
Molecular Structure, Spectroscopic and DFT Computational Studies of Arylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Results
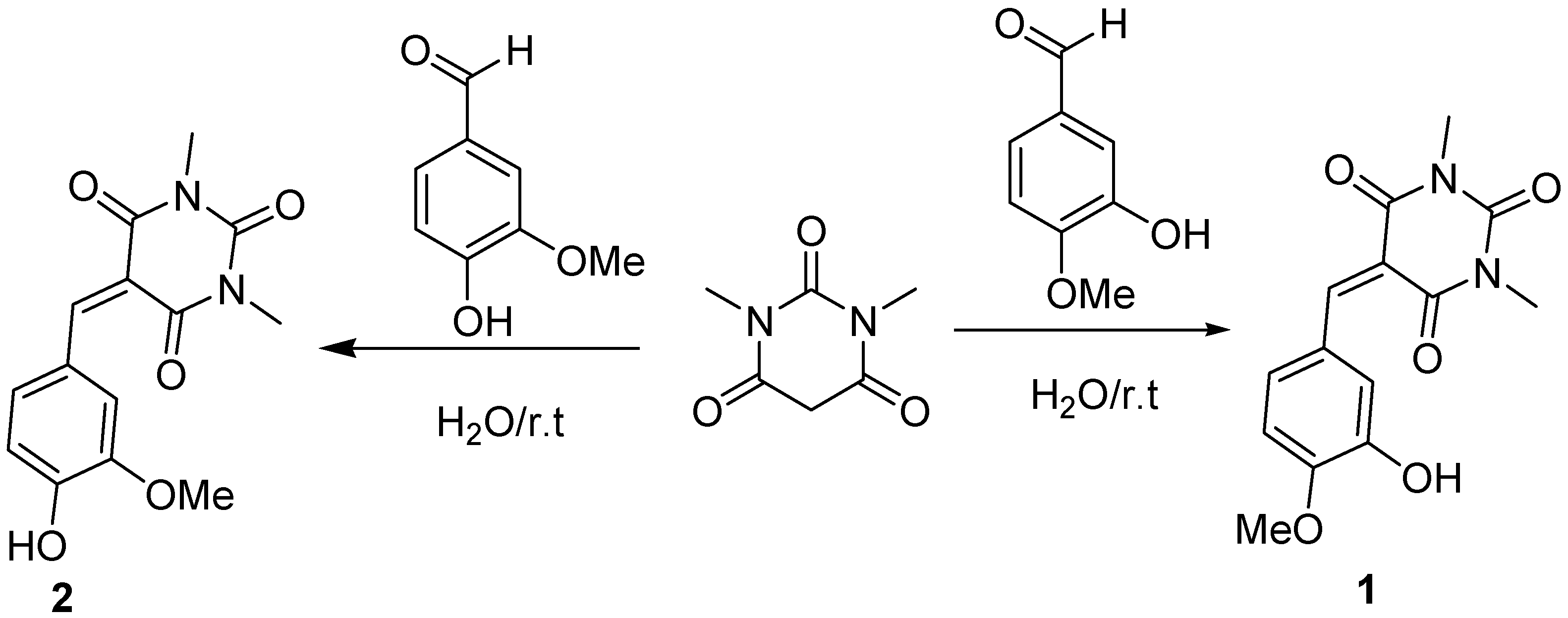
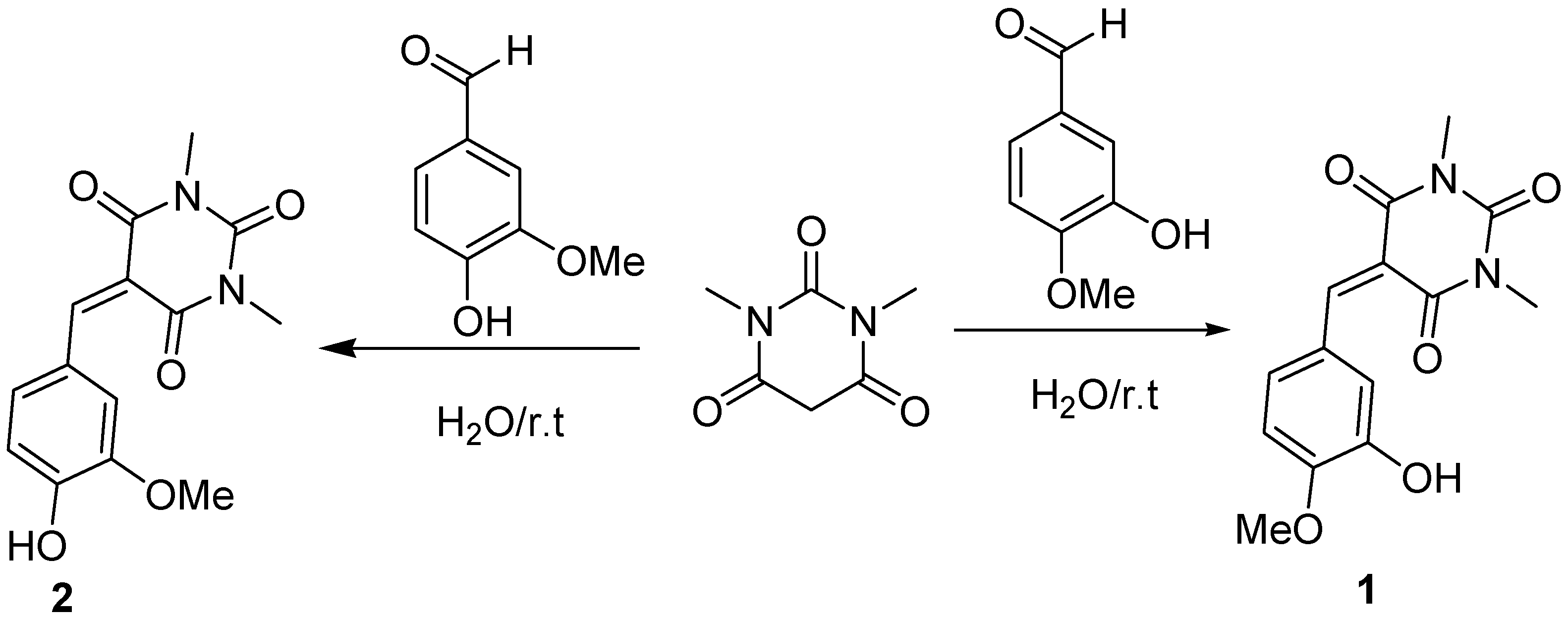
2.1. Chemistry
3. Discussion
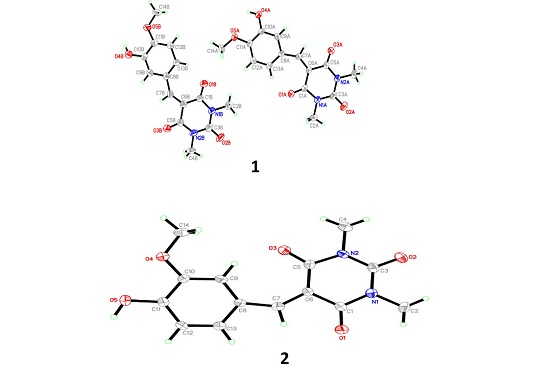
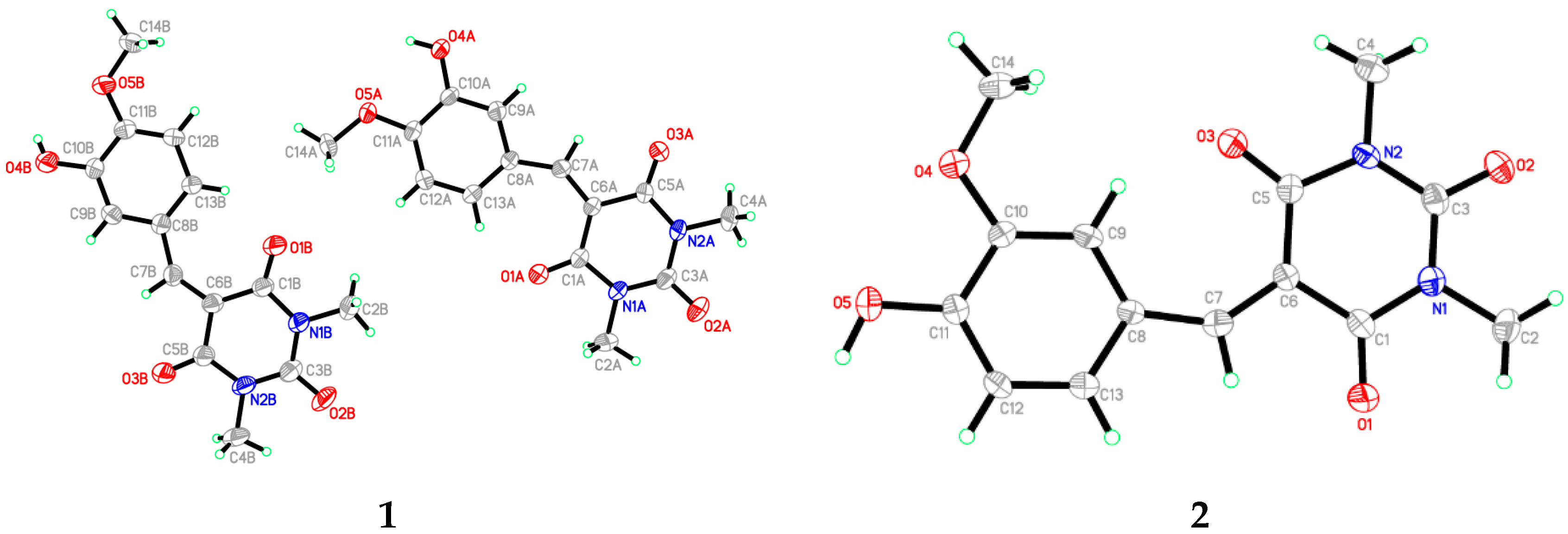
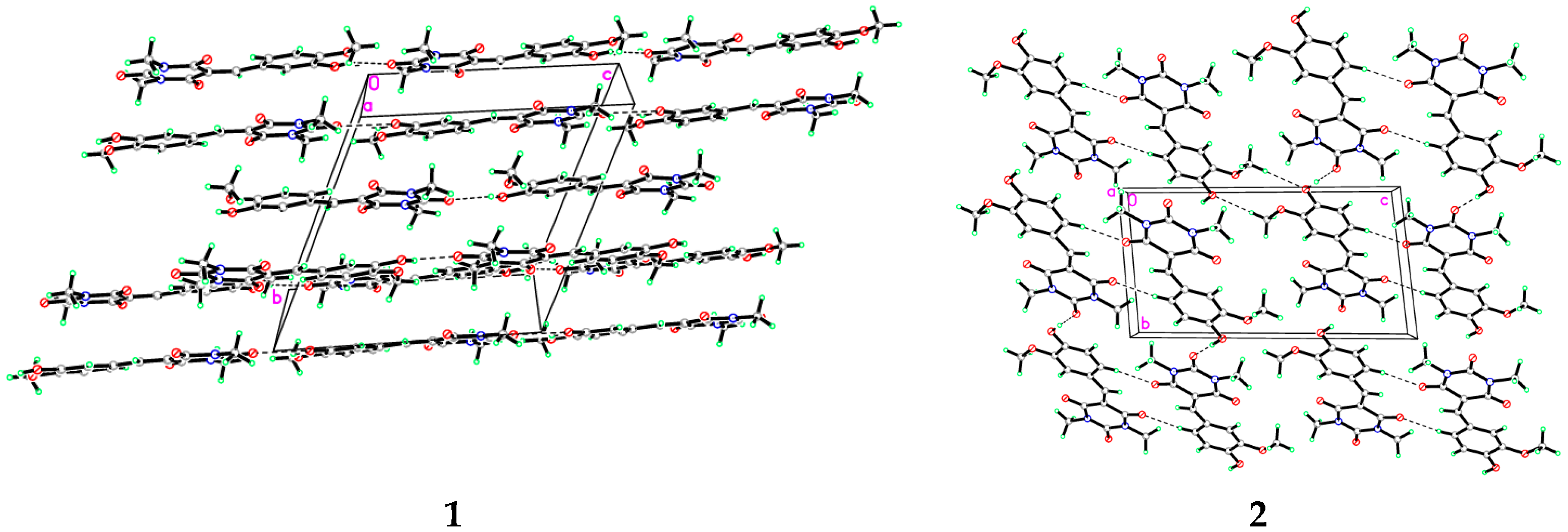
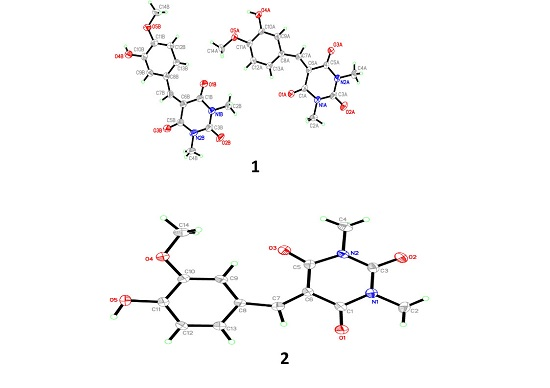
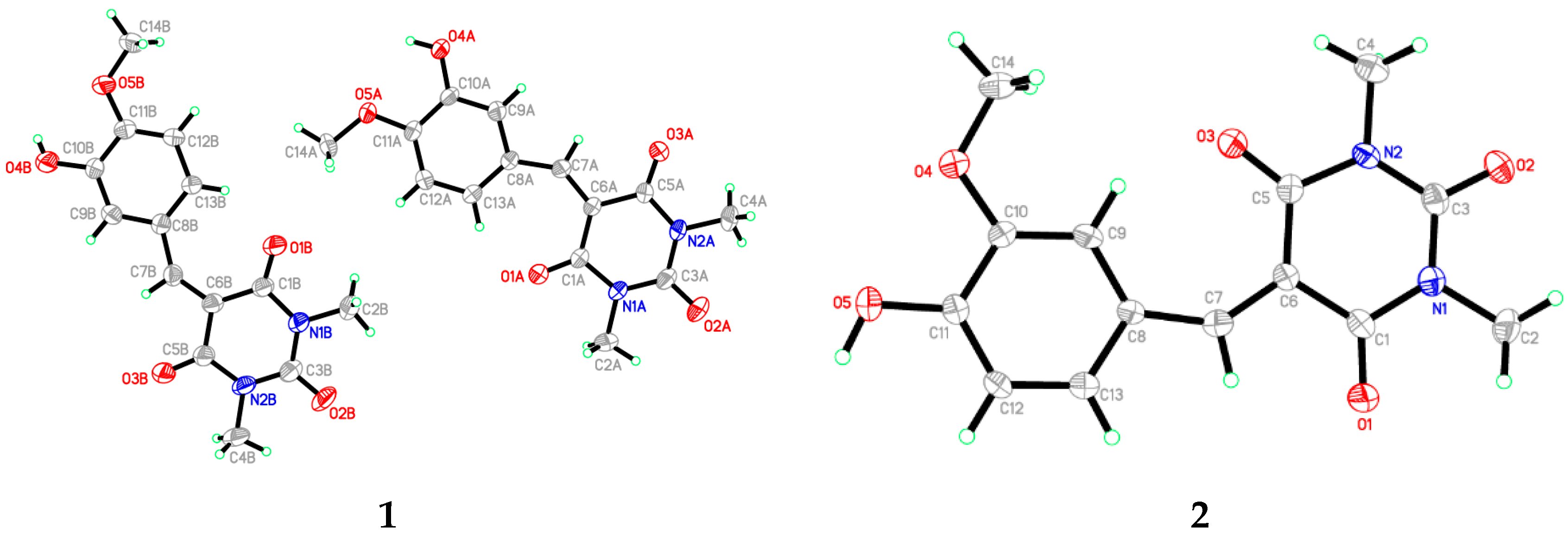
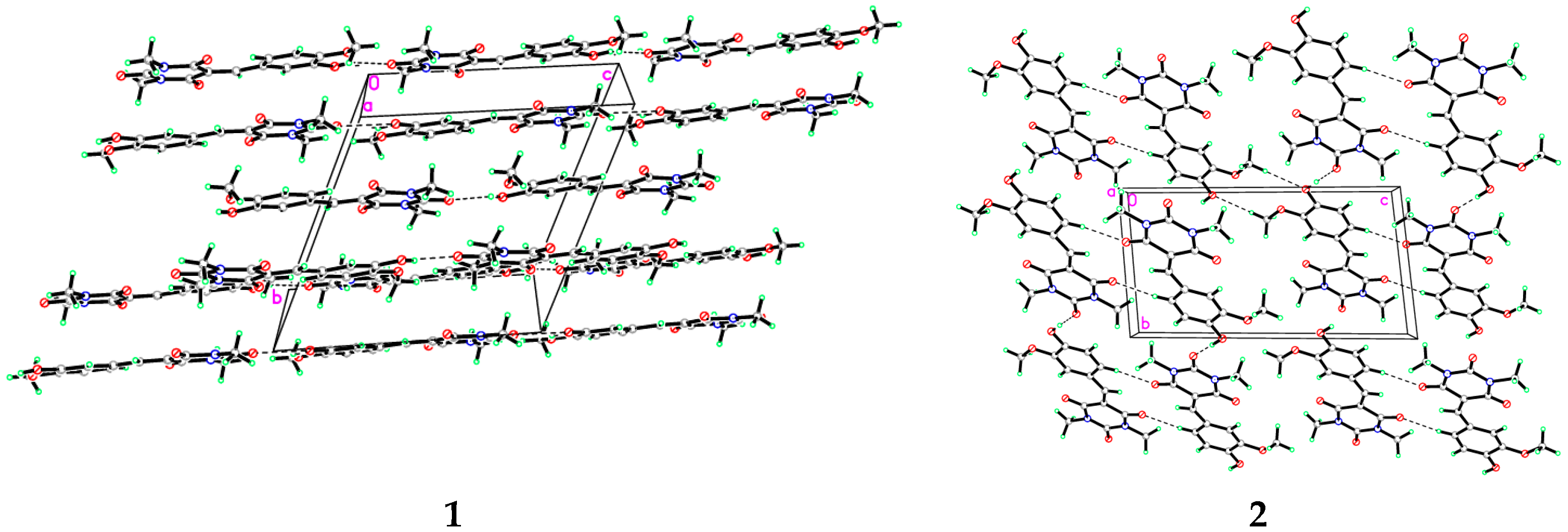
3.1. X-Ray Crystal Structure of 1 and 2
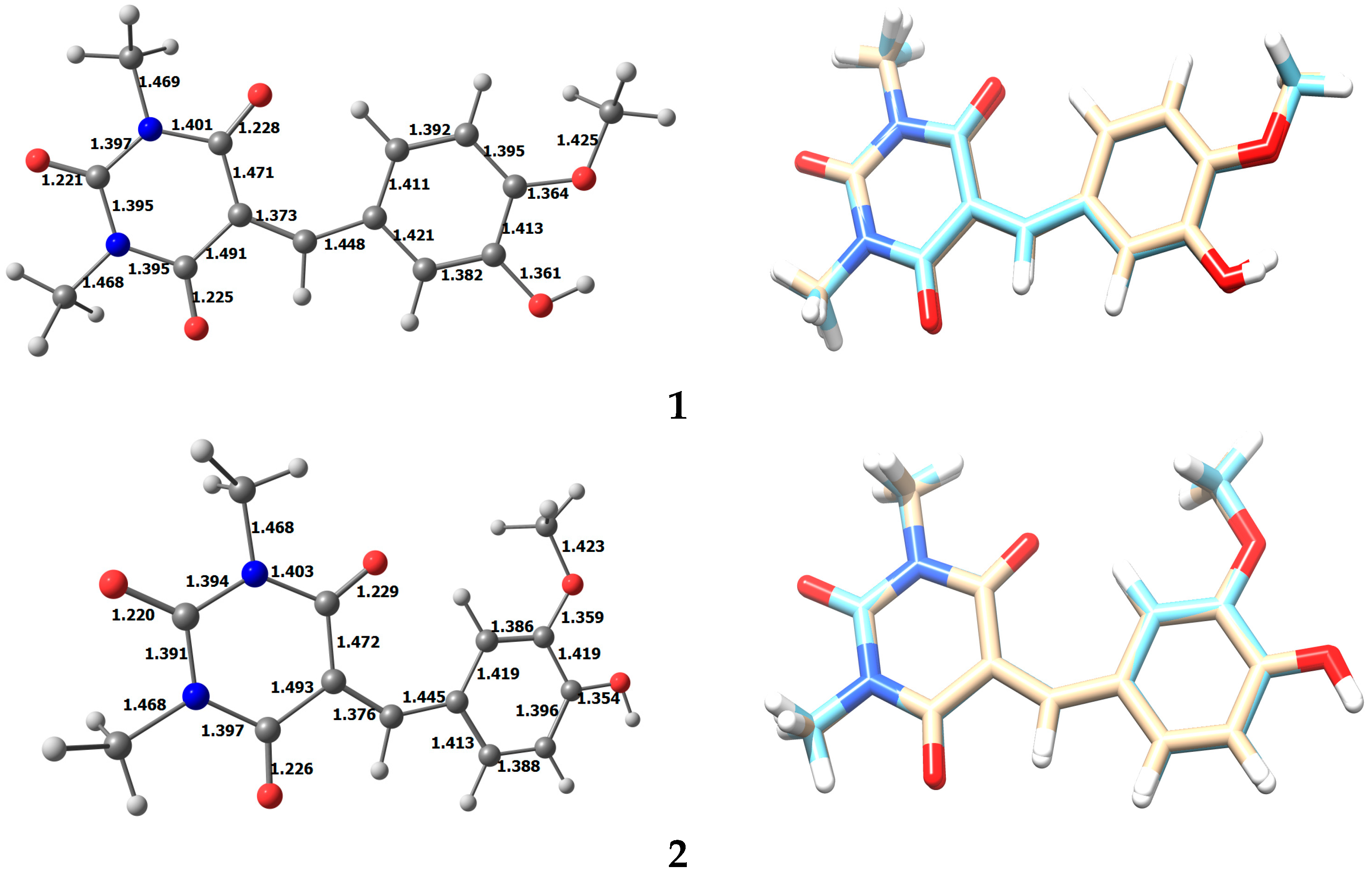
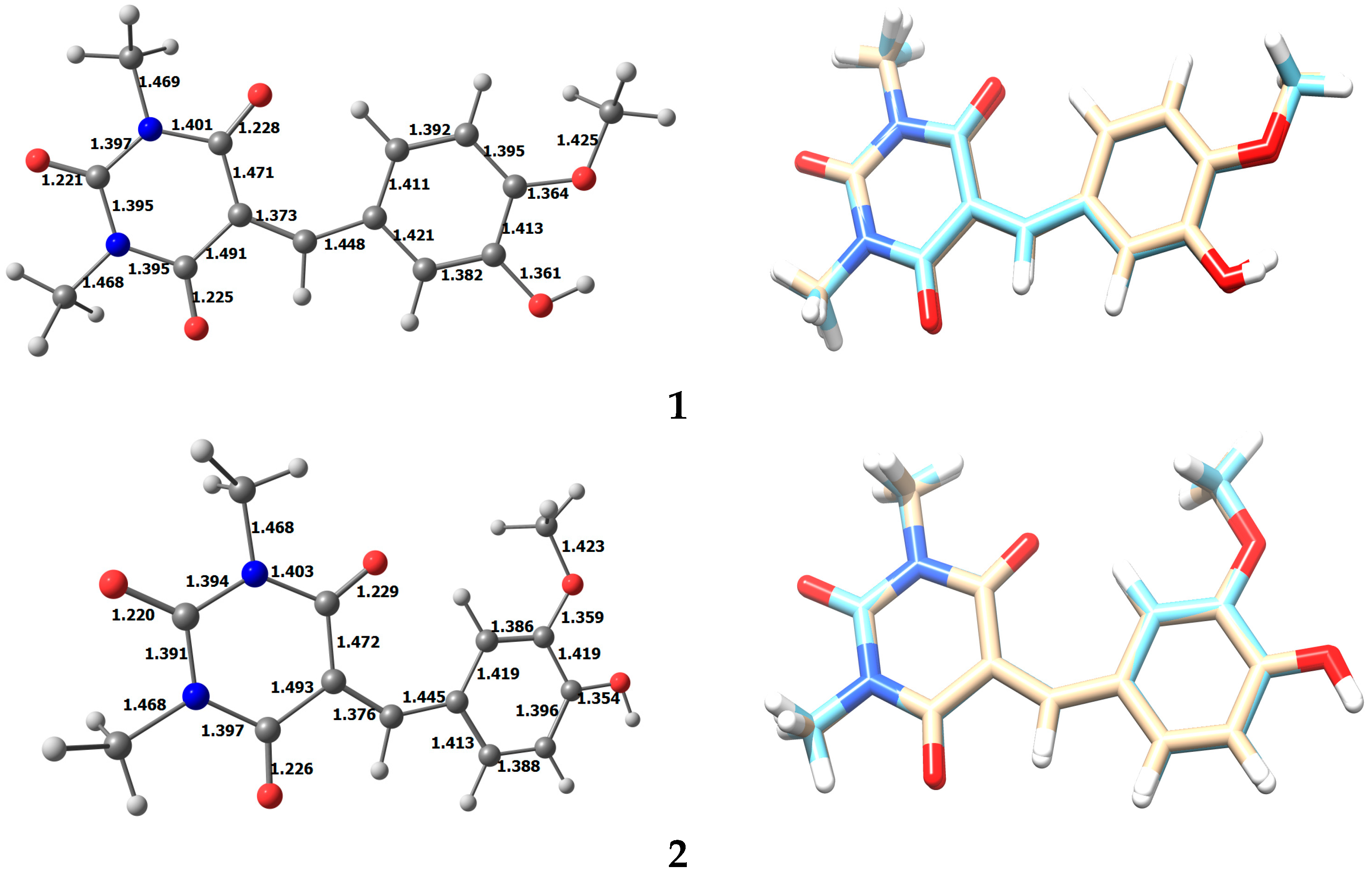
3.2. Optimized Structure of 1 and 2
3.3. Energy Analysis of the Two Isomers
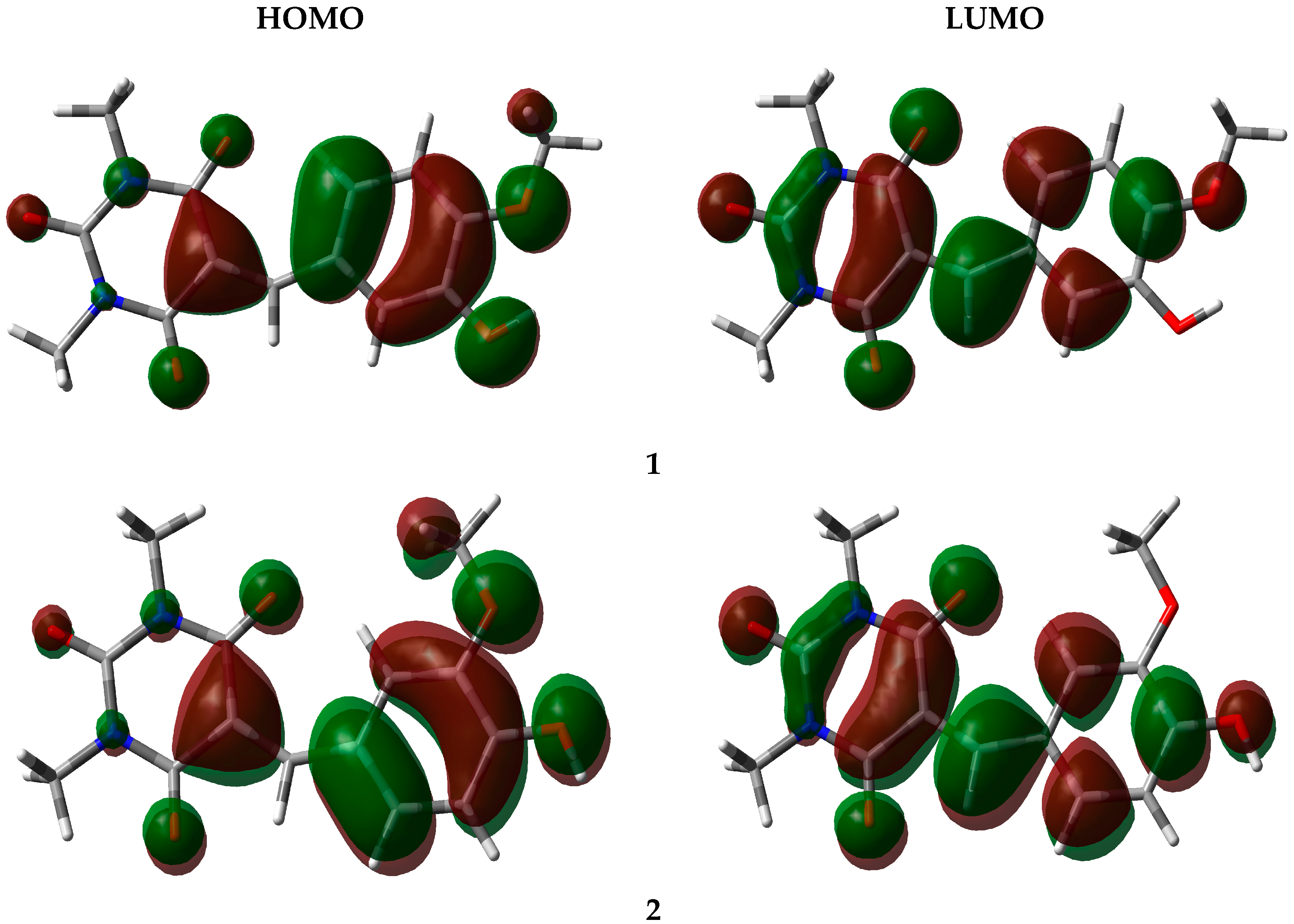
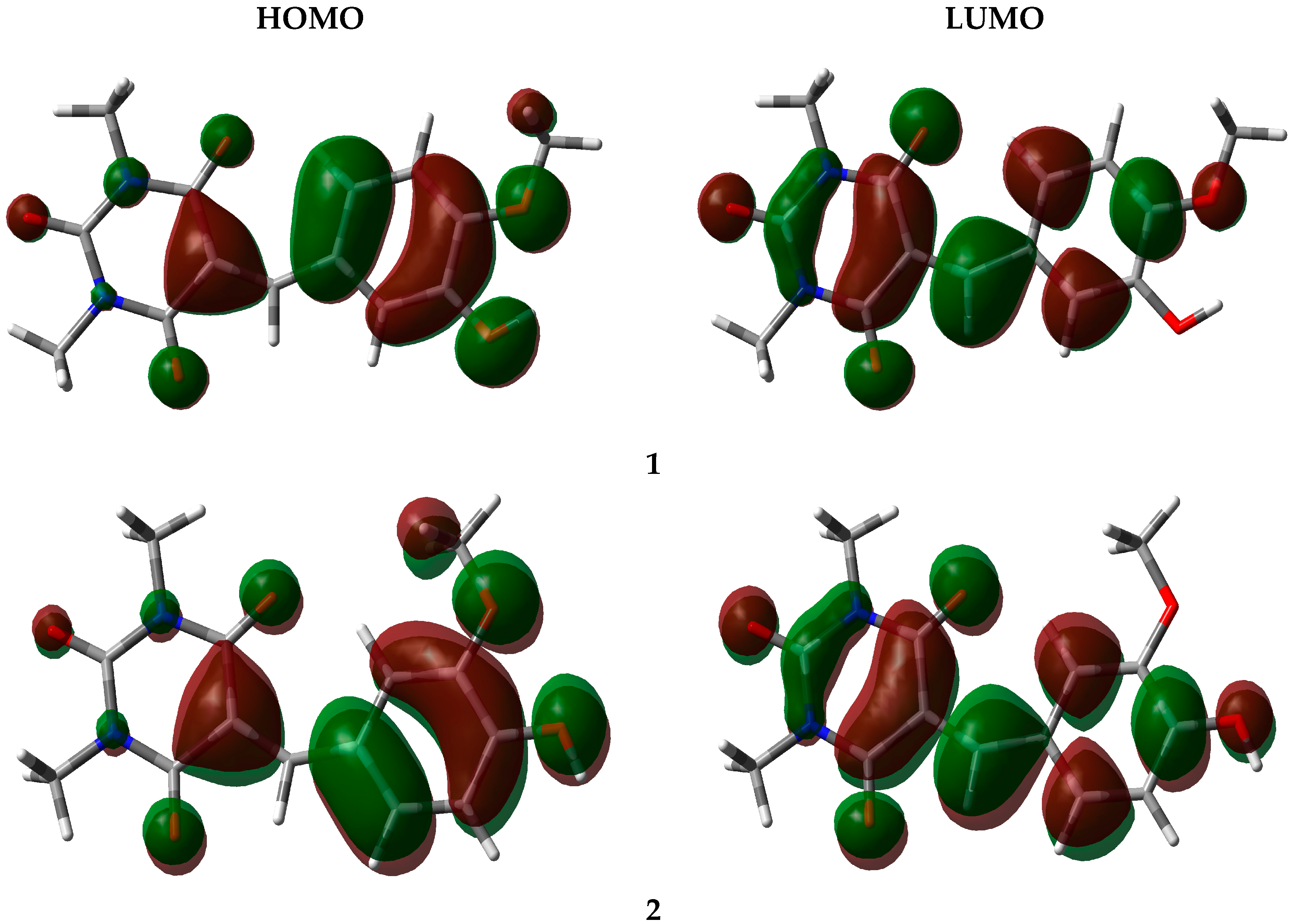
3.4. Frontier Orbital Energy Analysis
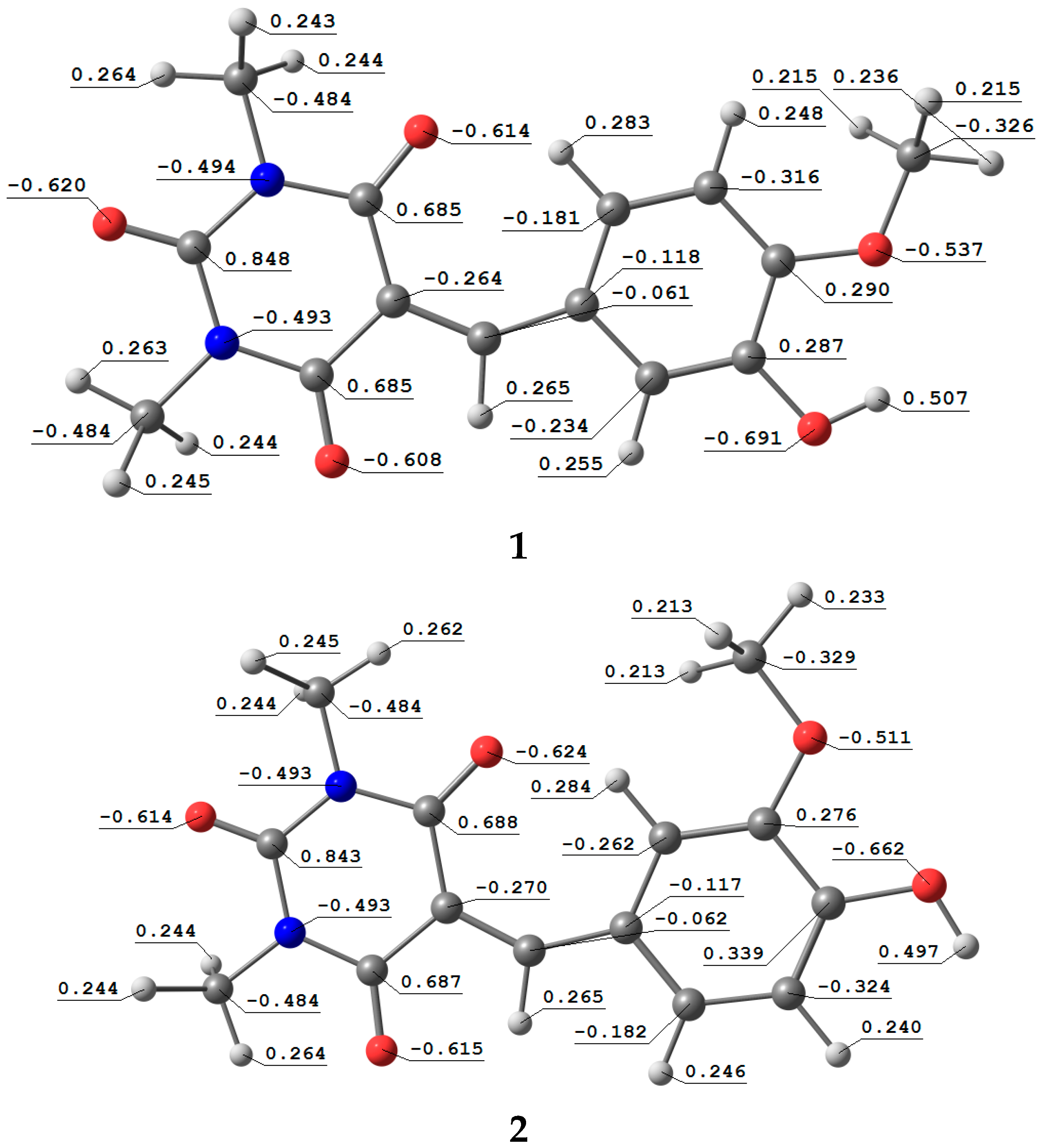
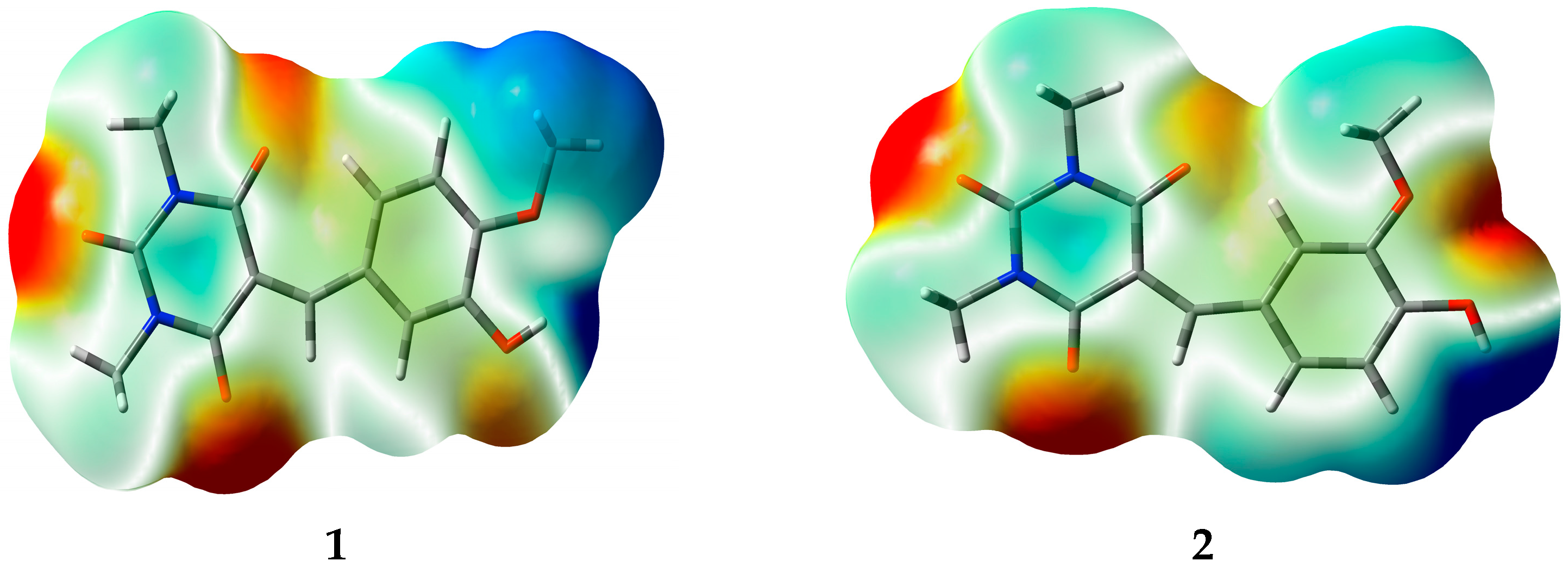
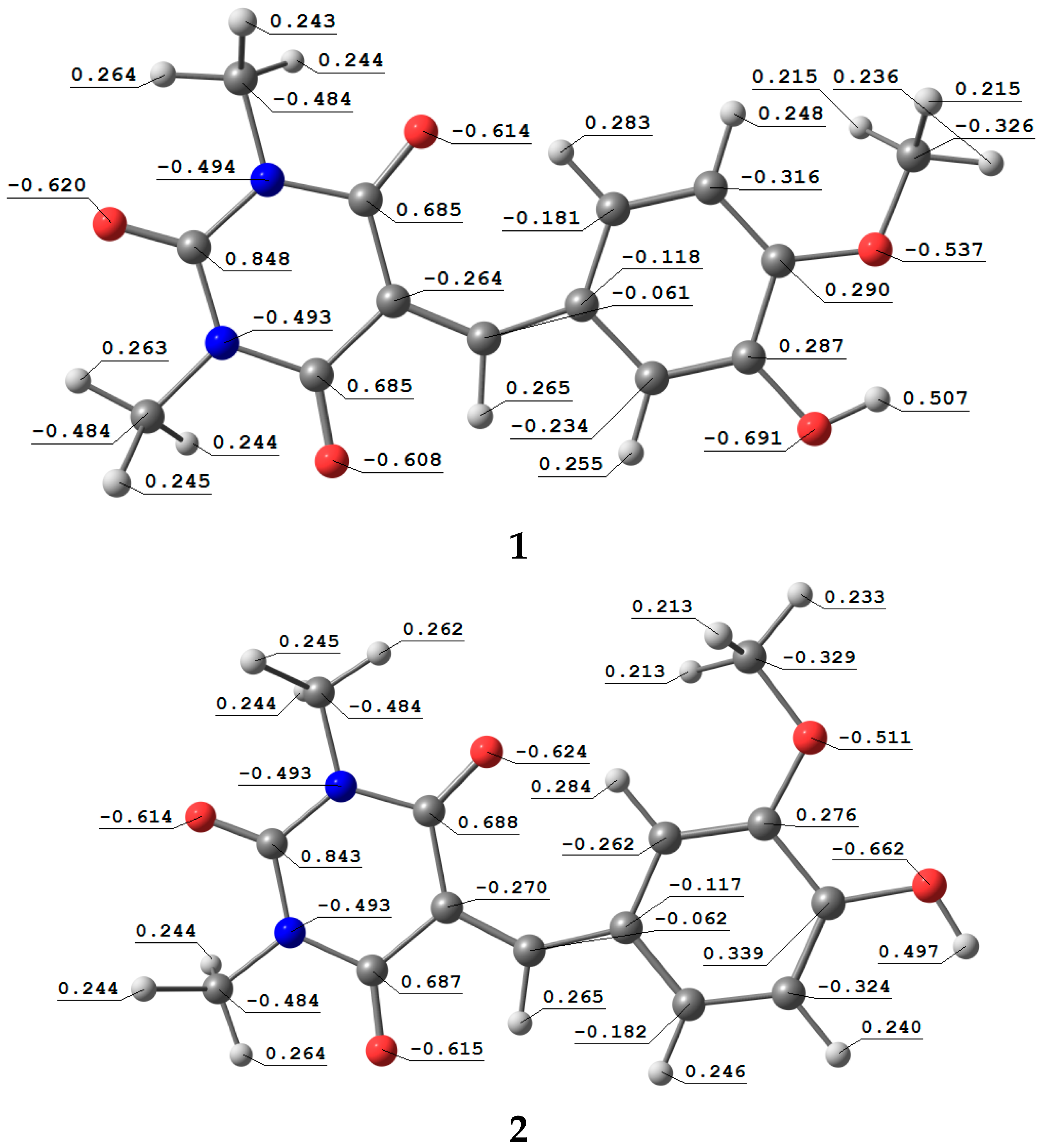
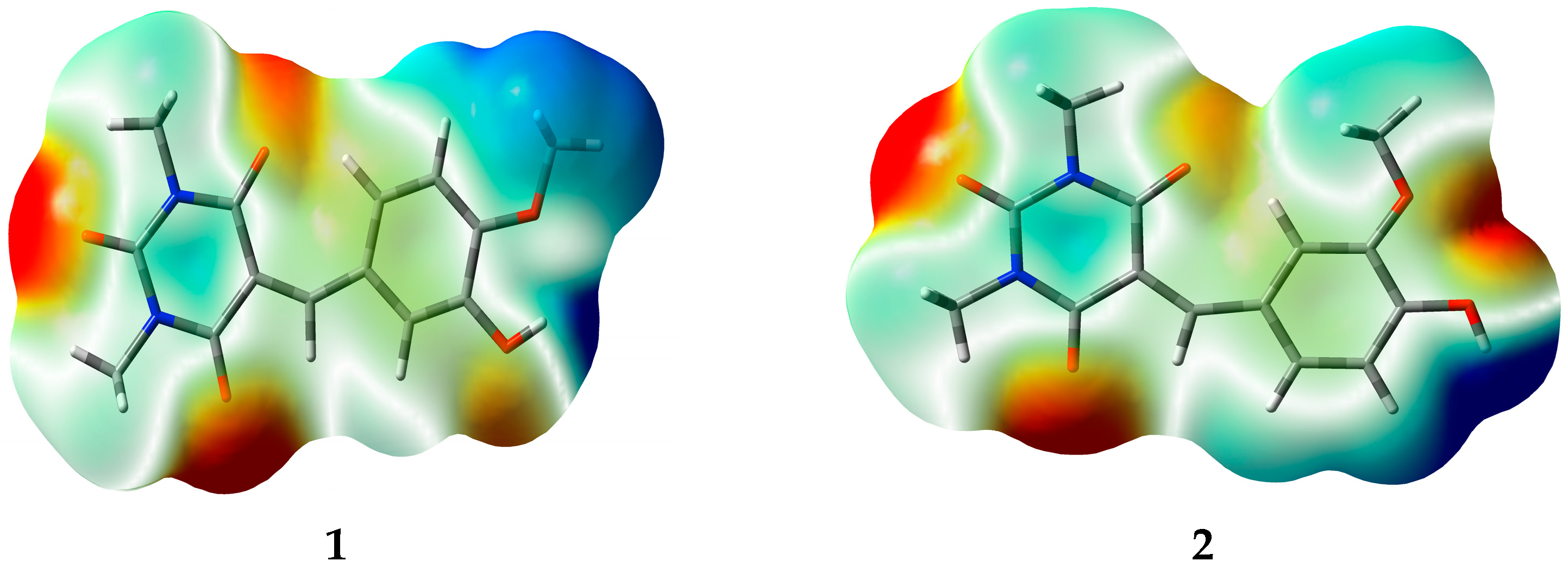
3.5. Natural Atomic Charges (QN) and Electrostatic Potential (ESP)
4. Materials and Methods
4.1. General Remarks
4.2. Procedure for the Synthesis of 1 and 2
4.3. Theoretical Calculations
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gomez, G.G.; Hutchison, R.B.; Kruse, C.A. Chemo-immunotherapy and chemo-adoptive immunotherapy of cancer. Cancer Treat. Rev. 2001, 27, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; O’Sullivan, S.; Harmon, S.; Keaveny, R.; Radomski, M.W.; Medina, C.; Gilmer, J.F. Design of barbiturate–nitrate hybrids that Inhibit MMP-9 activity and secretion. J. Med. Chem. 2012, 55, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Ghabbour, H.A.; Al-Majid, A.M.; Imad, R.; Javaid, K.; Shaikh, N.N.; Yousuf, S.; Choudhary, M.I.; Wadood, A. Synthesis, X-ray crystal structures, biological evaluation, and molecular docking studies of a series of barbiturate derivatives. J. Chem. 2016, 2016. [Google Scholar] [CrossRef]
- Ruble, J.C.; Hurd, A.R.; Johnson, T.A.; Sherry, D.A.; Barbachyn, M.R.; Toogood, P.L.; Bundy, G.L.; Graber, D.R.; Kamilar, G.M. Synthesis of (−)-PNU-286607 by asymmetric cyclization of alkylidene barbiturates. J. Am. Chem. Soc. 2009, 131, 3991–3997. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.R.; Mark, L.C.; Winkler, D.A.; Jones, G.P. Structure-activity relations of convulsant and anticonvulsant barbiturates: A computer-graphic-based pattern-recognition analysis. J. Med. Chem. 1983, 26, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Benmohamed, R.; Kim, J.; Arvanites, A.C.; Morimoto, R.I.; Ferrante, R.J.; Kirsch, D.R.; Silverman, R.B. Pyrimidine-2,4,6-trione derivatives and their inhibition of mutant SOD1-dependent protein aggregation. Toward a treatment for amyotrophic lateral sclerosis. J. Med. Chem. 2011, 54, 2409–2421. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Berezin, M.Y.; Guo, K.; Kao, J.; Achilefu, S. Near-infrared fluorescent pH-sensitive probes via unexpected barbituric acid mediated synthesis. Org. Lett. 2008, 11, 29–32. [Google Scholar] [CrossRef] [PubMed]
- McClenaghan, N.D.; Absalon, C.; Bassani, D.M. Facile synthesis of a fullerene-barbituric acid derivative and supramolecular catalysis of its photoinduced dimerization. J. Am. Chem. C. 2003, 125, 13004–13005. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, B.B.; Spiteller, M. Possible application of the organic barbiturates as NLO materials. Cryst. Growth Des. 2010, 10, 2470–2474. [Google Scholar] [CrossRef]
- Wasserscheid, P.; Joni, J. Handbook of Green Chemistry, Volume 5: Reactions in Water; Li, C.-J., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016. [Google Scholar]
- Barakat, A.; Islam, M.S.; Al-Majid, A.M.; Soliman, S.M.; Mabkhot, Y.N.; Al-Othman, Z.A.; Ghabbour, H.A.; Fun, H.K. Synthesis of novel 5-monoalkylbarbiturate derivatives: New access to 1,2-oxazepines. Tetrahedron Lett. 2015, 56, 6984–6987. [Google Scholar] [CrossRef]
- Barakat, A.; Al-Majid, A.M.; Al-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.K. An efficient and green procedure for synthesis of rhodanine derivatives by aldol-thia-Michael protocol using aqueous diethylamine medium. RSC Adv. 2014, 4, 4909–4916. [Google Scholar] [CrossRef]
- Al-Majid, A.M.; Barakat, A.; AL-Najjar, H.J.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.K. Tandem Aldol-Michael reactions in aqueous diethylamine medium: A greener and efficient approach to bis-pyrimidine derivatives. Int. J. Mol. Sci. 2013, 14, 23762–23773. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Najjar, H.J.; Al-Majid, A.M.; Soliman, S.M.; Mabkhot, Y.N.; Shaik, M.R.; Ghabbour, H.A.; Fun, H.K. Synthesis, NMR, FT-IR, X-ray structural characterization, DFT analysis and isomerism aspects of 5-(2,6-dichlorobenzylidene) pyrimidine-2,4,6(1H,3H,5H)-trione. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 147, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Al-Najjar, H.J.; Al-Majid, A.M.; Soliman, S.M.; Mabkhot, Y.N.; Ghabbour, H.A.; Fun, H.K. Synthesis and molecular characterization of 5,5′-((2,4-dichlorophenyl) methylene) bis (1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione). J. Mol. Struct. 2015, 1084, 207–215. [Google Scholar] [CrossRef]
- Al-Najjar, H.J.; Barakat, A.; Al-Majid, A.M.; Mabkhot, Y.N.; Weber, M.; Ghabbour, H.A.; Fun, H.K. A greener, efficient approach to Michael addition of barbituric acid to nitroalkene in aqueous diethylamine medium. Molecules 2014, 19, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Ghabbour, H.A.; Al-Majid, A.M.; El Ashry, E.S.H.; Warad, I.; Fun, H.K. Crystal structure of diethylammonium 5-((4-fluorophenyl)(6-hydroxy-1,3-dimethyl-2, 4-dioxo-1,2,3,4-tetrahydropyrimidin-5-yl)methyl)-1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-olate, C23H30FN5O6. Z. Krist. New Cryst. Struct. 2016, 231, 507–509. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL-PC; Version 5.1; Siemens Analytical Instruments Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Frisch, M.-J.; Trucks, G.-W.; Schlegel, H.-B.; Scuseria, G.-E.; Robb, M.-A.; Cheeseman, J.-R.; Montgomery, J.-A., Jr.; Vreven, T.; Kudin, K.-N.; Burant, J.-C.; et al. Gaussian 03Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |
|---|---|---|
| Chemical formula | C14H14N2O5 | C14H14N2O5 |
| M.W | 290.27 | 290.27 |
| T | 293 K | 296 K |
| λ (Mo Kα radiation) | 0.71073 Å | 0.71073 Å |
| Crystal system | Triclinic | Triclinic |
| Space group | P-1 | P-1 |
| Unit cell dimensions | a = 9.2265 (10) Å | a = 5.4539 (4) Å |
| b = 11.9757 (12) Å | b = 8.1536 (7) Å | |
| c = 12.8490 (13) Å | c = 15.1562 (12) Å | |
| α = 110.014 (3)° | α = 83.466 (3)° | |
| β = 101.912 (3)° | β = 83.950 (3)° | |
| γ = 90.188 (4)° | γ = 79.621 (3)° | |
| Volume | 1301.0 (2) Å3 | 656.16 (9) Å3 |
| Z | 4 | 2 |
| Density (calculated) | 1.482 Mg·m−3 | 1.469 Mg·m−3 |
| Absorption coefficient | 0.11 mm−1 | 0.11 mm−1 |
| F(000) | 608 | 304 |
| Crystal size | 0.35 × 0.23 × 0.13 mm | 0.19 × 0.13 × 0.08 mm |
| Theta range for data collection | 2.8°–24.9° | 2.6°–23.1° |
| Reflections collected/unique | 4554/3207 | 17059/1475 |
| Completeness to theta = 31.4° | 99.9 | 99.9 |
| Goodness-of-fit on F2 | 1.08 | 1.02 |
| Diffractometer | Bruker APEX-II CCD diffractometer | Bruker APEX-II CCD diffractometer |
| Absorption correction | multi-scan SADABS Bruker 2014 | multi-scan SADABS Bruker 2014 |
| Rint | 0.173 | 0.099 |
| R(F2 > 2σ(F2)) | 0.088 | 0.054 |
| wR(F2) = | 0.252 | 0.136 |
| Δρmax/Δρmin | 0.31 and −0.38 | 0.31 and −0.21 |
| CCDC number | 1484809 | 1484813 |
| O1A–C1A | 1.214 (7) | N1A–C2A | 1.469 (8) |
| O2A–C3A | 1.207 (6) | N1A–C3A | 1.363 (7) |
| O3A–C5A | 1.206 (7) | N1A–C1A | 1.403 (6) |
| O4A–C10A | 1.345 (7) | N2A–C4A | 1.470 (8) |
| O5A–C11A | 1.360 (7) | N2A–C3A | 1.388 (7) |
| O5A–C14A | 1.435 (8) | N2A–C5A | 1.389 (6) |
| O1B–C1B | 1.213 (7) | N1B–C3B | 1.375 (8) |
| O2B–C3B | 1.209 (6) | N1B–C1B | 1.395 (7) |
| O3B–C5B | 1.214 (8) | N1B–C2B | 1.465 (9) |
| O4B–C10B | 1.359 (8) | N2B–C4B | 1.477 (9) |
| O5B–C14B | 1.431 (9) | N2B–C5B | 1.389 (8) |
| O5B–C11B | 1.358 (6) | N2B–C3B | 1.365 (8) |
| C11A–O5A–C14A | 118.6 (5) | O3A–C5A–N2A | 119.2 (5) |
| C11B–O5B–C14B | 118.0 (5) | N2A–C5A–C6A | 117.2 (5) |
| C1A–N1A–C3A | 125.4 (5) | O3A–C5A–C6A | 123.6 (5) |
| C2A–N1A–C3A | 116.9 (4) | O4A–C10A–C11A | 122.6 (5) |
| C1A–N1A–C2A | 117.7 (5) | O4A–C10A–C9A | 117.9 (5) |
| C3A–N2A–C5A | 124.4 (5) | O5A–C11A–C12A | 125.1 (5) |
| C3A–N2A–C4A | 116.6 (4) | O5A–C11A–C10A | 115.0 (4) |
| C4A–N2A–C5A | 118.9 (5) | O1B–C1B–C6B | 125.2 (5) |
| C1B–N1B–C3B | 125.7 (5) | N1B–C1B–C6B | 116.2 (5) |
| C2B–N1B–C3B | 116.8 (5) | O1B–C1B–N1B | 118.7 (5) |
| C1B–N1B–C2B | 117.5 (5) | O2B–C3B–N2B | 121.5 (6) |
| C3B–N2B–C5B | 125.4 (5) | N1B–C3B–N2B | 117.3 (4) |
| C4B–N2B–C5B | 117.7 (5) | O2B–C3B–N1B | 121.2 (6) |
| C3B–N2B–C4B | 116.9 (5) | O3B–C5B–N2B | 119.9 (5) |
| O1A–C1A–N1A | 117.9 (5) | N2B–C5B–C6B | 116.8 (5) |
| N1A–C1A–C6A | 116.5 (5) | O3B–C5B–C6B | 123.4 (5) |
| O1A–C1A–C6A | 125.6 (5) | O4B–C10B–C9B | 118.2 (5) |
| O2A–C3A–N1A | 121.9 (5) | O4B–C10B–C11B | 121.8 (5) |
| O2A–C3A–N2A | 120.3 (5) | O5B–C11B–C10B | 115.4 (5) |
| N1A–C3A–N2A | 117.8 (4) | O5B–C11B–C12B | 124.6 (5) |
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| O4A–H4OA···O2A i | 0.87(5) | 1.96(5) | 2.744(6) | 150(4) |
| O4B–H4OB···O2B i | 0.70(8) | 2.10(9) | 2.765(6) | 161(11) |
| C9A–H9AA···O3A ii | 0.9300 | 2.5900 | 3.507(7) | 170.00 |
| C13A–H13A···O1A | 0.9300 | 2.1100 | 2.893(6) | 141.00 |
| C13B–H13B···O1B | 0.9300 | 2.0900 | 2.878 (6) | 142.00 |
| C9B–H9BA···O3B iii | 0.9300 | 2.5000 | 3.404 (8) | 165.00 |
| O1–C1 | 1.218 (4) | N1–C1 | 1.381 (4) |
| O2–C3 | 1.224 (3) | N1–C3 | 1.367 (4) |
| O3–C5 | 1.213 (3) | N1–C2 | 1.471 (4) |
| O4–C10 | 1.363 (3) | N2–C4 | 1.465 (4) |
| O4–C14 | 1.433 (4) | N2–C5 | 1.399 (4) |
| O5–C11 | 1.346 (3) | N2–C3 | 1.374 (3) |
| C10–O4–C14 | 117.4 (2) | O2–C3–N2 | 121.4 (2) |
| C1–N1–C3 | 124.7 (2) | N1–C3–N2 | 118.2 (2) |
| C2–N1–C3 | 117.2 (2) | O2–C3–N1 | 120.5 (2) |
| C1–N1–C2 | 118.1 (2) | O3–C5–N2 | 118.0 (3) |
| C3–N2–C5 | 124.3 (2) | N2–C5–C6 | 117.1 (2) |
| C4–N2–C5 | 118.1 (2) | O3–C5–C6 | 124.9 (3) |
| C3–N2–C4 | 117.6 (2) | O4–C10–C9 | 125.2 (2) |
| O1–C1–C6 | 123.7 (3) | O4–C10–C11 | 114.3 (2) |
| N1–C1–C6 | 117.0 (2) | O5–C11–C10 | 117.4 (2) |
| O1–C1–N1 | 119.4 (3) | O5–C11–C12 | 123.3 (2) |
| D–H···A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|
| O5–H1O5···O2 i | 0.95 (3) | 1.73 (3) | 2.679 (3) | 178 (3) |
| C4–H4C···O4 ii | 0.9800 | 2.5300 | 3.487 (4) | 166.66 |
| C9–H9A···O3 | 0.9500 | 2.0500 | 2.855 (4) | 141.00 |
| C13–H13A···O1 iii | 0.9500 | 2.3800 | 3.320 (3) | 172.00 |
| C14–H14A···O5 iv | 0.9800 | 2.5000 | 3.482 (4) | 175.00 |
| 1 | 2 | |
|---|---|---|
| Etot (a.u) | −1027.5976 | −1027.5931 |
| ZPVE (a.u) | 0.271445 | 0.270999 |
| Ecorr (a.u) | −1027.3262 | −1027.3221 |
| ∆Ea (Kcal/mol) | −2.5619393 | |
| H (a.u) | −1027.3056 | −1027.3013 |
| G (a.u) | −1027.3759 | −1027.3734 |
| Parameter | 1 | 2 |
|---|---|---|
| EHOMO | −5.97839 | −5.92832 |
| ELUMO | −2.40088 | −2.40877 |
| Gap | 3.57751 | 3.51955 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barakat, A.; Soliman, S.M.; Ghabbour, H.A.; Ali, M.; Al-Majid, A.M.; Shahidul Islam, M.; Ghfar, A.A. Molecular Structure, Spectroscopic and DFT Computational Studies of Arylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione. Crystals 2016, 6, 110. https://doi.org/10.3390/cryst6090110
Barakat A, Soliman SM, Ghabbour HA, Ali M, Al-Majid AM, Shahidul Islam M, Ghfar AA. Molecular Structure, Spectroscopic and DFT Computational Studies of Arylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione. Crystals. 2016; 6(9):110. https://doi.org/10.3390/cryst6090110
Chicago/Turabian StyleBarakat, Assem, Saied M. Soliman, Hazem A. Ghabbour, M. Ali, Abdullah Mohammed Al-Majid, Mohammad Shahidul Islam, and Ayman A. Ghfar. 2016. "Molecular Structure, Spectroscopic and DFT Computational Studies of Arylidene-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione" Crystals 6, no. 9: 110. https://doi.org/10.3390/cryst6090110