengths Ns and Nl, respectively, and φl represents the weight fraction of the longer chains in the system.
Table 2.
Simulation details for bidisperse polymer films 1.
| Ns = 10/Nl = 20 | |||
|---|---|---|---|
| Lx = Ly | H = Lz | H/Rgs,b | H/Rgl,b |
| 116 | 2 | 1.04 | 0.71 |
| 68 | 6 | 3.11 | 2.14 |
| 52 | 10 | 5.18 | 3.57 |
| 44 | 14 | 7.26 | 4.99 |
| 38 | 18 | 9.33 | 6.42 |
| 36 | 22 | 11.40 | 7.85 |
| 32 | 26 | 13.47 | 9.27 |
| 30 | 30 | 15.54 | 10.68 |
| 28 | 34 | 17.62 | 12.10 |
| 26 | 38 | 19.69 | 13.52 |
| Ns = 20/Nl = 150 | |||
| Lx = Ly | H = Lz | H/Rgs,b | H/Rgl,b |
| 330 | 2 | 0.71 | 0.25 |
| 190 | 6 | 2.14 | 0.74 |
| 146 | 10 | 3.57 | 1.24 |
| 124 | 14 | 4.99 | 1.74 |
| 110 | 18 | 6.42 | 2.23 |
| 100 | 22 | 7.85 | 2.73 |
| 90 | 26 | 9.27 | 3.23 |
| 84 | 30 | 10.68 | 3.72 |
| 80 | 34 | 12.10 | 4.22 |
| 76 | 38 | 13.52 | 4.72 |
| Ns = 150/Nl = 300 | |||
| Lx = Ly | H = Lz | H/Rgs,b | H/Rgl,b |
| 240 | 6 | 0.74 | 0.52 |
| 170 | 12 | 1.49 | 1.04 |
| 138 | 18 | 2.23 | 1.56 |
| 120 | 24 | 2.98 | 2.08 |
| 106 | 30 | 3.72 | 2.60 |
| 98 | 36 | 4.47 | 3.12 |
| 90 | 42 | 5.21 | 3.64 |
| 84 | 48 | 5.96 | 4.16 |
| 80 | 54 | 6.70 | 4.68 |
1Lx = Ly is the lateral length of the simulation box, H = Lz denotes the film thickness, H/Rgs,b and H/Rgl,b represent the film thicknesses normalized by the mean-square radii of gyration of the shorter and longer chains in the unconstrained monodisperse melts, respectively.
Table 3.
Simulation details and results for unconstrained monodisperse polymer melts 1.
| M | N | Lx = Ly = Lz | Rg,b | Dcm,b × 106 | 〈Z〉b |
|---|---|---|---|---|---|
| 1350 | 10 | 30 | 1.93 | 1220.50 | |
| 675 | 20 | 30 | 2.81 | 444.43 | |
| 213 | 150 | 40 | 8.06 | 12.05 | 4.7162 |
| 208 | 300 | 50 | 11.53 | 2.24 | 9.2341 |
1 M represents the number of polymer chains, N denotes the length of polymer chains, Lx = Ly = Lz is the length of the simulation box, Rg,b denotes the mean-square radius of gyration of polymers, Dcm,b is the center-of-mass diffusion coefficient, and 〈Z〉b represents the average number of entanglements per chain.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, S.; Chen, Q.; Ding, M.; Shi, T. Effect of Bidispersity on Dynamics of Confined Polymer Films. Polymers 2018, 10, 1327. https://doi.org/10.3390/polym10121327
AMA Style
Li S, Chen Q, Ding M, Shi T. Effect of Bidispersity on Dynamics of Confined Polymer Films. Polymers. 2018; 10(12):1327. https://doi.org/10.3390/polym10121327
Chicago/Turabian StyleLi, Sijia, Qiaoyue Chen, Mingming Ding, and Tongfei Shi. 2018. "Effect of Bidispersity on Dynamics of Confined Polymer Films" Polymers 10, no. 12: 1327. https://doi.org/10.3390/polym10121327
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.