In this work, we determined that the relative reactivities of
Tmc and
Lac in the copolymerization differ considerably from their reactivities in homopolymerizations, similarly as it was observed for ε-caprolactone/
Lac systems initiated with the same initiator
Ini [
32]. Significant discrepancies in reactivities of active centers differing in configuration of the used initiator were already observed in
Lac homopolymerization studies [
31]. It stems from different diastereomeric arrangements at the end of growing chains formed by asymmetric
Lac terminal unit (
S configuration) and residue from
R- or
S-
Ini. Results of our reference
Lac homopolymerizations, performed for [
Lac]
0 = 1.2 mol·L
−1, are shown in
Supporting Information, confirming large differences in rates of polymerization. On the other hand, one cannot expect any differences in
Tmc homopolymerization rates, what was confirmed experimentally (initial rate coefficients equal to about 0.088 and 0.093 (±5%) L·mol
−1·s
−1, for polymerizations initiated with
R-
Ini and
S-
Ini, respectively). Therefore, we could expect that the copolymerization reactivity ratios change by altering the active-center initiator-residue configuration, what can result in quite different copolymer structures. In fact, the differences in reactivity ratios were larger than expected by us.
This striking phenomenon is of general importance, since it provides a useful tool for tuning the resultant copolymer microstructure and properties.
3.1. Outlook of General Features of Copolymerization Kinetics
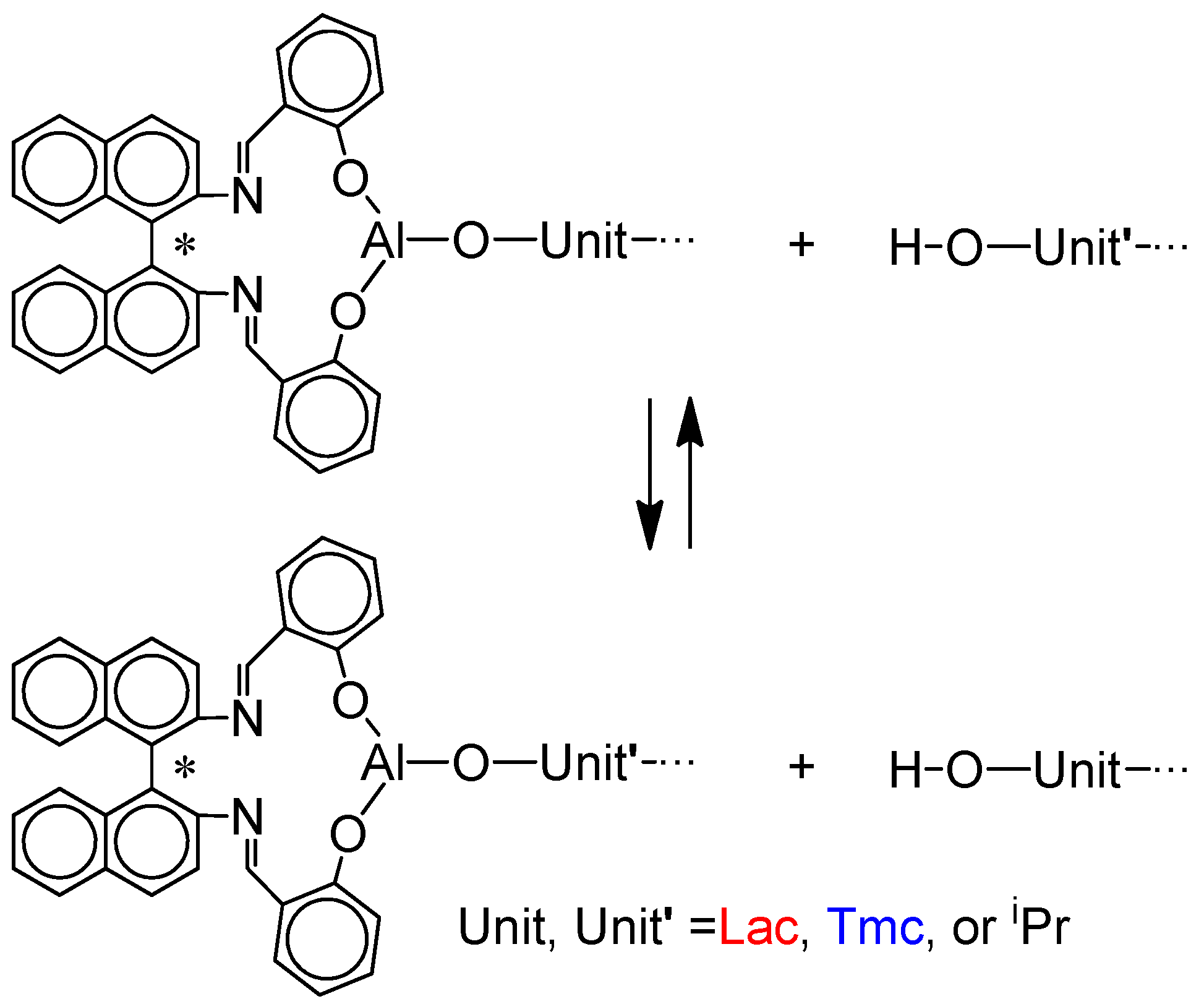
The propagation and depropagation reactions, describing the studied copolymerization, are shown in
Scheme 4.
One can note that diastereomeric arrangements imply differences in propagation and depropagation rate constants, in relation to configuration of used initiator, with the exception of the last reversible reaction in the
Scheme 4: homopropagation and corresponding depropagation of
Tmc (apparently no diastereometry, one can expect identical reactivity of enantiomeric
Tmc-
Res *).
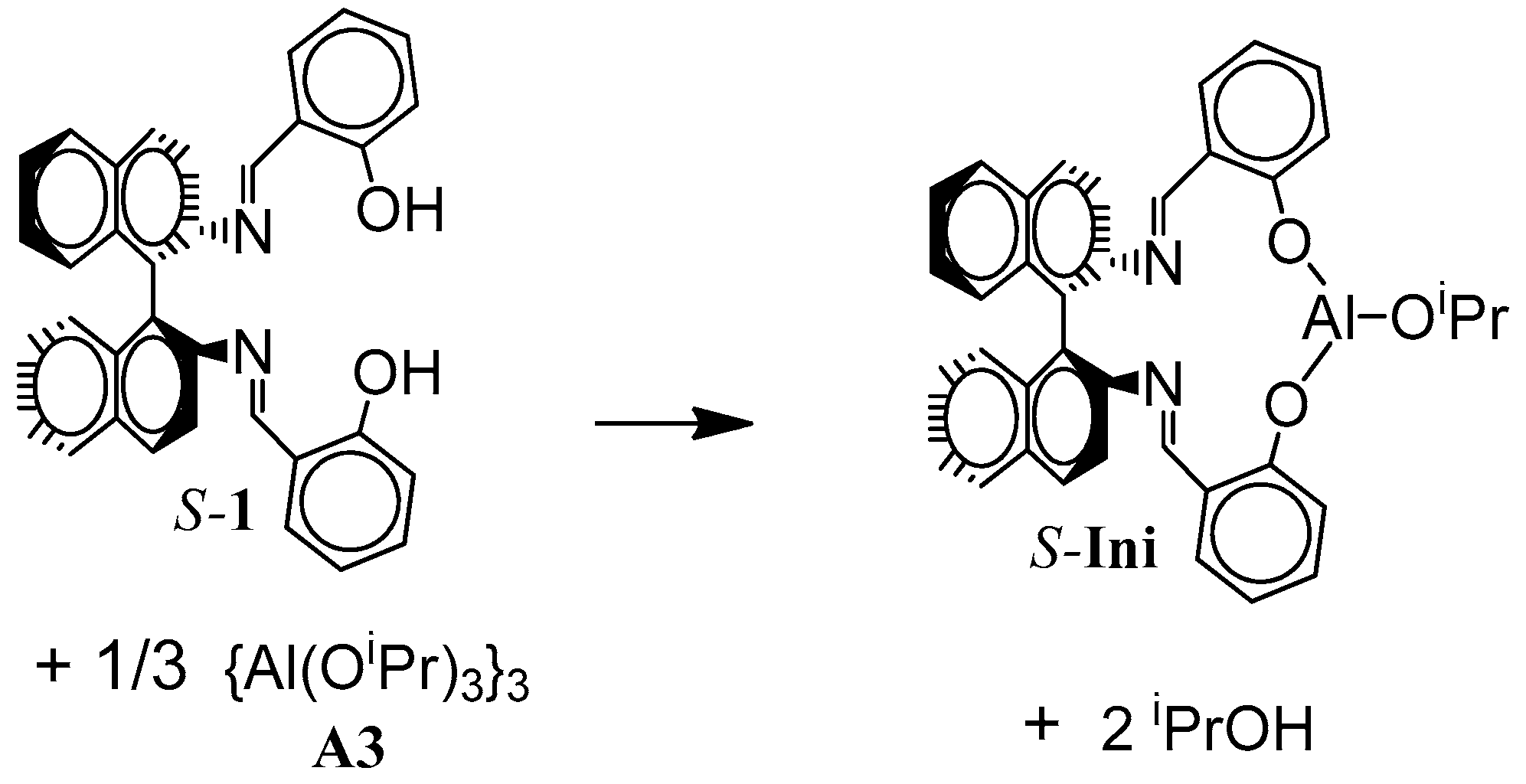
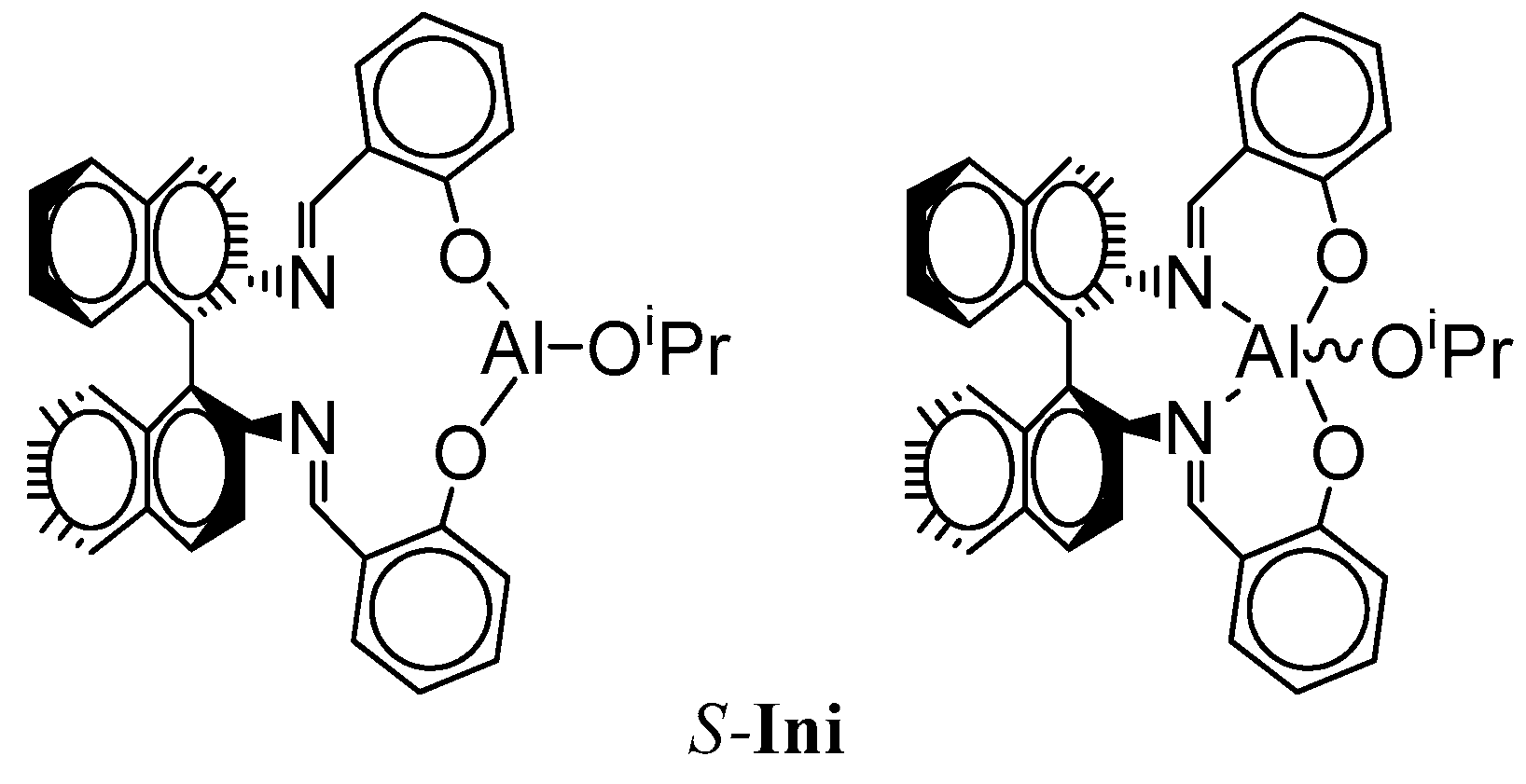
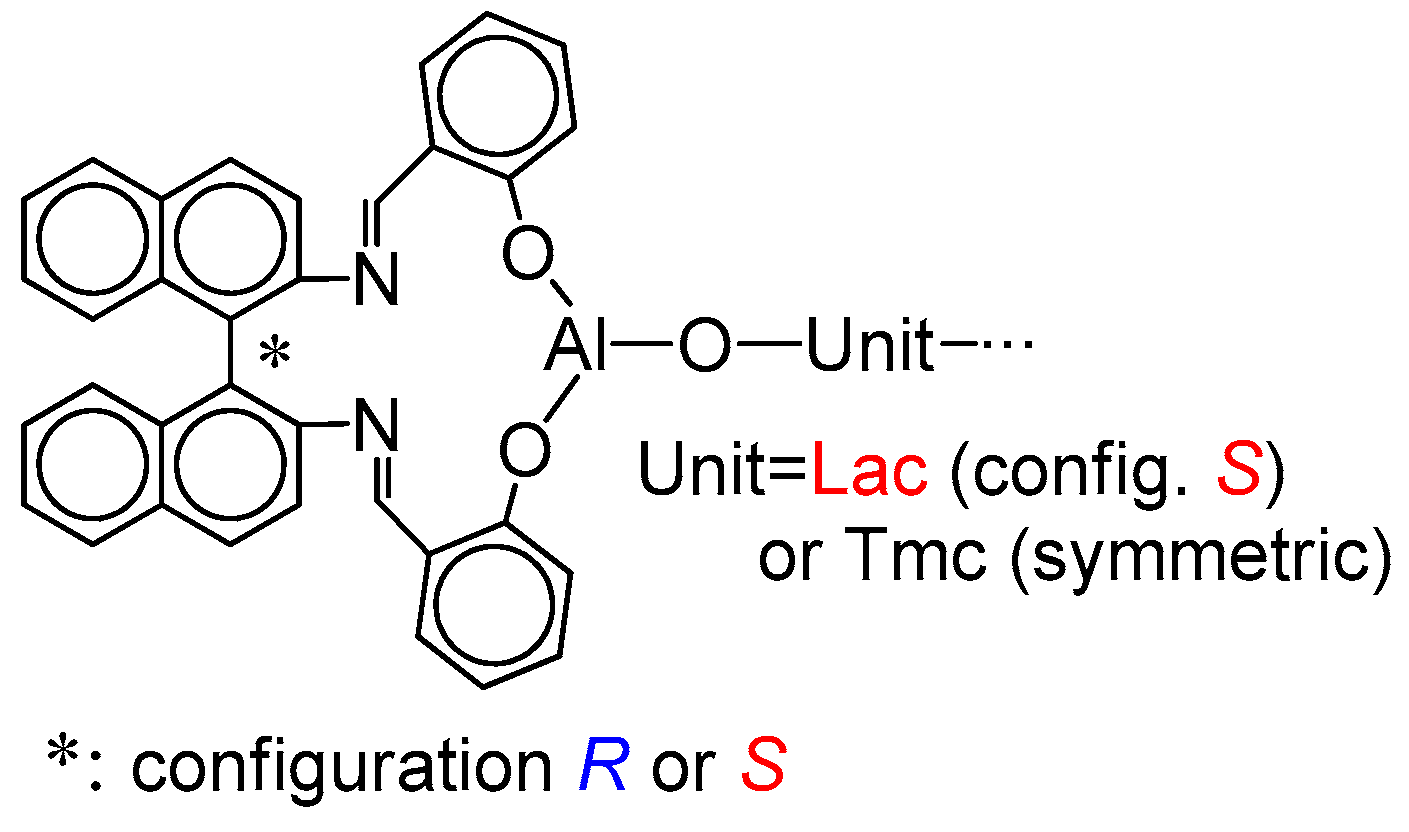
The structure of the active species is shown in
Scheme 5. If the copolymer unit is symmetric (
Tmc), one can expect that the rate of insertion of alike monomer molecule into Al-O-Unit bond does not depend on configuration of initiator residue
Res at the chain-end. On the other hand, addition of asymmetric monomer molecule (
Lac) depends on configuration of
Res because we can have two different diastereomeric arrangements here. Similarly, when Unit is asymmetric (
Lac), addition of any of comonomers (
Lac or
Tmc) depends on configuration of
Res (reactions involving diastereomeric active species).
Thus, depending on the configuration of Ini, copolymerization can proceed in a different way.
In principle, copolymerization kinetics can be monitored by any method giving access to comonomer conversions. Unfortunately, spectroscopic methods we considered (UV, IR,
1H NMR) could not be used effectively because of the lack of sufficiently separated signals of the comonomers and the copolymer. Only
13C NMR spectra could give the corresponding information. Unfortunately, due to technical problems (taking samples from the reaction mixture avoiding its contamination, followed by isolation of product) only a few kinetic data points could be obtained. Much more convenient methods seemed polarimetry for following conversion of
Lac and dilatometry for following conversion of
Tmc (polymerization of
Lac results in no change of the reaction system volume), following the approach, applied by Florczak and Duda in analysis of copolymerization kinetics of
Lac with ε-caprolactone [
32].
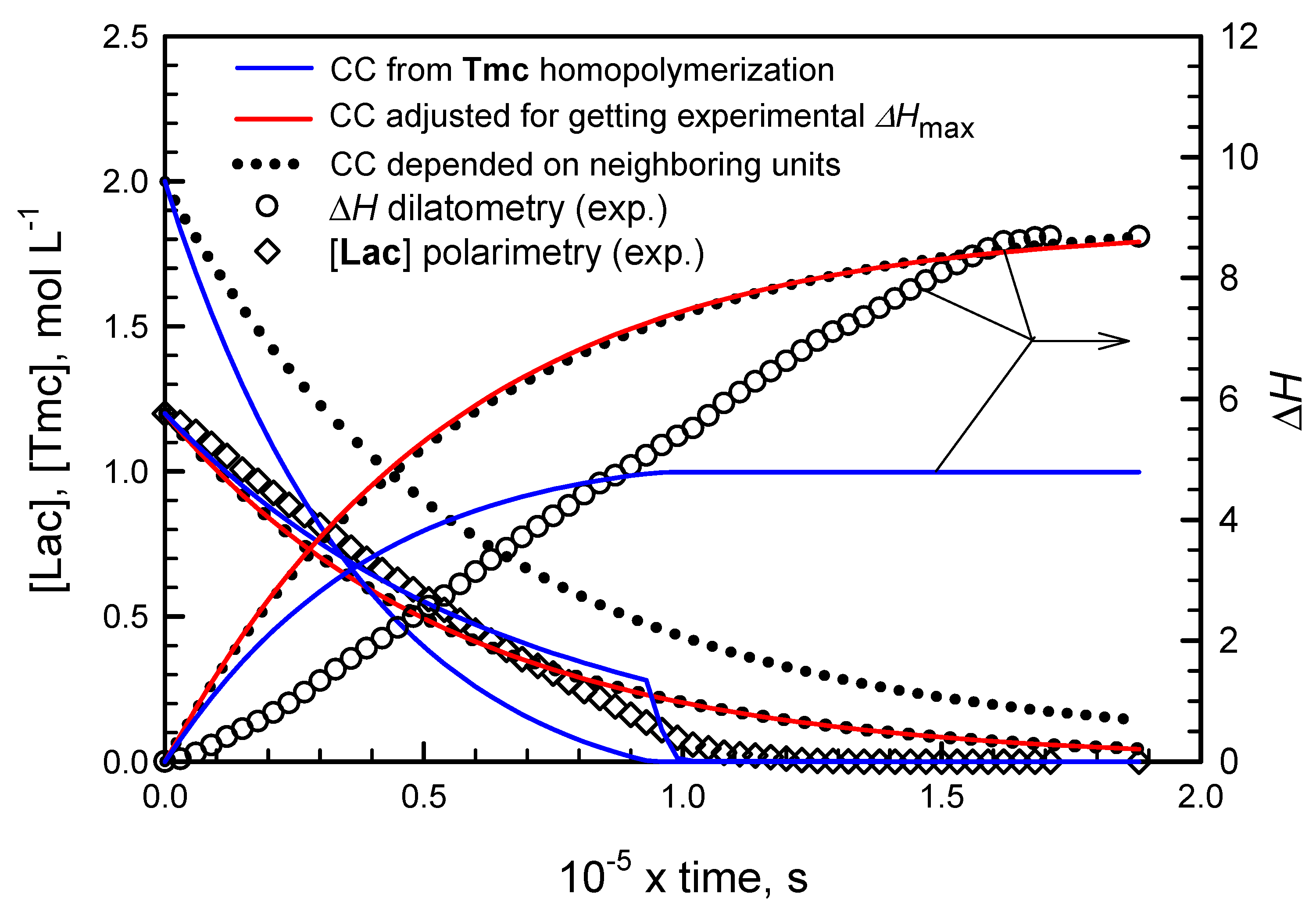
However, it appeared that following conversion of Tmc with dilatometry was not straightforward. We have observed that changes of the system volume were significantly larger than expected on the basis of contraction coefficients determined from homopolymerization of Tmc. Moreover, conversion of Tmc determined from dilatometry in a standard way was significantly different from that obtained from 13C NMR, available for a few reaction times of one copolymerization system, initiated with the mixture of R- and S-Ini (cf. below in the corresponding section).
Consequently, we came to conclusion that contraction coefficients for Tmc and, possibly, also for Lac units, depend on the type of triad in which the given unit occupies the central place. Thus, in order to be able to use dilatometry for following the Tmc conversion, we had to determine, or at least estimate, three values of contraction coefficients for any of comonomers, e.g., for A unit coefficients for homotriad AAA, heterotriad BAB, and the average value for asymmetric triads AAB and BAA. The average value for asymmetric triads is sufficient because in copolymer of sufficiently long chains the numbers of triads AAB and BAA are virtually equal.
However, in order to estimate these parameters directly we would have to have a sufficiently large number of experimental data describing the relationship between comonomer conversions and copolymer microstructure (triad level), and volume contraction corresponding to the given samples.
Unfortunately,
13C NMR did not give the sufficient information about triad (nor dyad) contributions, because some triads, assigned according to Dobrzynski and Kasperczyk [
23], overlap (e.g., of triads Tmc
LLTmc, LacL
LTmc, and Tmc
LLLac: LL means here
Lac unit, composed of two lactic units L, in bold are marked the lactic units relevant to overlapping signals, cf.
Table 1).
Signals of copolymer structures stemming from segmental exchange (e.g., of the isolated lactic unit TmcLTmc) were observed by us only in spectra of systems kept more than 24 h after completing copolymerization which confirmed our assumption that reshuffling can be neglected.
We managed to estimate the contraction coefficients from analysis of copolymerization kinetics. Unfortunately, some assumptions, leading eventually to estimates valid only for the assumed model of copolymerization, had to be adopted. These assumptions were as follows:
These assumptions allowed us to describe copolymerization systems entirely by the kinetic
Scheme 4, not accounting OH-terminated chains. Such a simple model of copolymerization appeared to be useful if copolymerization main features, such as copolymer composition and microstructure, are concerned. However, when more detailed features, like molar mass distribution are to be analyzed, the copolymerization model including the rate of exchange of OH-terminated chains with ones bearing active species, has to be used, as shown below in the paper.
Nevertheless, kinetics of irreversible copolymerizations following
Scheme 4 (with all depropagation rate constant equal to zero) can be predicted from integration of the corresponding kinetic differential equations. On the other hand, Monte Carlo simulations, taking into account depropagations, could confirm validity of neglecting them, as well as could give access to detailed description of copolymer microstructure.
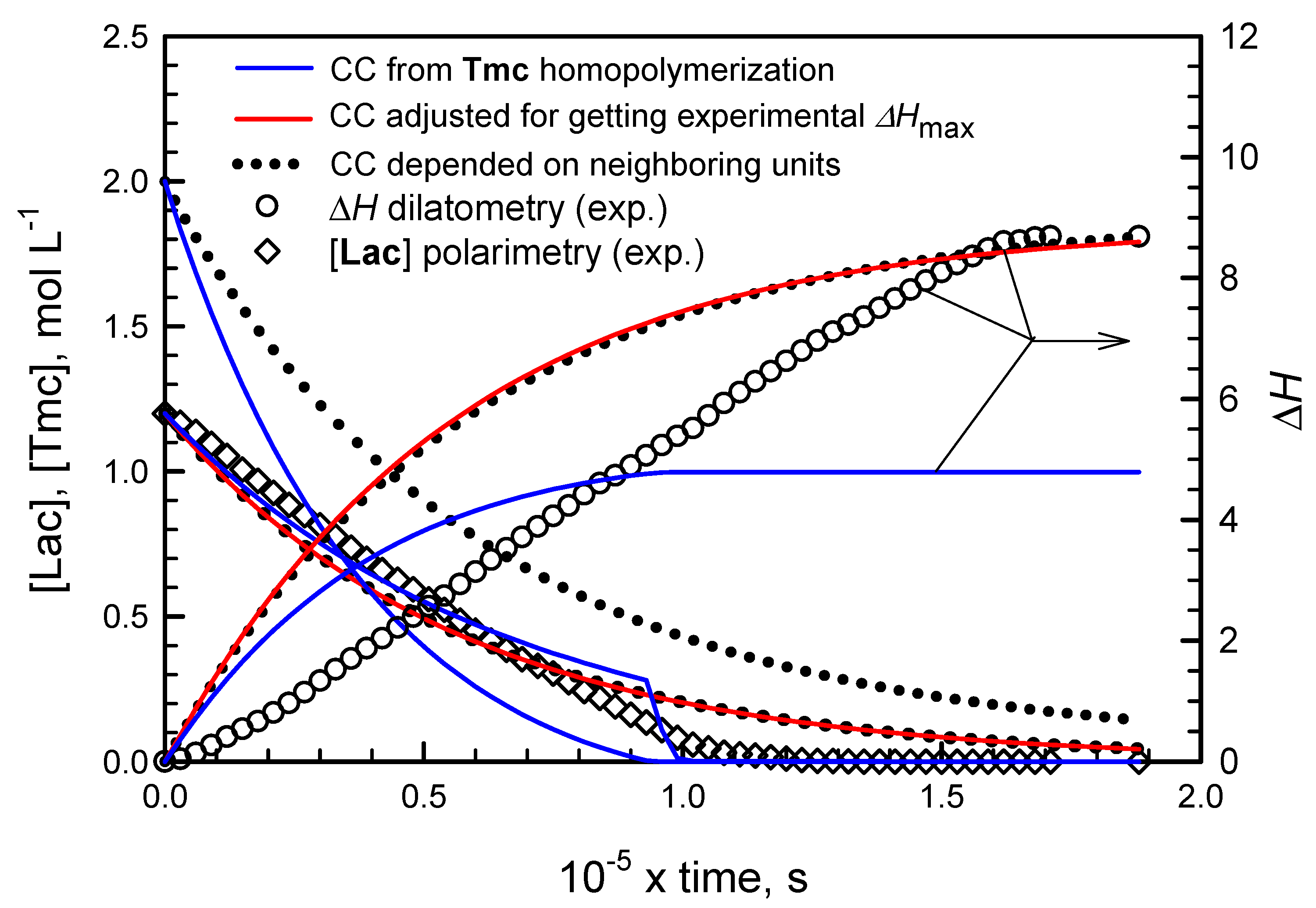
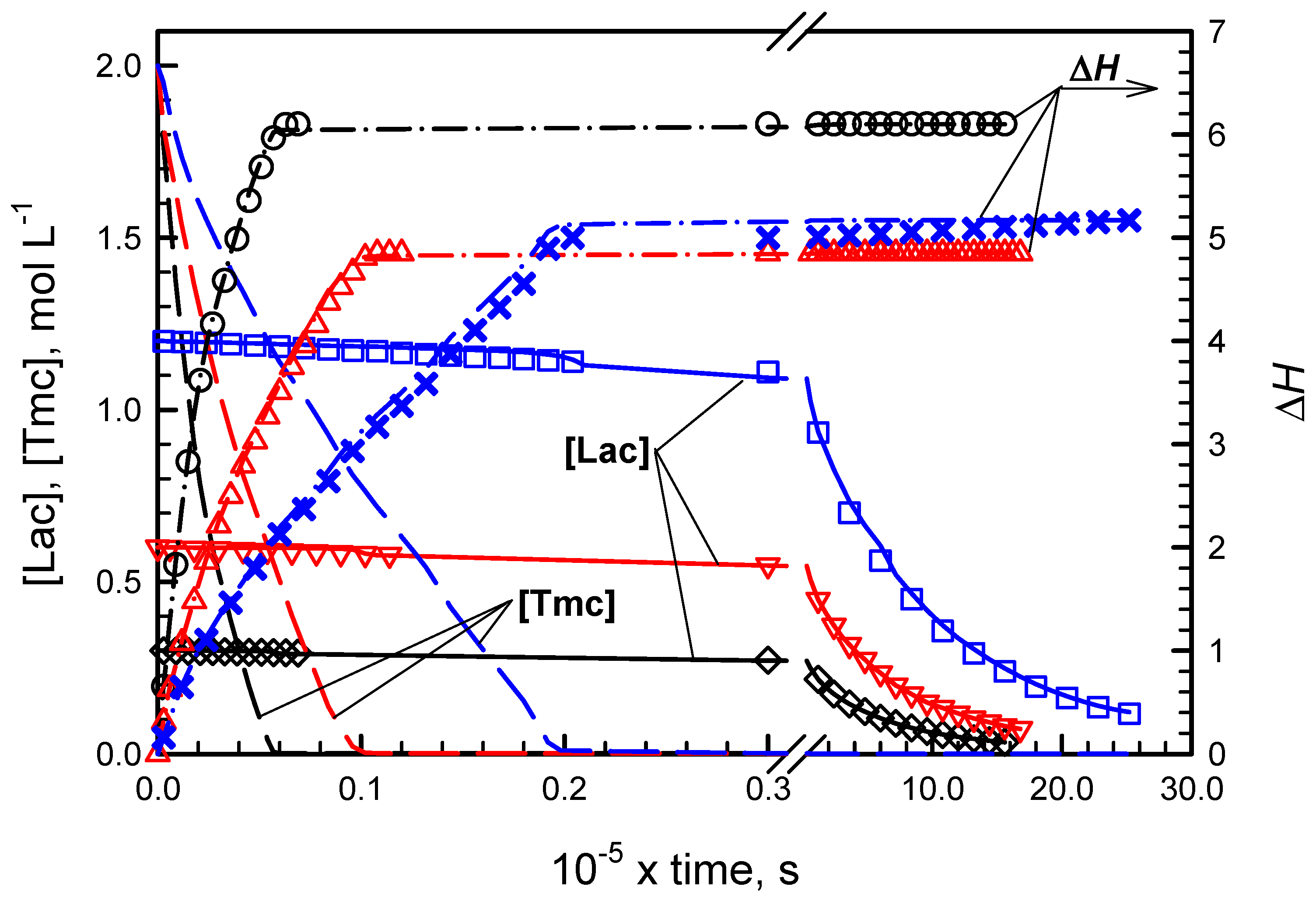
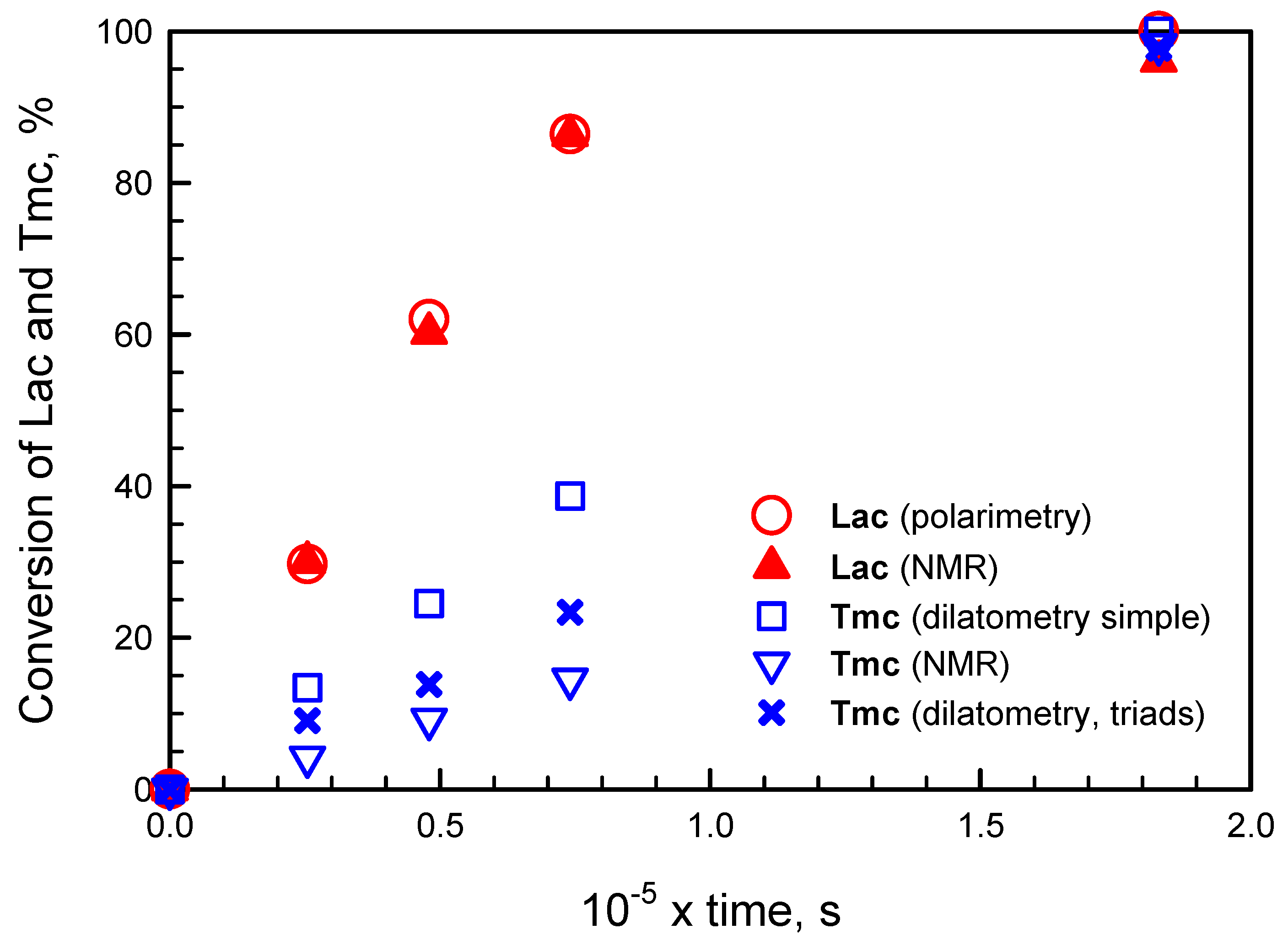
Applying numerical integration of differential kinetic equations we could predict evolution of copolymerization in time. However, attempts to fit rate coefficients failed, indicating that comonomer consumption rates seemed not to decrease with comonomer concentrations as expected. Rates of consumption of comonomers were initially lower than those predicted from simulations and eventually higher: the apparent rate coefficients seemed to increase with conversion. An example of such fitting is given in
Figure 1. More details concerning this type of kinetic analysis are given in
Supporting Information.
The presented plots suggest that apparent rate coefficients change with conversion, being initially lower than obtained from parameter fitting (experimental slope for evolution of [Lac] initially lower than obtained in simulations) and, at the end of comonomer consumption, the rate coefficients seem to be higher than obtained from simulations (the corresponding slope for experimental points for reaction times longer than 1000 min is higher than that of fitted plots).
Important is also observation that volume contraction coefficients for copolymer units differ from those observed in homopolymerizations. Either contraction coefficients for
Tmc unit depend on other units neighboring the given one, or the same can be said about
Lac units (contraction coefficients for some triads with
Lac located in the middle not equal to zero as in homopolymerization), or contraction coefficients for both types of copolymer units depend on copolymer microstructure. Our fitting analysis (see below and
Supporting Information) suggest that the third possibility is true.
Analyzing simulated kinetic curves in comparison to experimental ones we deduced that the reaction medium, continuously changing while concentrations of comonomers and copolymer units evolve, makes the rate coefficients depend on conversion. It can be understood as both comonomer molecules and copolymer units solvate active species in varying proportions at different conversions. As Lac molecule and copolymer units are asymmetric one cannot exclude some diastereomeric effect making the apparent rate coefficients dependent on conversion.
In order to get a relatively simple kinetic model of copolymerization consistent with experimental data we have assumed that the relative changes of apparent rate coefficients with conversion are approximately identical for all reactions, changing simultaneously, resulting in constant ratios of rate constants. The analyzed models of copolymerization and methods of fitting apparent relative rate coefficients to experimental data are described in detail in
Supporting Information. Here we only indicate the main result of this analysis. Namely, if the abovementioned assumption is valid then the kinetics presented in conversion scale is characterized by kinetic parameters independent of conversion or reaction time. These kinetic parameters are ratios of instantaneous (changing) rate coefficients of reactions operating in the system. One can choose different ratios to describe the analyzed copolymerizations but our choice was given as below.
Initially, we related all rate constants to the
Tmc homopropagation rate constant
kTT (see
Scheme 4), chosen as the rate constant presumably independent of configuration of
Ini and denoted the corresponding ratios
zXY =
kXY/
kTT, where X, Y is L and/or T, which stand for
Lac or
Tmc comonomer/unit.
However, one can easily find that one can relate these
zXY parameters (XY different than LL) with
zLL and standard parameters such as reactivity ratios and, for depropagation rate constants, additionally with the equilibrium constants. These relationships are presented in the equation set (1). Starting from this equation set we use letters L and T, while denoting with
Lac and
Tmc comonomers/comonomer units, respectively. Besides, in all equation sets we use for these letters red and blue color, respectively. The same colors are used in some Figures and plots describing copolymer units or blocks related to the studied comonomers.
Thus, we could formulate the kinetic differential equations in conversion scale, with only the limited number of the mentioned relative kinetic parameters:
zLL and reactivity ratios
rL and
rT (and for not negligible depropagations also the equilibrium constants), taking into account the following relationships (formulated here for irreversible copolymerization):
where [ ] in Equation (2) corresponds to concentration of any reagent, species, or copolymer sequence in copolymer. For instance, the corresponding equations formulated for evolution of concentrations of
Lac (L) monomer and LT dyads are given by equation set (3):
Applying the formulated set of kinetic differential equations in conversion scale (see
Supporting Information) to experimental data allowed us to predict main features of copolymers obtained in the studied systems.
3.2. Dependence of Copolymer Structure on Configuration of Ini
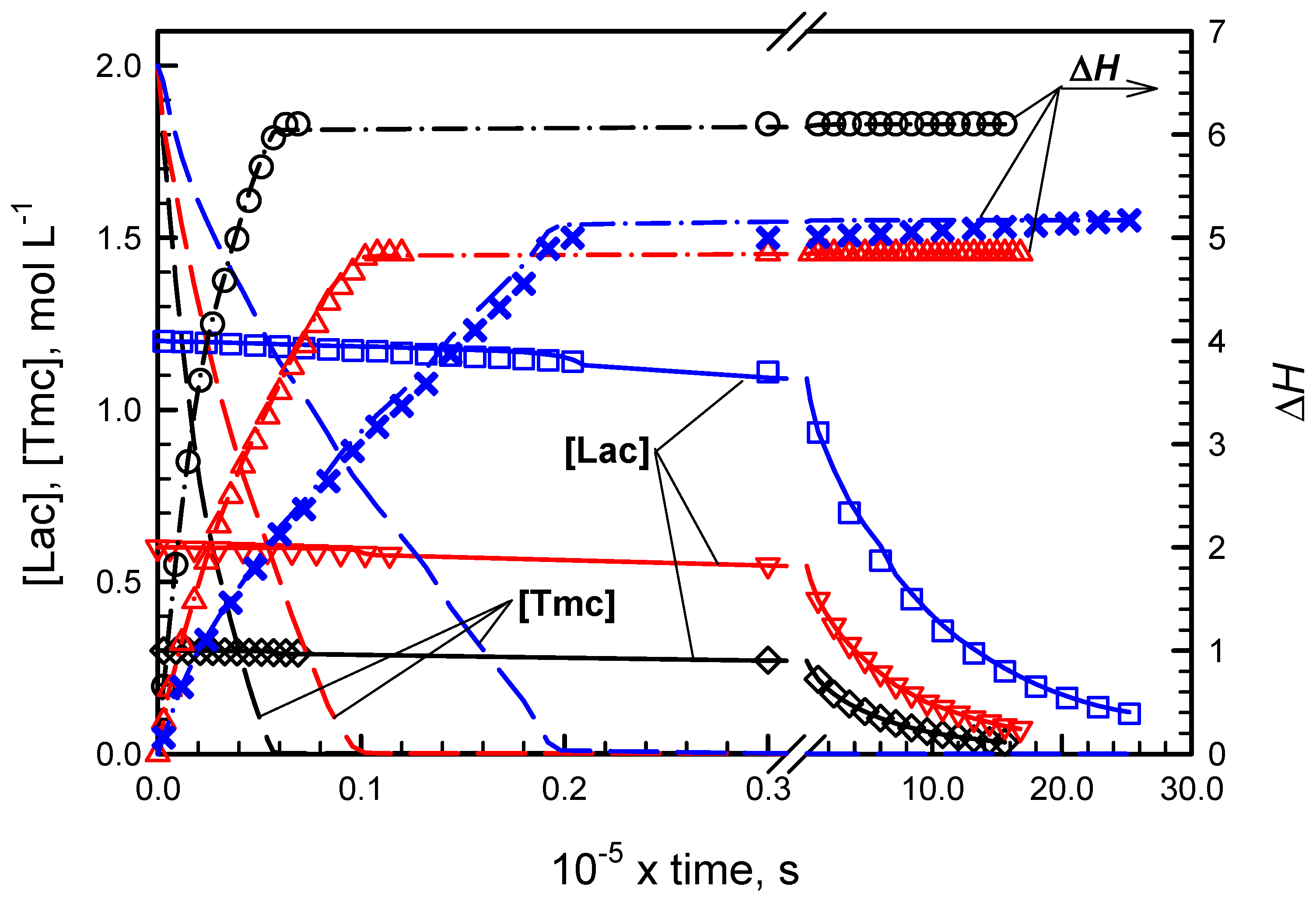
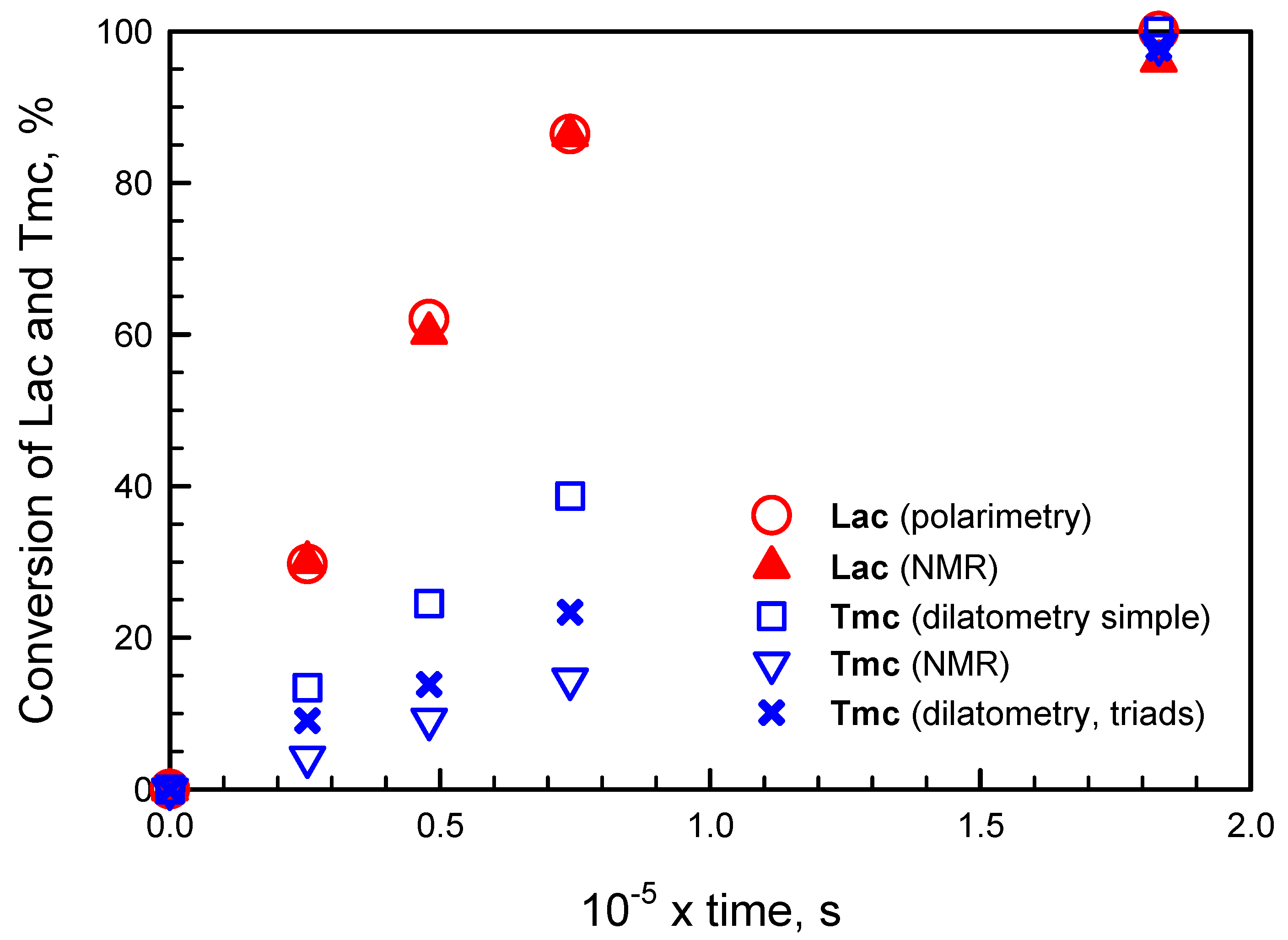
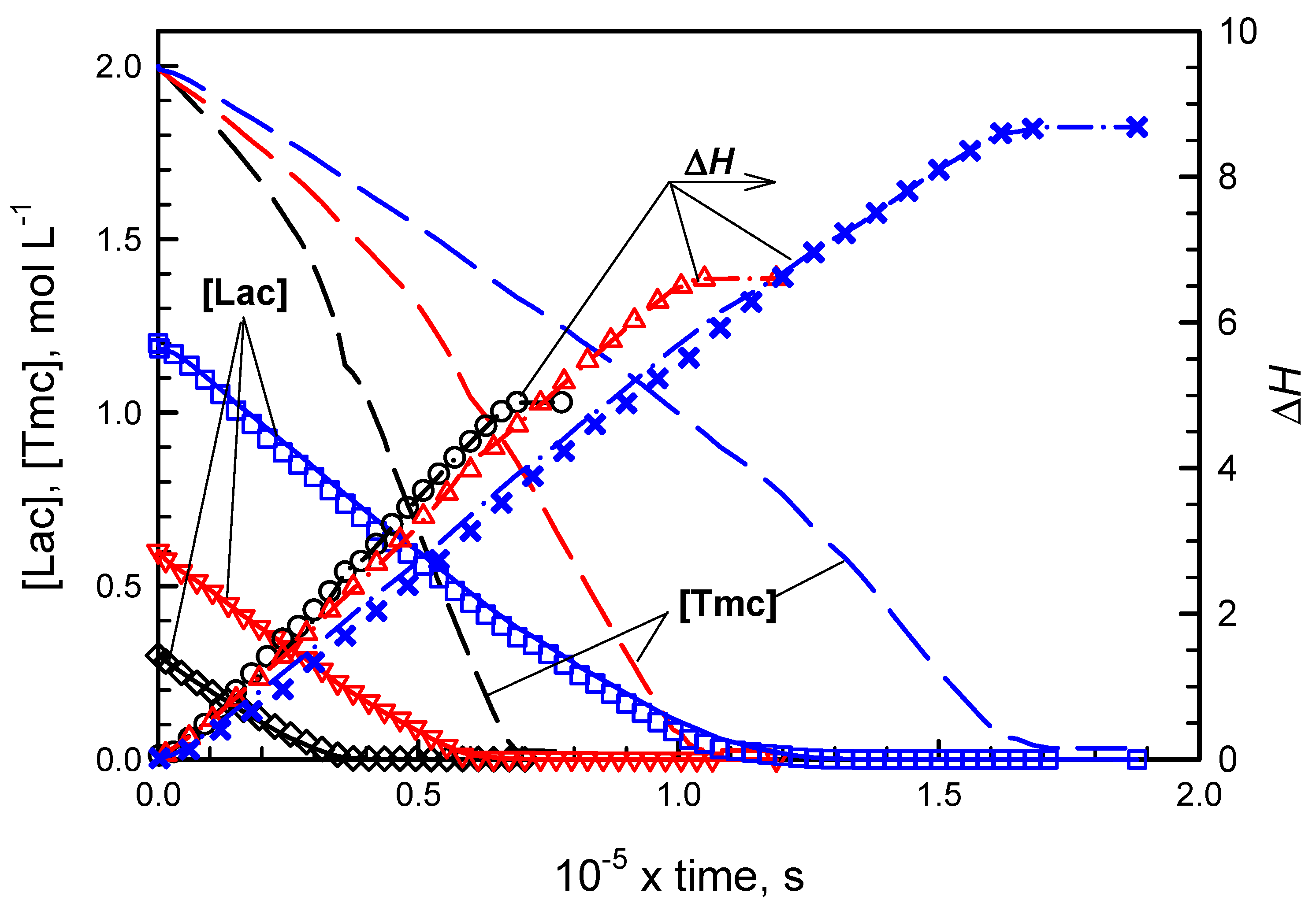
Figure 2 and
Figure 3 present conversions of comonomers in copolymerizations initiated with
R- and
S-
Ini, respectively. Conversions of
Lac were detected directly due to polarimetric measurements while conversions of
Tmc were obtained from the elaborated kinetic analysis presented in
Supporting Information. The kinetic and contraction parameters were fitted to describe simultaneously three copolymerization experiments initiated with the given enantiomer of
Ini, differing with the initial
Lac concentrations.
While in systems with R initiator enantiomer, initially mostly Tmc is consumed, giving presumably the product resembling diblock copolymer, in systems with S-Ini we obtained product containing in the initial parts of chains both comonomers in similar quantities. The chains are terminated eventually with Tmc blocks due to the fact that Lac was consumed before Tmc, used in significant excess. Kinetic analysis confirms this description of products giving both reactivity ratios much higher than unity in R-Ini systems and the product of reactivity ratios lower than unity in S-Ini systems.
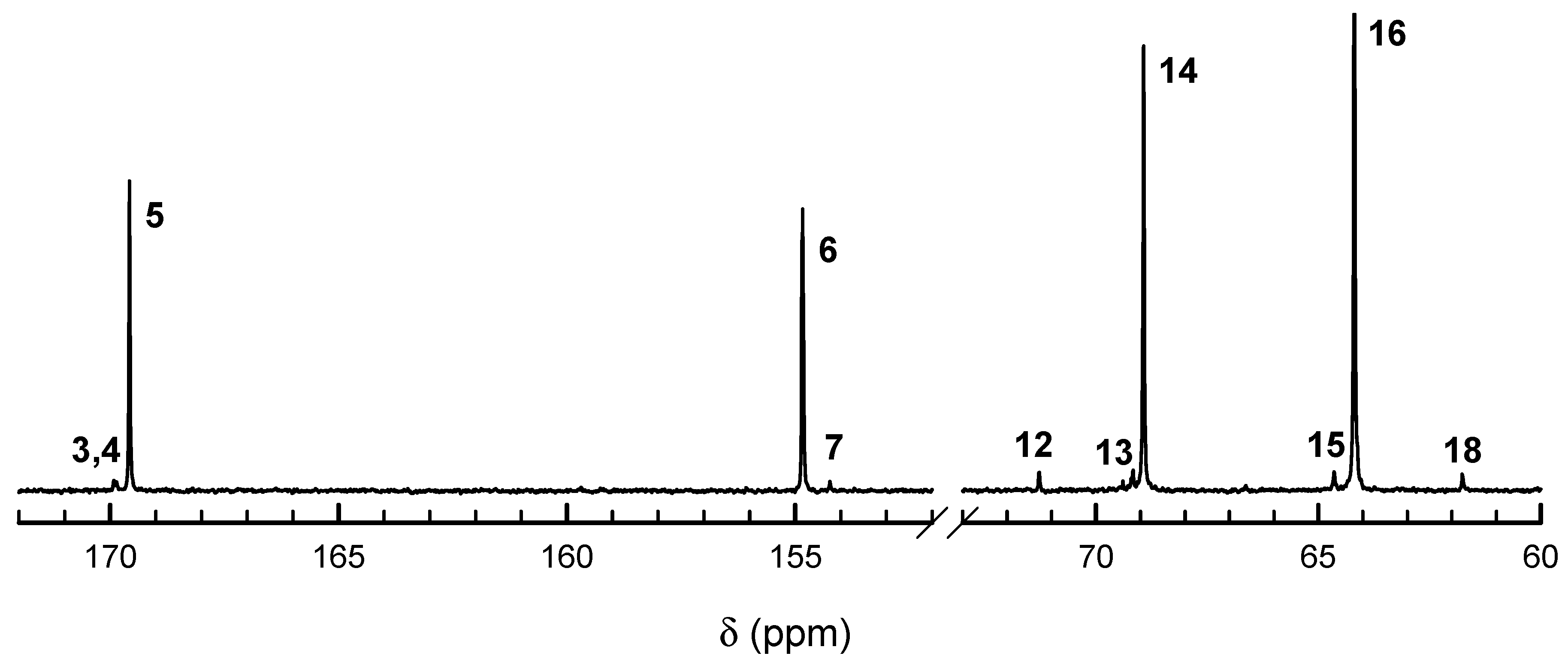
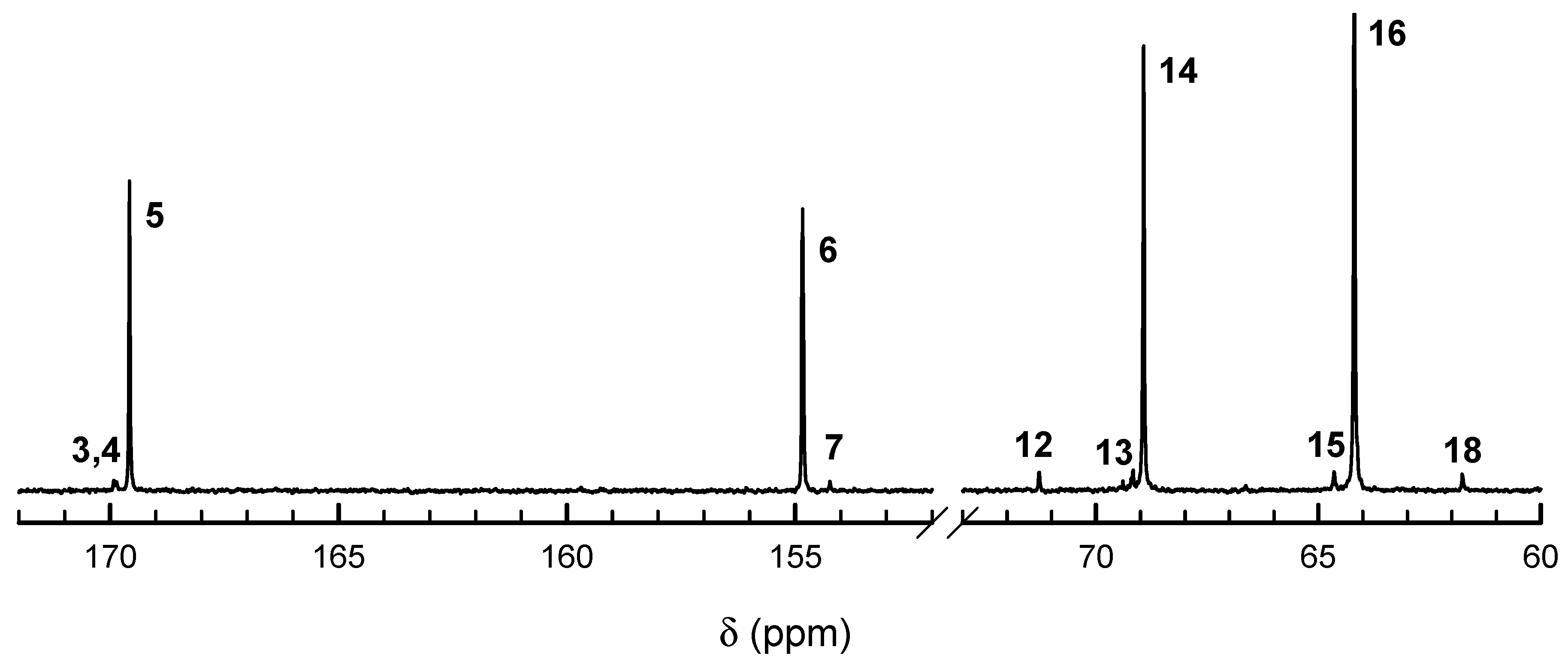
13C NMR spectra (
Figure 4 and
Figure 5) also confirm the above description of copolymers, indicating the large excess of homodyads in products initiated with
R-
Ini and a low quantity of homodyads LacLac in copolymer initiated with
S-
Ini, what indicates some tendency to alternacy, as expected from the estimated product of reactivity ratios being significantly lower than unity. Assignment of
13C NMR signals is given in
Table 1.
Our kinetic analysis allowing us to estimate the reactivity ratios for the studied systems is described in details in
Supporting Information. It was based on fitting of the simulated evolution of the studied systems, described by the formulated differential kinetic equations with the relative kinetic parameters, including reactivity ratios, to experimental evolution of the studied systems.
Main results of the primary kinetic analysis are given in
Table 2 while the
Figure 2 and
Figure 3 present experimental and computed from kinetic analysis evolution of copolymerizations initiated with
R and
S enantiomers of
Ini.
Rather unexpected was the finding that the difference between Tmc reactivity ratios in relation to configuration of Ini (rT(R) > rT(S)) is as large as three orders of magnitude. It stems probably from strong steric hindrance of R-Ini residue of active species while the transition state for Lac addition to Tmc * is formed. The corresponding hindrance for Tmc addition is smaller because of the lack of methyl groups, present in Lac. On the other hand, the smaller difference observed for Lac reactivity ratios (rL(R) > rL(S)) and indicating that addition of Lac to Lac * is faster than addition of Tmc for both configurations of Ini, is probably due to relatively large differences in activation enthalpies of the corresponding reactions.
It is important to indicate here that our fitting computations could not estimate the
zLL parameter in copolymerizations initiated with
S-
Ini, nor initiated with the mixture of
Ini enantiomers. It is so, because of the systems quickly attaining steady state conditions maintaining the proportion of active species. These steady state conditions, applied also while deriving Mayo-Lewis equations, are generally accepted while analyzing copolymerization kinetics. However, when at least one of the reactivity ratios is very high, attaining of the steady state conditions can require quite large conversions. For such systems, here observed for
R-
Ini initiated
Lac/
Tmc copolymerization, the Mayo-Lewis equations can be regarded as giving only crude estimates for relative rates of comonomer consumptions. In fact, the
zLL parameter in ‘normal’ copolymerizations determines the steady state condition ratio of concentrations of
Lac * and
Tmc * active species, not influencing the relative rates of comonomer consumption. The estimation of the
zLL =
kLL/
kTT parameter can be done only applying some specific methods, for instance analyzing in details the molar mass distribution in relation to chain compositions [
37].
Thus, in our simulations, when fitting the relative kinetic parameters for
S-
Ini systems by minimization of the defined objective function (see
Supporting Information), we observed, as expected, independence of the fitting results for
rL and
rT, for any assumed value for
zLL in the range between 10
−2 and 10
2 (not checked outside this range).
On the other hand, due to non-steady state conditions up to high conversions, while fitting relative parameters for
R-
Ini systems we could attain minima of the objective function (see
Supporting Information), giving different results for
rL and
rT, for any assumed value of
zLL. Besides, also due to slowly attaining the steady state conditions, the fitting results depended on the assumed proportion of initiating unimers. The observed minima were on quite similar levels for
zLL in the range between 10
−3 and 0.5, being on apparently higher levels outside this range. Thus, our estimates of the reactivity ratios
rL and
rT and of the relative rates of comonomer consumption for
R-
Ini systems, given in the
Table 2, are rather crude. They were obtained for
zLL equal to 4 × 10
−3, giving only slightly lower the objective function minimum than observed for different
zLL in the indicated range. Moreover, these estimates were obtained assuming initiation exclusively with
Tmc unimers. The last assumption was made because of the observed large differences of rates of homopolymerization of
Tmc and
Lac initiated with
R-
Ini,
Tmc polymerizing much faster, see
Supporting Information.
The results presented in
Table 2 indicate tremendous differences between copolymerization systems differing only in configuration of asymmetric bulky initiator. While
R-
Ini gives copolymer of virtually oligoblock structure: initially mostly only
Tmc is consumed, forming the corresponding blocks, and next the blocks of poly-
Lac are formed. The product of reactivity ratios is very high, indicating that practically negligible inserts of
Lac units in the initial, mostly homo-
Tmc parts of chains, are short blocks of
Lac rather than separate single
Lac units. Similarly, at the end parts of chains, being approximately the
Lac homo-blocks, one can expect only infrequent inserts of short homo-blocks of
Tmc. However, due to a very high
rL, similarly as of
rT, and
kLL <<
kTT, (
zLL estimated to be about 10
−2) some homo-
Lac chains, if formed in the initial part of copolymerization, grow slower than chains formed from
Tmc unimers. Some of these chains can survive till the end of copolymerizations, forming small, not negligible for systems with not sufficiently high
DPn, fractions of homo
Lac polymer, differing in the average molar mass from copolymer chains. One cannot also exclude formation of a fraction of homo-
Tmc polymer in some copolymerization systems. This characteristics of
R-
Ini copolymerization systems results in dispersity of product significantly higher than expected for random copolymerizations proceeding without side reactions like, for instance, segmental exchange or cyclizations. Even if one assumed that the system is initiated only with
Tmc unimers (as done by us in simulations giving
rL and
rT values for
R-
Ini systems in
Table 1), dispersity can be quite high, due to slow transformation of
Tmc * chains into
Lac * chains. This phenomenon resembles slow initiation, which also leads to broadened dispersity.
On the other hand, using as initiator the S-Ini results in initial rates of consumption of both comonomers not differing much and in some tendency to alternacy (product of reactivity ratios about 0.1). Due to the excess of Tmc in all studied copolymerization systems one can expect copolymer chains ended with homoblocks of Tmc containing some small amount of inserts of separated units of Lac.
Our Monte Carlo simulations confirm the above description of copolymer chains, deduced from the estimated reactivity ratios for products obtained in the studied systems.
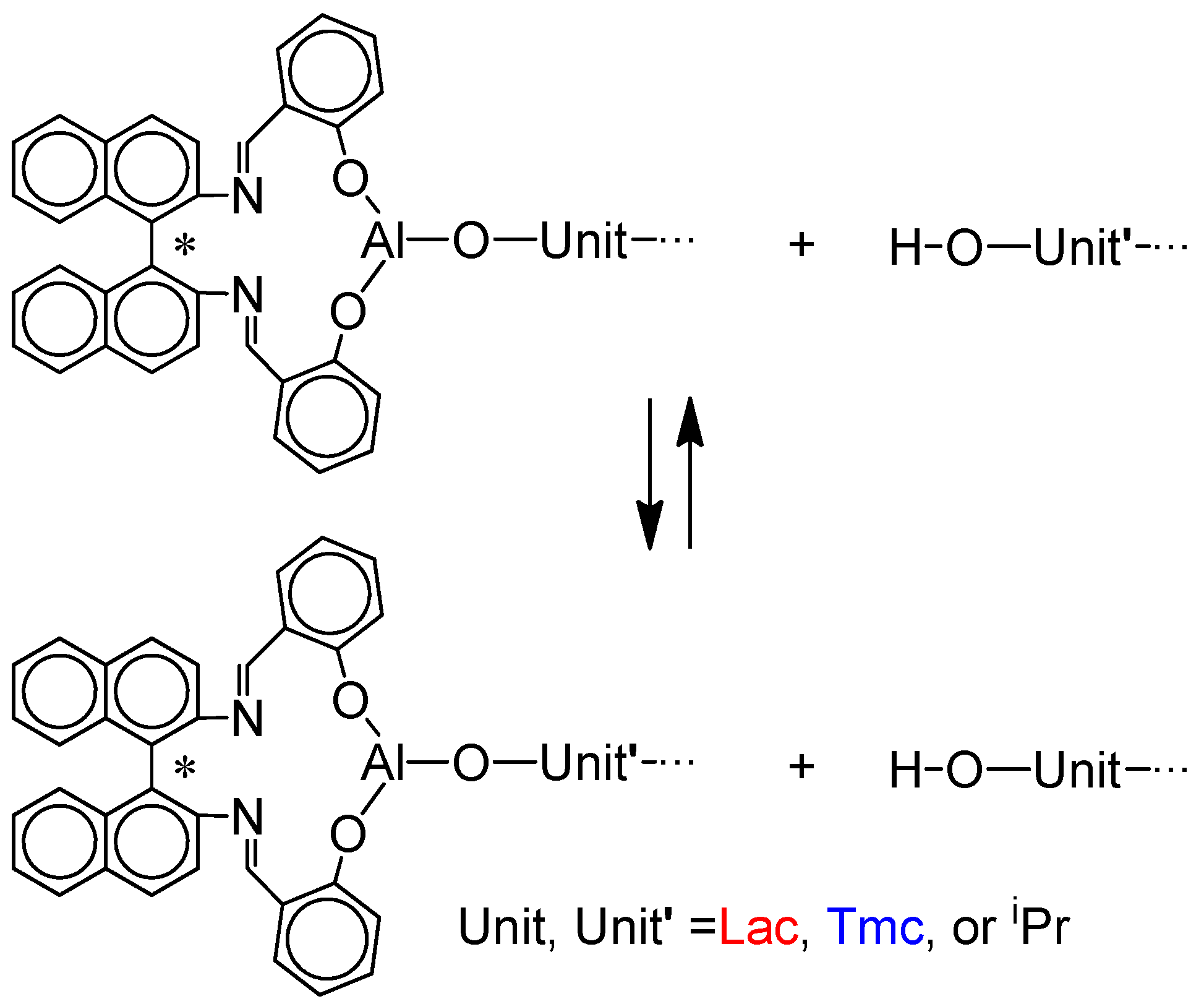
However, there was one experimental inconsistency in numerical simulations with experimental data. Namely, dispersity of copolymer chains initiated with
S-
Ini (fast inter-transformations of
Lac * and
Tmc * active species), are significantly higher than expected from simulations assuming, as mentioned above, fast exchange of active chain-ends with hydroxyl terminated ones. A relatively large dispersity (above 1.5 for higher conversions) was successfully explained by the rate of the exchange reactions involving OH terminated chains (
Scheme 6) being not sufficiently high. Therefore, the copolymerization systems do not behave exactly, as expected from simulations assuming the discussed until now model. Verifying this hypothesis with Monte Carlo simulations, we came to the conclusion that the hydroxyl terminated chains are probably transformed into living chains with rates much lower than propagation, resulting in broadening of the molar mass distribution. Numerical simulations, described in
Supporting Information, taking into account the slow chain-transfer processes, allowed us to estimate the average relative rate constants of chain-exchange in analyzed copolymerizations
kex/
kTT as well as the effective ratio of homopropagation rate constants
kLLR/
kLLS, important for systems initiated with the mixture of
Ini enantiomers, as described below.
Consequently, the presented below results take into account the chain-end exchange reactions in all studied systems.
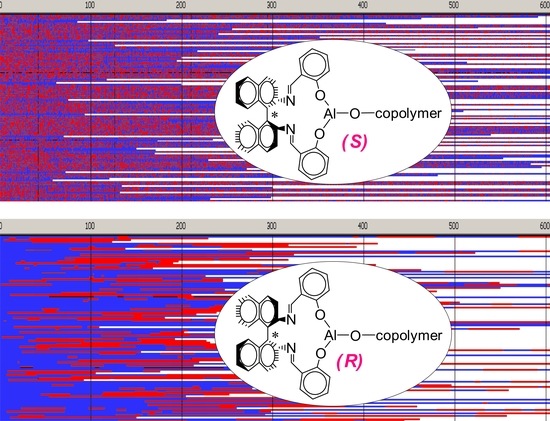
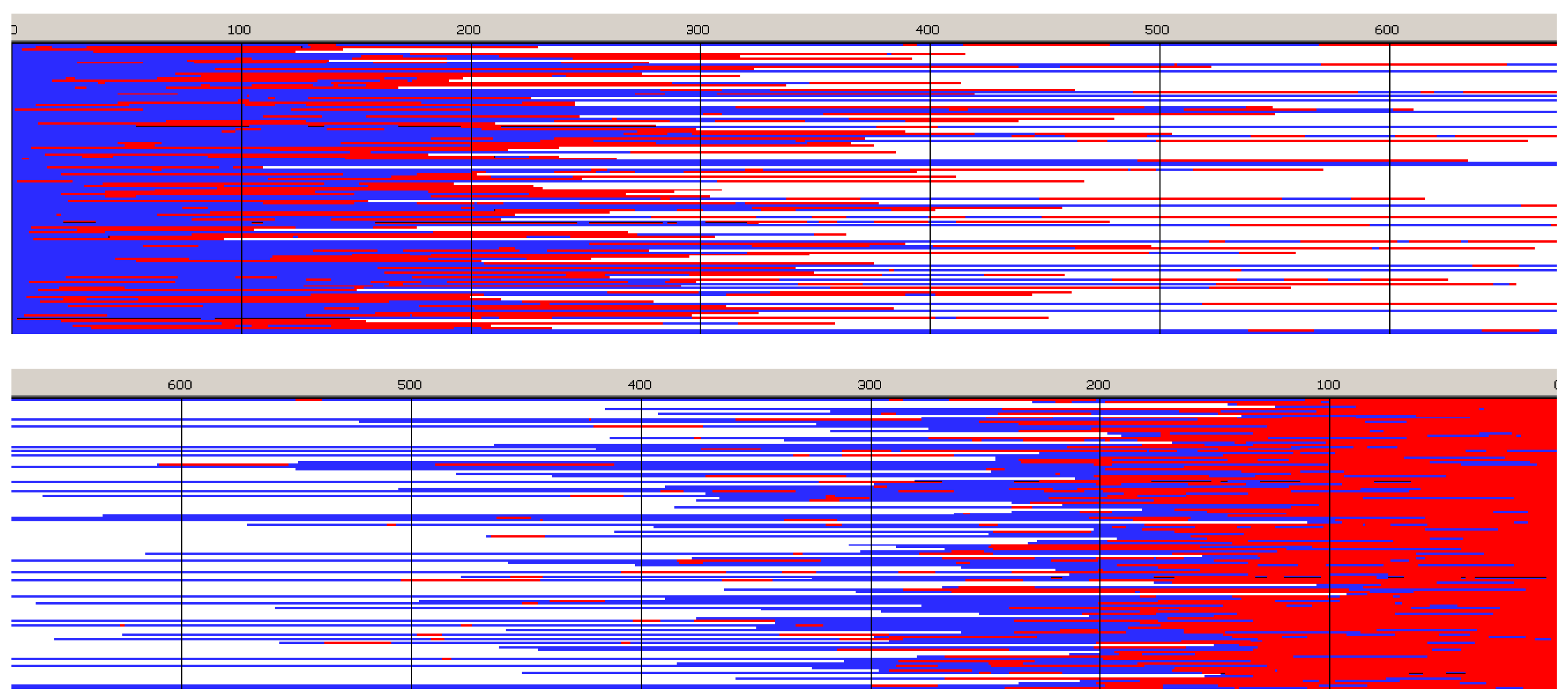
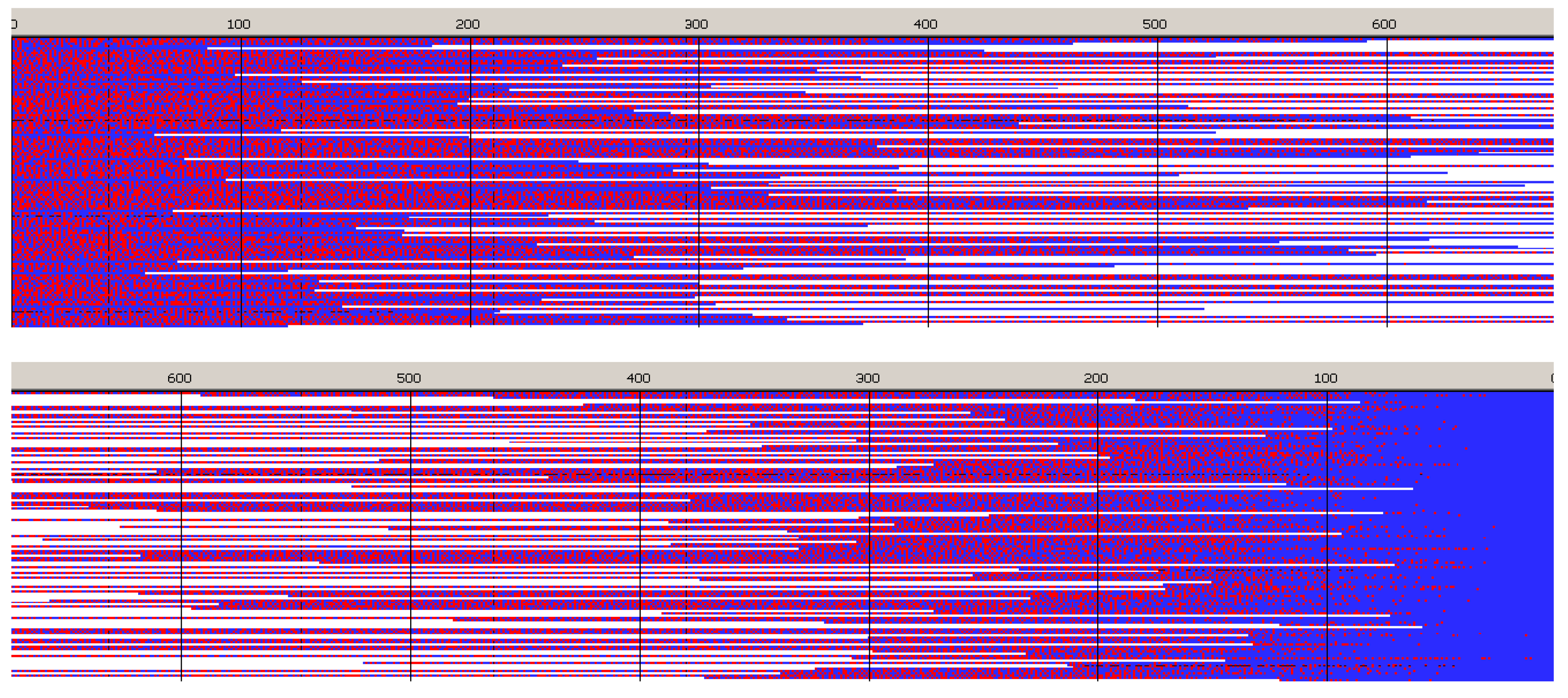
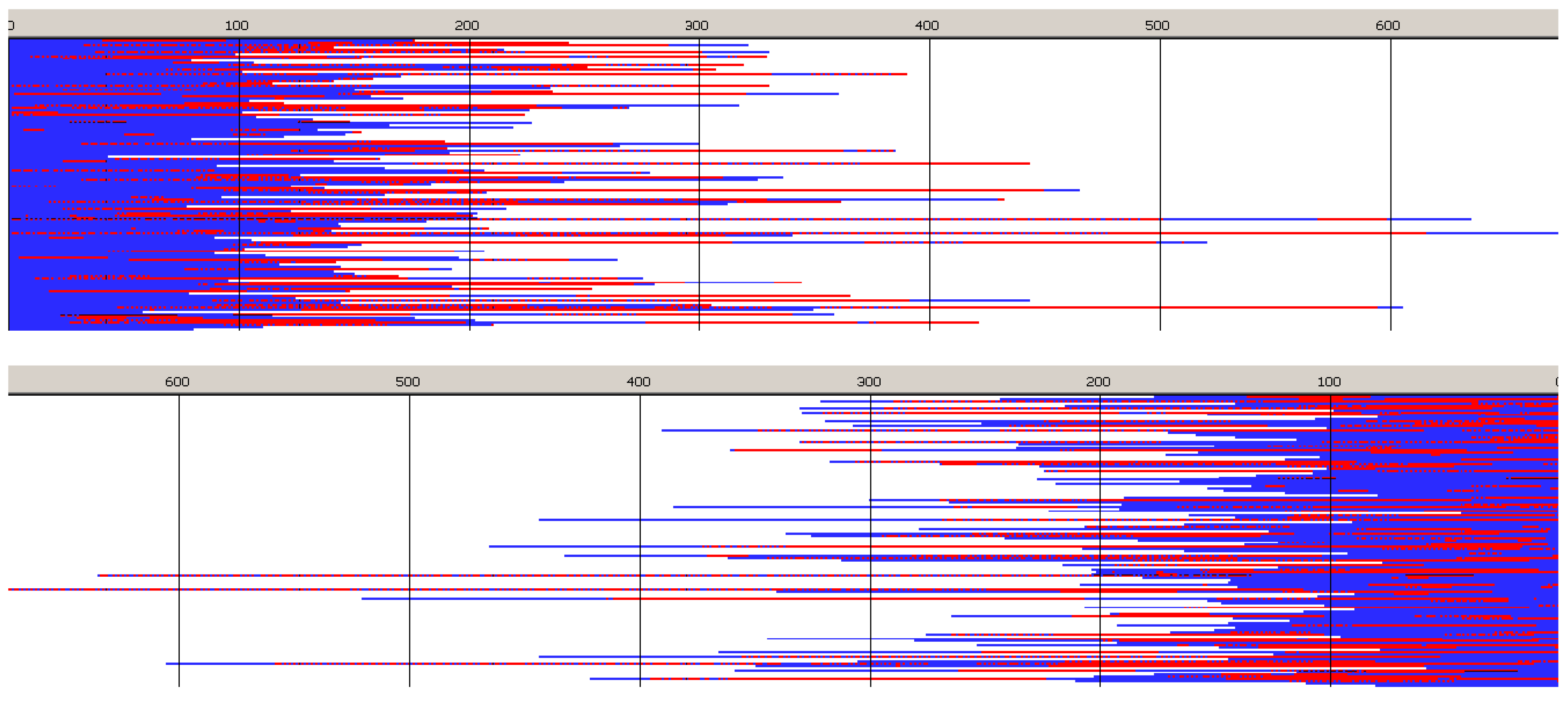
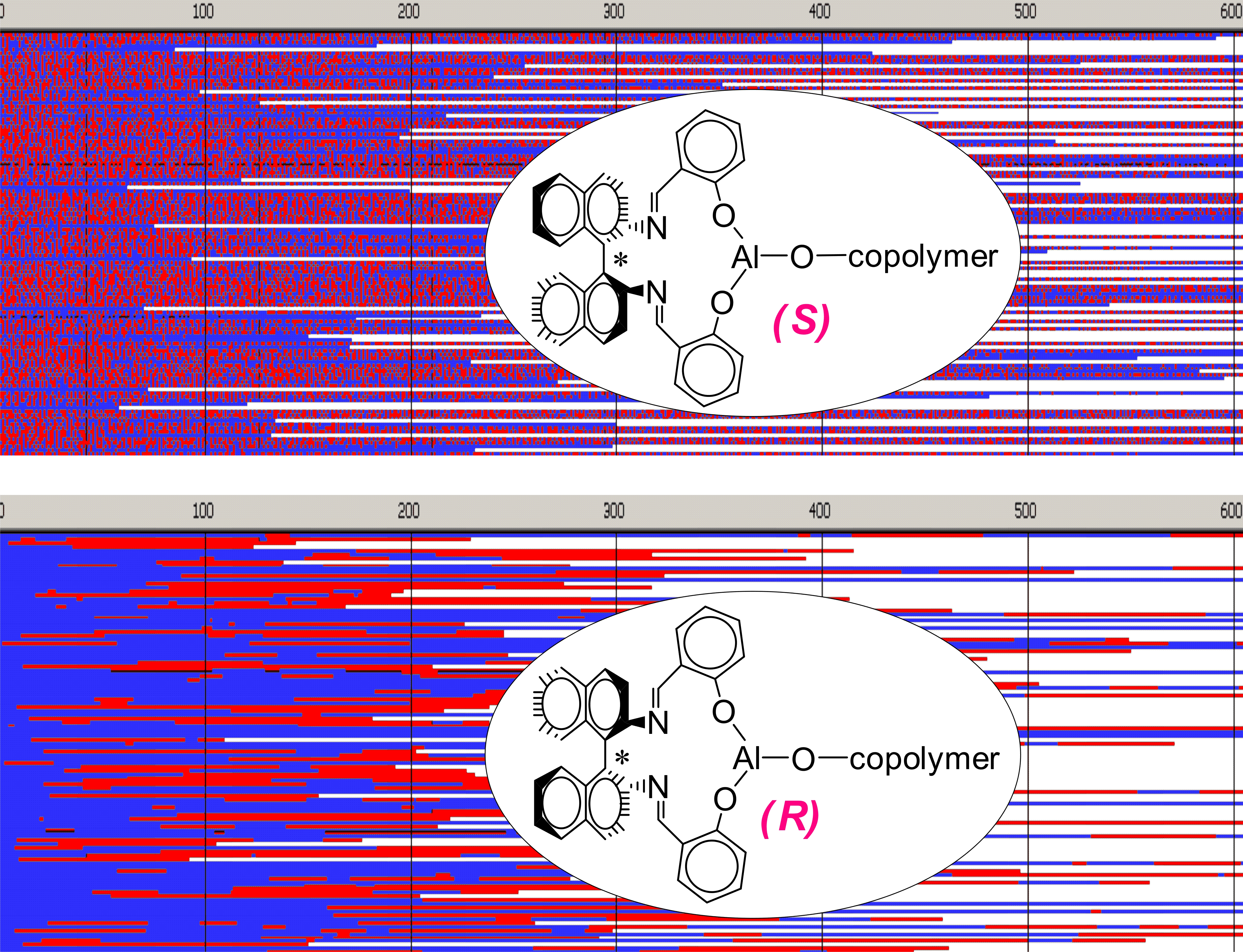
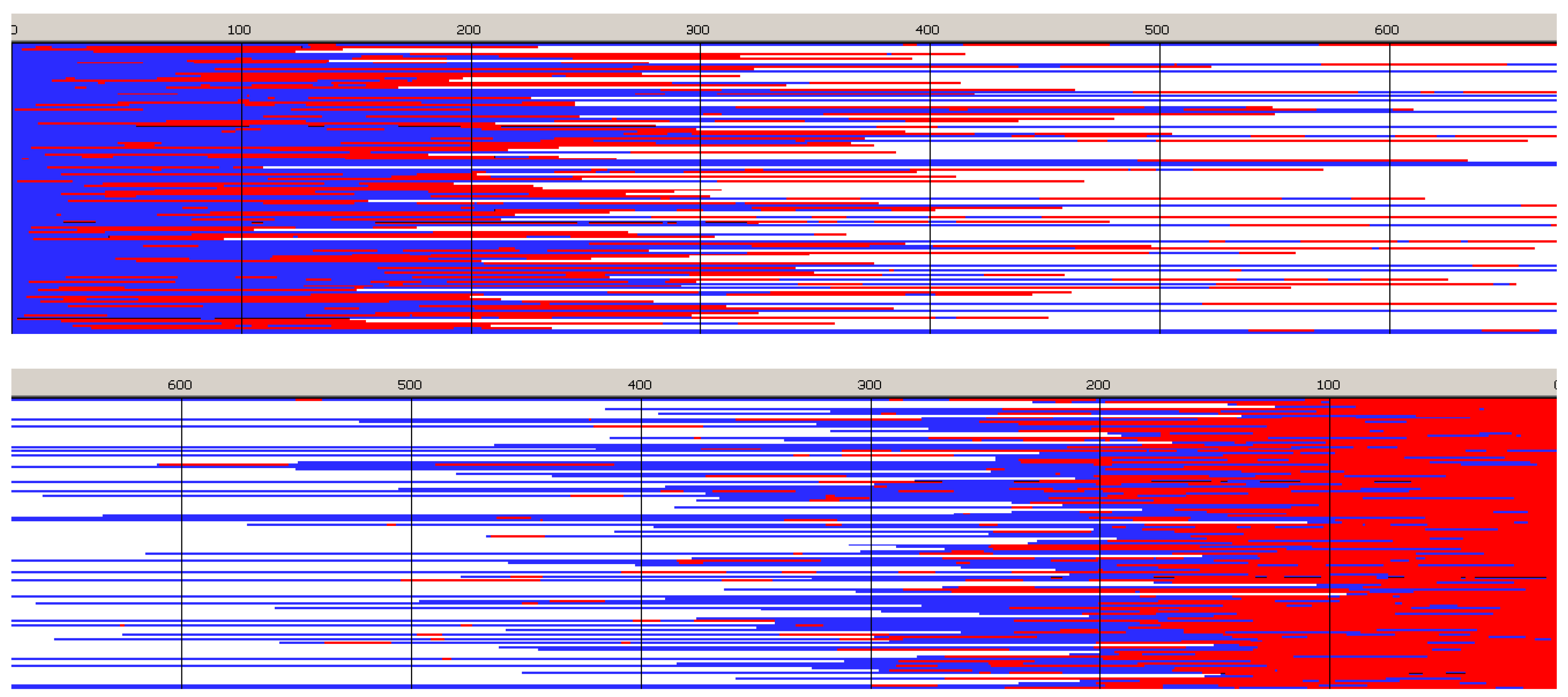
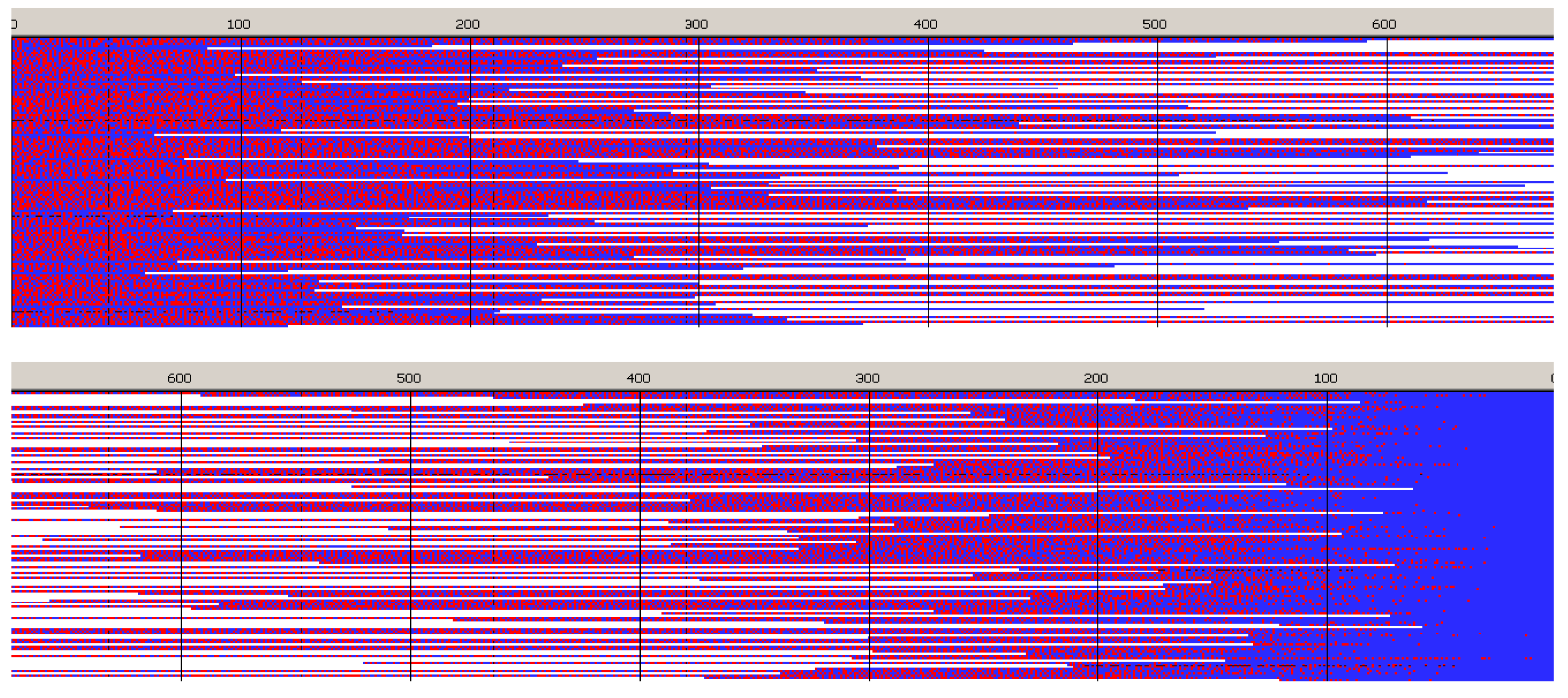
Figure 6,
Figure 7,
Figure 8 and
Figure 9 present structures of copolymer chains formed in systems initiated by
S- or
R- enantiomer of
Ini, simulated by MC method neglecting depropagation reactions. Initial concentrations of reagents were the same: [
Tmc]
0 = 2, [
Lac]
0 = 1.2 mol·L
−1, and [
Ini]
0 = 2 × 10
−3 mol·L
−1 (+[
iPrOH]
0 = 4 × 10
−3 mol·L
−1 due to in situ synthesis of
Ini). The only difference consisted in initiation by
Tmc * unimer in case of
R- enantiomer and mixture of
Tmc * and
Lac * unimers (proportionally to comonomer concentrations) in case of
S-enantiomer. Reactivity ratios used in each simulation are given in
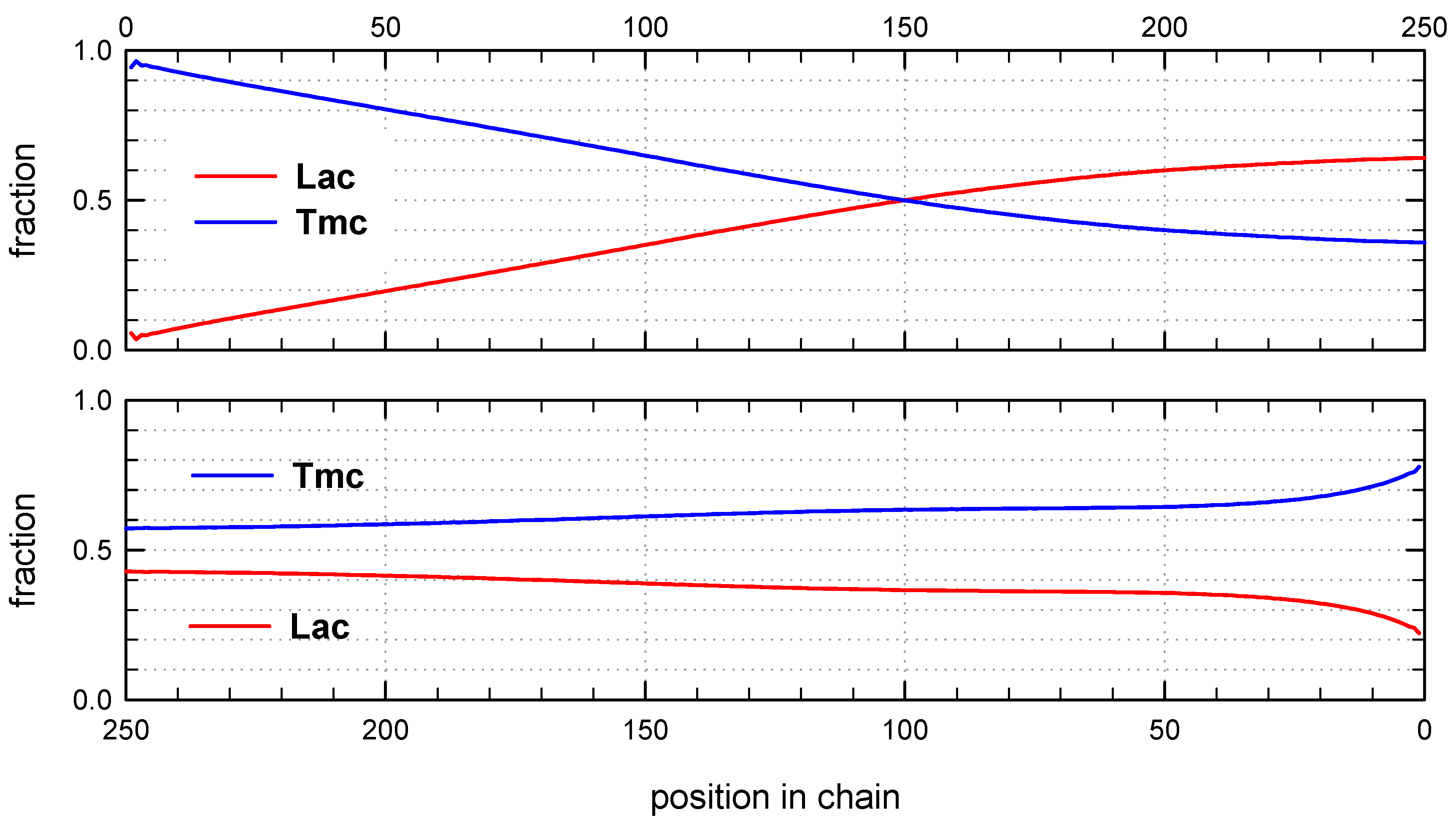
Table 2. Top boxes: plots prepared using unit positions numerated starting from chain beginning; bottom boxes: starting units numeration from chain-end.
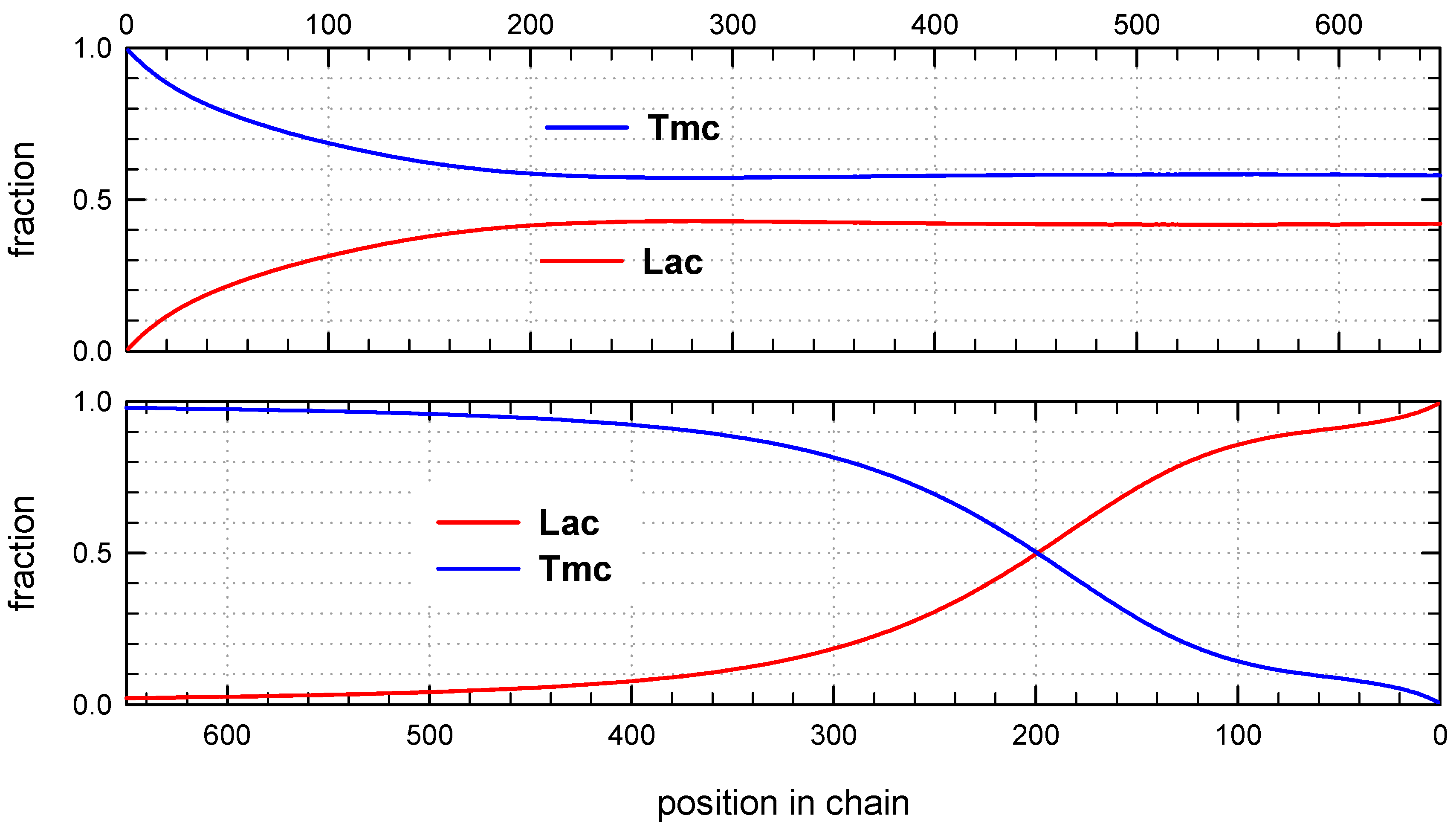
Copolymer formed with
R-
Ini and initiated with
Tmc * unimers (
Figure 6) is a block copolymer, containing on the average 5.2 blocks in a chain. As it was initiated with
Tmc unimers chains start with
Tmc blocks and, due to faster consumption of
Tmc, chains are terminated with
Lac blocks.
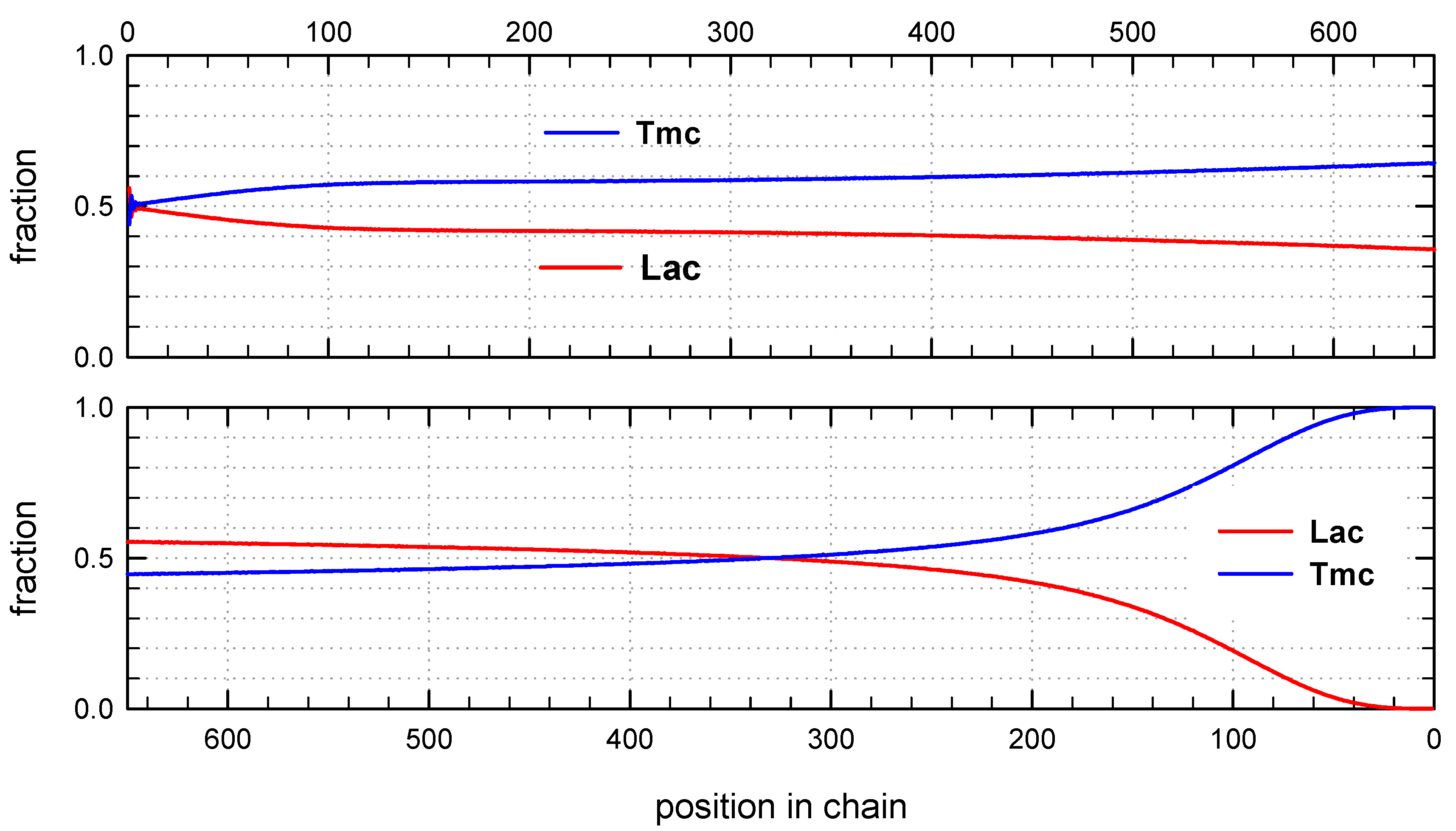
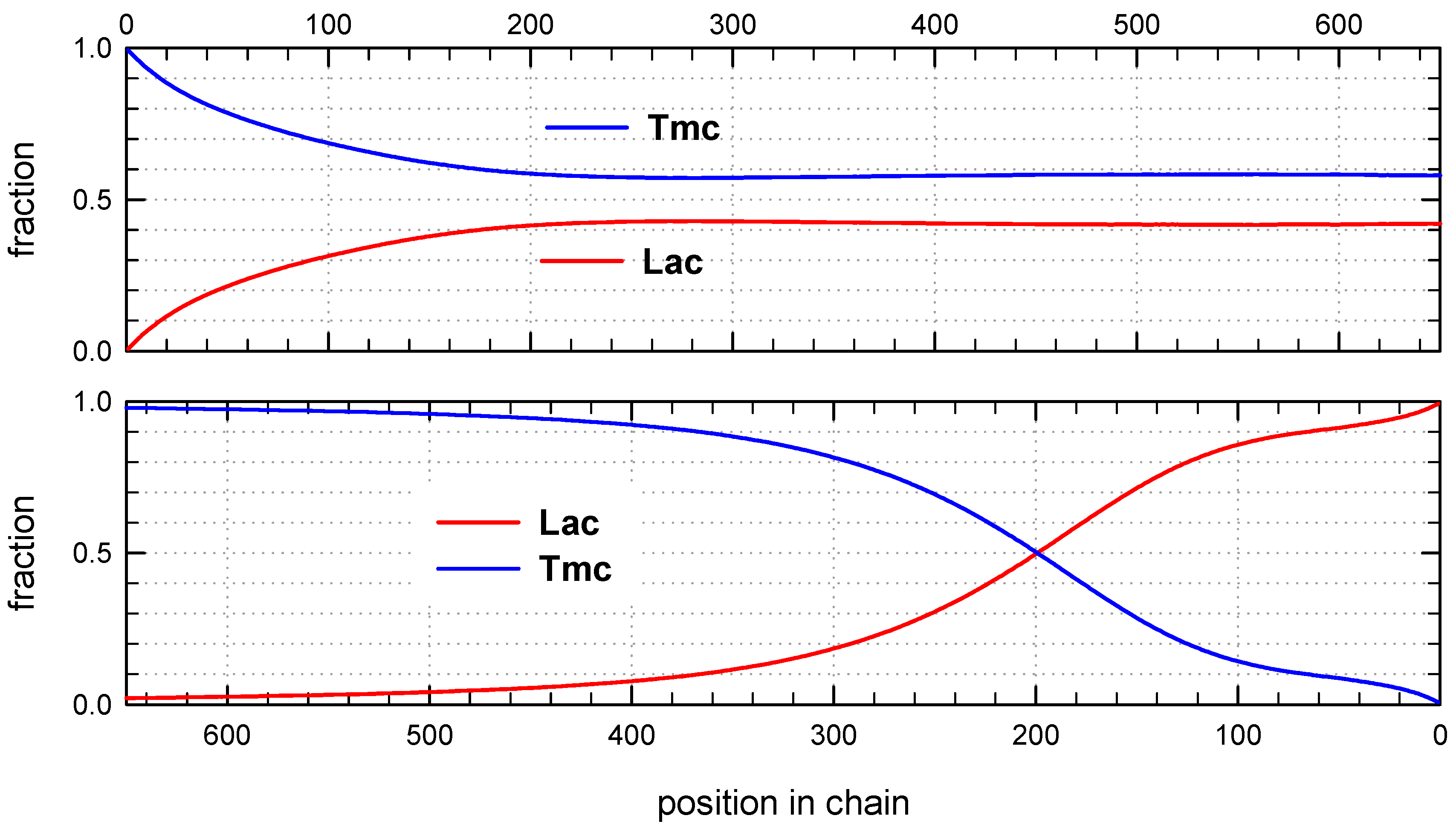
Monte Carlo simulations allow to present also the average composition of copolymers along average chain (computed for the whole set of chains) as well as similar distribution of homo and hetero dyads. The corresponding plots are shown in
Figure 8 and
Figure 9.
The corresponding plots differ, being dependent on numeration of unit positions, starting either from the chain beginning or chain-end. Due to rather broad molar mass distribution these differences are quite large. If position of unit is counted from chain beginning the fractions of copolymer units at more distant positions come to plateau and practically does not change up to chain positions at least about twice DPn. On the other hand, if unit positions are counted from chain-ends, one can clearly see a gradient-like feature of copolymer chains.
Distribution of units in copolymer formed with
S-
Ini and initiated with the mixture of
Lac-S-Ini * and
Tmc-S-Ini * unimers, in proportion corresponding to initial concentration of comonomers (1.2:2) (
Figure 7) differs significantly from system initiated by
Tmc-
R-
Ini * unimers.
Initial parts of chains, exceeding up to about 70% of chain length, contain statistical distribution of comonomers, apparently with close to each other proportion of comonomer units, no gradient of comonomer composition along individual chains in these regions were visible. However, when we analyze positions of units and sequences in the whole set of copolymer chains, the contributions of
Lac and
Tmc units (
Figure 9) change slightly steadily with unit position, indicating gradient feature. The shapes of the corresponding plots differ, depending on that if unit positions are numerated starting from the chain beginning or chain-end, similarly as it was observed for
R-
Ini system.
In the
Supporting Information one can find the similar plots presenting distribution of dyads and the average lengths of homoblocks along chain for systems initiated with both
Ini enantiomers.
The presented copolymer structures
Figure 6 and
Figure 7 were prepared choosing the exchange relative parameter
zex (listed in the Figure captions) to get dispersity close to the observed in experiments. One can observe that
zex for
R- and
S-
Ini systems differ significantly. It stems probably not only from different reactivities of
Lac-
R-
Res* and
Lac-
S-
Res* species (diastereomers) but also from the simplifying assumption (see
Supporting Information) that all relative kinetic exchange parameters are equal, independently on copolymer units neighboring OH group or
R/
S-
Ini residue of active centers.
The rate of chain-end exchange is more important in copolymerization system initiated with the mixture of R and S enantiomers of Ini. It is so because beyond determining copolymer dispersity it predetermines also the effective rate of exchange of the Ini residues of different configuration at active chain-ends and consequently the chain reactivity ratios, establishing copolymer microstructure. Any growing chain can, if the exchange is sufficiently fast, change configuration of its active species, with frequency dependent on the discussed relative rate coefficient zex.
3.3. Copolymerization Initiated with the Mixture of Enantiomers of Ini
The observed differences in features of copolymerization systems initiated with R-Ini and S-Ini suggests that one can control, to some extent, copolymerization features by using instead of one enantiomer of the studied asymmetric bulky initiator Ini the mixture of its enantiomers in variable proportions.
We performed such an experiment choosing the proportion of initiators R-Ini:S-Ini equal to 94:6. The large excess of R enantiomer was adopted because the rate of copolymerization initiated with R-Ini, leading to long homo-blocks, is much lower than that initiated with S-Ini and, on the other hand, the latter leads to statistical, almost alternating structures. Thus, the S enantiomer, although being in minority but with relatively fast cross-propagation rate constants, can effectively change the type of active species from Lac * to Tmc * and vice versa. The expected result was to obtain relatively homogeneous multi-block chains.
Copolymerization results were only partly as expected. Although the copolymerization rate and proportion of hetero-dyads were higher than in
R-
Ini copolymerization (for
13C NMR see the
Supporting Information) and the average number of copolymer blocks increased, a gradient-like feature is still visible in Monte Carlo simulations. It was explained by rather slow, not sufficiently fast, as we expected, exchange of
Lac-
R-
Res * active species (characterized by high reactivity ratio) into
Lac-
S-
Res * active species (via
Lac-OH terminating chains, acting as intermediates). If this process was fast enough,
Lac-
S-
Res * could fast attach
Tmc comonomer (low reactivity ratio), forming
Tmc-
S-
Res* terminated chain, which can readily attach
Lac. Similarly,
Tmc-
R-
Res * active species (high reactivity ratio, slow addition of
Lac comonomer unit), apparently not as fast as we expected, can be transformed (also via OH terminated intermediate) into
Tmc-
S-
Res * attaching next relatively quickly
Lac, forming
Lac-
S-
Res * species, already discussed. Consequently, contribution of heterodyads, as observed in
13C NMR spectrum (
Supporting Information), is higher than in copolymer formed with
R-
Ini, but instead of approximately homogeneous unit distribution, one can rather expect regions in one chain differing in microstructure: those formed with
R-
Ini * and ones formed with
S-
Ini* active species
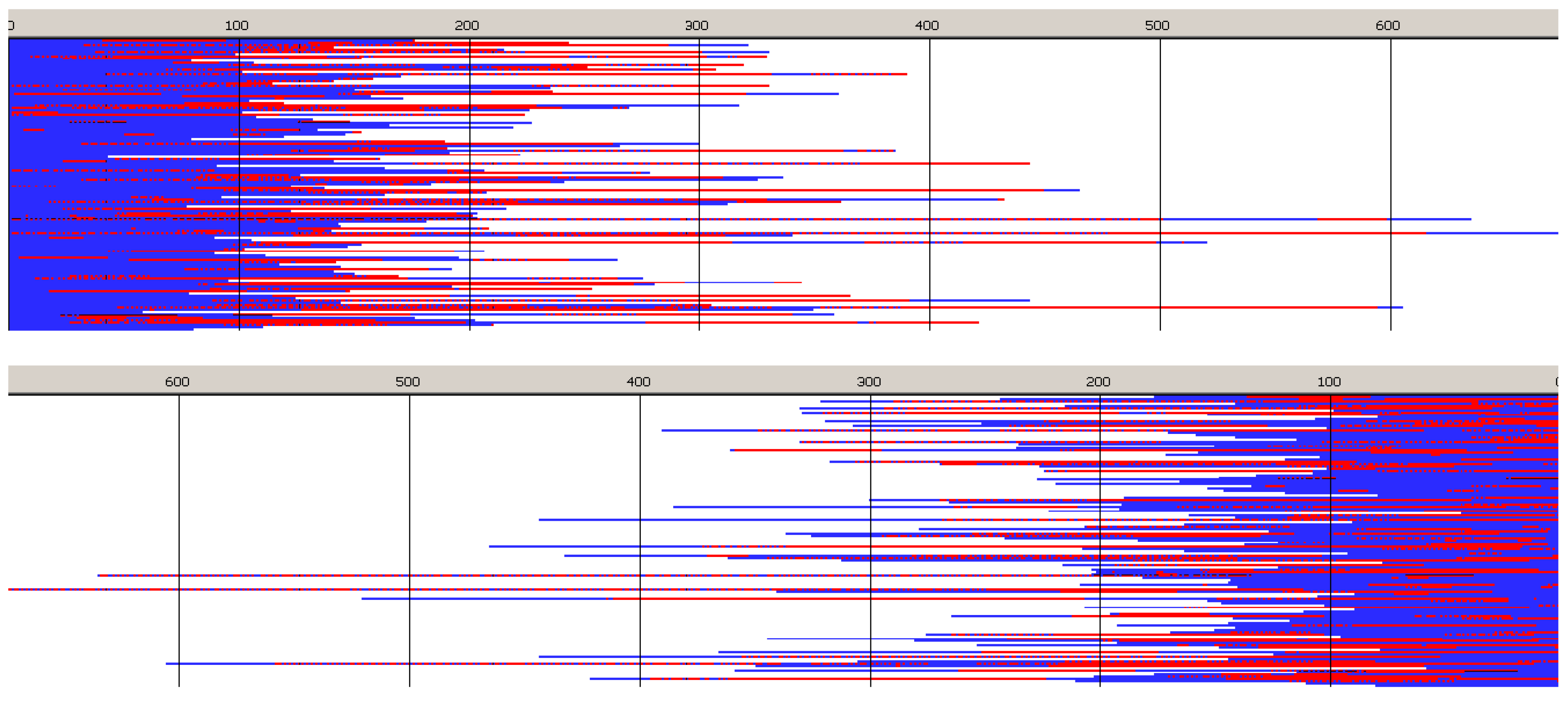
Monte Carlo simulations, presented in
Figure 10 and
Figure 11 (and those in
Supporting Information) confirm the described briefly structure of the copolymer. The average number of blocks is significantly higher (25.3) than estimated for copolymerization initiated with
R-
Ini (5.2). Unfortunately, due to insufficiently fast exchange of
R and
S active species one can easily find (
Figure 10) segments of copolymer chains containing a statistical distribution of copolymer chains. Consequently, dispersity of block-lengths is high at any chain position (see
Supporting Information), being the highest at the beginning of chains (about 10 for
Lac, and about 16 for
Tmc blocks) and the lowest at chain positions close to active species (about four-five for both types of blocks). In the
Supporting Information one can find also the plots presenting the computed distribution of dyads and the average lengths of homoblocks along chain and the discussion concerning the average homoblock lengths along copolymer chains. The average homoblock lengths can be calculated not only in dependence on chain position, but also on the way the homoblocks are selected for computing their average
DP.
Analyzing this copolymerization and performing fitting computations, we got to a rather unexpected result. Namely, the reasonably good fitting of kinetic parameters for the chain-end R/S exchanging systems was achieved only when we have removed restriction of equal Tmc homopropagation rate constants on active species terminated with initiator residue Res of different configuration. It can be explained by solvation of Tmc-Res * active species (of both Ini configurations) by asymmetric Lac comonomer molecules and corresponding copolymer units. Depending on configuration of Res presumably different average numbers of asymmetric Lac monomers/copolymer units solvate active species, and consequently also different numbers of Tmc molecules can be present in the corresponding solvation spheres. Thus, different spatial molecular arrangements can be expected around Tmc-R-Res * and Tmc-S-Res * active species. In fact, taking into account asymmetric solvating entities one can consider these active species with their solvation spheres as different environments or arrangements, what results in their different reactivities, and consequently different Tmc homopropagation rate constants kTTR ≠ kTTS. These differences in solvation spheres can be of two kinds: Tmc-R-Res * (solvated) and Tmc-S-Res * (solvated) can be diastereomeric if the same numbers of Lac monomer molecules and copolymer units are present in them, or they can differ in numbers of Tmc molecules if enantiomeric Tmc-Res * active species differ in accepting in their solvation spheres asymmetric Lac molecules, competing with symmetric Tmc molecules.
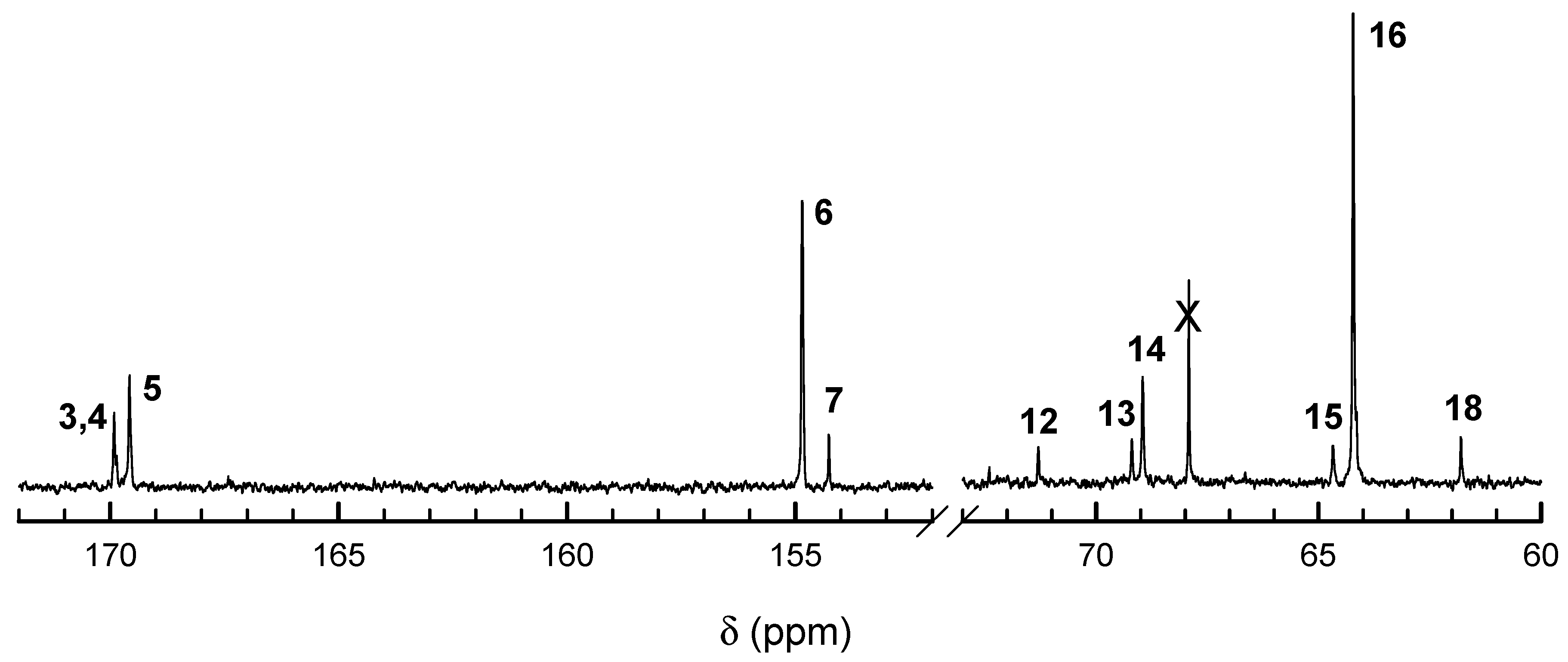
We believe that this presumption based on our simulations (that
kTTR ≠
kTTS) is sound because not only the corresponding fitting to experimental [
Lac] and Δ
H is the best but also it gives the closest agreement with experimental [
Tmc], determined with
13C NMR (
Figure 12).
Although this correlation between [
Tmc]exp and [
Tmc]
calc obtained applying the devised method with triad dependence of volume contraction coefficients CC is not very good, we think that the observed differences stem from our approximations concerning the assumed model of copolymerization. Namely, the assumed very similar, parallel changes of rate coefficients with conversion and also the approximations concerning volume contraction coefficients may be responsible for the observed discrepancy. We think that the largest errors in estimation of
Tmc concentrations are introduced by the limitation of the number of CC coefficients to be fitted, done by assumption that CC
AAB + CC
BAA = CC
AAA + CC
BAB (see
Supplementary Materials).




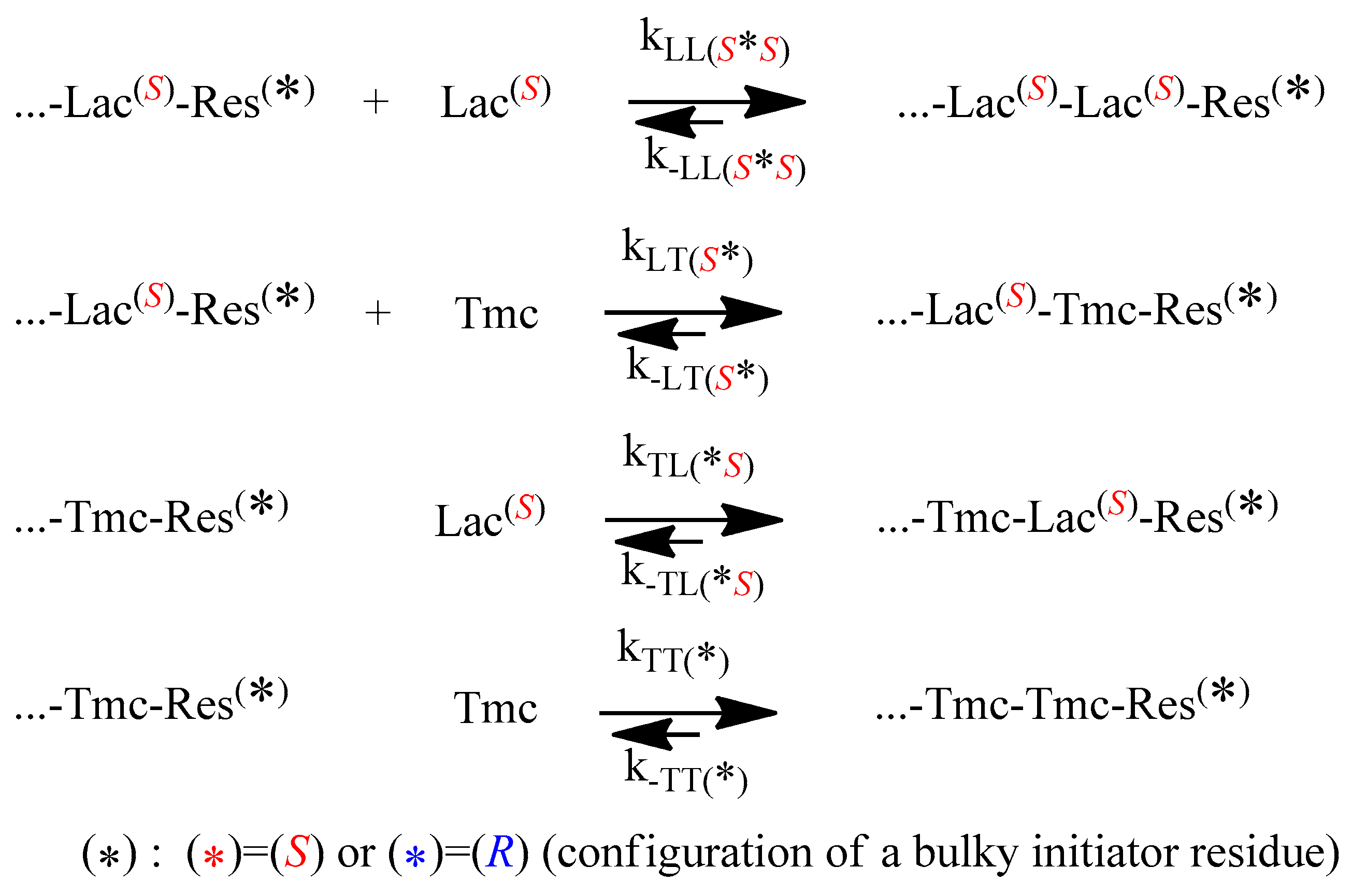




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
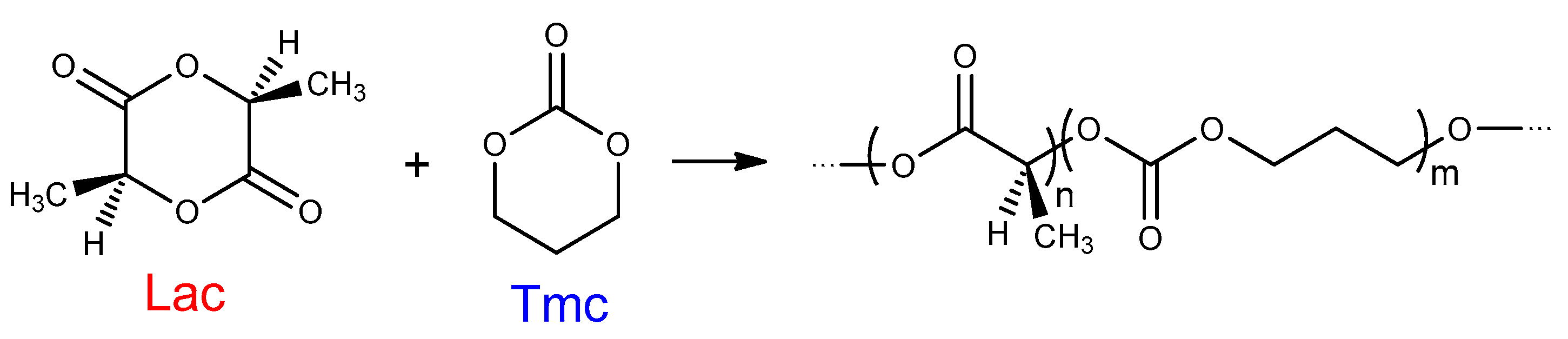{kind=link}
{kind=link}
{kind=link}
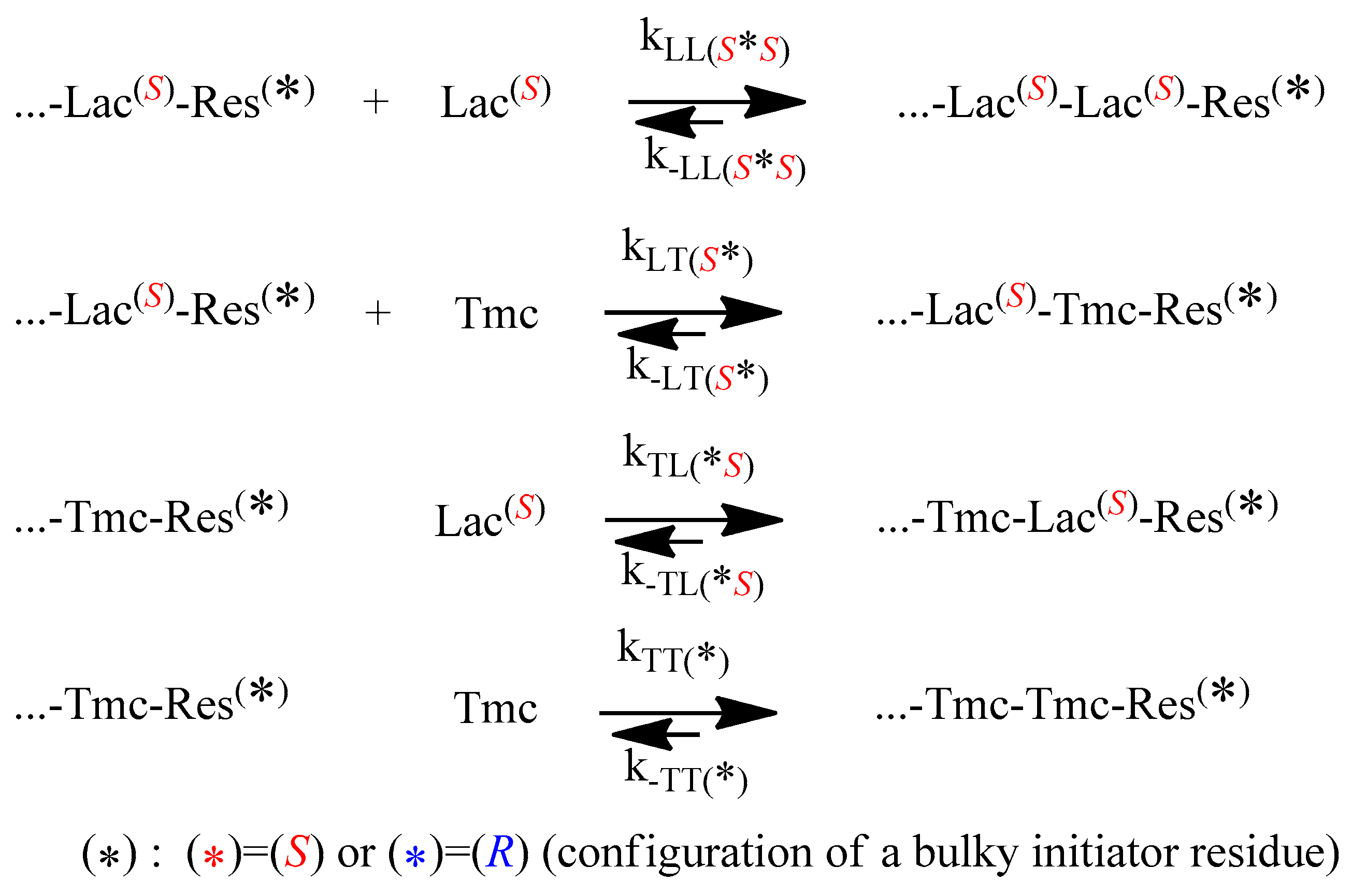{kind=link}
{kind=link}
{kind=link}