1. Introduction
Silica is a non-carbon filler that is an extremely important reinforcing filler in the rubber industry [
1]. Previous studies have confirmed that silica combines good mechanical performance [
2], high wet grip resistance [
3] and low rolling resistance [
4] for silica/rubber composites. At present, silica/rubber composites are commonly used for producing “green tires” [
5,
6], which have low rolling resistance, resulting in reducing the vehicle’s fuel consumption.
As an inorganic particle, silica has many hydroxyl groups (–OH) on its surface [
7], causing the hydrophilic nature of the silica particle [
8,
9,
10]. Therefore, the silica particle is less compatible with a hydrophobic polymer, such as rubber [
11]. However, the silica surface can be modified due to the numerous reactive hydroxyl groups [
12,
13]. Silica modification is an effective method for improving the compatibility between silica and rubber. Treatment with reactive silane coupling agent (SCA) is one of the major methods used in silica modification [
14,
15]. In principle, SCA possesses a readily hydrolyzable alkoxy group that reacts with the hydroxyl groups on the silica surface to form a stable siloxane linkage [
16]. In addition, some surface active agents (SAA), such as poly ethylene glycol, triethanolamine and cetyltrimethylammonium bromide, are commonly used in silica modification. SAA can absorb on the silica surface, resulting in covering the silica surface and reducing the amount of the exposed hydroxyl groups.
Natural rubber (NR), which contains 93%–95%
cis-1,4-polyisoprene, is an essential biosynthesized polymer [
17]. It is naturally found in the form of a colloidal system known as NR latex, in which rubber particles are dispersed in an aqueous medium [
18,
19]. Therefore, the latex compounding method is used to prepare silica/NR composites to address the problems of low efficiency in the outdated mechanical blending method [
20,
21].
Preparing a silica/NR slurry with good silica dispersion and stable NR particles is key for the latex compounding method mentioned above. The silica in this slurry should be completely co-coagulated with the rubber when preparing silica/NR master batches, which is a floc containing silica and rubber. As these materials work at a high frequency of motion, silica/rubber composites should have excellent dynamic performance, which is not required in most kinds of polymer composites. Therefore, the silica in the master batches should have the potential to form a chemical interaction with NR, because the internal friction of silica/NR composites, which is the major factor in dynamic performance such as rolling resistance and wet grip of tires, can be reduced by a chemical interaction between silica and rubber. Preparing modified silica, using a proper strategy to achieve an ideal organic silica surface, is the most practical process to meet the above requirements.
In previous research, sulfide-containing SCAs were used for silica modification when preparing silica/rubber composites using the latex compounding method [
21,
22,
23,
24]. In principle, sulfide-containing SCAs react with silica as mentioned above, resulting in improving the dispersion of silica in a latex system and rubber matrices. Meanwhile, sulfide-containing SCAs can react with the double bonds in rubber molecules by their sulfide group. Therefore, the structure by which rubber molecules and silica particles are linked by the SCA is formed between silica and rubber by the help of the sulfide-containing SCAs through a “coupling bridge” [
25]. Previous researchers indicated that the “coupling bridge”, which is a typical chemical interface between silica and rubber, benefits the mechanical and dynamic properties of silica/rubber composites [
26]. However, the hydroxyl groups on the silica react with the hydroxyl groups of hydrolyzed SCA in silica modified by SCA in the aqueous phase [
27], meaning the hydrolysis of SCA is a precursor for silica modification. Polycondensation also occurs among the hydroxyl groups of hydrolyzed SCA, producing the polycondensates of SCA, resulting in the aggregation of several SCA molecules [
25]. Therefore, using only SCA in silica modification is not adequately efficient. Only the chemical interface existing between silica and rubber is detrimental to the stretching of the rubber molecular chain under external force [
28,
29,
30]. Moreover, the reaction between rubber and sulfide-containing SCA, known as “scorchy” behavior, is inevitable during the process [
31], even though the mixing time and temperature are precisely controlled. Therefore, using sulfide-containing SCA only for silica modification is not an ideal method for preparing silica/rubber composites.
Parts of SAAs directly modify silica in the aqueous phase without polycondensation, and not all SAAs react with rubber under all conditions. Therefore, a physical interface between silica and rubber can only be formed with the help of SAA [
32,
33,
34]. However, the dynamic performance of silica/rubber composites is very poor in the absence of a chemical interface between silica and rubber [
8,
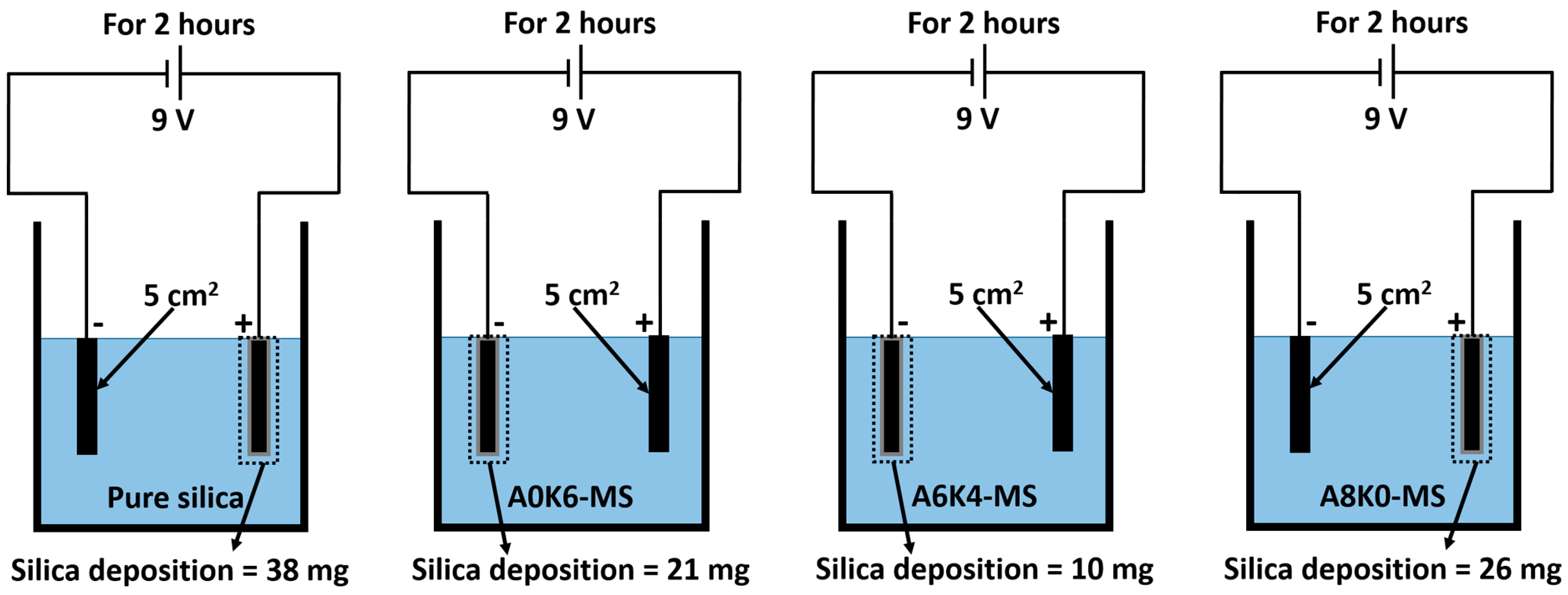
28]. In our previous research, silica modified by SAA was not completely co-coagulated with the rubber when preparing silica rubber master batches using the latex compounding method.
For rubber composites prepared for “green tires”, the dynamic performance is extremely important. Therefore, for this research, a chemical interface between silica and rubber was formed. Preparing high performance silica/rubber composites for “green tires” using the latex compounding method is a more complex and restrictive study than the preparation of other polymer composites. Using different modifiers to form chemical and physical interfaces between silica and rubber is a simple and feasible method to achieve this goal.
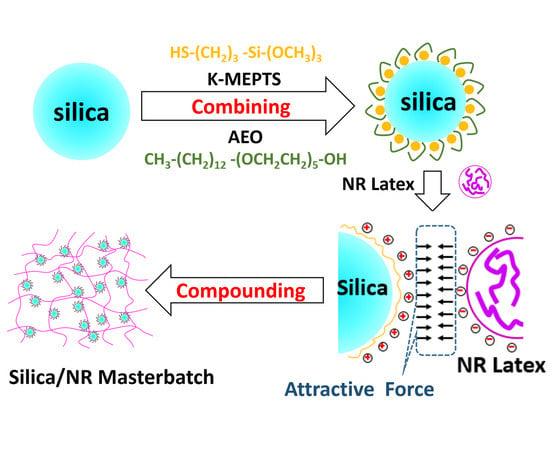
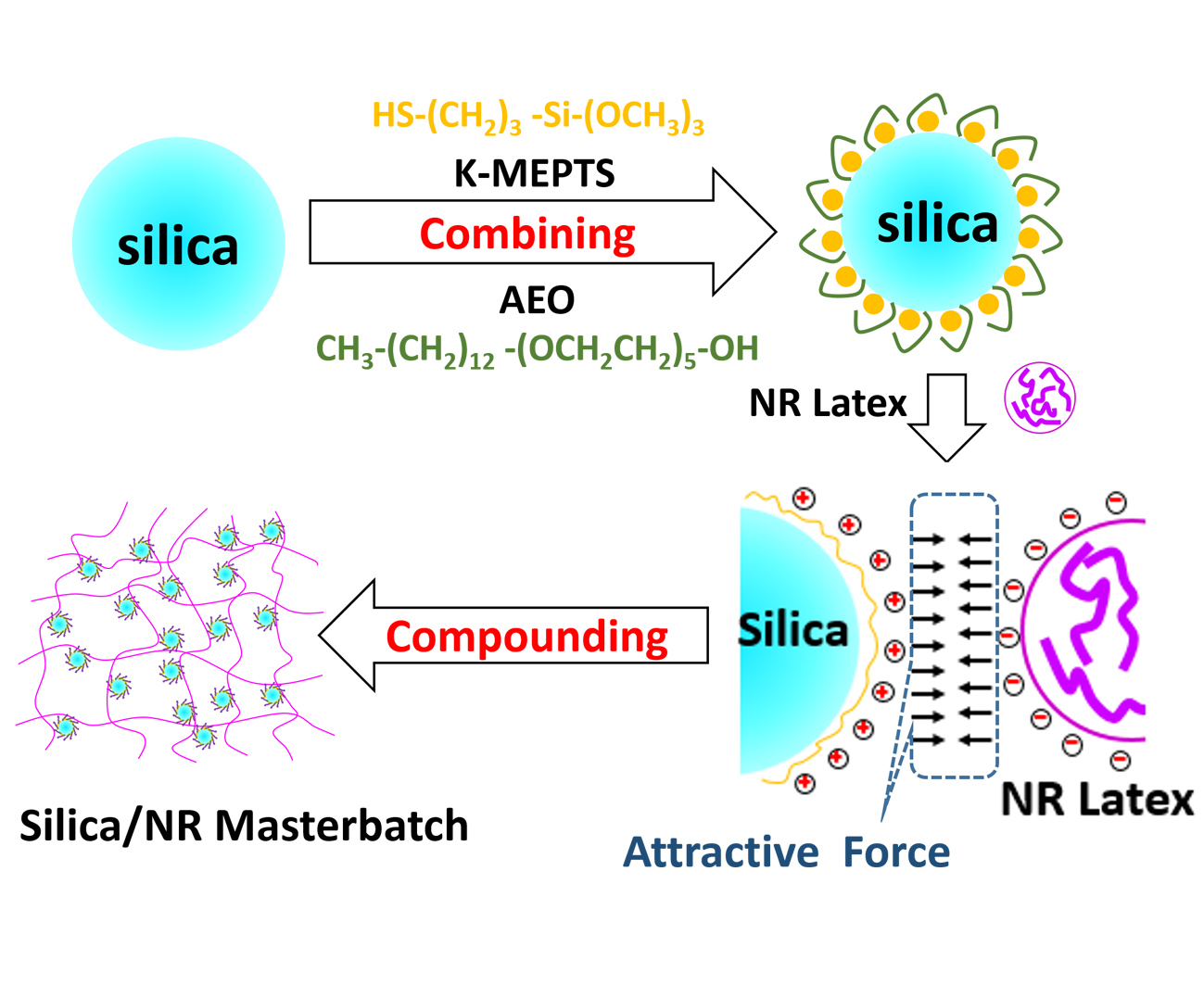
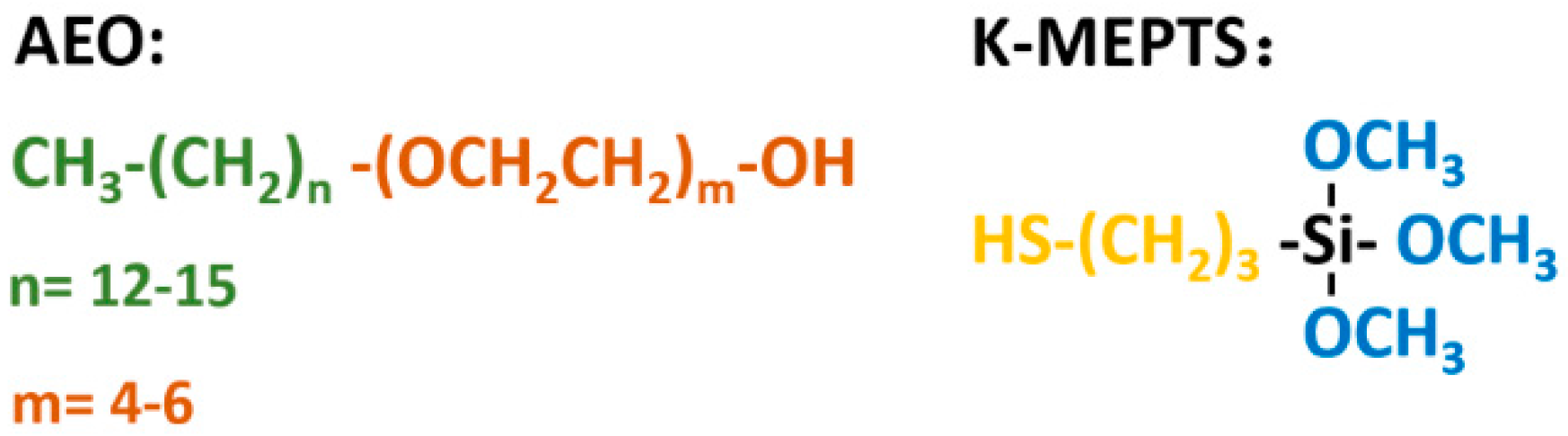
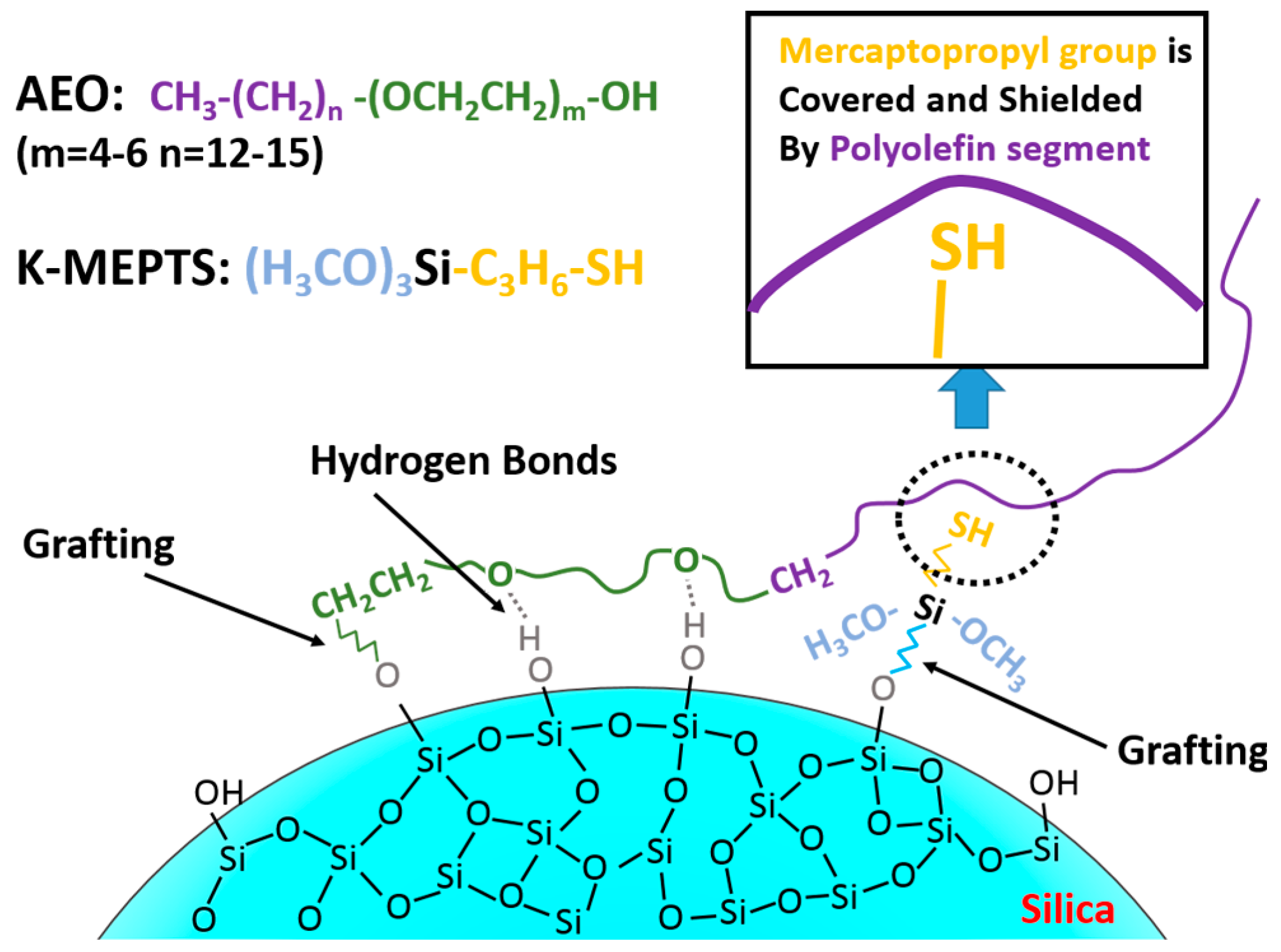
In this research, the silica/NR master batches were prepared using the latex compounding method. Alcohol polyoxyethylene ether (AEO) [
35]), a widely-used nonionic SAA, and 3-mercaptopropyltriethoxysilane (K-MEPTS) [
36], which was commercially developed and widely used in the rubber industry, were selected as modifiers to be used together in silica modification in the aqueous phase (as shown in
Figure 1). This is a novel strategy proposed for silica modification for preparing silica/rubber master batches. The magnitude of the chemical and physical interface between silica and rubber is varied by adjusting the amount of AEO and K-MEPTS used in silica modification. In our research, pure and different modified silica were characterized by Fourier transform infrared spectroscopy (FT-IR), thermal gravimetric analysis (TGA), and Raman spectroscopy. The interaction between K-MEPTS and AEO was confirmed through the results of these characterizations. Pure and different modified silica were used in preparing silica/NR master batches by the latex compounding method. In this part of the research, the role played by K-MEPTS and AEO using the co-coagulation of silica and NR was confirmed. Finally, the properties of silica/NR composites containing different modified silica were compared. The effect of the chemical interface between silica and rubber, formed by K-MEPTS, and the physical interface between silica and rubber, formed by AEO, on the performance of silica/NR composites was investigated.
2. Experimental Materials and Methods
2.1. Materials
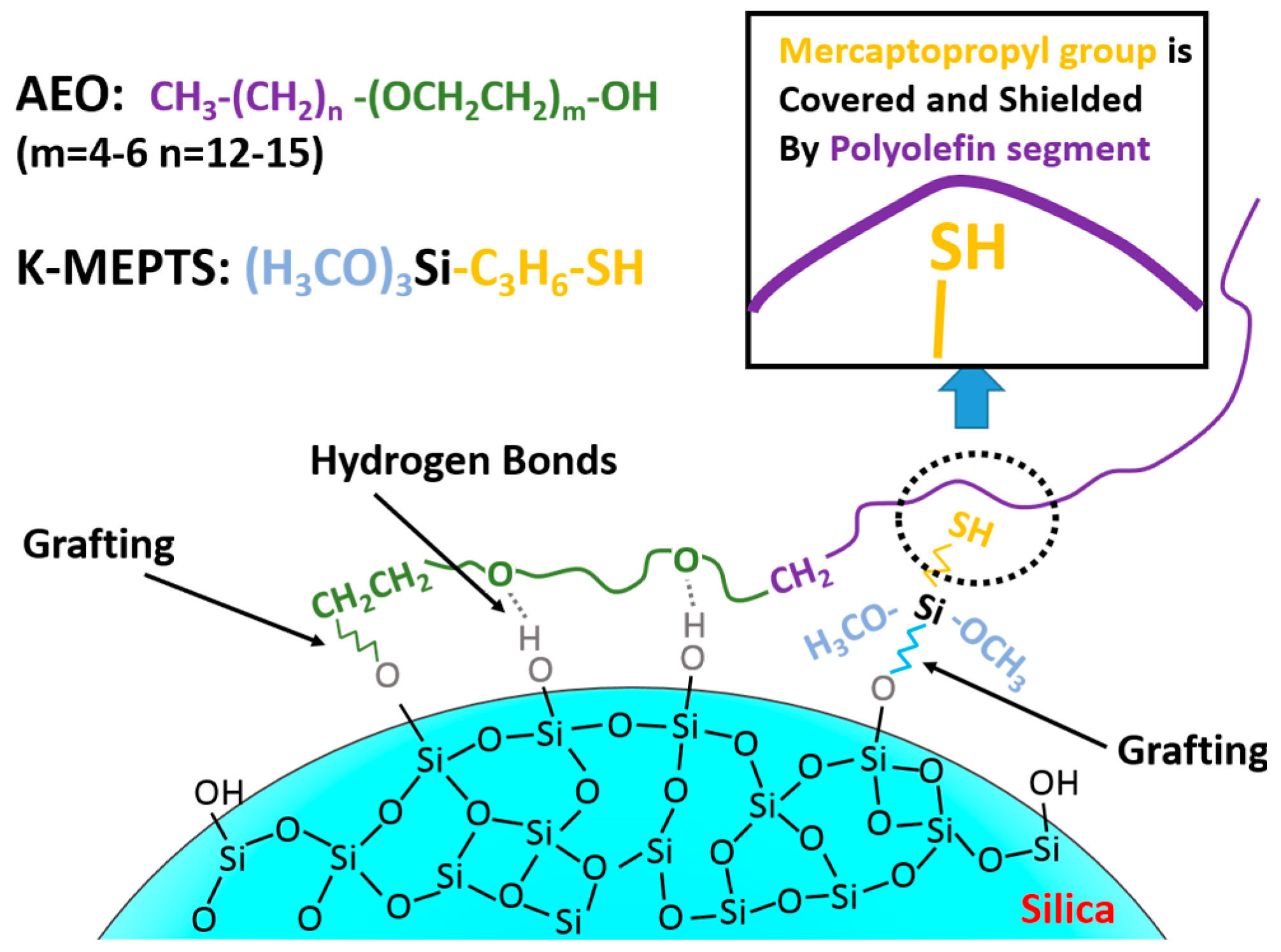
High-ammonia NR latex with 60% total solid content was purchased from Hainan Rubber Industry Group Co., Ltd. (Hainan, China). Precipitated silica water slurry of K-160 (nanoparticle size: 20–30 nm, Brunauer-Emmett-Teller (BET) specific surface: 160.06 m2/g) was produced by Wilmar China (Jiamusi, China). AEO (average molecular weight: 421 g/mol), which possesses a terminal hydroxyl group that can react with hydroxyl groups on the silica surface and possesses a long molecular chain that consists of polyolefin and polyether, was prepared by BASF SE. K-MEPTS (molecular weight: 196 g/mol) was obtained from Nanjing Capatue Chemical Co., Ltd. (Nanjing, China). The rest of the required materials were commercially available.
2.2. Preparation of Modified Silica
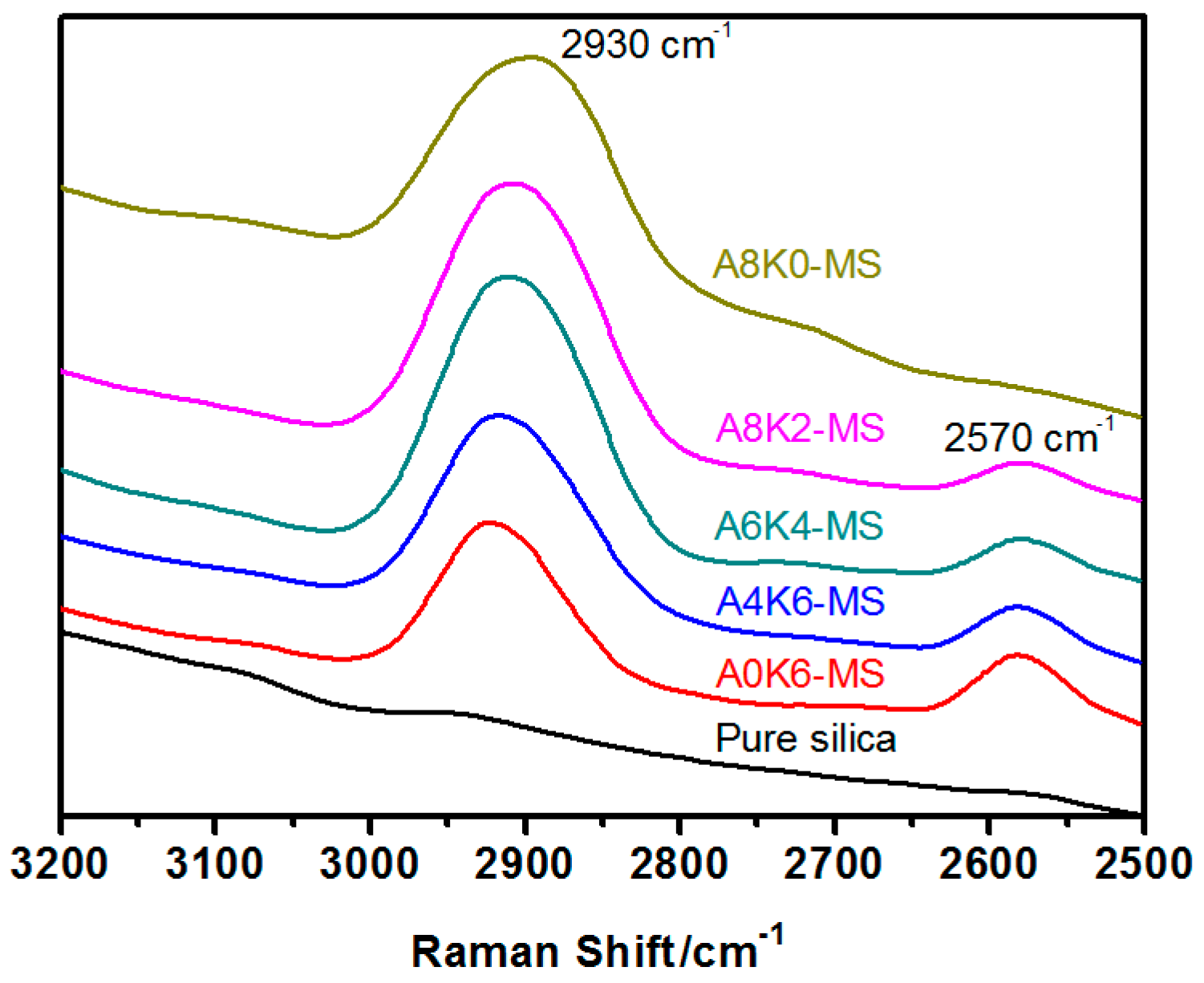
The silica slurry solid content was measured to dilute to a 10% concentration (e.g., 100 g dry weight of silica for every 1000 g silica slurry) by adding water into the raw precipitated silica slurry. The silica water slurry was subjected to high-speed stirring (800 rpm) for 30 min to obtain a stable suspension. Five beakers, numbered 1–5, were prepared, and 1000 g of silica slurry were transferred to each beaker. All silica slurries were heated to a temperature of 70 °C under high-speed stirring (800 rpm). K-MEPTS (6, 6, 4, and 2 g) was added into Beakers 1–4, and AEO (4, 6, 8 and 8 g) was added into Beakers 2–5, respectively. The slurry was stirred for 0.5 h, and the modified silica slurry was then obtained. According to the amount of AEO and K-MEPTS added in the silica modification, we labelled the modified silica in Beakers 1–5 as A0K6-MS, A4K6-MS, A6K4-MS, A8K2-MS and A8K0-MS, respectively. The amounts of K-MEPTS and AEO used in the different modified silica are listed in
Table 1.
Part of the pure silica and modified silica powder was obtained by drying the corresponding modified silica slurry. Pure silica and modified silica powders were extracted in a Soxhlet extractor using ethanol for 24 h, 15 min for each reflux, to remove un-grafted AEO and K-MEPTS. Then, all extracted silica powders were dried in the same oven at 70 °C for 24 h. These silica powders were prepared for characterization using FT-IR, TGA and Raman spectroscopy.
2.3. Preparation of Master Batches
The modified silica slurry was cooled to room temperature and blended with the NR latex. The solid content of the NR latex was confirmed in advance, and the weight ratio of silica to NR was 50:100 (e.g., 50 g of silica nanoparticles for every 100 g solid content of NR). Then, the mixture of silica and NR latex was stirred for 0.5 h and coagulated with 3% formic acid solution. Finally, the flocs were washed with water 6 times and then dehydrated in a drying oven at 60 °C for 36 h to obtain silica/NR master batches. The master batches prepared with A0K6-MS, A4K6-MS, A6K4-MS, A8K2-MS and A8K0-MS were called A0K6-MB, A4K6-MB, A6K4-MB, A8K2-MB and A8K0-MB, respectively.
2.4. Preparation of Silica/NR Composites
The formulation of silica/NR compounds is shown in
Table 2. Silica/NR compounds were obtained through three stages of mixing. First, the master batches were masticated for 2 min in an internal mixer equipped with an oil circulating system to maintain the processing temperature at 55 °C. Then, zinc oxide, stearic acid and
N-1,3-dimethylbutyl-
N′-phenyl-
p-phenylenediamine were added to the master batches successively. Second, compounds were kneaded for 5 min in the same internal mixer at 150 °C to further promote the reaction between silica and modifier and then naturally cooled to room temperature. Finally,
N-cyclohexyl-2-benzothiazole-sulfenamide, diphenyl guanidine and sulfur were uniformly blended in sequence with the cooled compound in a 6-inch mill (Shanghai Rubber Machinery Works No. 1, Shanghai, China) at room temperature. The total mixing time was no more than 15 min. The silica/NR compounds that contained A4K6-MB, A6K4-MB and A8K2-MB were denoted as A4K6-C, A6K4-C and A8K2-C, respectively.
The scorch time (T10) and optimum cure time (T90) of the compound were measured using a disc vulcameter. The compounds were vulcanized at 143 °C according to their optimum cure time (T90) in a standard mold to produce the silica/NR vulcanizates, which were stored at room temperature for at least 24 h before determining the performance.
2.5. Characterizations
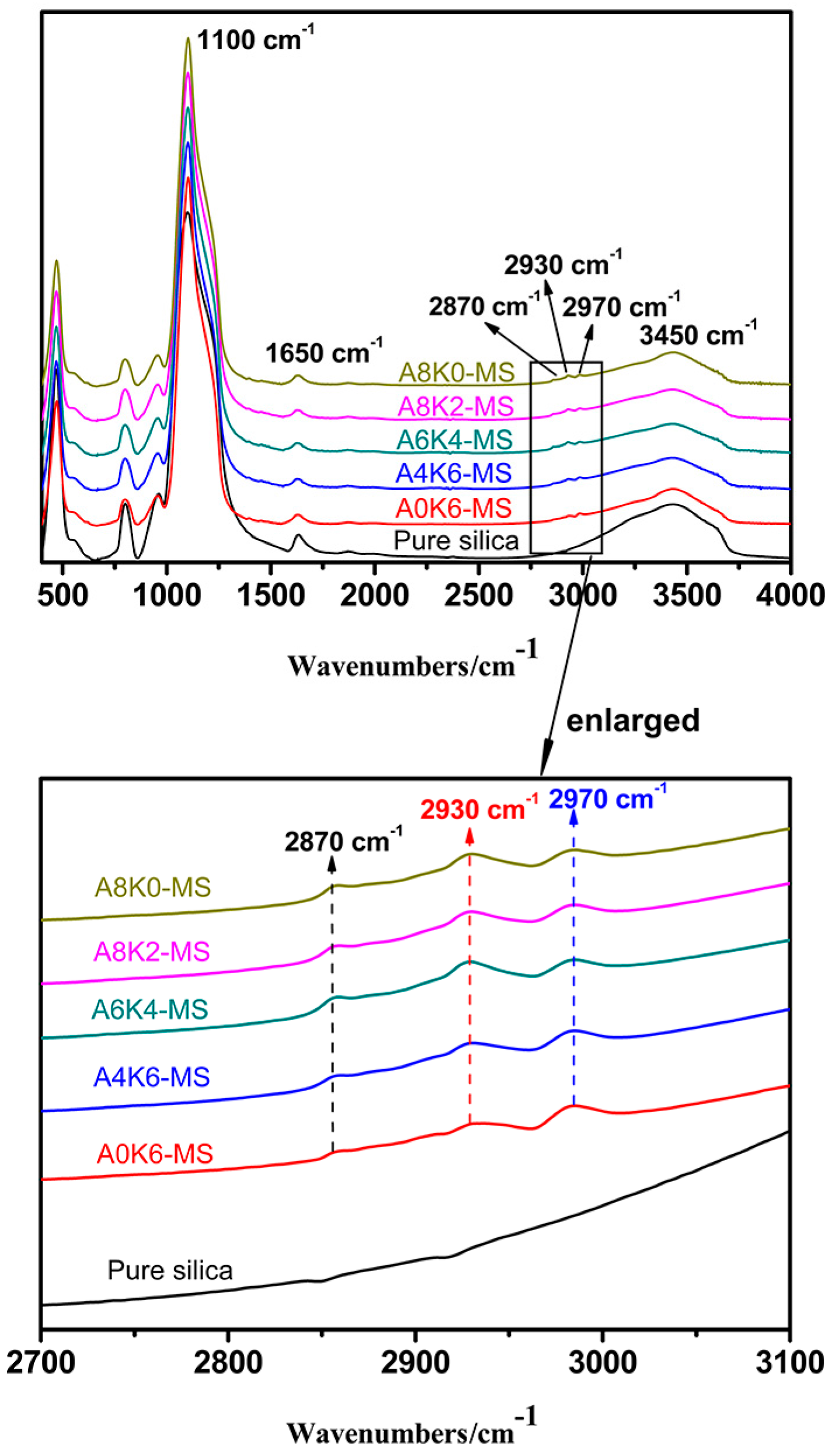
The groups of pure and modified silica were characterized by Fourier transform infrared spectroscopy (FT-IR; Bruker Optik GmbH Co., Tensor 27, Ettlingen, Germany), using the absorption mode under a wave ranging from 4000–400 cm−1 with a resolution of 4 cm−1. The samples were pressed into pellets together with potassium bromide.
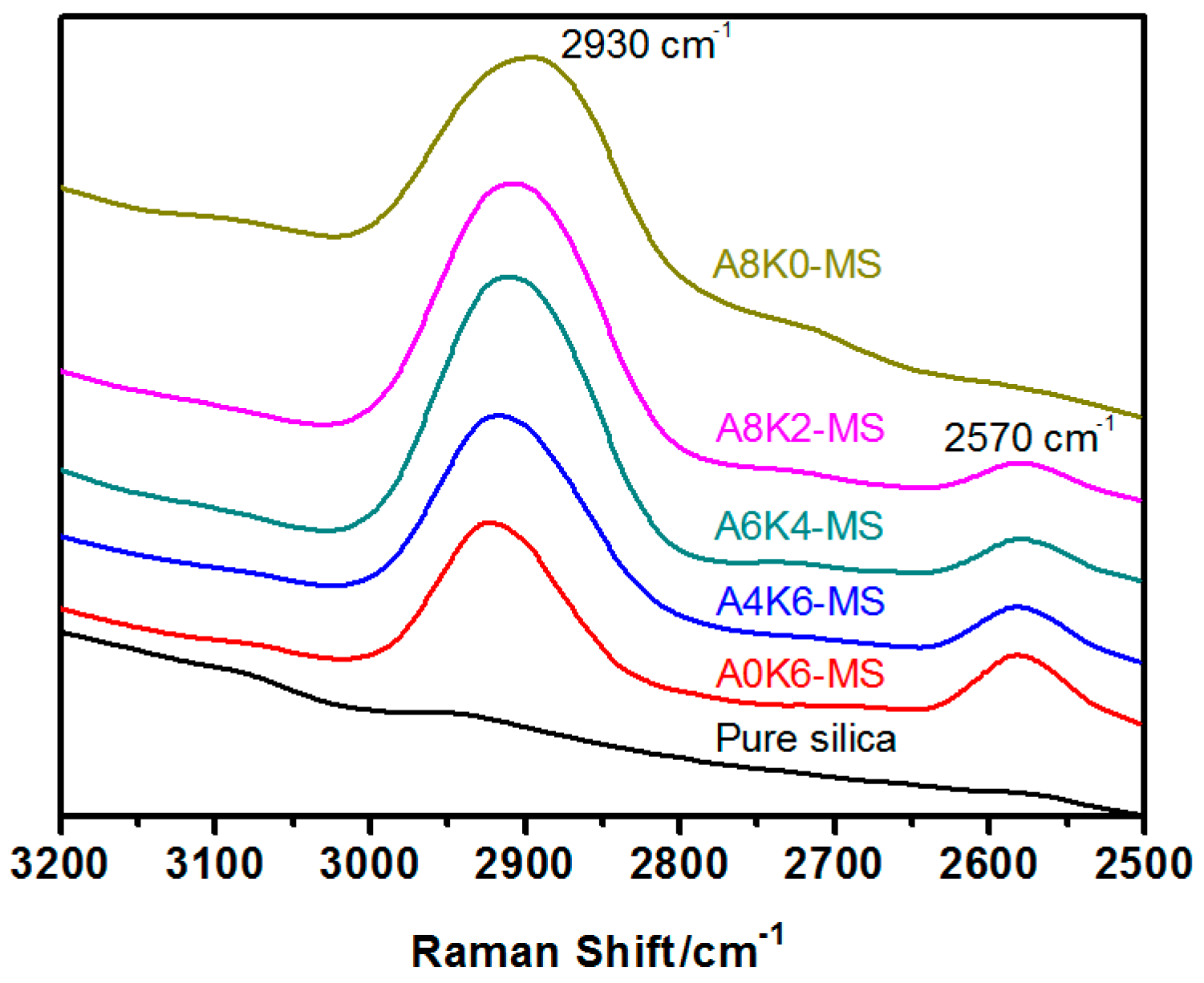
Raman spectra of pure and modified silica samples were recorded from 3200–2500 cm−1 on an inVia confocal Raman spectrometer (Renishaw PLC, Gloucestershire, UK) using a 514-nm laser beam. The power of a 514-nm argon ion excitation laser at the source is approximately 50 mW (highest power) and 20 mW at the surface of the sample. The Raman spectra of the samples were obtained from pressed solid samples in a sealed capillary tube.
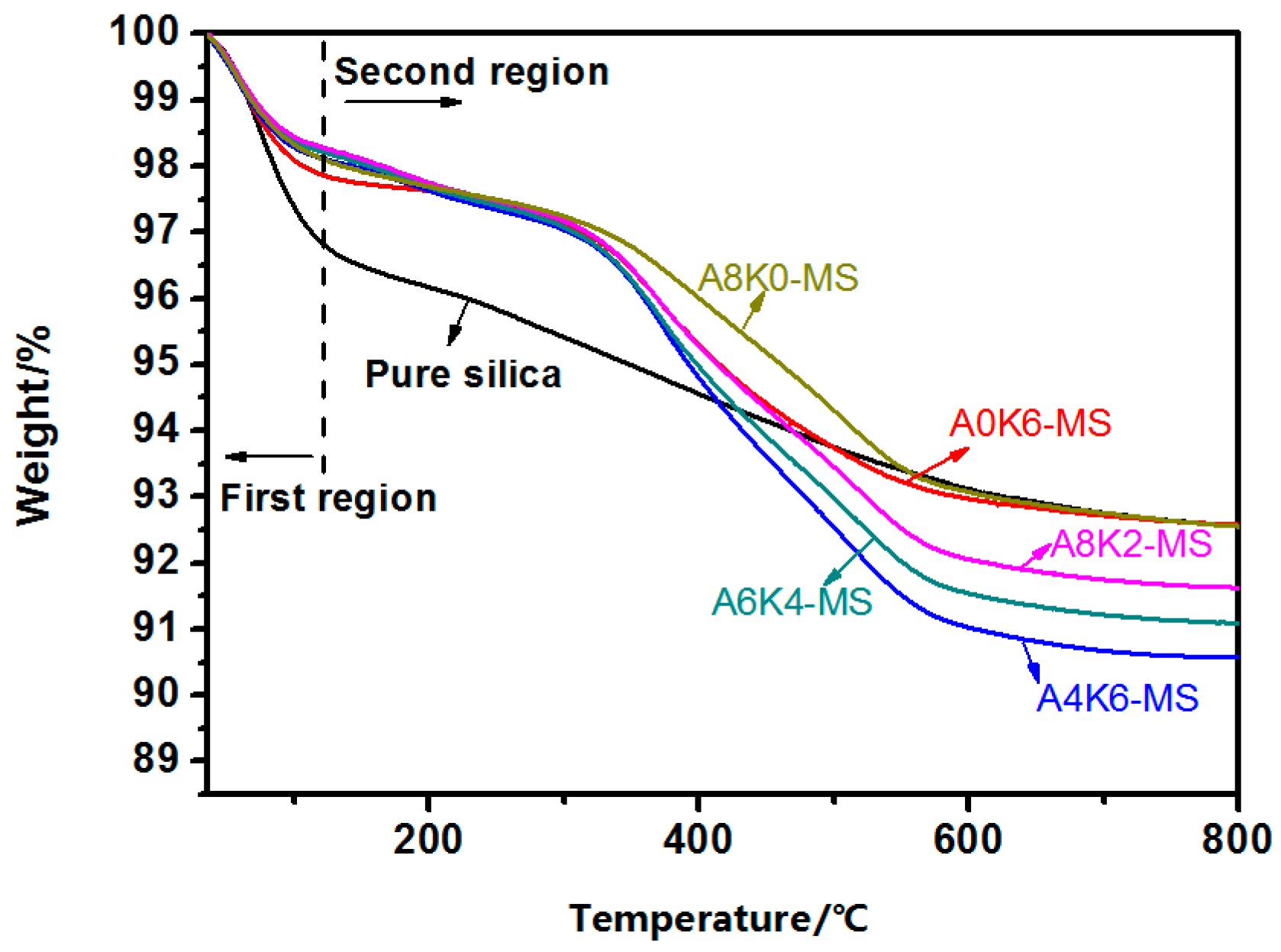
Weight loss measurements of pure and modified silica and the master batches were performed on a thermal gravimetric analyzer (TGA) STARe system (Mettler-Toledo Co., Greifensee, Switzerland) in a nitrogen atmosphere. Samples for the TGA tests were heated at a heating rate of 10 °C/min. The residual weight of master batches, NR, silica, K-MEPTS and AEO were recorded as Rm, Rr, Rs, R5 and RA, respectively.
The filler dispersion in silica/NR master batches and silica/NR composites was observed under a Tecnai G2 20 transmission electron microscope (TEM, FEI Co., Hillsboro, OR, USA) with an accelerating voltage of 200 kV. Thin sections for TEM observations were cut by a microtome at −100 °C and collected on copper grids.
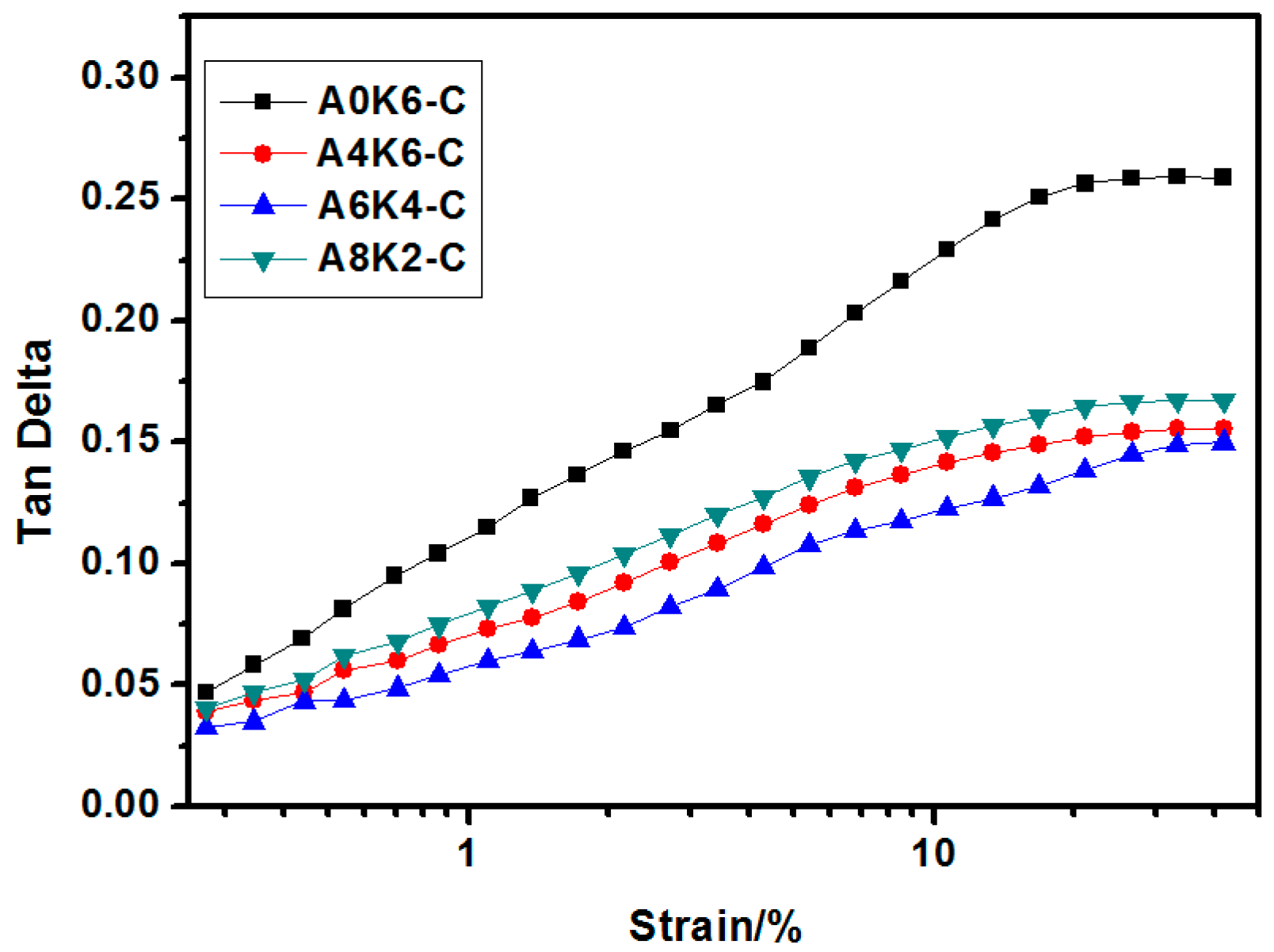
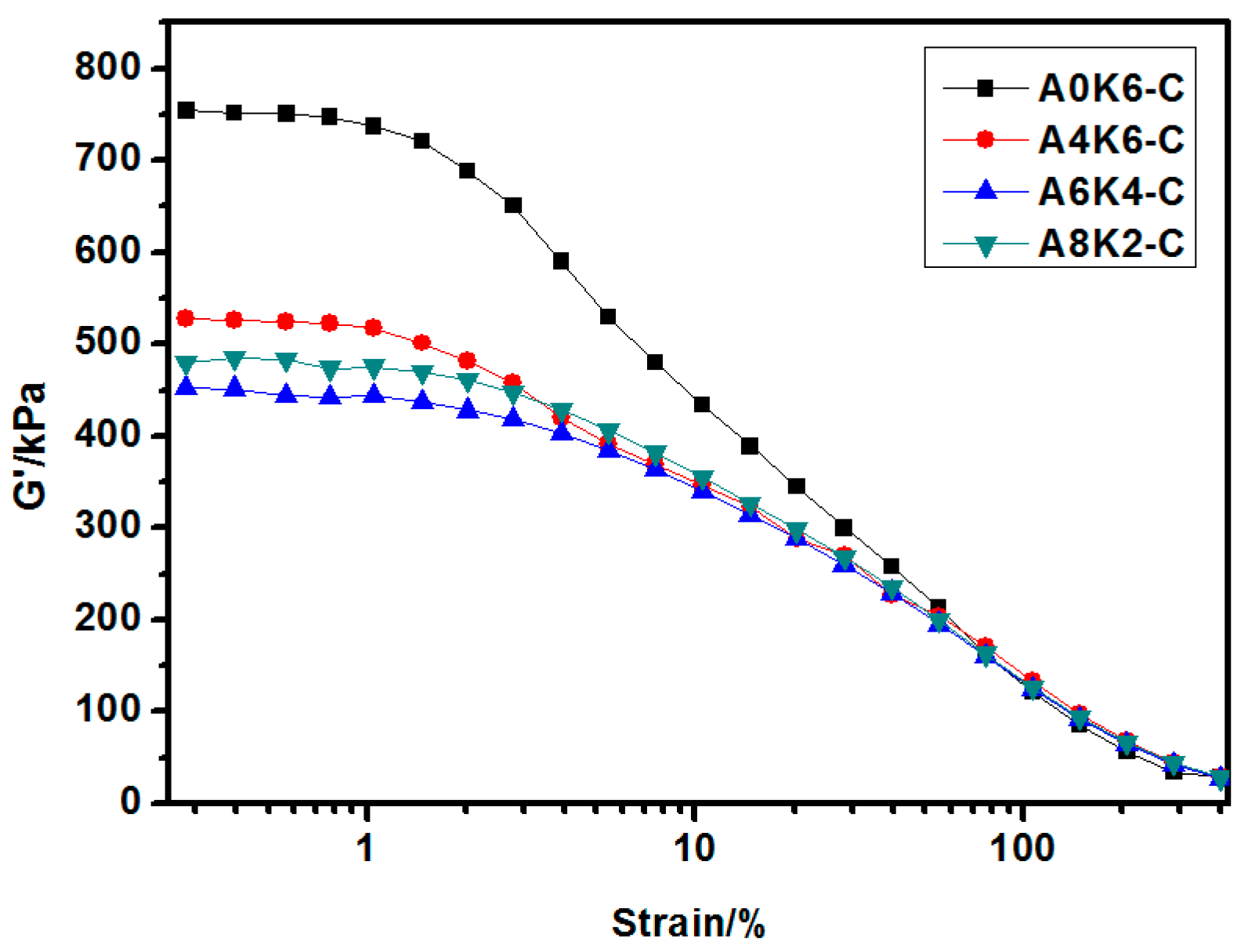
The dynamic rheological performances of silica/NR compounds and silica/NR composites were analyzed using RPA2000 (Alpha Technologies Co., Ltd., Akron, OH, USA) at 60 °C. For the rubber compounds, the strain varied from 0.1%–400% at the test frequency of 1 Hz. For the rubber vulcanizates, the strain varied from 0.1%–40% at the test frequency of 1 Hz. The test of each specimen was repeated 3 times.
The vulcanization characteristics of silica/NR compounds were measured at 143 °C using a P3555B2 disc vulkameter (Beijing Huanfeng Chemical Machinery Trial Plant, Beijing, China). The test of each specimen was repeated 3 times.
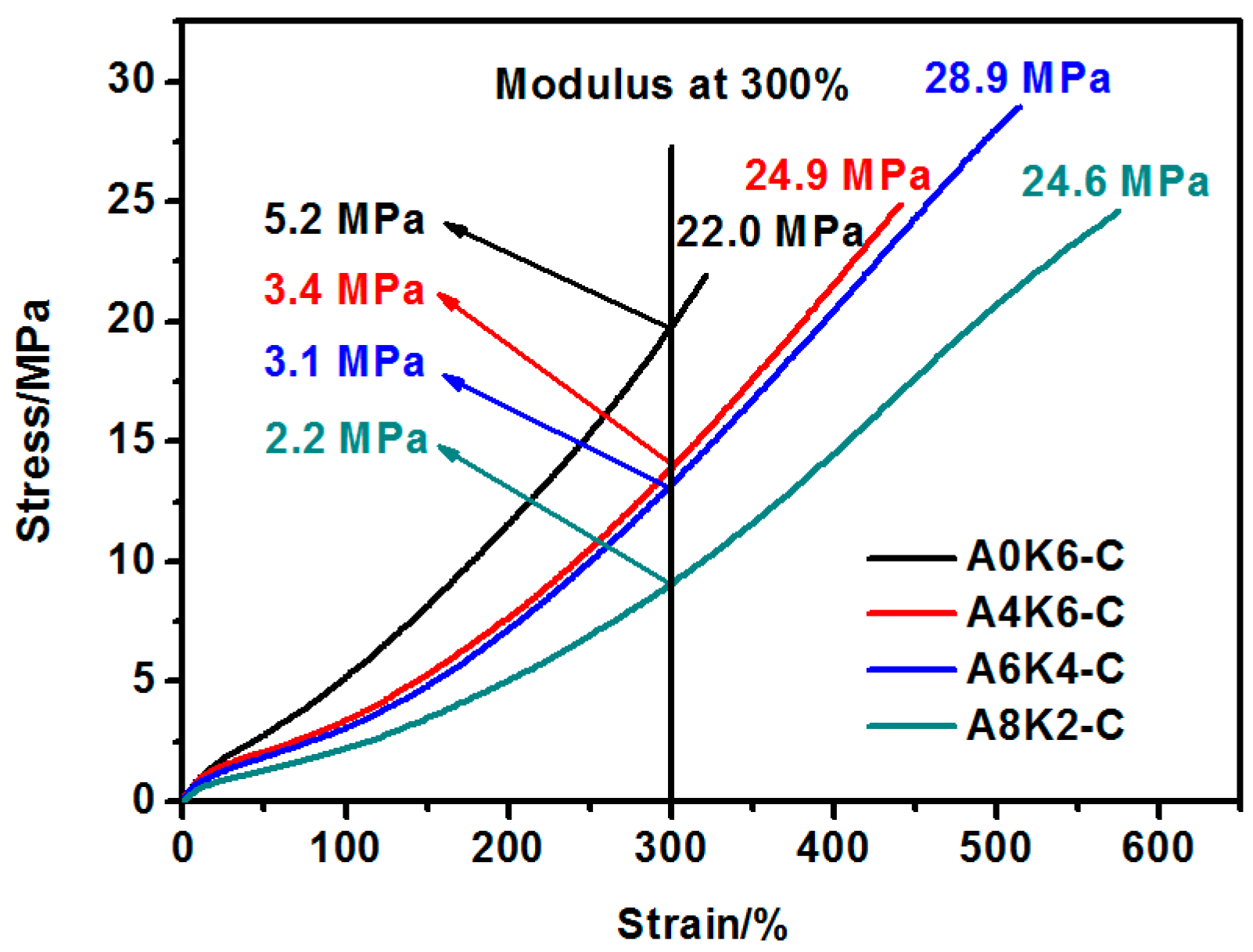
The mechanical performance of the silica/NR composites were investigated according to ASTM D638 specifications using a CMT4104 electrical tensile tester (Shenzhen SANS Test Machine Co., Shenzhen, China) with an across-head speed of 500 mm/min. The test of each specimen was repeated 5 times.
4. Conclusions
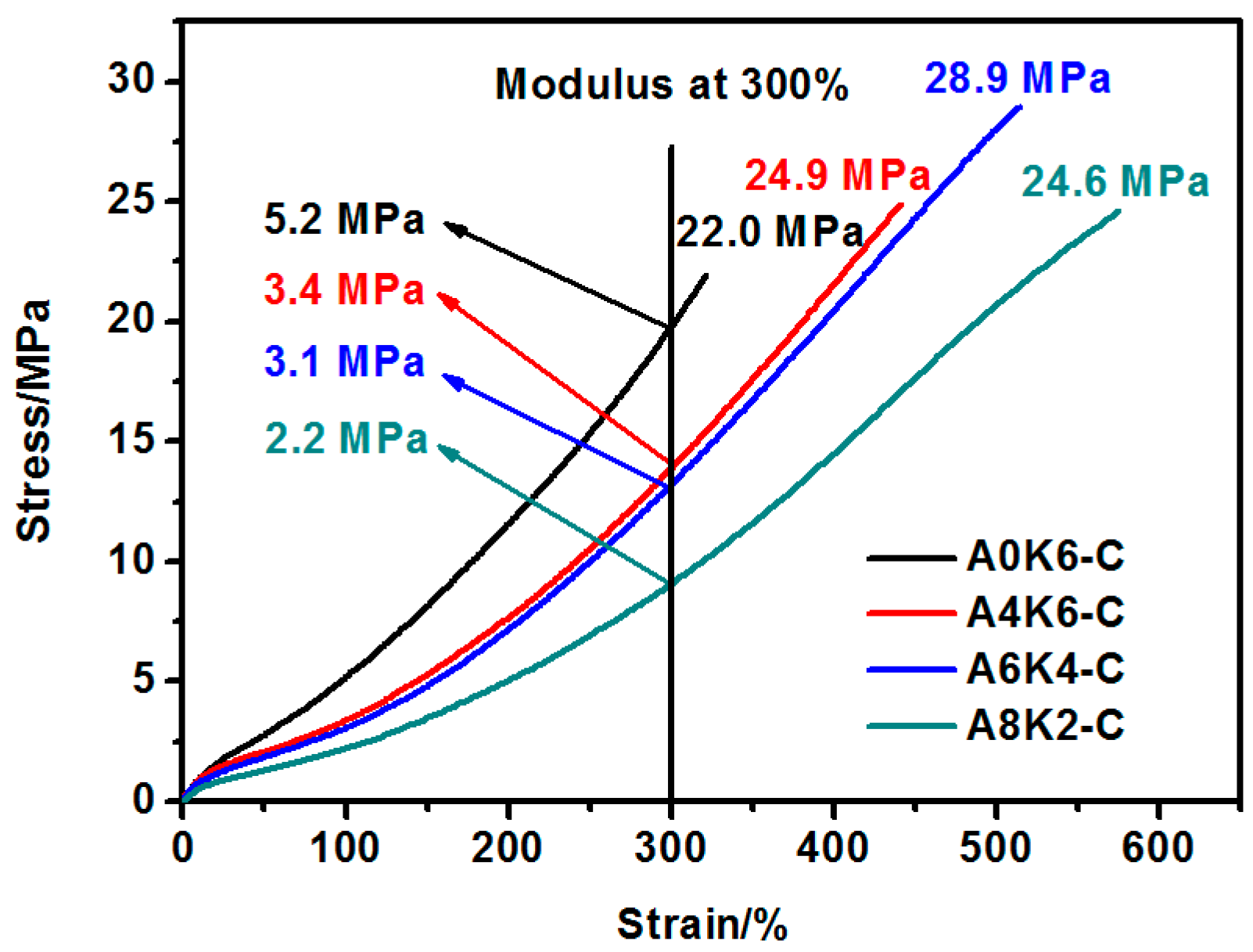
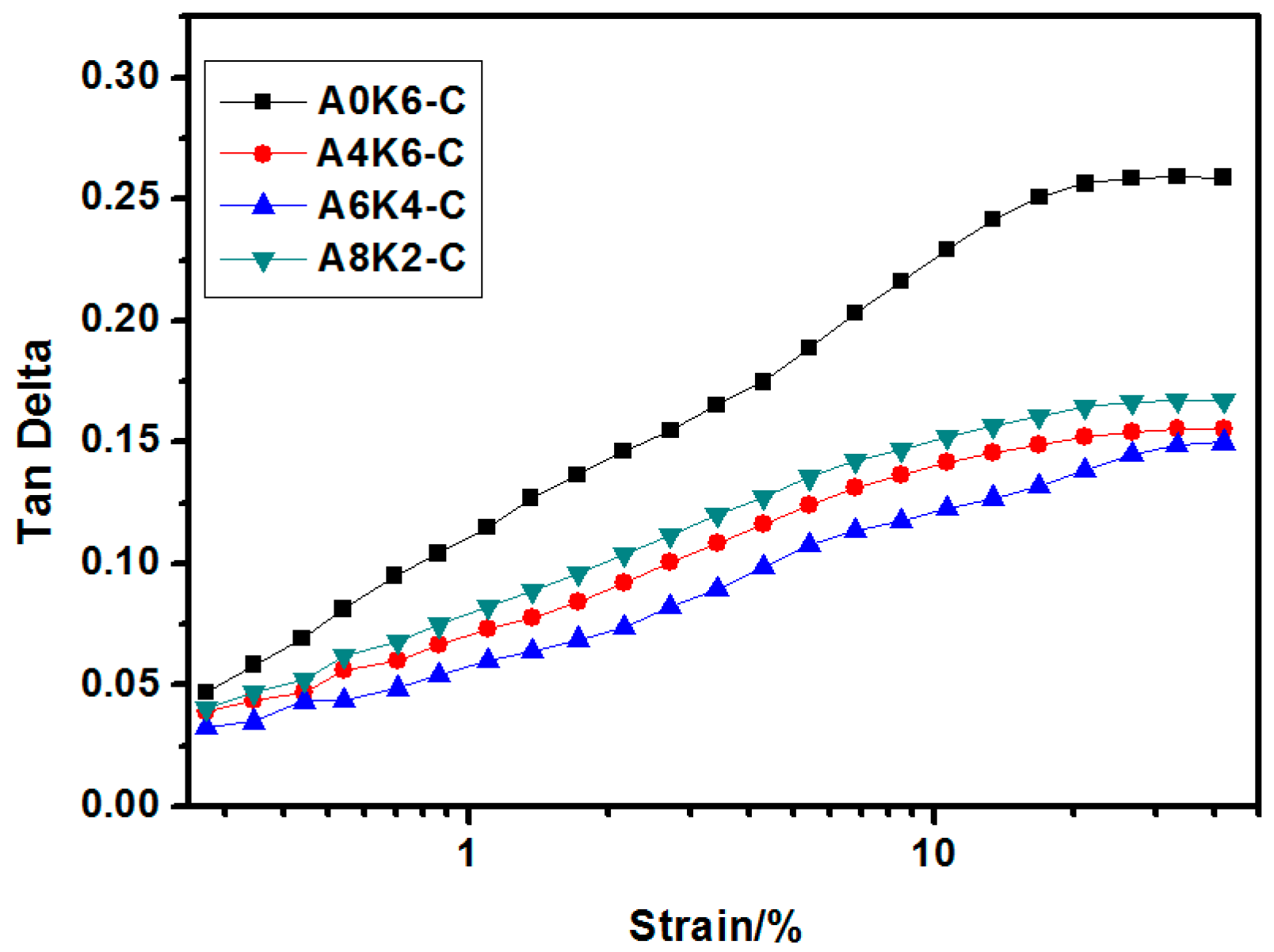
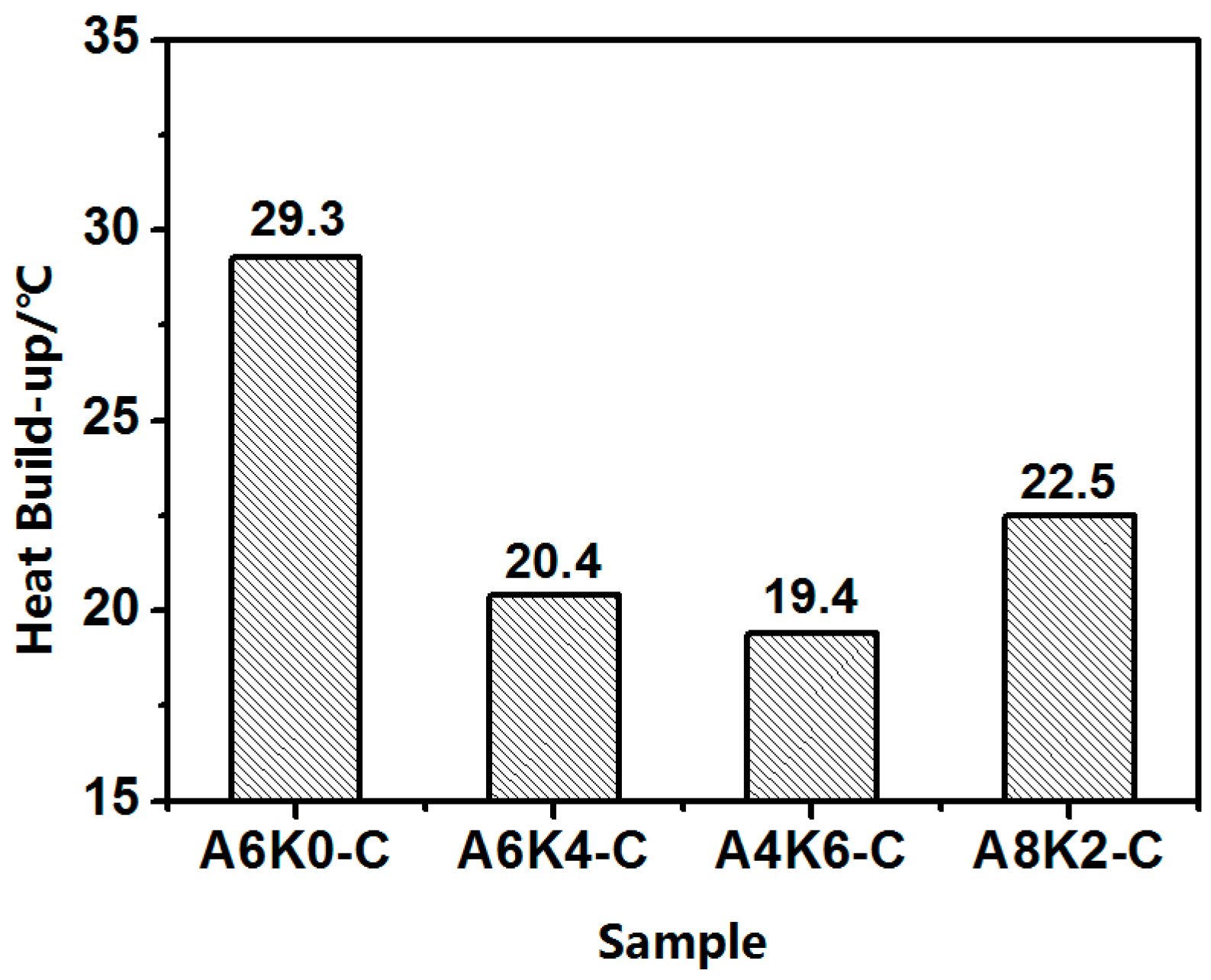
In this research, we proposed an extremely effective strategy of using both AEO and K-MEPTS together for silica modification, by preparing silica/NR using the latex compounding method. As we designed, tailored silica was dispersed in the aqueous phase and completely co-coagulated with the NR. The silica/NR composites prepared by these master batches had ideal dynamic and mechanical performance, especially for A6K4-C, which was a composite containing silica modified with 6% AEO and 4% K-MEPTS (weight ratio of modifier to silica). This research provides a significant potential for preparing high performance silica/NR composites in a user-friendly method.
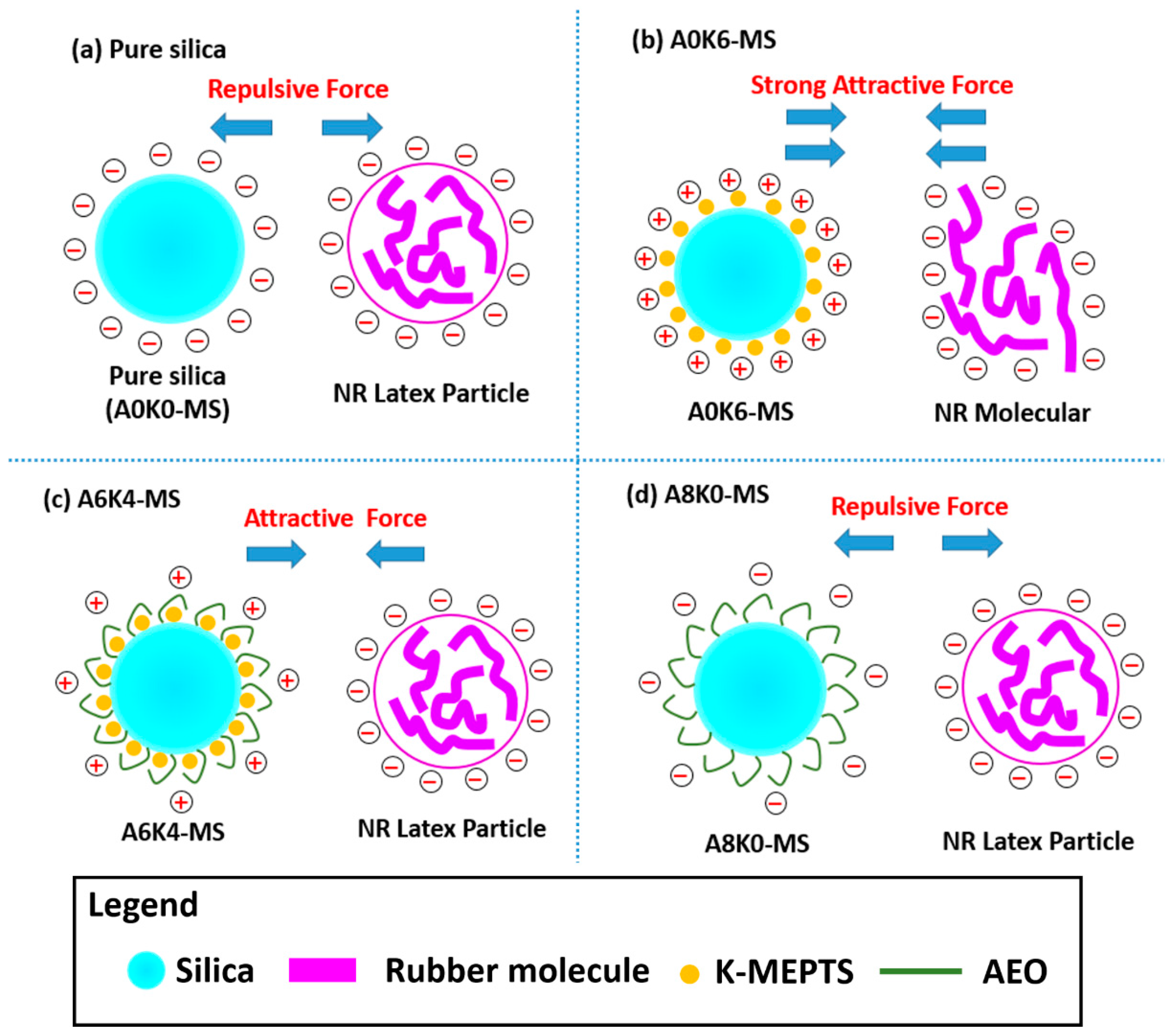
Furthermore, we investigated the effect of several interactions on the performance of silica/rubber composites. We confirmed that both AEO and K-MEPTS grafted onto the silica surface, and AEO shielded K-MEPTS, resulting in weakening the chargeability and activity of the mercaptopropyl group on K-MEPTS. This interaction between these two modifiers contributed to solving the problem of huge silica loss and “scorchy” behavior in the preparation of silica/rubber composites. This is a novel and practical interaction confirmed in this work. In silica/NR composites, AEO formed a physical interface between silica and rubber, resulting in reducing the aggregation of silica and improving the silica dispersion in the rubber matrix. Moreover, K-MEPTS formed a chemical interface between silica and rubber, resulting in strengthening the connection between silica and rubber and reducing the mutual friction between silica particles. Finally, AEO and K-MEPTS acted synergistically to improve the mechanical and dynamic performances of silica/NR composites.
Overall, the novel strategy outlined in this article provides a new strategy for the preparation of rubber materials with excellent performance. We hope that the preparation of silica/NR master batches using this strategy will present practical and profound applications in the rubber industry.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}