Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
3. Results
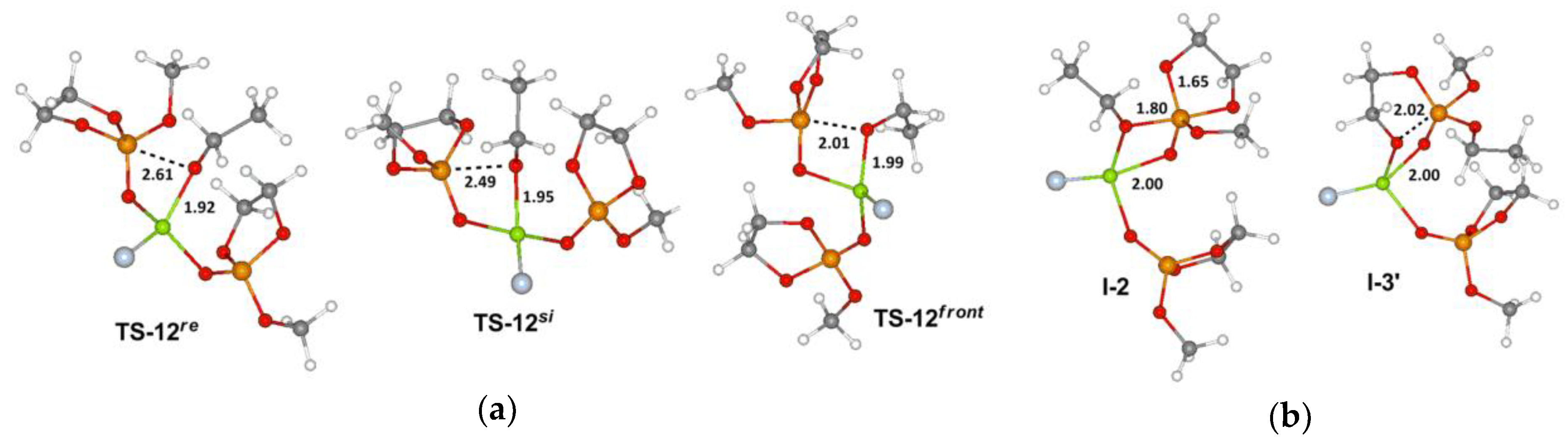
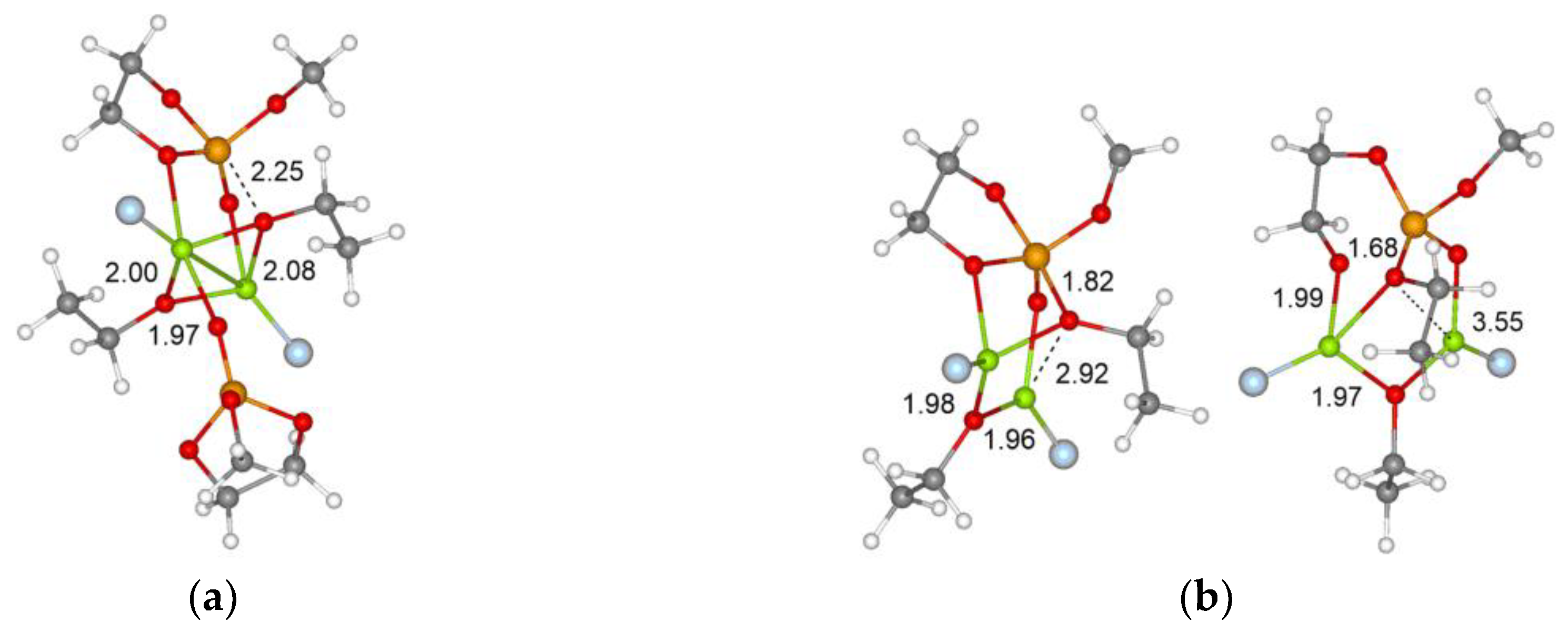
3.1. DFT Modeling of the Formation of the Catalytic Species
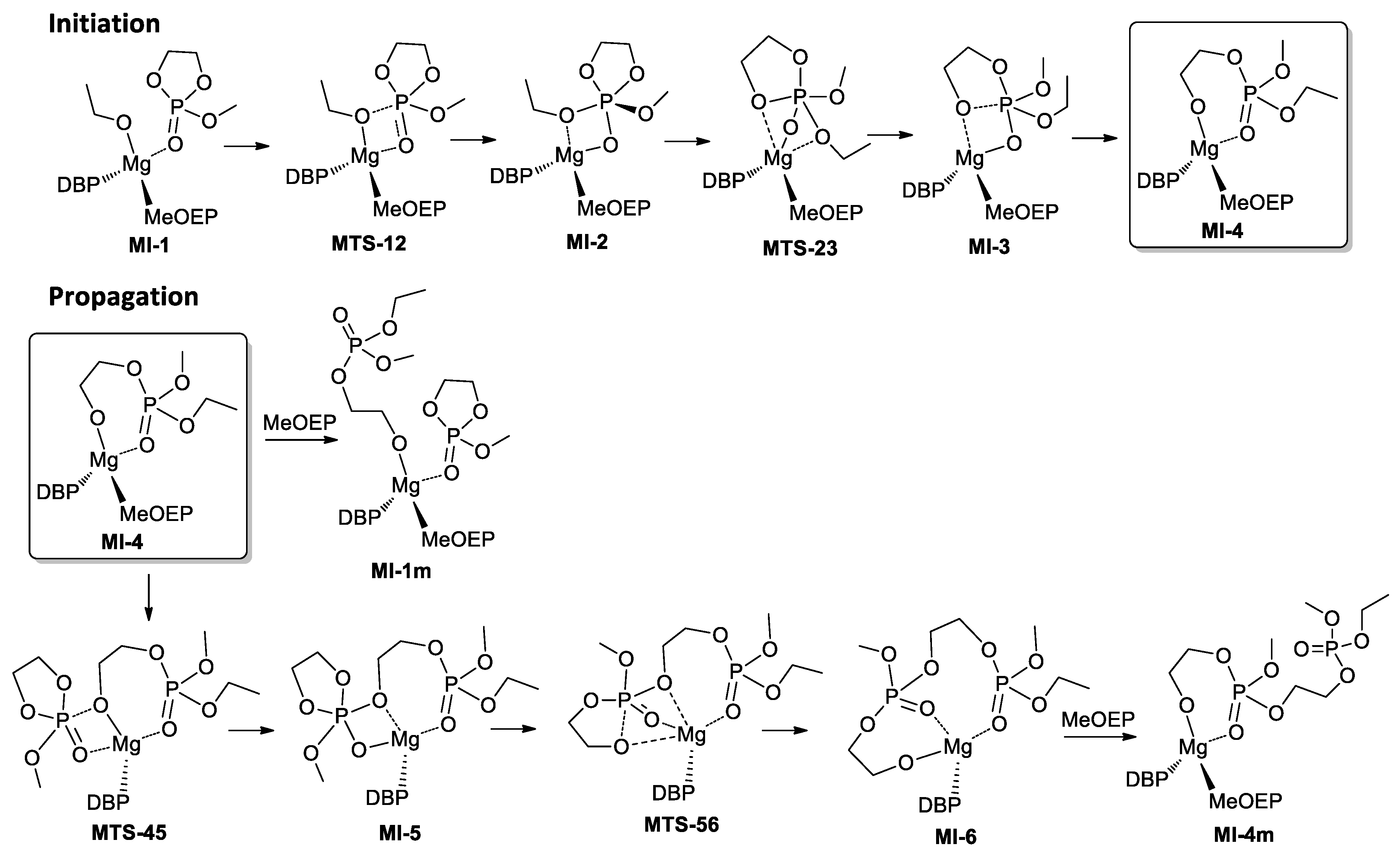
3.2. DFT Modeling of the Mononuclear ROP Mechanism
3.2.1. Initiation Stage
3.2.2. Propagation Stage
3.3. DFT Modeling of the Binuclear ROP Mechanism
3.3.1. Initiation Stage
3.3.2. Propagation Stage
3.4. Polymerization Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, Y.-C.; Yuan, Y.-Y.; Du, J.-Z.; Yang, X.-Z.; Wang, J. Recent progress in polyphosphoesters: From controlled synthesis to biomedical applications. Macromol. Biosci. 2009, 9, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Z.E.; Jérôme, C. Polyphosphoesters: New trends in synthesis and drug delivery applications. Macromol. Biosci. 2016, 16, 1745–1761. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.N.; Tee, H.T.; Velencoso, M.M.; Wurm, F.R. Main-chain poly(phosphoester)s: History, syntheses, degradation, bio-and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122. [Google Scholar] [CrossRef]
- Steinbach, T.; Wurm, F.R. Poly(phosphoester)s: A new platform for degradable polymers. Angew. Chem. Int. Ed. 2015, 54, 6098–6108. [Google Scholar] [CrossRef] [PubMed]
- Du, J.-Z.; Du, X.-J.; Mao, C.-Q.; Wang, J. Tailor-made dual pH-sensitive polymer–doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, J.; Mao, H.Q.; Leong, K.W. Polyphosphoesters in drug and gene delivery. Adv. Drug Deliv. Rev. 2003, 55, 483–499. [Google Scholar] [CrossRef]
- Wu, X.L.; Kim, J.H.; Koo, H.; Bae, S.M.; Shin, H.; Kim, M.S.; Lee, B.-H.; Park, R.-W.; Kim, I.-S.; Choi, K.; et al. Tumor-targeting peptide conjugated pH-responsive micelles as a potential drug carrier for cancer therapy. Bioconjugate Chem. 2010, 21, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotech. 2016, 11, 372–377. [Google Scholar] [CrossRef]
- Steinbach, T.; Wurm, F.R. Degradable polyphosphoester-protein conjugates: “PPEylation” of proteins. Biomacromolecules 2016, 17, 3338–3346. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Yamaguchi, E. Synthesis of well-defined thermoresponsive polyphosphoester macroinitiators using organocatalysts. Macromolecules 2010, 43, 2664–2666. [Google Scholar] [CrossRef]
- Clément, B.; Grignard, B.; Koole, L.; Jérome, C.; Lecomte, P. Metal-free strategies for the synthesis of functional and well-defined polyphosphoesters. Macromolecules 2012, 45, 4476–4486. [Google Scholar] [CrossRef]
- Stukenbroeker, T.S.; Solis-Ibarra, D.; Waymouth, R.M. Synthesis and topological trapping of cyclic poly(alkylene phosphates). Macromolecules 2014, 47, 8224–8230. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, H.; Shen, Y.; Zhang, F.; Seetho, K.; Zou, J.; Taylor, J.-S.A.; Dove, A.P.; Wooley, K.L. A Simple and efficient synthesis of an acid-labile polyphosphoramidate by organobase-catalyzed ring-opening polymerization and transformation to polyphosphoester ionomers by acid treatment. Macromolecules 2013, 46, 5141–5149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, A.; Zou, J.; Lin, L.Y.; Wooley, K.L. Facile synthesis of clickable, water-soluble, and degradable polyphosphoesters. ACS Macro Lett. 2012, 1, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.K.; Steinbach, T.; Wurm, F.R. Multifunctional poly(phosphoester)s with two orthogonal protective groups. RSC Adv. 2015, 5, 42881–42888. [Google Scholar] [CrossRef] [Green Version]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Controlled ring-opening polymerisation of cyclic phosphates, phosphonates and phosphoramidates catalysed by hereroleptic BHT-alkoxy magnesium complexes. Polym. Chem. 2017, 8, 6806–6816. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Kosarev, M.A.; Tavtorkin, A.N.; Minyaev, M.E.; Roznyatovsky, V.A.; Ivchenko, P.V. Synthesis and ring-opening polymerization of glycidyl ethylene phosphate with a formation of linear and branched polyphosphates. Mendeleev Commun. 2018, 28, 155–157. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Komarov, P.D.; Tavtorkin, A.N.; Minyaev, M.E.; Kosarev, M.A.; Ivchenko, P.V. Synthesis in aqueous media of poly(ethylene phosphoric acids) by mild thermolysis of homopolymers and block copolymers based on tert-butyl ethylene phosphate. Eur. Polym. J. 2018, 106, 249–256. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Tavtorkin, A.N.; Ivchenko, P.V.; Borisov, R.S.; Churakov, A.V. Monomeric and dimeric magnesium mono-BHT complexes as effective ROP catalysts. Catal. Commun. 2016, 87, 106–111. [Google Scholar] [CrossRef]
- Nifant’ev, I.E.; Shlyakhtin, A.V.; Bagrov, V.V.; Minyaev, M.E.; Churakov, A.V.; Karchevsky, S.G.; Birin, K.P.; Ivchenko, P.V. Mono-BHT heteroleptic magnesium complexes: synthesis, molecular structure and catalytic behavior in the ring-opening polymerization of cyclic esters. Dalton Trans. 2017, 46, 12132–12146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: a quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.W.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.K.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Sosa, C.; Andzelm, J.; Elkin, B.C.; Wimmer, E.; Dobbs, K.D.; Dixon, D.A. A local density functional study of the structure and vibrational frequencies of molecular transition-metal compounds. J. Phys. Chem. 1992, 96, 6630–6636. [Google Scholar] [CrossRef]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Steitz, T.A.; Lipscomb, W.N. Molecular structure of methyl ethylene phosphate. J. Am. Chem. Soc. 1965, 87, 2488–2489. [Google Scholar] [CrossRef]
- Paier, J.; Marsman, M.; Kresse, G. Why does the B3LYP hybrid functional fail for metals? J. Chem. Phys. 2007, 127, 024103. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, M.G.; Bushmarinov, I.S.; Sun, J.; Perdew, J.P.; Lyssenko, K.A. Density functional theory is straying from the path toward the exact functional. Science 2017, 355, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Kabb, C.P.; Dai, Y.; Hill, M.R.; Ghiviriga, I.; Bapat, A.P.; Sumerlin, B.S. Macromolecular metamorphosis via stimulus-induced transformations of polymer architecture. Nat. Chem. 2017, 9, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT investigations on the ring-opening polymerization of substituted cyclic carbonates catalyzed by zinc-{β-diketiminate} complexes. Polym. Chem. 2015, 6, 3336–3352. [Google Scholar] [CrossRef] [Green Version]
- Del Rosal, I.; Brignou, P.; Guillaume, S.M.; Carpentier, J.-F.; Maron, L. DFT investigations on the ring-opening polymerization of cyclic carbonates catalyzed by zinc-{β-diiminate} complexes. Polym. Chem. 2011, 2, 2564–2573. [Google Scholar] [CrossRef]
- Susperregui, N.; Kramer, M.U.; Okuda, J.; Maron, L. Theoretical Study on the Ring-Opening Polymerization of ε-Caprolactone by [YMeX(THF)5]+ with X = BH4, NMe2. Organometallics 2011, 30, 1326–1333. [Google Scholar] [CrossRef]
- Fang, J.; Yu, I.; Mehrkhodavandi, P.; Maron, L. Theoretical investigation of lactide ring-opening polymerization induced by a dinuclear indium catalyst. Organometallics 2013, 32, 6950–6956. [Google Scholar] [CrossRef]
- Kuzdrowska, M.; Annunziata, L.; Marks, S.; Schmid, M.; Jaffredo, C.G.; Roesky, P.W.; Guillaume, S.M.; Maron, L. Organometallic calcium and strontium borohydrides as initiators for the polymerization of ε-caprolactone and l-lactide: combined experimental and computational investigations. Dalton Trans. 2013, 42, 9352–9360. [Google Scholar] [CrossRef] [PubMed]
- Yurieva, A.G.; Poleshchuk, O.Kh.; Filimonov, V.D. Comparative analysis of a full-electron basis set and pseudopotential for the iodine atom in DFT quantum-chemical calculations of iodine-containing compounds. J. Struct. Chem. 2008, 49, 548–552. [Google Scholar] [CrossRef]
- Abdel-Rhmana, M.H.; Hassanian, M.M.; El-Asmy, A.A. Spectral and structural density functional theory on 4–ethyl and 4–(p-tolyl)-1–(pyridin-2–yl)thiosemicarbazides and their Pd(II) complexes. J. Mol. Struct. 2012, 1019, 110–119. [Google Scholar] [CrossRef]
- Bañuelos-Hernández, A.E.; Mendoza-Espinoza, J.A.; Pereda-Miranda, R.; Cerda-García-Rojas, C.M. Studies of (−)-pironetin binding to α-tubulin: conformation, docking, and molecular dynamics. J. Org. Chem. 2014, 79, 3752–3764. [Google Scholar] [CrossRef]
- Save, M.; Schappacher, M.; Soum, A. Controlled ring-opening polymerization of lactones and lactides initiated by lanthanum isopropoxide, 1. General aspects and kinetics. Macromol. Chem. Phys. 2002, 203, 889–899. [Google Scholar] [CrossRef]
- Libiszowski, J.; Kałużynski, K.; Penczek, S. Polymerization of cyclic esters of phosphoric acid. VI. Poly(alkyl ethylene phosphates). Polymerization of 2–alkoxy-2–oxo-1,3,2–dioxaphospholans and structure of polymers. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 1275–1283. [Google Scholar] [CrossRef]
- Sosnowski, S.; Libiszowski, J.; Słomkowski, S.; Penczek, S. Thermodynamics of the polymerization of ethylene methyl phosphate. Makromol. Chem. Rapid Commun. 1984, 5, 239–244. [Google Scholar] [CrossRef]
- Fliedel, C.; Vila-Viçosa, D.; Calhorda, M.J.; Dagorne, S.; Avilés, T. Dinuclear zinc–N-heterocyclic carbene complexes for either the controlled ring-opening polymerization of lactide or the controlled degradation of polylactide under mild conditions. Chem. Cat. Chem. 2014, 6, 1357–1367. [Google Scholar] [CrossRef]
- Ivchenko, P.V.; Shlyakhtin, A.V.; Nifant’ev, I.E. Ring-opening polymerization of glycolide and rac-lactide, catalyzed by aryloxy magnesium complexes: DFT study of reaction profile and stereocontrol mechanism. Mendeleev Commun. 2017, 27, 278–280. [Google Scholar] [CrossRef]
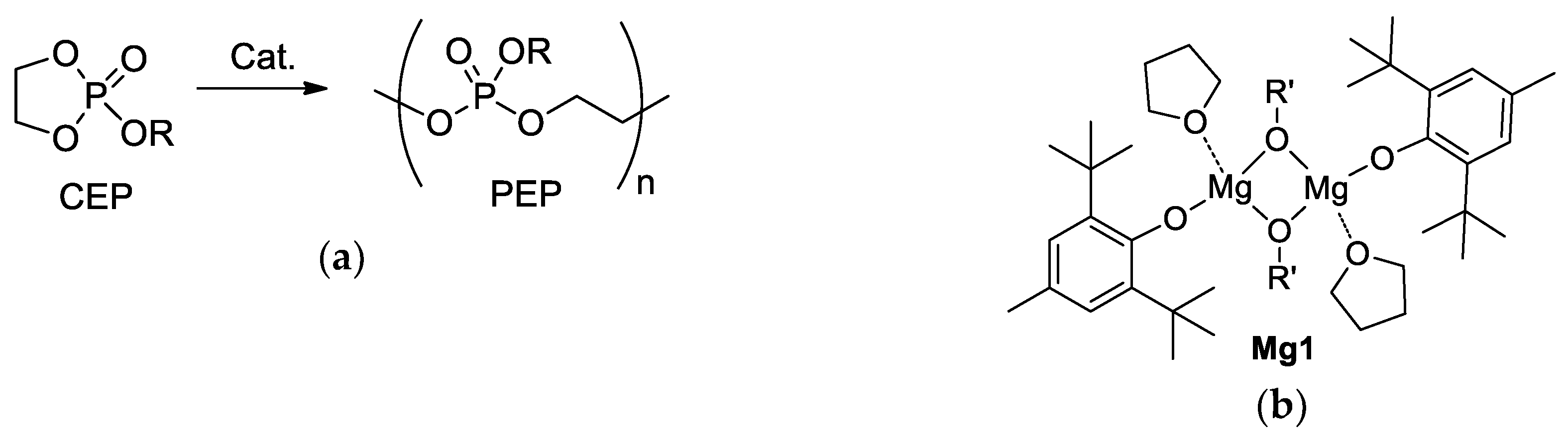





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | MI-1 | MTS-12 | MI-2 | MTS-23 | MI-3 | MI-4 | MI-1m |
| ΔGMI-1, kcal/mol | 0.0 | 6.6 | −2.8 | 8.5 | −3.7 | −7.0 | −2.3 |
| ΔHMI-1, kcal/mol | 0.0 | 3.5 | −6.0 | 3.6 | −7.1 | −8.9 | −12.4 |
| Molecule | MTS-45 | MI-5 | MI-5m | MTS-56 | MI-6 | MI-6m | MI-4m |
| ΔGMI-1, kcal/mol | −6.8 | −7.6 | −0.2 | 2.7 | −11.4 | −1.7 | −5.7 |
| ΔHMI-1, kcal/mol | −10.2 | −9.9 | −17.7 | −2.9 | −13.8 | −17.6 | −16.8 |
| Molecule | DI-1 | DI-1i | DTS-12 | DTS-12i | DTS-12r | DI-2i | DI-2r |
| ΔGMI-1, kcal/mol | 0.0 | −3.2 | 13.1 | 12.5 | 17.4 | 10.7 | 9.5 |
| ΔHMI-1, kcal/mol | 0.0 | 13.4 | 9.4 | 23.8 | 28.6 | 21.5 | 22.8 |
| Molecule | DTS-23i | DI-3i | DI-3 | DTS-34a | DTS-34b | DI-4a | DI-4b |
| ΔGMI-1, kcal/mol | 10.7 | −6.1 | −14.7 | 3.8 | 8.4 | 2.7 | 6.5 |
| ΔHMI-1, kcal/mol | 21.5 | 6.8 | −13.1 | 0.9 | 5.1 | −0.8 | 5.3 |
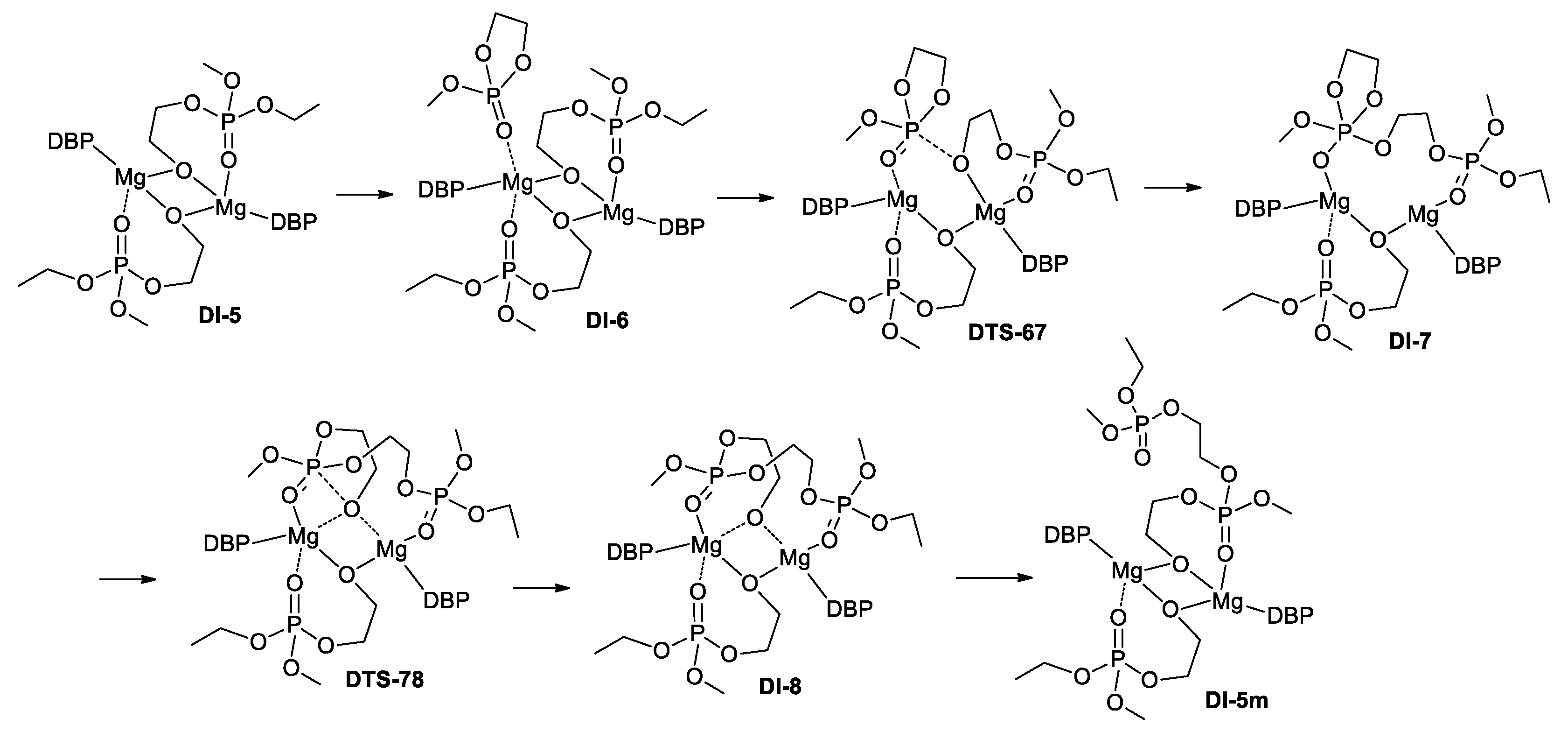
| Molecule | DI-5 | DI-6 | DTS-67 | DI-7 | DTS-78 | DI-8 | DI-5m |
| ΔGMI-1, kcal/mol | −21.1 | −9.6 | −1.8 | −9.9 | 4.1 | 4.1 | −16.6 |
| ΔHMI-1, kcal/mol | −19.2 | −21.6 | −15.5 | −25.1 | −13.4 | −10.0 | −24.1 |
| Run | Monomer | Reac. time, (a) min | Reac. T, °C | Conv., % (b) | Mntheo × 103 (c) | MnNMR × 103 (d) | MnSEC × 103 | ÐM | ΔG≠ kcal/mol |
|---|---|---|---|---|---|---|---|---|---|
| 1 | εCL | 10 | 5 | 54 | 6.3 | 6.8 | 6.44 (e) | 1.18 | 21.7 (f) |
| 2 | rac-LA | 10 | 5 | 67 | 9.8 | 10.1 | 9.65 (e) | 1.24 | 21.6 (f) |
| 3a | L-LA | 10 | 5 | 9 | 1.5 | n.d. (g) | n.d. | n.d. | 31.2 (f) |
| 3b | L-LA | 45 | 5 | 37 | 5.4 | 5.7 | 5.55 (e) | 1.21 | 29.4 (f) |
| 3c | L-LA | 240 | 5 | 98 | 14.5 | 15.3 | 15.28 (e) | 1.20 | 29.4 (f) |
| 4 | MeOEP | 10 | 5 | >99 | 13.9 | 13.7 | 9.8 (h) | 1.35 | ~14 |
| 5 | MeOEP | 10 | −20 | 96 | 13.9 | 13.2 | 10.0 (h) | 1.32 | ~14 |
| 6 | MeOEP | 10 | −50 | 81 | 11.2 | 11.4 | 8.8 (h) | 1.26 | ~14 |
| 7 | MeOEP/rac-LA | 10 | 5 | >99/82 | 13.2 | 15.9 | 19.5 (h) | 1.62 | ~14/18.2 (i) |
| 8 | MeOEP/L-LA | 10 | 5 | >99/62 | 10.3 | 12.5 | 15.7 (h) | 1.71 | ~14/22.7 (i) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nifant’ev, I.; Shlyakhtin, A.; Kosarev, M.; Karchevsky, S.; Ivchenko, P. Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers 2018, 10, 1105. https://doi.org/10.3390/polym10101105
Nifant’ev I, Shlyakhtin A, Kosarev M, Karchevsky S, Ivchenko P. Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data. Polymers. 2018; 10(10):1105. https://doi.org/10.3390/polym10101105
Chicago/Turabian StyleNifant’ev, Ilya, Andrey Shlyakhtin, Maxim Kosarev, Stanislav Karchevsky, and Pavel Ivchenko. 2018. "Mechanistic Insights of BHT-Mg-Catalyzed Ethylene Phosphate’s Coordination Ring-Opening Polymerization: DFT Modeling and Experimental Data" Polymers 10, no. 10: 1105. https://doi.org/10.3390/polym10101105