
The Effect of Ligand Design on Metal Ion Spin State—Lessons from Spin Crossover Complexes


Abstract
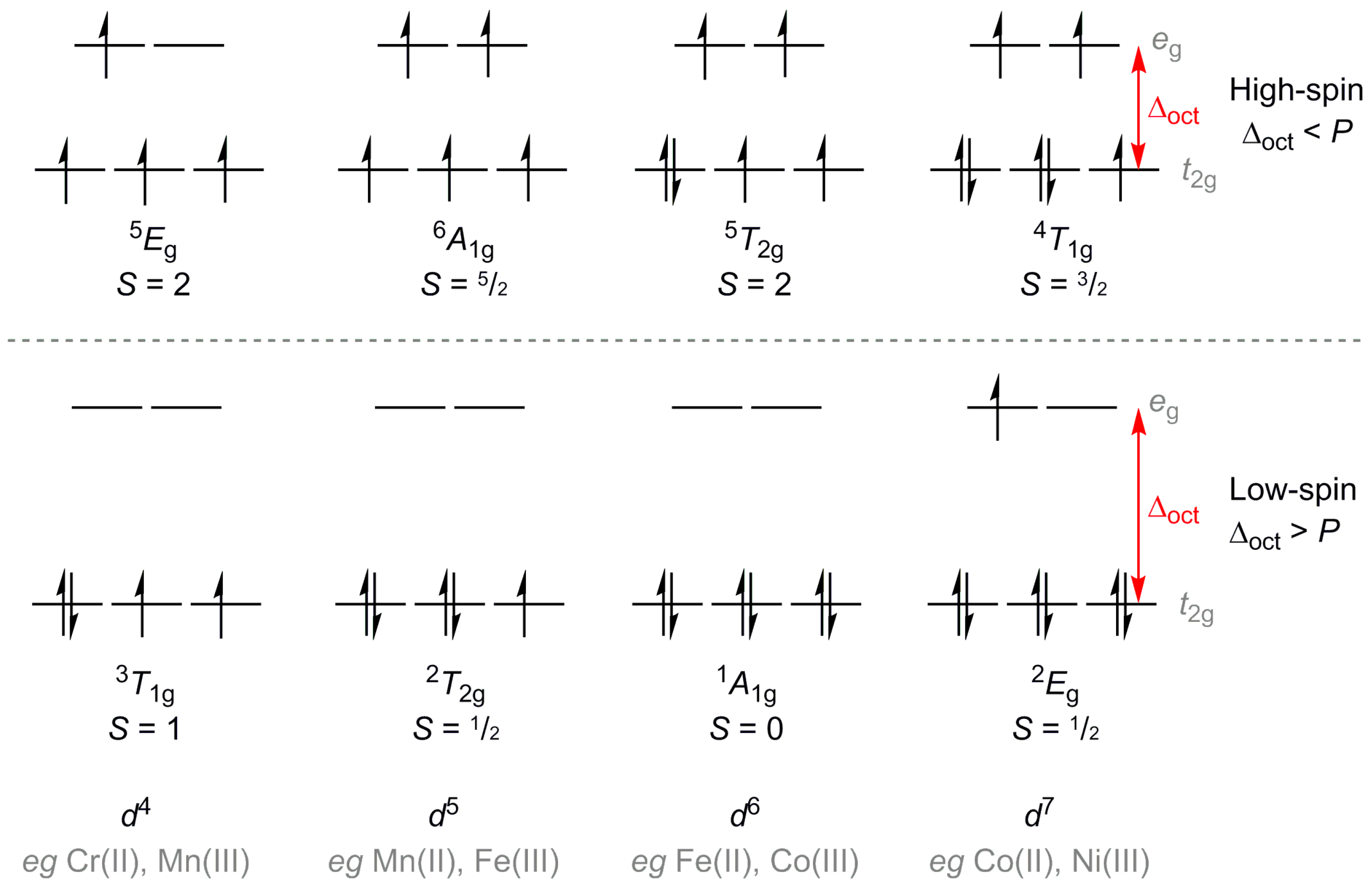
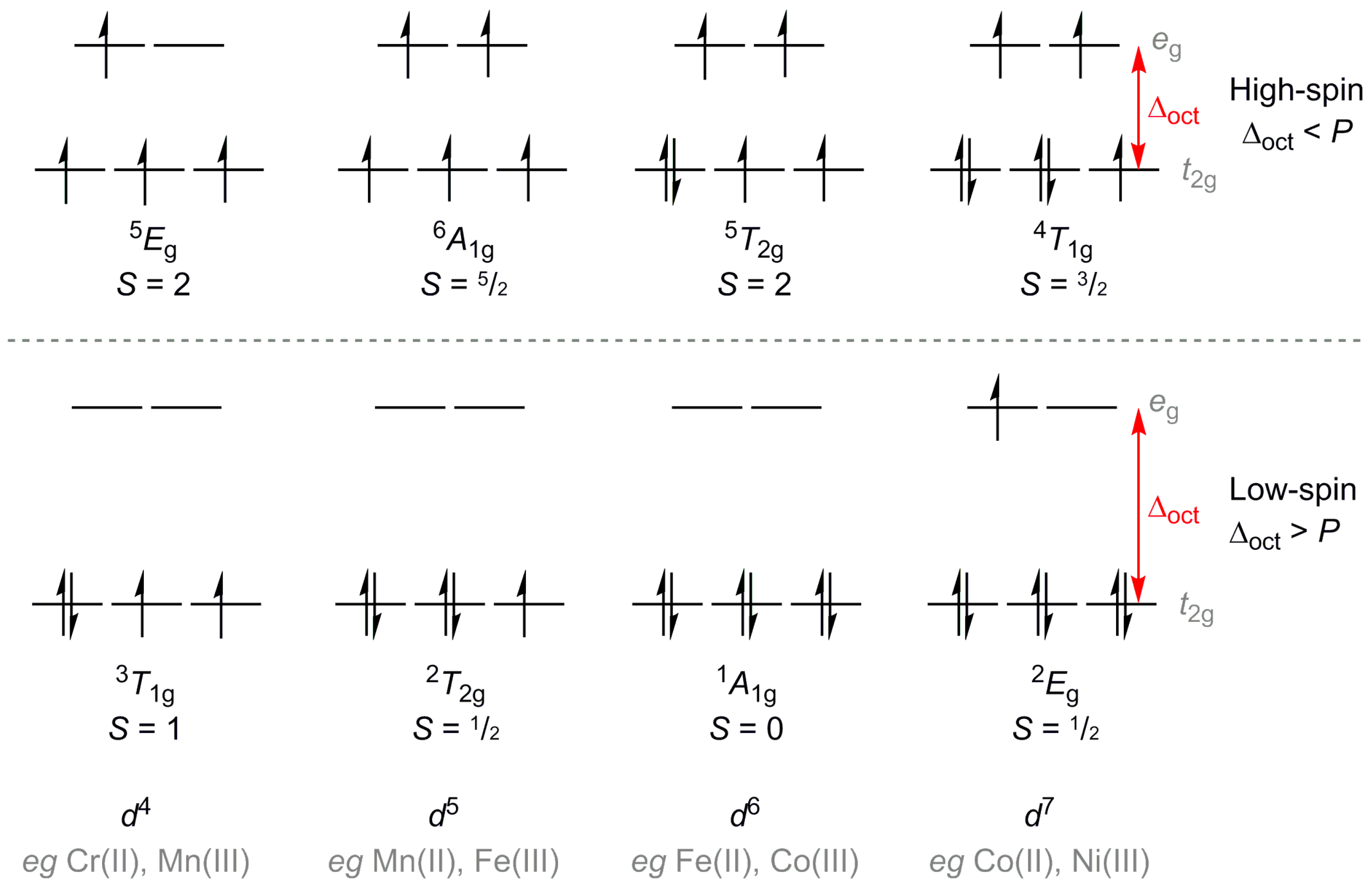
:1. Introduction
2. Results
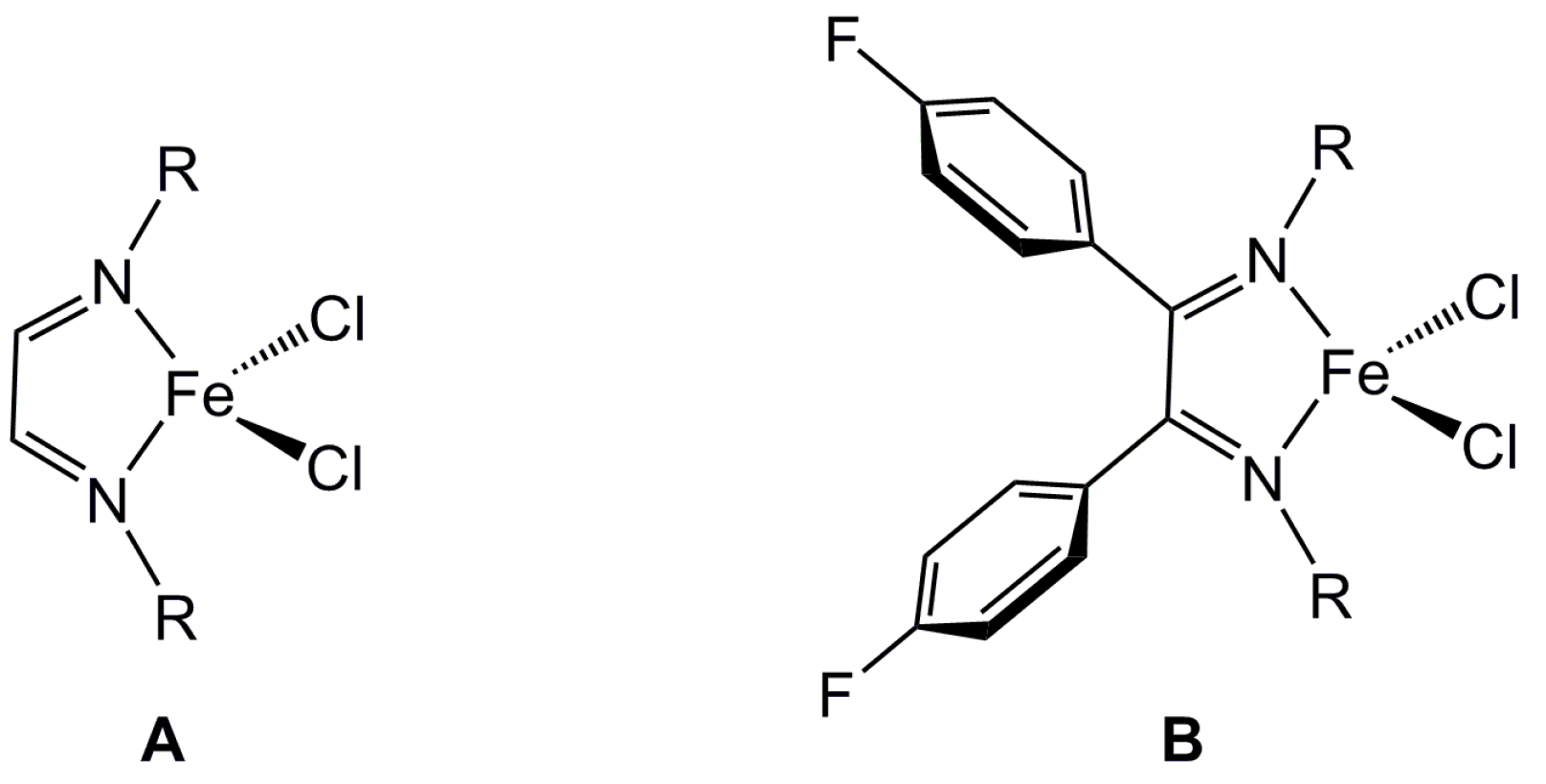
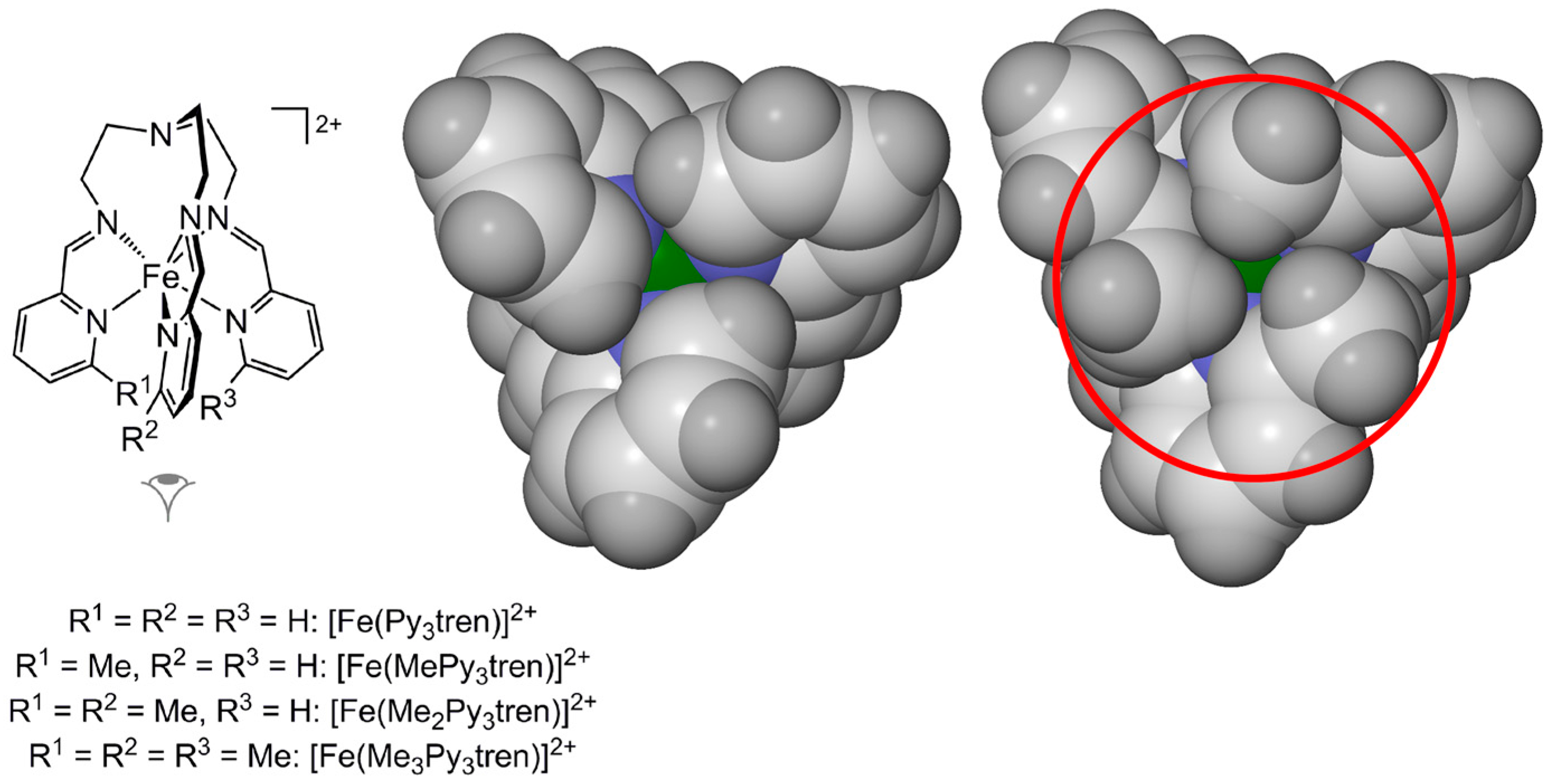
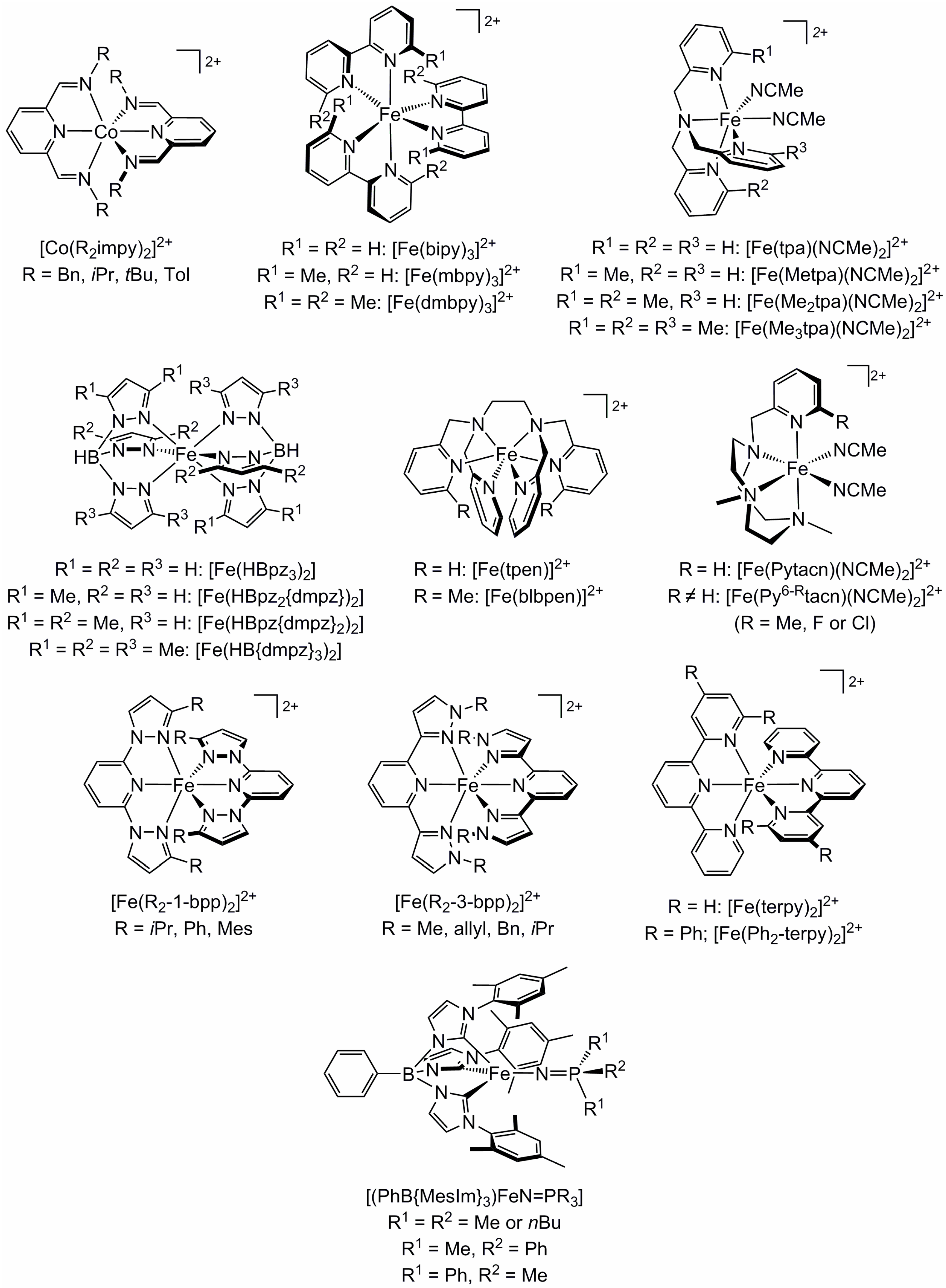
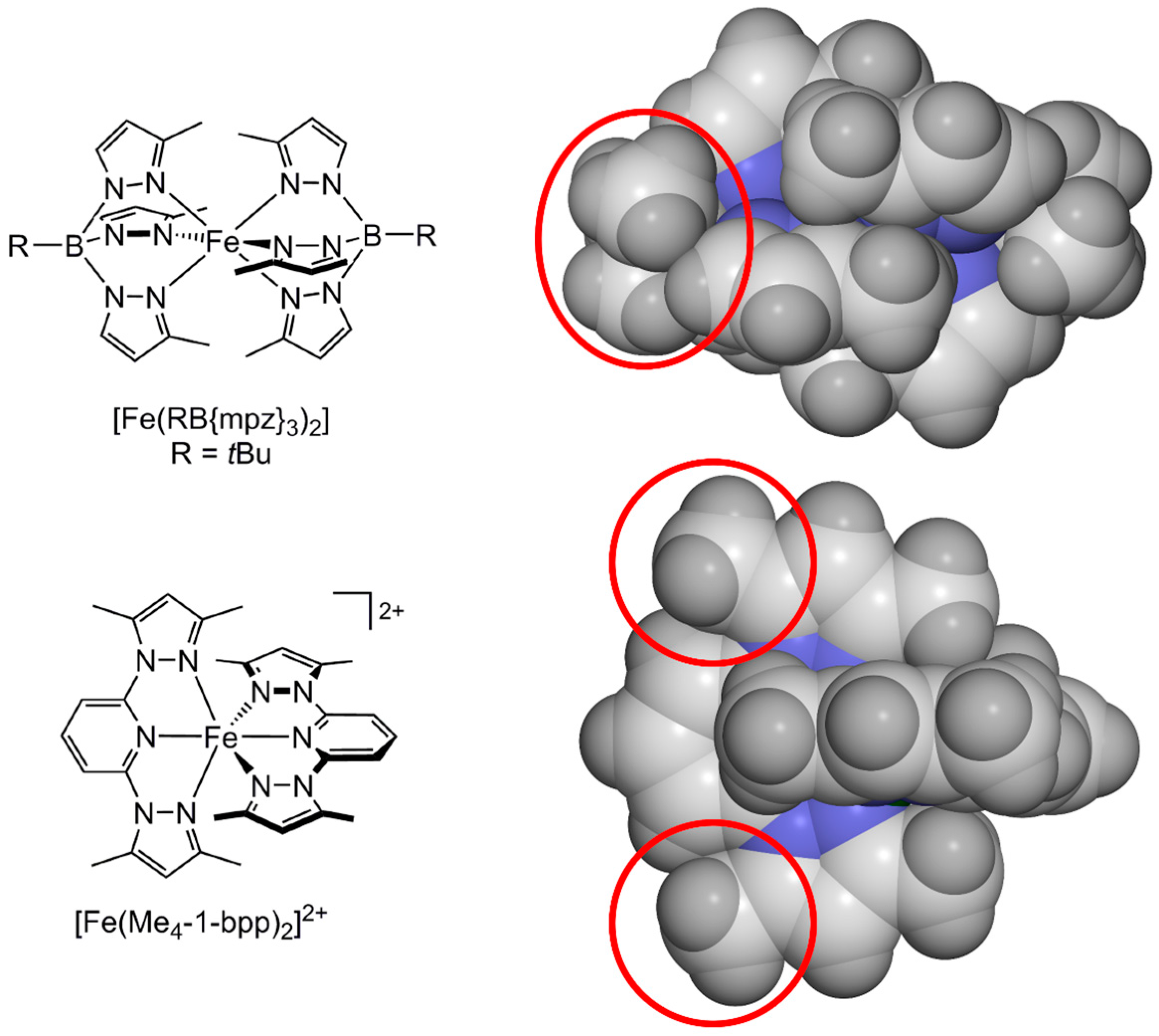
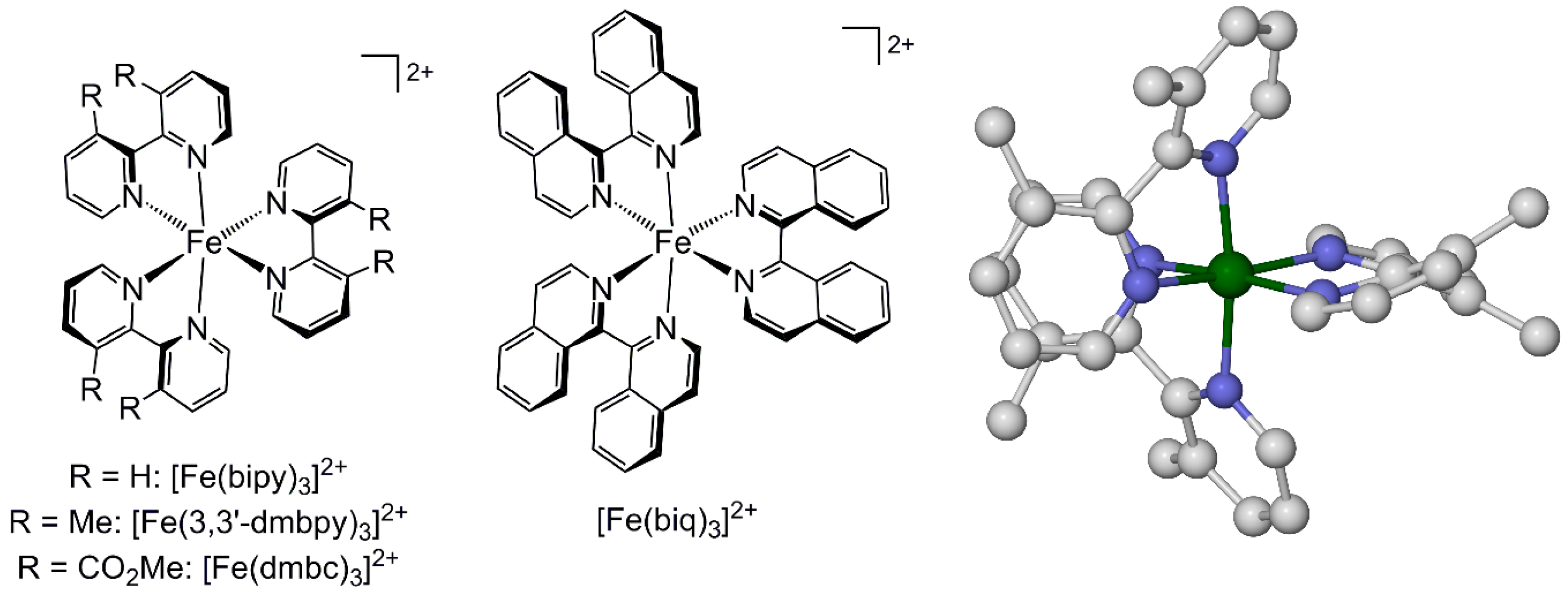
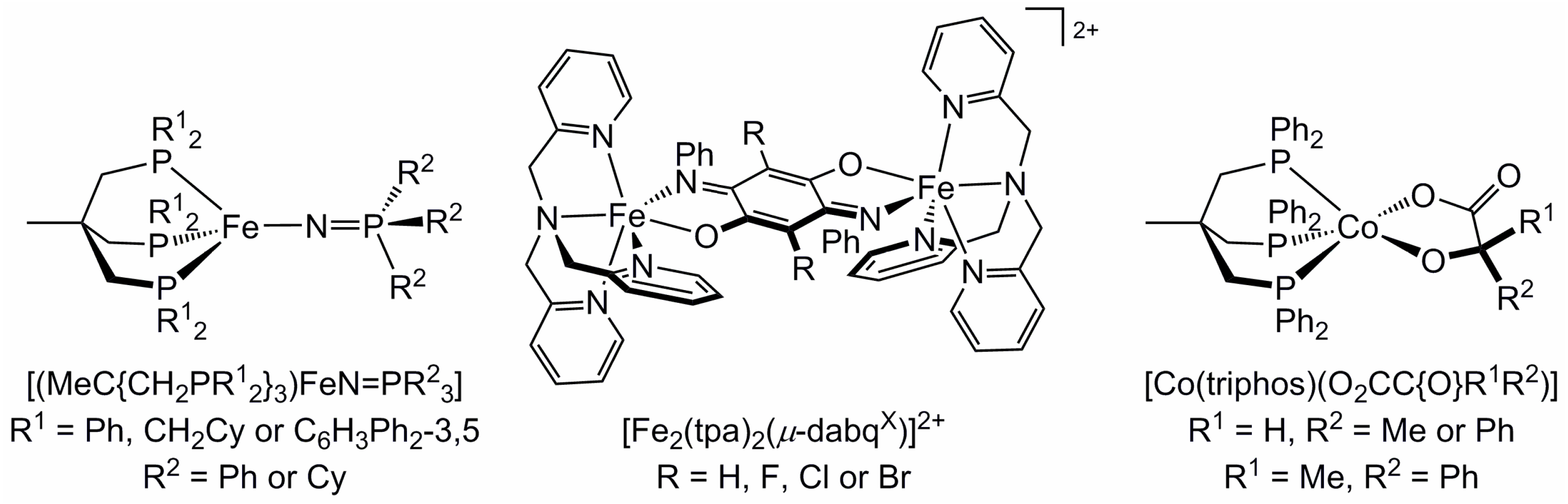
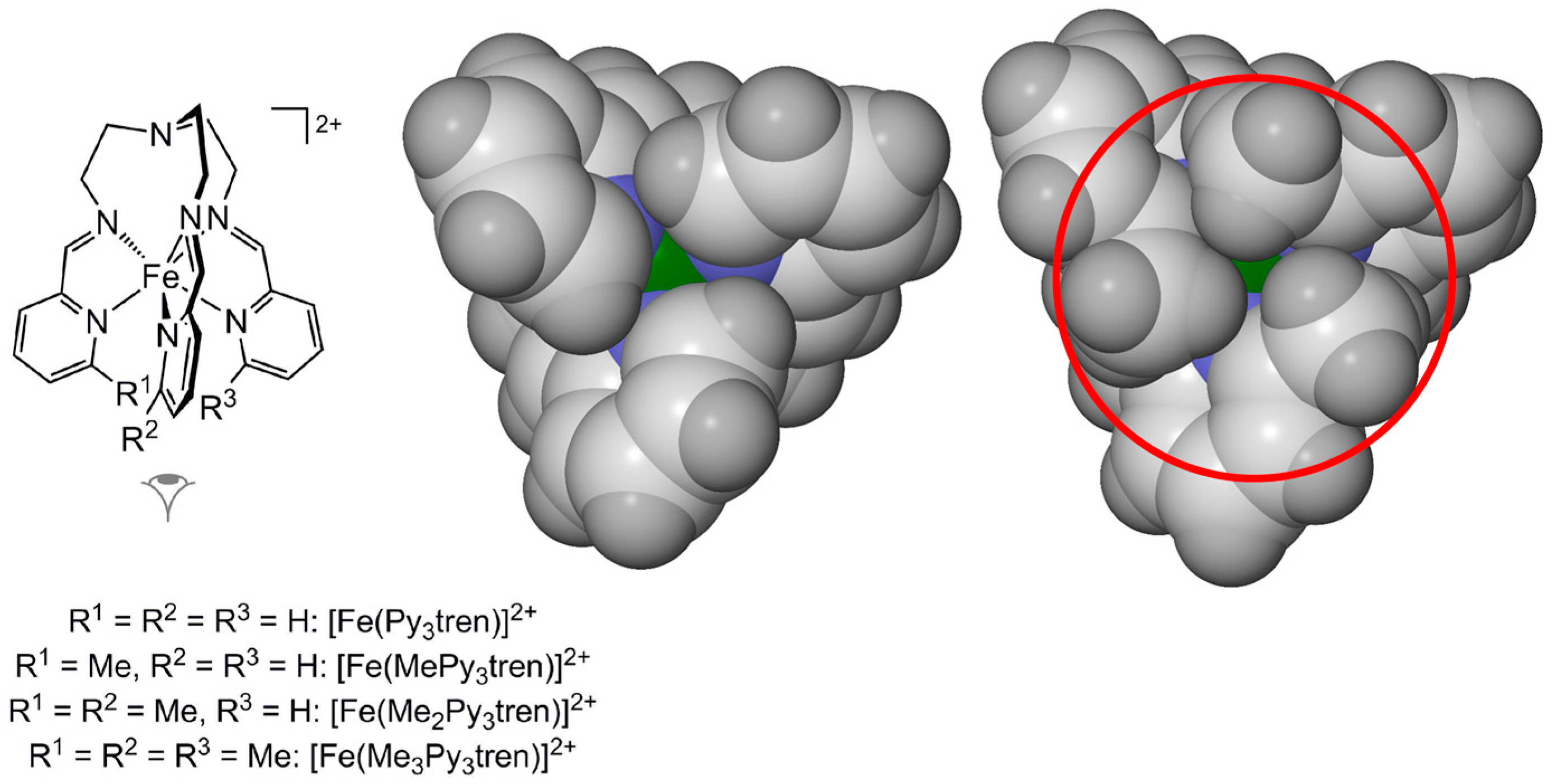
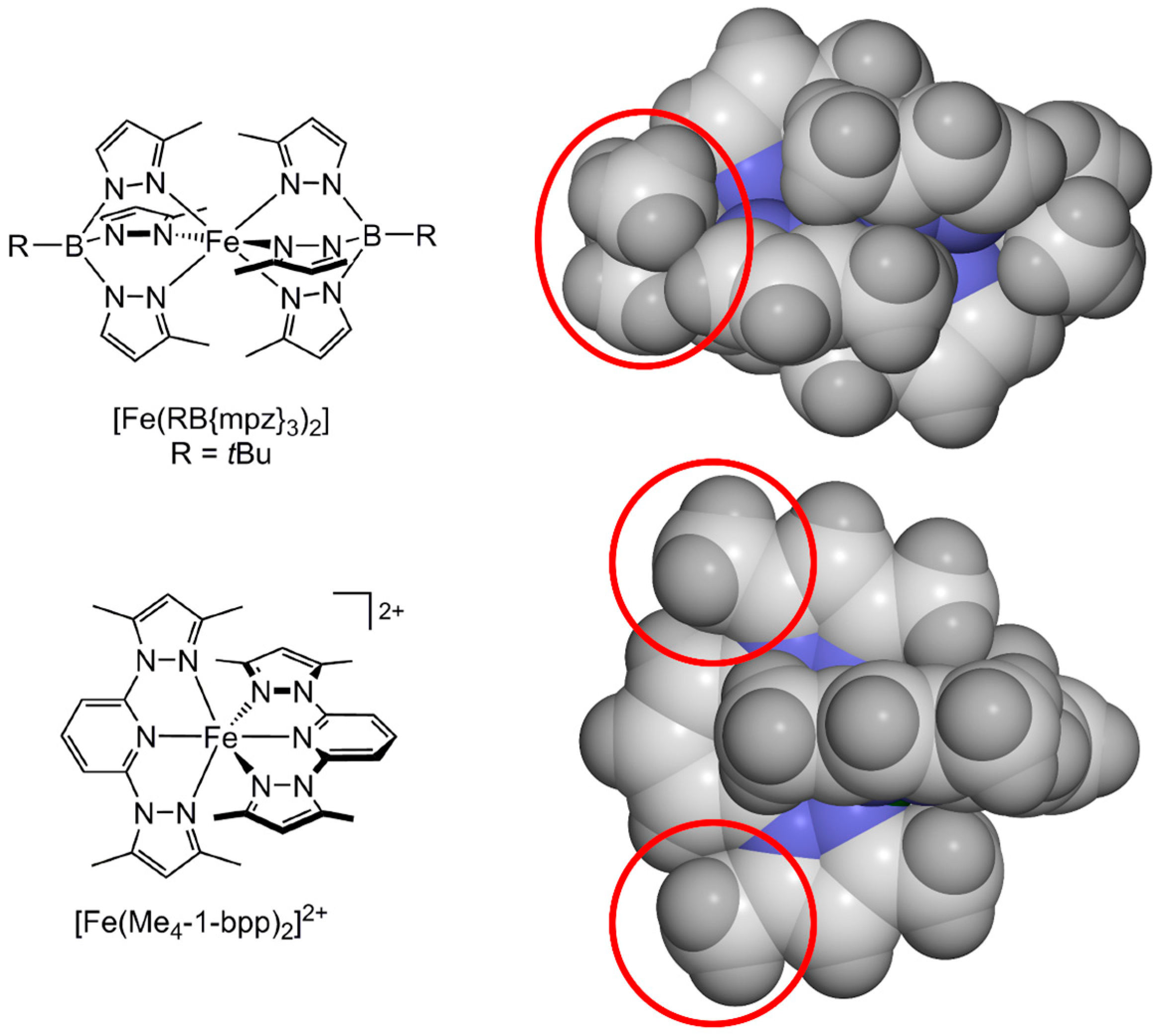
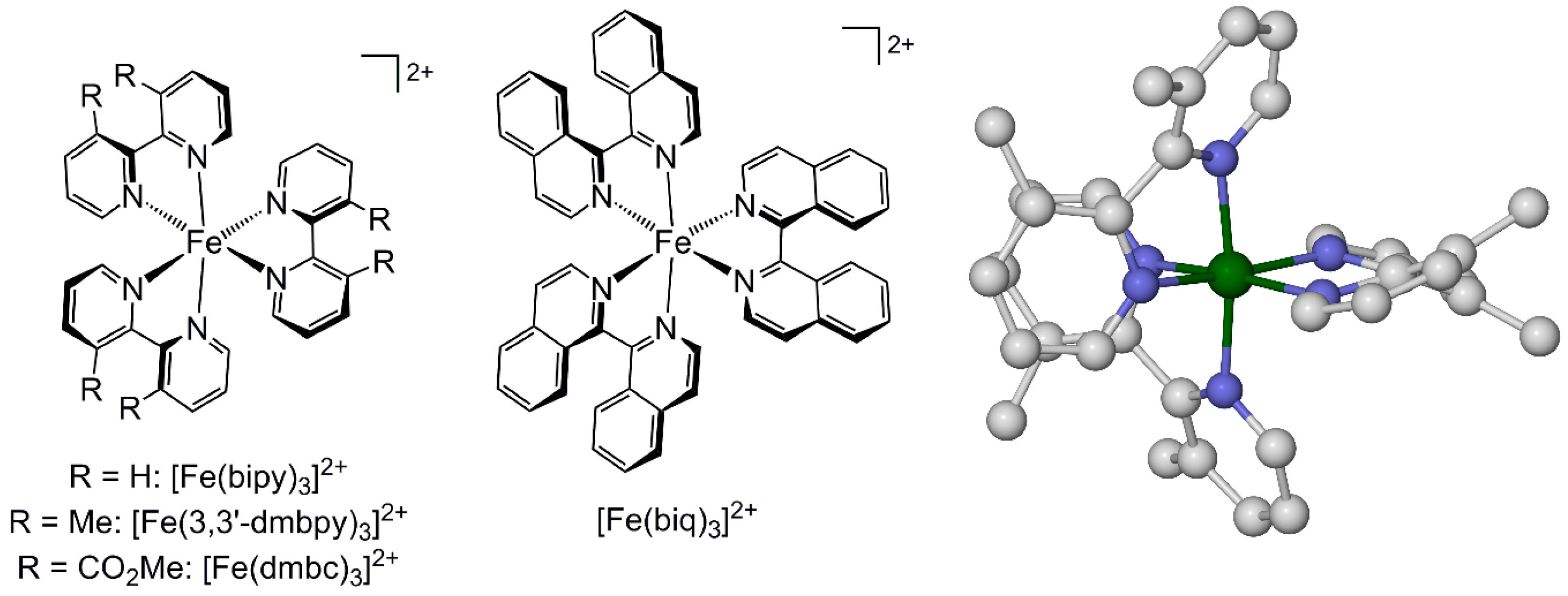
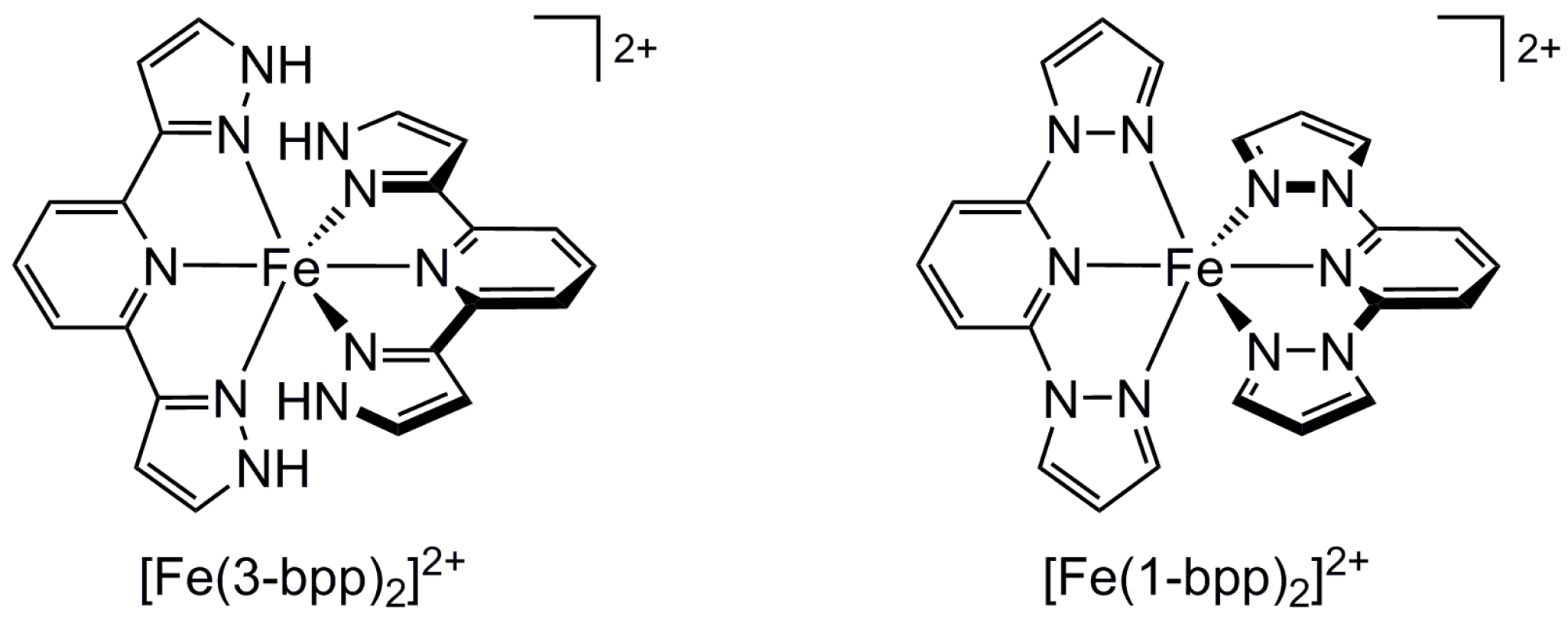
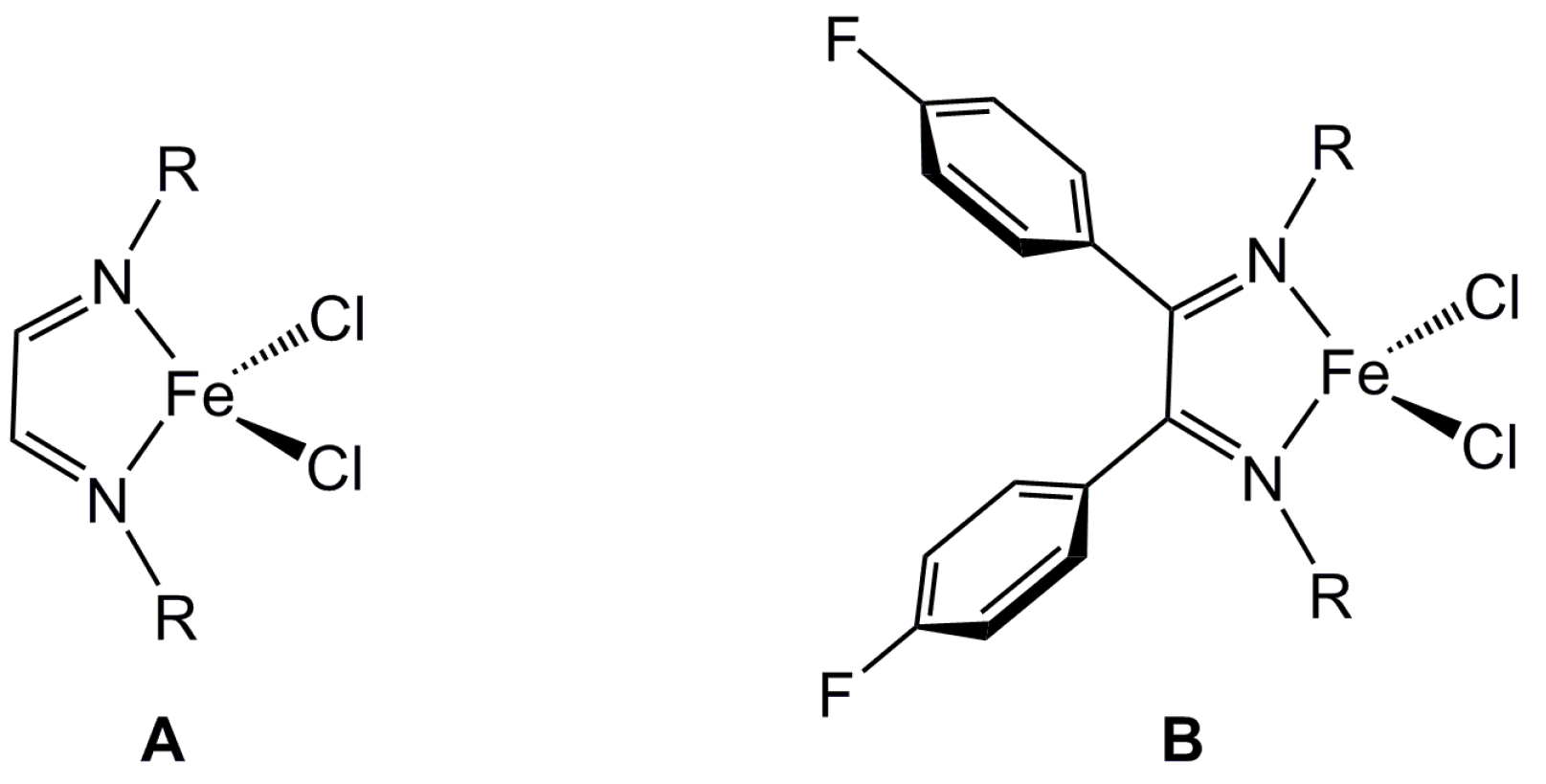
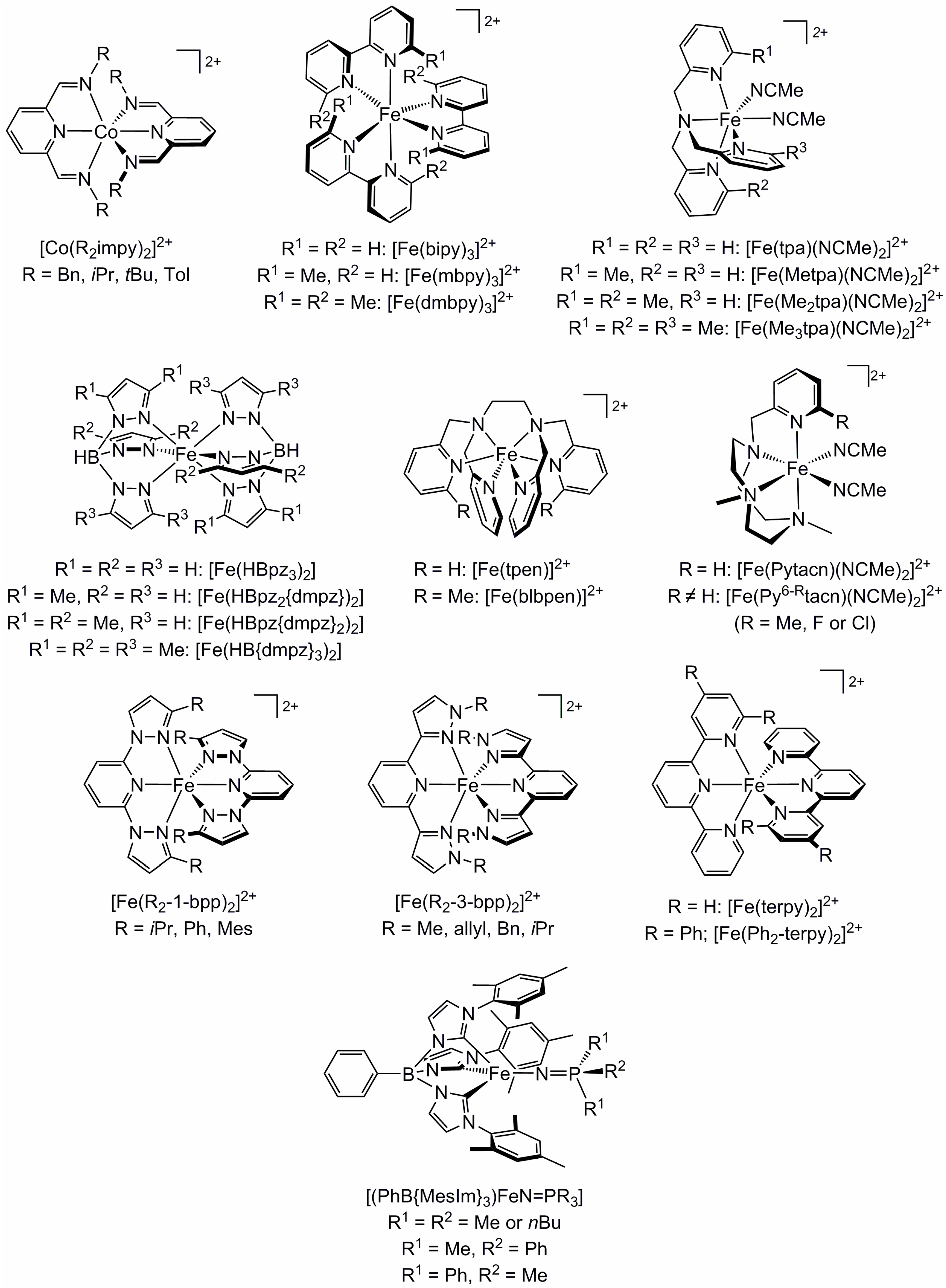
2.1. Steric Influence of Ligand Substituents on Metal Ion Spin States
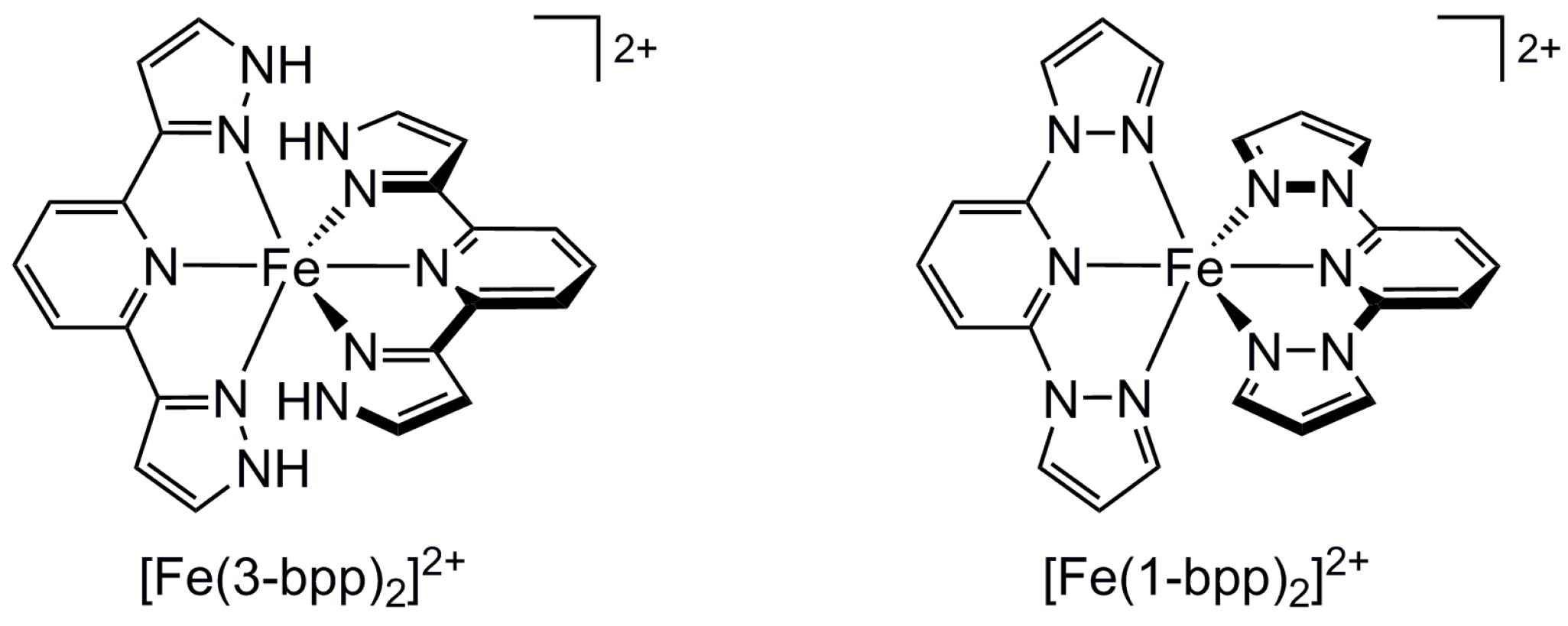
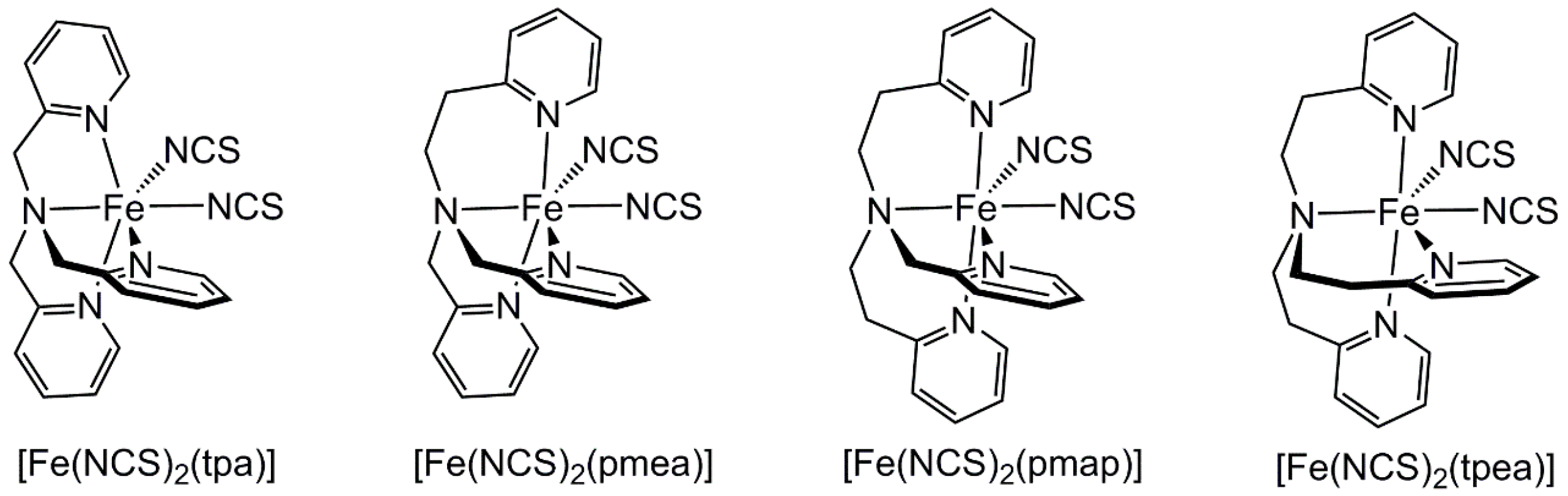
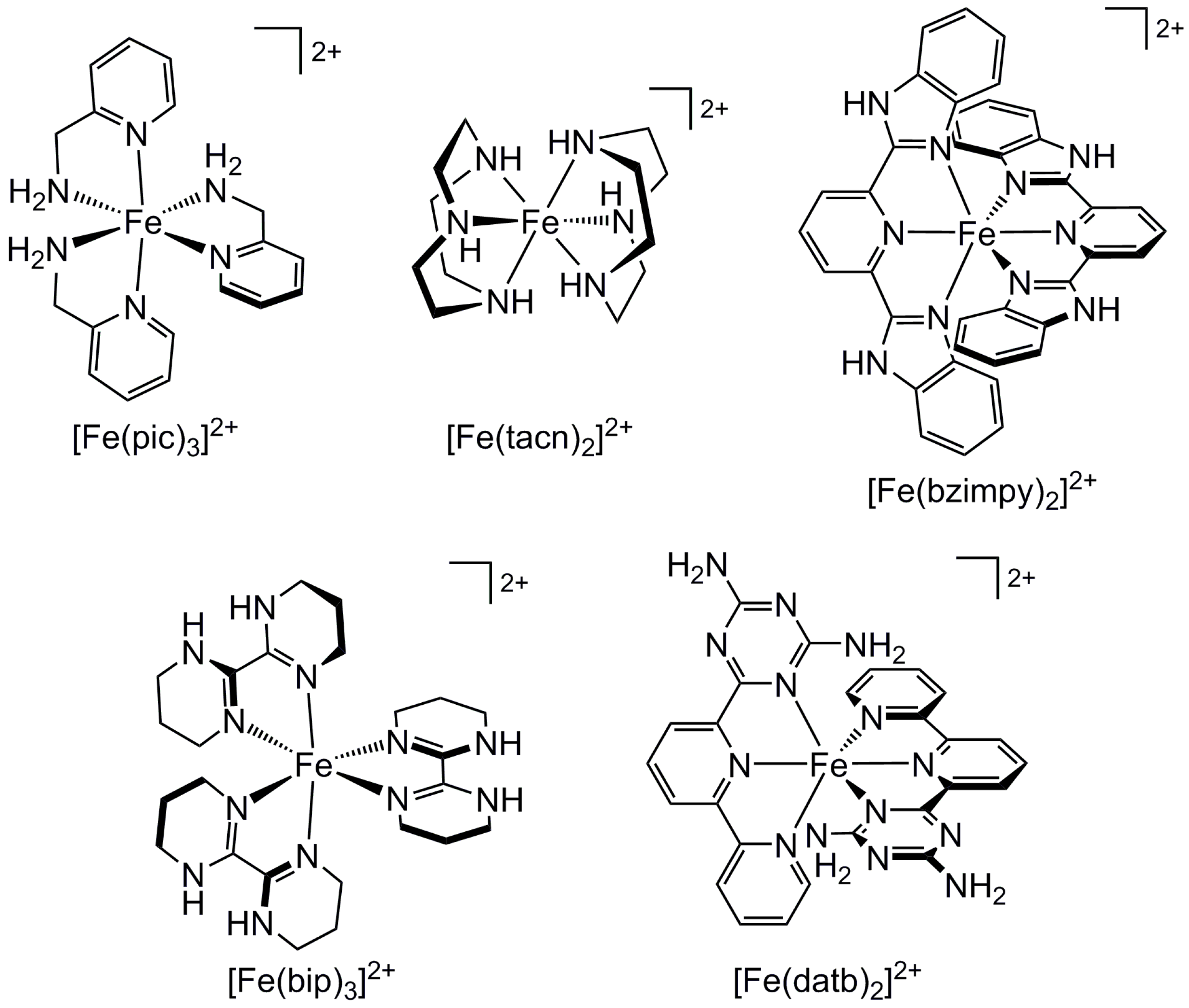
2.2. Influence of Ligand Conformation on Metal Ion Spin States
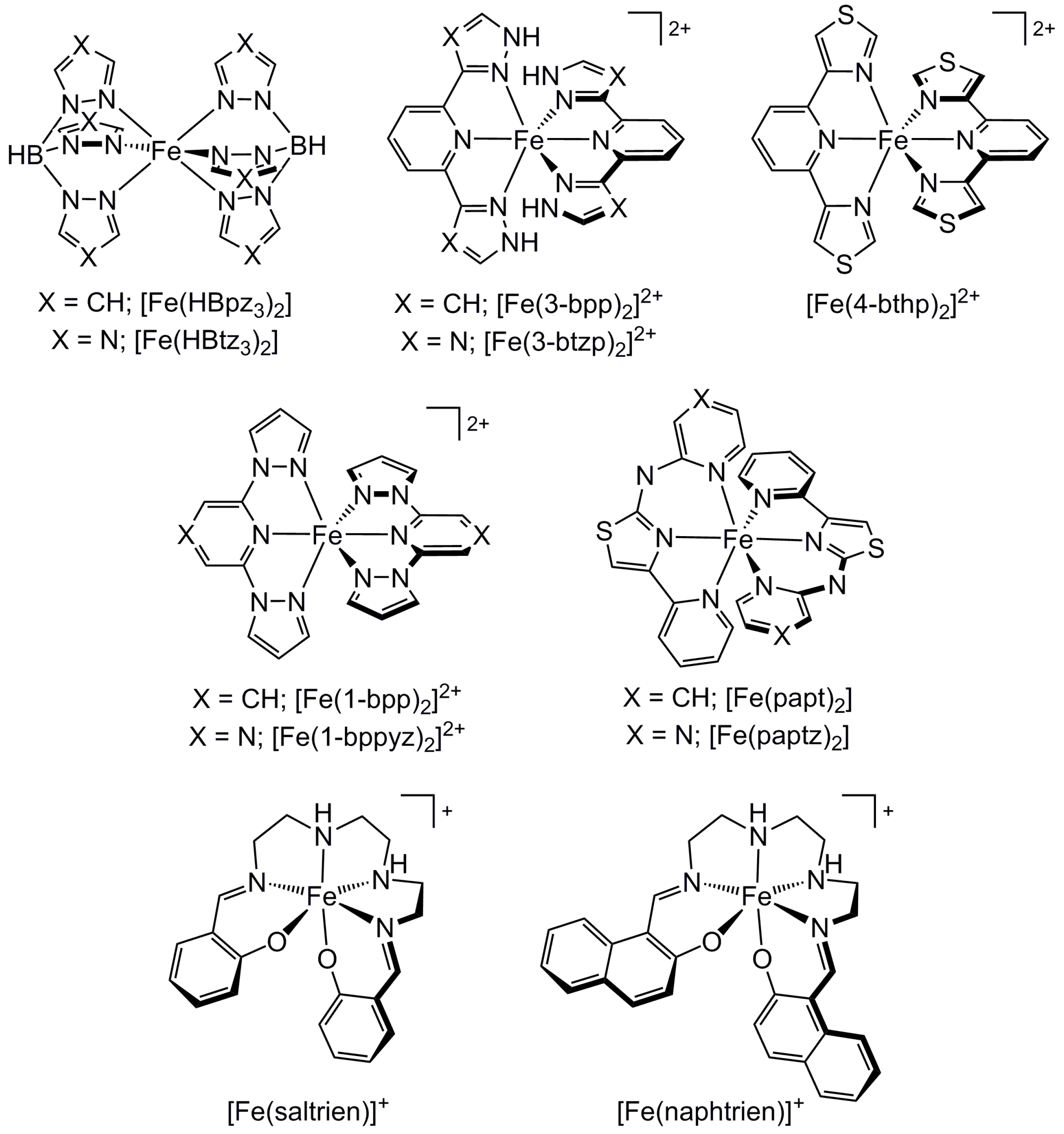
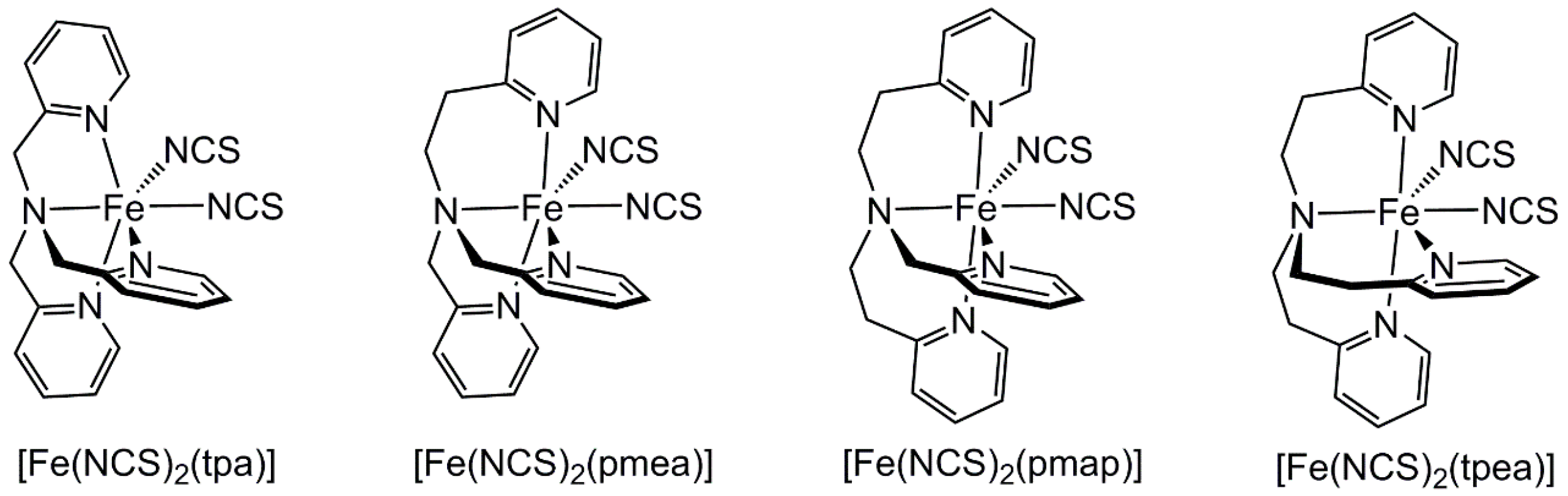
2.3. Influence of Chelate and Macrocycle Ring Size on Metal Ion Spin States
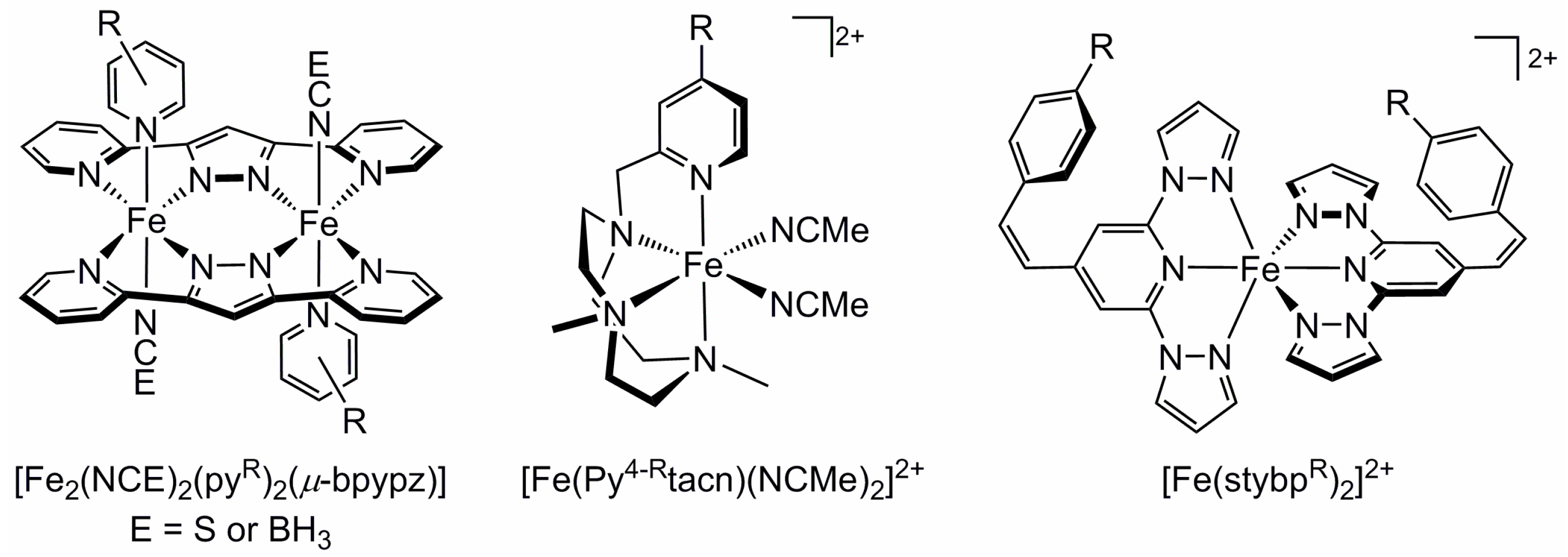
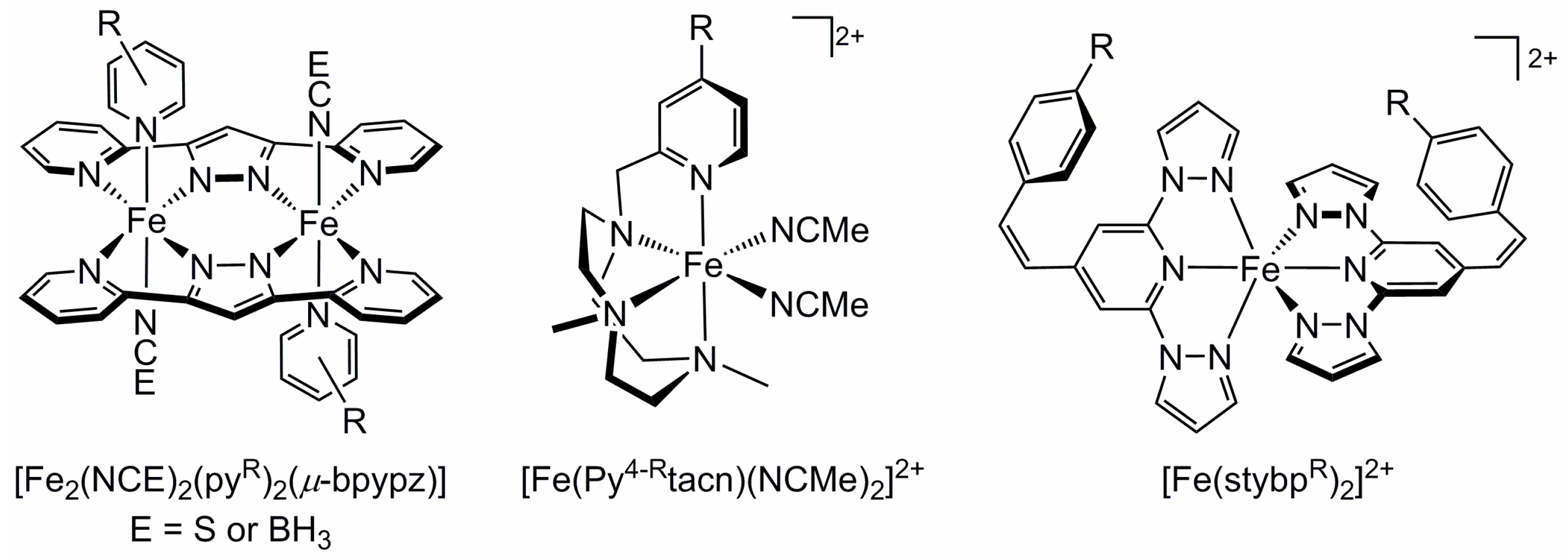
2.4. Influence of Ligand Donor Types on Metal Ion Spin State
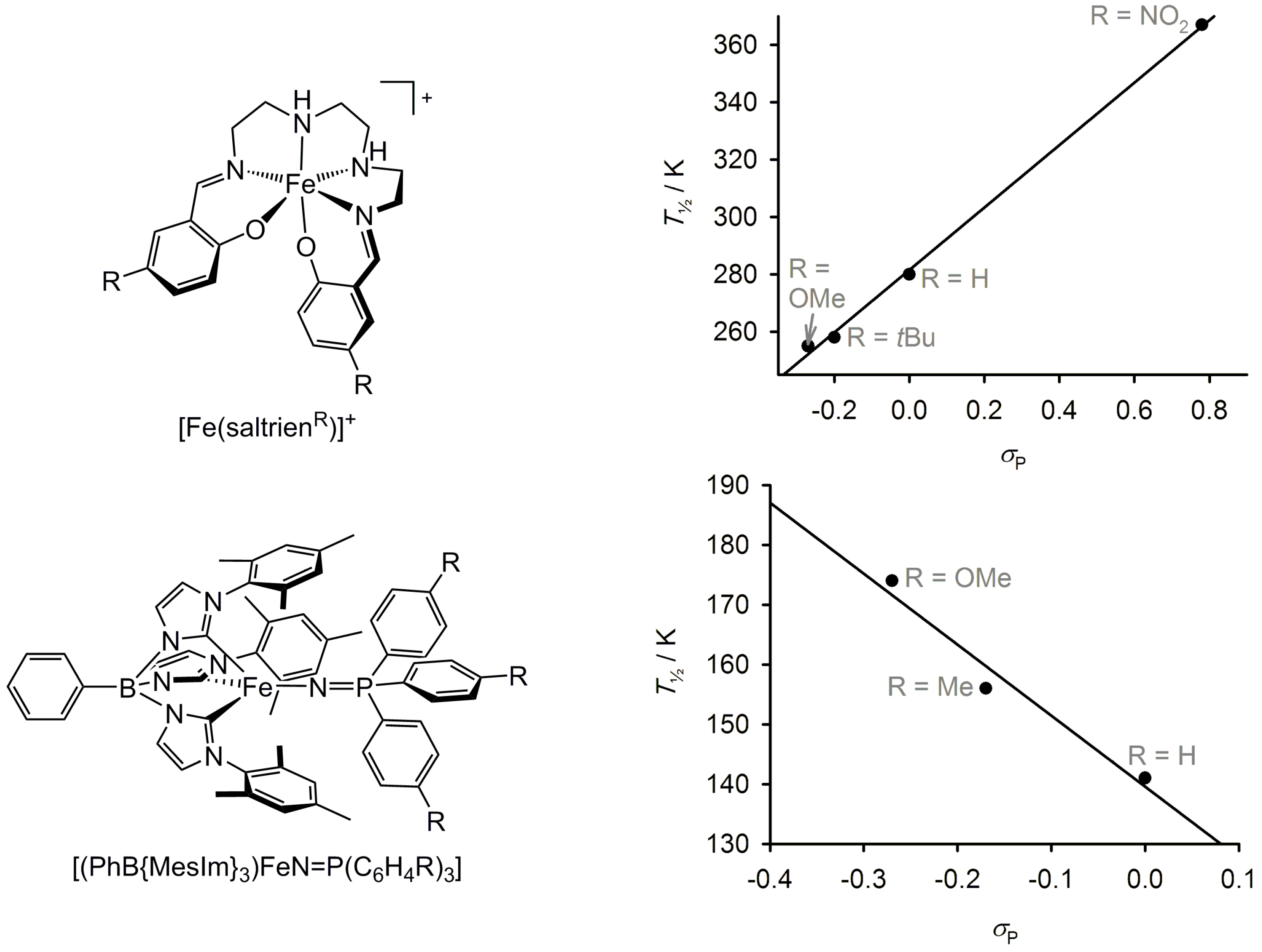
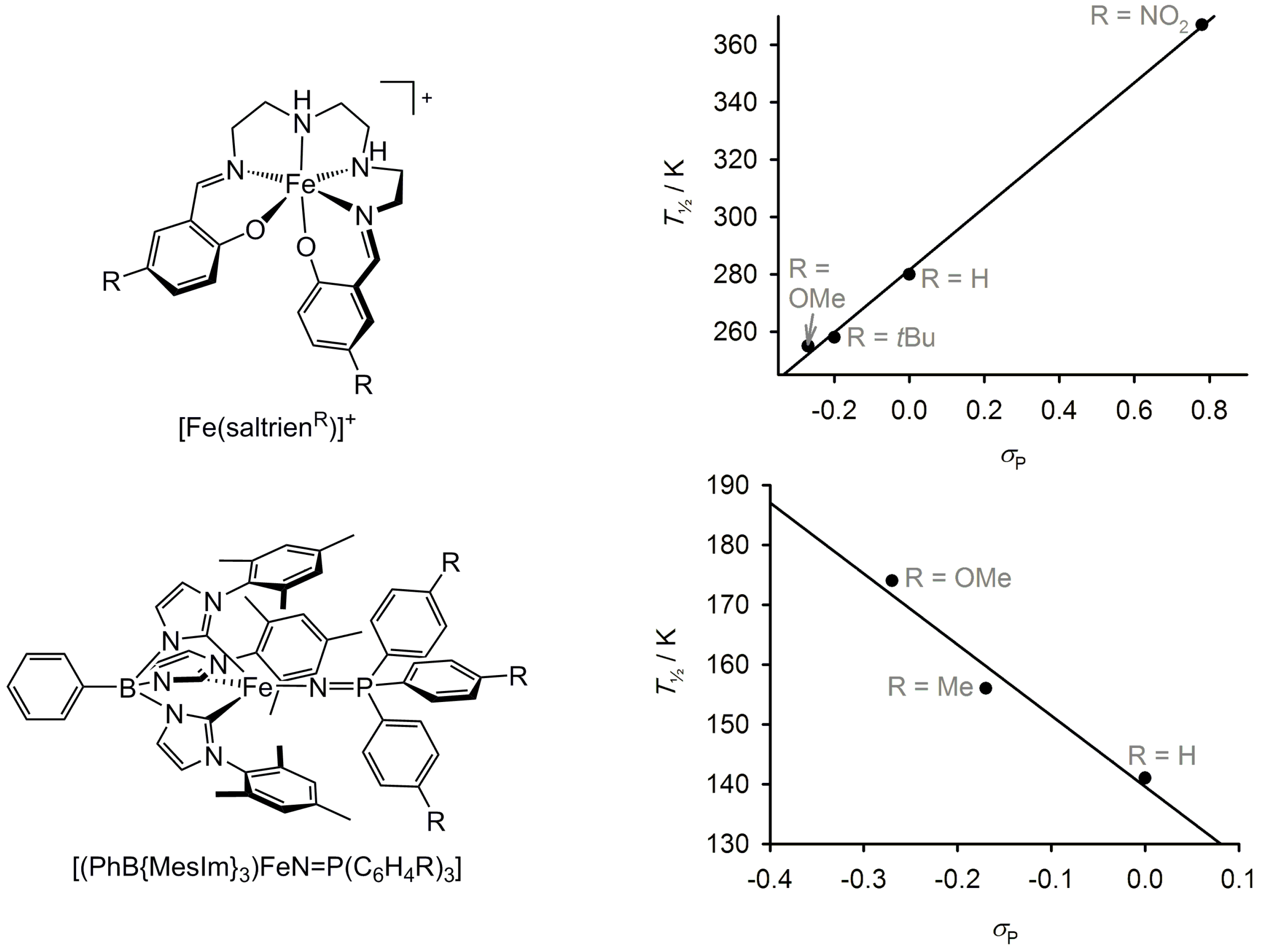
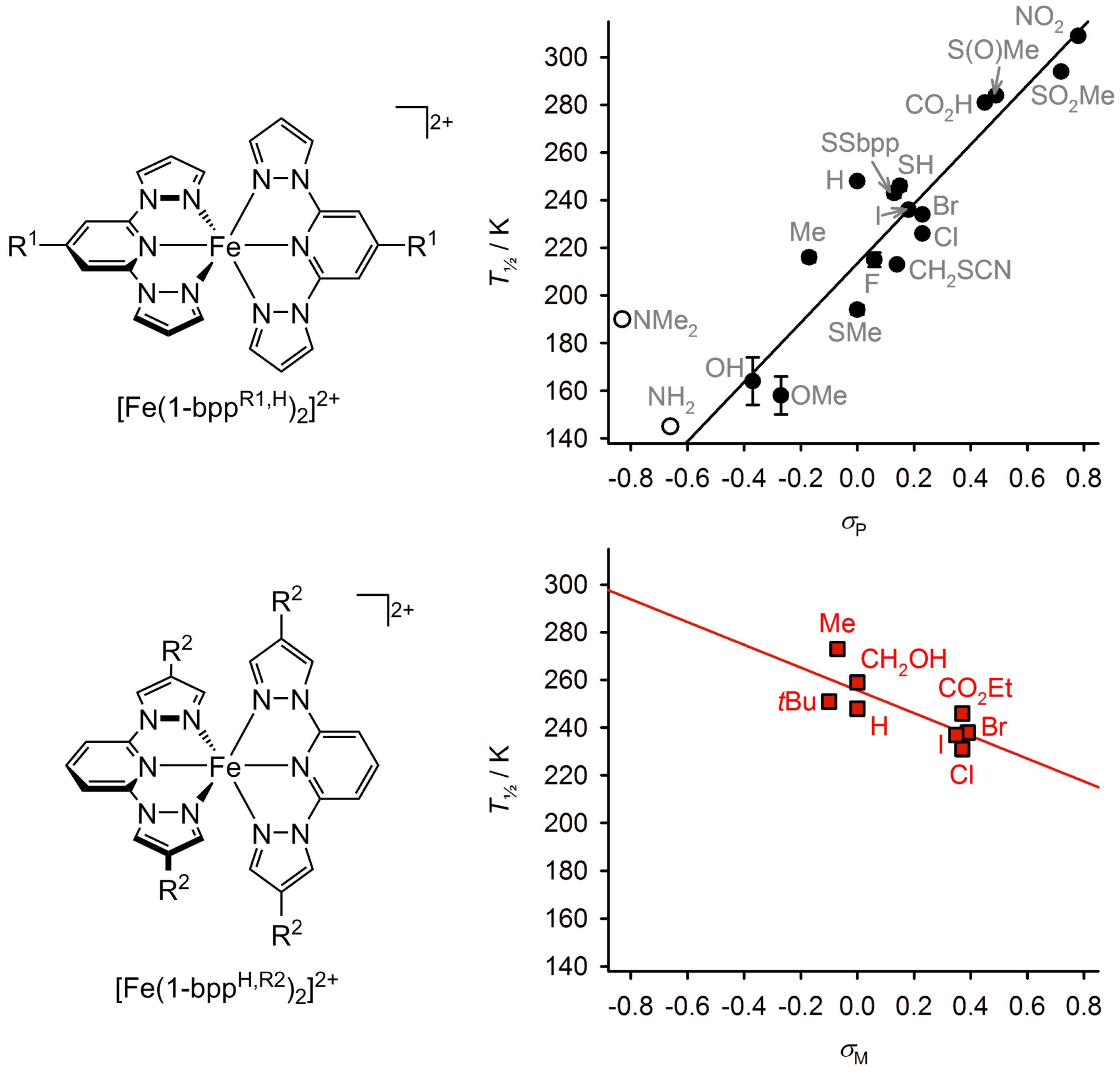
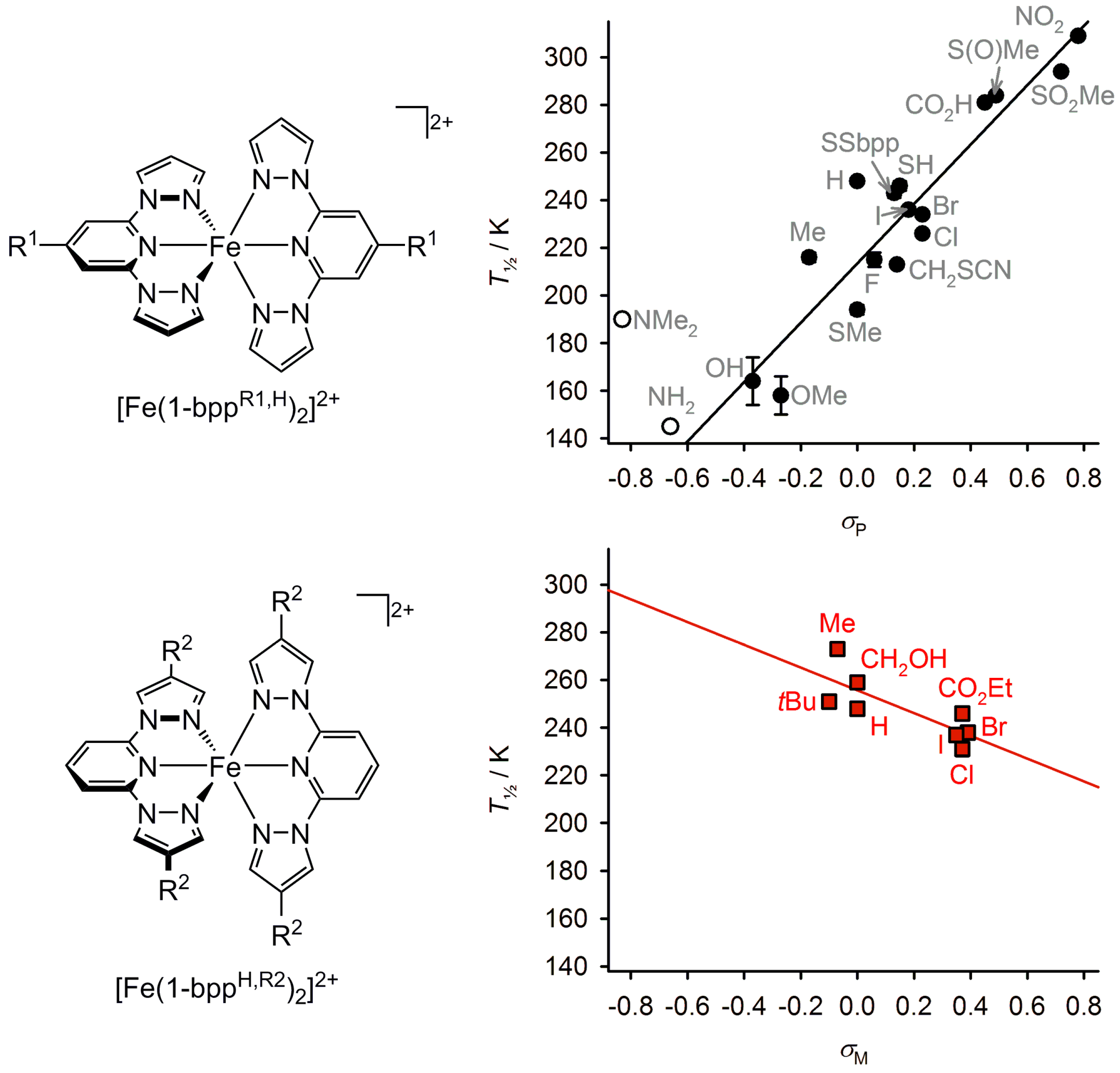
2.5. Electronic Influence of Ligand Substituents on Metal Ion Spin State—Inductive vs. Resonance Effects
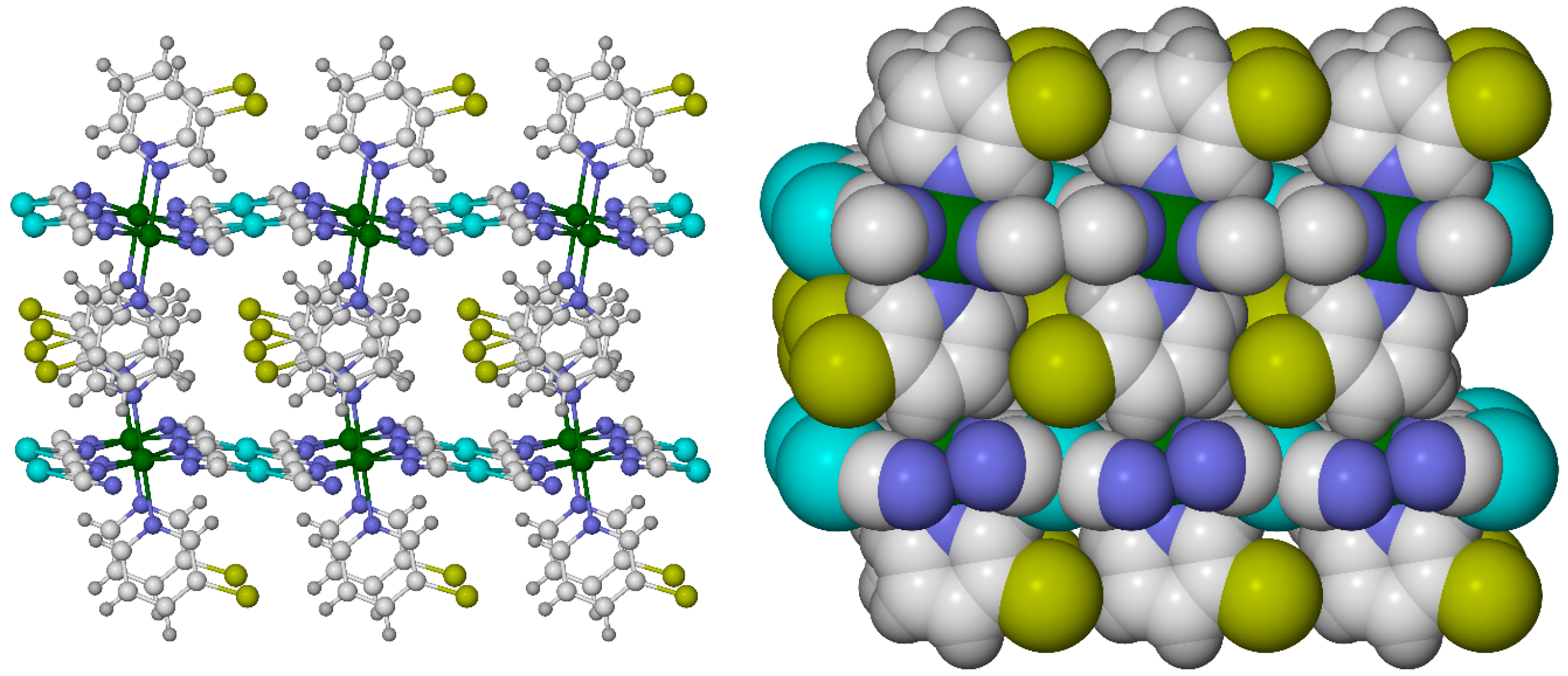
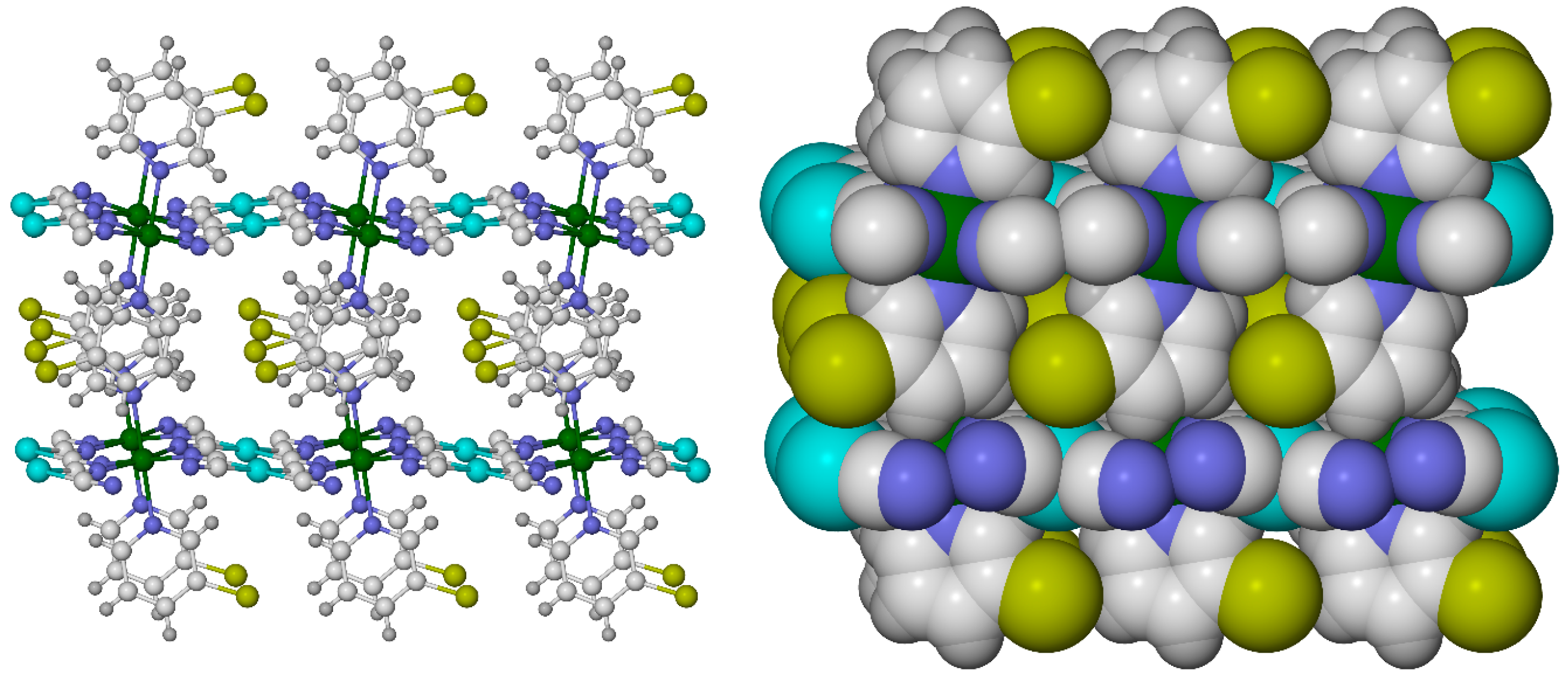
2.6. Effect of Secondary Bonding Interactions on Metal Ion Spin State—Hydrogen Bonding
3. Materials and Methods
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-bpp | 2,6-di{pyrazol-1-yl}pyridine |
| 1-bppyz | 2,6-di{pyrazol-1-yl}pyrazine |
| 3-bpp | 2,6-di{1H-pyrazol-3-yl}pyridine |
| 3-btzp | 2,6-di{1H-1,2,4-triazol-3-yl}pyridine |
| 3,3′-dmbpy | 3,3′-dimethyl-2,2′-bipyridine |
| 4-bthp | 2,6-di{thiazol-4-yl}pyridine |
| BArF‒ | tetrakis(3,5-di{trifluoromethyl}phenyl)borate |
| Bip | 2,2′-bi(1,4,5,6-tetrahydropyrimidine) |
| bipy | 2,2′-bipyridine |
| biq | 2,2′-bi(iso)quinoline |
| blbpen | N N′-bis(6-methylpyrid-2-ylmethyl)-N,N′-bis(pyrid-2-ylmethyl)-1,2-diaminoethane |
| Bn | benzyl |
| bpypzH | 3,5-di(pyrid-2-yl)-1H-pyrazole |
| bzimpy | 2,6-bis(1H-benzimidazol-2-yl)pyridine |
| Cy | cyclohexyl |
| dabqH2 | 2,5-(phenylamino)-1,4-benzoquinone |
| dmbc | dimethyl 2,2'-bipyridine-3,3'-dicarboxylate |
| dmbpy | 6,6′-dimethyl-2,2′-bipyridine |
| dmpz | 3,5-dimethylpyrazol-1-yl |
| impy | 2,6-bis(carbaldimino)pyridine |
| iPr | isopropyl |
| mbpy | 6-methyl-2,2′-bipyridine |
| Me | methyl |
| MeCN | acetonitrile |
| MePy3tren | bis(4-{pyrid-2-yl}-3-aza-3-butenyl)(4-{6-methylpyrid-2-yl}-3-aza-3-butenyl)amine |
| Me2Py3tren | (4-{pyrid-2-yl}-3-aza-3-butenyl)-bis-(4-{6-methylpyrid-2-yl}-3-aza-3-butenyl)amine |
| Me3Py3tren | tris(4-{6-methylpyrid-2-yl}-3-aza-3-butenyl)amine |
| MesIm | 3-(2,4,6-trimethylphenyl)imidazol-1-yl |
| Metpa | bis(pyrid-2-ylmethyl)({6-methylpyrid-2-yl}methyl)amine |
| Me2tpa | (pyrid-2-ylmethyl)-bis({6-methylpyrid-2-yl}methyl)amine |
| Me3tpa | tris({6-methylpyrid-2-yl}methyl)amine |
| Mes | mesityl, 2,4,6-triphenylphenyl |
| Mpz | 3-methylpyrazol-1-yl |
| naphtrienH2 | 1,8-bis(2-hydroxynaphthaldiminato)-3,6-diazaoctane |
| paptH | 2-(pyrid-2-y1amino)-4-(pyridin-2-yl)thiazole |
| Ph | phenyl |
| Pic | 2-(aminomethyl)pyridine, 2-picolylamine |
| Pmap | (pyrid-2-ylmethyl)-bis(2-{pyrid-2-yl}ethyl)amine |
| Pmea | bis(pyrid-2-ylmethyl)(2-{pyrid-2-yl}ethyl)amine |
| Pytacn | 1-(pyrid-2-ylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane |
| Py3tren | tris(4-{pyrid-2-yl}-3-aza-3-butenyl)amine |
| Py | pyridine |
| pz | pyrazol-1-yl |
| pzaptH | 2-(pyrazin-2-y1amino)-4-(pyridin-2-yl)thiazole |
| saltenH2 | 1,7-bis(salicylaldiminato)-4-azaheptane |
| saltrienH2 | 1,8-bis(salicylaldiminato)-3,6-diazaoctane |
| stybp | 4-(cis-styryl)-2,6-di(pyrazol-1-yl)pyridine |
| SCO | Spin crossover |
| Tacn | 1,4,7-triazacyclononane |
| tBu | tertbutyl |
| thf | etrahydrofuran |
| Tol | 4-methylphenyl, 4-tolyl |
| Tpa | tris(pyrid-2-ylmethyl)amine |
| tpea | tris(2-{pyrid-2-yl}ethyl)amine |
| tpen | N,N,N′,N′-tetrakis-(pyrid-2-ylmethyl)-1,2-diaminoethane |
| triphos | 1,1,1-tris(diphenylphosphinomethyl)ethane. |
References
- Weller, M.; Overton, T.; Rourke, J.; Armstrong, F. Inorganic Chemistry, 6th ed.; Oxford University Press: Oxford, UK, 2014; pp. 515–549. [Google Scholar]
- O’Connor, C.J. Magnetochemistry—Theory and experiment. Prog. Inorg. Chem. 1982, 29, 203–283. [Google Scholar]
- Swart, M.; Costas, M. (Eds.) Spin States in Biochemistry and Inorganic Chemistry: Influence on Structure and Reactivity; John Wiley & Sons: Chichester, UK, 2016; p. 465.
- Andris, E.; Jašík, J.; Gomez, L.; Costas, M.; Roithová, J. Spectroscopic characterization and reactivity of triplet and quintet iron(IV)-oxo complexes in the gas phase. Angew. Chem. Int. Ed. 2016, 55, 3637–3641. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Spin-crossover in [Fe(3-bpp)2][BF4]2 in different solvents—A dramatic stabilisation of the low-spin state in water. Dalton Trans. 2011, 40, 12021–12024. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.-R.; Park, J.G.; Haney, C.R.; Harris, T.D. Spin crossover iron(II) complexes as PARACEST MRI thermometers. Chem. Sci. 2014, 5, 2461–2465. [Google Scholar] [CrossRef]
- Harvey, J.N.; Poli, R.; Smith, K.M. Understanding the reactivity of transition metal complexes involving multiple spin states. Coord. Chem. Rev. 2003, 238–239, 347–361. [Google Scholar] [CrossRef]
- Ye, S.; Geng, C.-Y.; Shaik, S.; Neese, F. Electronic structure analysis of multistate reactivity in transition metal catalyzed reactions: The case of C–H bond activation by non-heme iron(IV)-oxo cores. Phys. Chem. Chem. Phys. 2013, 15, 8017–8030. [Google Scholar] [CrossRef] [PubMed]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Lee, Y.-M.; Fukuzumi, S. Tuning reactivity and mechanism in oxidation reactions by mononuclear nonheme iron(IV)-oxo complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.; Que, L., Jr. Toward the synthesis of more reactive S = 2 non-heme oxo-iron(IV) complexes. Acc. Chem. Res. 2015, 48, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.L. Distinctive reaction pathways at base metals in high-spin organometallic catalysts. Acc. Chem. Res. 2015, 48, 1696–1702. [Google Scholar] [CrossRef] [PubMed]
- Shaver, M.P.; Allan, L.E.N.; Rzepa, H.S.; Gibson, V.C. Correlation of metal spin state with catalytic reactivity: Polymerizations mediated by α-diimine–iron complexes. Angew. Chem. Int. Ed. 2006, 45, 1241–1244. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.P.; Swart, M. Subtle effects control the polymerisation mechanism in α-diimine iron catalysts. Dalton Trans. 2011, 40, 8419–8428. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials—Properties and Applications; John Wiley & Sons: Chichester, UK, 2013; p. 568.
- Bousseksou, A.; Molnár, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Gütlich, P.; Gaspar, A.B.; Garcia, Y. Spin state switching in iron coordination compounds. Beilstein J. Org. Chem. 2013, 9, 342–391. [Google Scholar] [CrossRef] [PubMed]
- Toftlund, H. Spin equilibrium in solutions. Monatsh. Chem. 2001, 132, 1269–1277. [Google Scholar] [CrossRef]
- Halcrow, M.A. Structure: Function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119–4142. [Google Scholar] [CrossRef] [PubMed]
- Kahn, O.; Krober, J.; Jay, C. Spin transition molecular materials for displays and data recording. Adv. Mater. 1992, 4, 718–728. [Google Scholar] [CrossRef]
- Manrique-Juárez, M.D.; Rat, S.; Salmon, L.; Molnár, G.; Quintero, C.M.; Nicu, L.; Shepherd, H.J.; Bousseksou, A. Switchable molecule-based materials for micro- and nanoscale actuating applications: Achievements and prospects. Coord. Chem. Rev. 2016, 308, 395–408. [Google Scholar] [CrossRef]
- Cavallini, M. Status and perspectives in thin films and patterning of spin crossover compounds. Phys. Chem. Chem. Phys. 2012, 14, 11867–11876. [Google Scholar] [CrossRef] [PubMed]
- Hassan, N.; Koudriavtsev, A.B.; Linert, W. Isoequilibrium relationships and cooperative effects in spin-state transitions in solution. Pure Appl. Chem. 2008, 80, 1281–1292. [Google Scholar] [CrossRef]
- König, E. Structural changes accompanying continuous and discontinuous spin-state transitions. Prog. Inorg. Chem. 1987, 35, 527–622. [Google Scholar]
- Hoselton, M.A.; Wilson, L.J.; Drago, R.S. Substituent effects on the spin equilibrium observed with hexadentate ligands on iron(II). J. Am. Chem. Soc. 1975, 97, 1722–1729. [Google Scholar] [CrossRef]
- Seredyuk, M.; Gaspar, A.B.; Ksenofontov, V.; Galyametdinov, Y.; Kusz, J.; Gütlich, P. Does the solid-liquid crystal phase transition provoke the spin-state change in spin-crossover metallomesogens? J. Am. Chem. Soc. 2008, 130, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.; Luckett, C.; May, L.; Beatty, A.M.; Scheidt, W.R. Synthesis and characterization of tripodal iron(II) complexes prepared from 2-pyridinecarboxaldehyde and 1-methyl-2-imidazolecarboxaldehyde: Stabilization of iron(II) cations with N6 donor sets. Inorg. Chim. Acta 2004, 357, 2390–2396. [Google Scholar] [CrossRef]
- Seredyuk, M.; Gaspar, A.B.; Kusz, J.; Bednarek, G.; Gütlich, P. Variable-temperature X-ray crystal structure determinations of {Fe[tren(6-Mepy)3]}(ClO4)2 and {Zn[tren(6-Mepy)3]}(ClO4)2 compounds: Correlation of the structural data with magnetic and Mössbauer spectroscopy data. J. Appl. Cryst. 2007, 40, 1135–1145. [Google Scholar] [CrossRef]
- Simmons, M.G.; Wilson, L.J. Magnetic and spin lifetime studies in solution of a ΔS = 1 spin-equilibrium process for some six-coordinate bis(N-R-2,6-pyridinedicarboxaldimine)cobalt(II) complexes. Inorg. Chem. 1977, 16, 126–130. [Google Scholar] [CrossRef]
- Onggo, D.; Hook, J.M.; Rae, A.D.; Goodwin, H.A. The influence of steric effects in substituted 2,2′-bipyridine on the spin state of iron(II) in [FeN6]2+ systems. Inorg. Chim. Acta 1990, 173, 19–30. [Google Scholar] [CrossRef]
- Onggo, D.; Goodwin, H.A. Steric effects of the spin state of iron(II) in complexes of substituted bipyridine derivatives. Aust. J. Chem. 1991, 44, 1539–1551. [Google Scholar] [CrossRef]
- Zang, Y.; Kim, J.; Dong, Y.; Wilkinson, E.C.; Appelman, E.H.; Que, L., Jr. Models for nonheme iron intermediates: Structural basis for tuning the spin states of Fe(tpa) complexes. J. Am. Chem. Soc. 1997, 119, 4197–4205. [Google Scholar] [CrossRef]
- Jesson, J.P.; Trofimenko, S.; Eaton, D.R. Spin equilibria in octahedral iron(II) poly(1-pyrazolyl) borates. J. Am. Chem. Soc. 1967, 89, 3158–3164. [Google Scholar] [CrossRef]
- Beattie, J.K.; Binstead, R.A.; West, R.J. Intersystem crossing observed by ultrasonic relaxation of the singlet-quintet spin equilibrium of iron(II) complexes in solution. J. Am. Chem. Soc. 1978, 100, 3044–3050. [Google Scholar] [CrossRef]
- Buchen, T.; Gütlich, P. Substituent effects on the spin equilibrium in iron(II) pyrazolylborate complexes. Inorg. Chim. Acta 1995, 231, 221–223. [Google Scholar] [CrossRef]
- Toftlund, H. Spin equilibria in octahedral iron(II) complexes. Coord. Chem. Rev. 1989, 94, 67–108. [Google Scholar] [CrossRef]
- Prat, I.; Company, A.; Corona, T.; Parella, T.; Ribas, X.; Costas, M. Assessing the impact of electronic and steric tuning of the ligand in the spin state and catalytic oxidation ability of the FeII(Pytacn) family of complexes. Inorg. Chem. 2013, 52, 9229–9244. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.M.; McAllister, J.A.; Kilner, C.A.; Thornton-Pett, M.; Bridgeman, A.J.; Halcrow, M.A. Stereochemical effects on the spin-state transition shown by salts of [FeL2] [L = 2,6-di(pyrazol-1-yl)-pyridine]. J. Chem. Soc. Dalton Trans. 2002, 548–554. [Google Scholar] [CrossRef]
- Holland, J.M.; Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. Control of the spin state of Fe(II) 2,6-di(pyrazol-1-yl)pyridine complexes by distal ligand substitution. Inorg. Chem. Commun. 2002, 5, 328–332. [Google Scholar] [CrossRef]
- Roberts, T.D.; Little, M.A.; Kershaw Cook, L.J.; Barrett, S.A.; Tuna, F.; Halcrow, M.A. Iron(II) complexes of 2,6-di(1-alkylpyrazol-3-yl)pyridine derivatives—The influence of distal substituents on the spin state of the iron center. Polyhedron 2013, 64, 4–12. [Google Scholar] [CrossRef]
- Constable, E.C.; Baum, G.; Bill, E.; Dyson, R.; van Eldik, R.; Fenske, D.; Kaderli, S.; Morris, D.; Neubrand, A.; Neuberger, M.; et al. Control of iron(II) spin states in 2,2′:6′,2′′-terpyridine complexes through ligand substitution. Chem. Eur. J. 1999, 5, 498–508. [Google Scholar] [CrossRef]
- Lin, H.-J.; Siretanu, D.; Dickie, D.A.; Subedi, D.; Scepaniak, J.J.; Mitcov, D.; Clérac, R.; Smith, J.M. Steric and electronic control of the spin state in three-fold symmetric, four-coordinate iron(II) complexes. J. Am. Chem. Soc. 2014, 136, 13326–13332. [Google Scholar] [CrossRef] [PubMed]
- Cirera, J.; Ruiz, E. Theoretical modeling of the ligand-tuning effect over the transition temperature in four-coordinated FeII molecules. Inorg. Chem. 2016, 55, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Jesson, J.P.; Weiher, J.F.; Trofimenko, S. Mössbauer and magnetic susceptibility investigation of the 5T2–1A1 crossover in some octahedral ferrous complexes in the solid state. J. Chem. Phys. 1968, 48, 2058–2066. [Google Scholar] [CrossRef]
- Oliver, J.D.; Mullica, D.F.; Hutchinson, B.B.; Milligan, W.O. Iron-nitrogen bond lengths in low-spin and high-spin iron(II) complexes with poly(pyrazolyl) borate ligands. Inorg. Chem. 1980, 19, 165–169. [Google Scholar] [CrossRef]
- Elhaïk, J.; Evans, D.J.; Kilner, C.A.; Halcrow, M.A. A structural, magnetic and Mössbauer spectroscopic study of an unusual angular Jahn-Teller distortion in a series of high-spin iron(II) complexes. Dalton Trans. 2005, 9, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-H.; Yang, L.-J.; Liu, Q.-L.; Ling, Y.; Wang, W.; Sun, B.-W. Lattice water molecules tuned spin-crossover for an iron(II) complex with thermal hysteresis. Dalton Trans. 2014, 43, 16937–16942. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, D.M.; Armstrong, W.H. M{hydrotris(3-phenylpyrazol-l-yl)borate}2: Sterically encumbered iron(II) and manganese(II) complexes. Inorg. Chem. 1990, 29, 3607–3612. [Google Scholar] [CrossRef]
- Cecchi, P.; Berrettoni, M.; Giorgetti, M.; Lobbia, G.G.; Calogero, S.; Stievano, L. The effect of the 3-trifluoromethyl substituent in polypyrazolylborato complexes on the iron(II) spin state; X-ray diffraction and absorption and Mössbauer studies. Inorg. Chim. Acta 2001, 318, 67–76. [Google Scholar] [CrossRef]
- Gruden-Pavlović, M.; Stepanović, S.; Perić, M.; Güell, M.; Swart, M. A density functional study of the spin state energetics of polypyrazolylborato complexes of first-row transition metals. Phys. Chem. Chem. Phys. 2014, 16, 14514–14522. [Google Scholar] [CrossRef] [PubMed]
- Pelascini, F.; Wesolek, M.; Peruch, F.; De Cian, A.; Kyritsaka, N.; Lutz, P.J.; Kress, J. Iron complexes of terdentate nitrogen ligands: Formation and X-ray structure of three new dicationic complexes. Polyhedron 2004, 23, 3193–3199. [Google Scholar] [CrossRef]
- Brauchli, S.Y.; Constable, E.C.; Harris, K.; Häussinger, D.; Housecroft, C.E.; Rösel, P.J.; Zampese, J.A. Towards catenanes using π-stacking interactions and their influence on the spin-state of a bis(2,2′:6′,2′′-terpyridine)iron(II) domain. Dalton Trans. 2010, 39, 10739–10748. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. The spin-states and spin-transitions of mononuclear iron(II) complexes of nitrogen-donor ligands. Polyhedron 2007, 26, 3523–3576. [Google Scholar] [CrossRef]
- Kisslinger, S.; Kelm, H.; Zheng, S.; Beitat, A.; Würtele, C.; Wortmann, R.; Bonnet, S.; Herres-Pawlis, S.; Krüger, H.-J.; Schindler, S. Synthesis and characterization of iron(II) thiocyanate complexes with derivatives of the tris(pyridine-2-ylmethyl)amine (tmpa) ligand. Z. Anorg. Allg. Chem. 2012, 638, 2069–2077. [Google Scholar] [CrossRef]
- Hamon, P.; Thépot, J.-Y.; le Floch, M.; Boulon, M.-E.; Cador, O.; Golhen, S.; Ouahab, L.; Fadel, L.; Saillard, J.-Y.; Hamon, J.-R. Dramatic remote substitutent effects on the electronic spin state of bis(scorpionate) iron(II) complexes. Angew. Chem. Int. Ed. 2008, 47, 8687–8691. [Google Scholar] [CrossRef] [PubMed]
- Halcrow, M.A. Iron(II) complexes of 2,6-di(pyrazol-1-yl)pyridines—A versatile system for spin-crossover research. Coord. Chem. Rev. 2009, 253, 2493–2514. [Google Scholar] [CrossRef]
- Mohammed, R. Iron Complexes of New Dipyrazolylpyridine Derivatives for Spin-Crossover Applications. Ph.D. Thesis, University of Leeds, Leeds, UK, 2012. [Google Scholar]
- Muñoz, M.C.; Gaspar, A.B.; Galet, A.; Real, J.A. Spin-crossover behavior in cyanide-bridged iron(II)-silver(I) bimetallic 2D Hofmann-like metal-organic frameworks. Inorg. Chem. 2007, 46, 8182–8192. [Google Scholar] [CrossRef] [PubMed]
- Agustí, G.; Muñoz, M.C.; Gaspar, A.B.; Real, J.A. Spin-crossover behavior in cyanide-bridged iron(II)-gold(I) bimetallic 2D Hofmann-like metal-organic frameworks. Inorg. Chem. 2008, 47, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Agustí, G.; Muñoz, M.C.; Gaspar, A.B.; Real, J.A. Spin-crossover behavior in cyanide-bridged iron(II)-copper(I) bimetallic 1–3D metal-organic frameworks. Inorg. Chem. 2009, 48, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Martínez, V.; Gaspar, A.B.; Muñoz, M.C.; Bukin, G.V.; Levchenko, G.; Real, J.A. Synthesis and characterisation of a new series of bistable iron(II) spin-crossover 2D metal-organic frameworks. Chem. Eur. J. 2009, 15, 10960–10971. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.C.; Goodwin, H.A.; Onggo, D. Steric influences on the ground state of iron(II) in the tris(3,3′-dimethyl-2,2′-bipyridine)iron(II) ion. Aust. J. Chem. 1988, 41, 1157–1169. [Google Scholar] [CrossRef]
- Arcis-Castíllo, Z.; Zheng, S.; Siegler, M.A.; Roubeau, O.; Bedoui, S.; Bonnet, S. Tuning the transition temperature and cooperativity of bapbpy-based mononuclear spin-crossover compounds: Interplay between molecular and crystal engineering. Chem. Eur. J. 2011, 17, 14826–14836. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Reintjens, N.R.M.; Siegler, M.A.; Roubeau, O.; Bouwman, E.; Rudavskyi, A.; Havenith, R.W.A.; Bonnet, S. Stabilization of the low-spin state in a mononuclear iron(II) complex and high-temperature cooperative spin crossover mediated by hydrogen bonding. Chem. Eur. J. 2016, 22, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Reger, D.L.; Little, C.A.; Smith, M.D.; Rheingold, A.L.; Lam, K.-C.; Concolino, T.L.; Long, G.J.; Hermann, R.P.; Grandjean, F. Synthetic, structural, magnetic, and Mössbauer spectral study of [Fe[HC(3,5-Me2pz)3]2}I2 and its spin-state crossover behavior. Eur. J. Inorg. Chem. 2002, 2002, 1190–1197. [Google Scholar] [CrossRef]
- Reger, D.L.; Gardinier, J.R.; Elgin, J.D.; Smith, M.D.; Hautot, D.; Long, G.J.; Grandjean, F. Structure-function correlations in iron(II) tris(pyrazolyl)borate spin-state crossover complexes. Inorg. Chem. 2006, 45, 8862–8875. [Google Scholar] [CrossRef] [PubMed]
- Elhaïk, J.; Kilner, C.A.; Halcrow, M.A. Structural diversity in iron(II) complexes of 2,6-di(pyrazol-1-yl)- pyridine and 2,6-di(3-methylpyrazol-1-yl)pyridine. Dalton Trans. 2006, 6, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, R.; Barrett, S.A.; Kilner, C.A.; Halcrow, M.A. The influence of ligand conformation on the thermal spin transitions in iron(III) saltrien complexes. Dalton Trans. 2008, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Kershaw Cook, L.J.; Tuna, F.; Halcrow, M.A. Iron(II) and cobalt(II) complexes of tris-azinyl analogues of 2,2′:6′,2′′-terpyridine. Dalton Trans. 2013, 42, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Sertphon, D.; Harding, D.J.; Harding, P.; Murray, K.S.; Moubaraki, B.; Adams, H. Steric trapping of the high spin state in FeIII quinolylsalicylaldimine complexes. Aust. J. Chem. 2014, 67, 1574–1580. [Google Scholar] [CrossRef]
- Matouzenko, G.S.; Borshch, S.A.; Jeanneau, E.; Bushuev, M.B. Spin crossover in a family of iron(II) complexes with hexadentate ligands: Ligand strain as a factor determining the transition temperature. Chem. Eur. J. 2009, 15, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Leibold, M.; Kisslinger, S.; Heinemann, F.W.; Hampel, F.; Ichiyanagi, Y.; Klein, M.; Homenya, P.; Renz, F.; Toftlund, H.; Brehm, G.; et al. Effect of chelate ring size in iron(II) isothiocyanato complexes with tetradentate tripyridyl-alkylamine ligands on spin crossover properties. Z. Anorg. Allg. Chem. 2016, 642, 85–94. [Google Scholar] [CrossRef]
- Wei, R.-J.; Li, B.; Tao, J.; Huang, R.-B.; Zheng, L.-S.; Zheng, Z. Making spin-crossover crystals by successive polymorphic transformations. Inorg. Chem. 2011, 50, 1170–1172. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wei, R.-J.; Tao, J.; Huang, R.-B.; Zheng, L.-S. Pressure effects on a spin-crossover monomeric compound [Fe(pmea)(SCN)2] (pmea = bis[(2-pyridyl)methyl]-2-(2-pyridyl)ethylamine). Inorg. Chem. 2010, 49, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Nihei, M.; Shiga, T.; Maeda, Y.; Oshio, H. Spin crossover iron(III) complexes. Coord. Chem. Rev. 2007, 251, 2606–2621. [Google Scholar] [CrossRef]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin crossover iron(III) complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Watkins, D.D.; Riley, D.P.; Stone, J.A.; Busch, D.H. Iron(II) complexes with unsubstituted saturated tetraaza macrocyclic ligands of varying ring size. Inorg. Chem. 1976, 15, 387–393. [Google Scholar] [CrossRef]
- Christiansen, L.; Hendrickson, D.N.; Toftlund, H.; Wilson, S.R.; Xie, C.-L. Synthesis and structure of metal complexes of triaza macrocycles with three pendant pyridylmethyl arms. Inorg. Chem. 1986, 25, 2813–2818. [Google Scholar] [CrossRef]
- Al-Obaidi, A.H.R.; McGarvey, J.J.; Taylor, K.P.; Bell, S.E.J.; Jensen, K.B.; Toftlund, H. Observation of biphasic kinetics in light-induced spin-state crossover in an iron(II) complex in solution. J. Chem. Soc. Chem. Commun. 1993, 536–538. [Google Scholar] [CrossRef]
- Janiak, C.; Scharmann, T.G.; Bräuniger, T.; Holubová, J.; Nádvorník, M. Spin crossover studies on bis{hydro-tris(1,2,4-triazolyl)borato}iron(II). Z. Anorg. Allg. Chem. 1998, 624, 769–774. [Google Scholar] [CrossRef]
- Catalan, J.; Abboud, J.L.M.; Elguero, J. Basicity and acidity of azoles. Adv. Heterocycl. Chem. 1987, 41, 187–274. [Google Scholar]
- Sugiyarto, K.H.; Craig, D.C.; Rae, A.D.; Goodwin, H.A. Structural, magnetic and Mössbauer spectral studies of salts of bis[2,6-bis(pyrazol-3-yl) pyridine] iron(II)—A spin crossover system. Aust. J. Chem. 1994, 47, 869–890. [Google Scholar] [CrossRef]
- Sugiyarto, K.H.; Craig, D.C.; Rae, A.D.; Goodwin, H.A. Structural and electronic properties of iron(II) and nickel(II) complexes of 2,6-bis(triazol-3-yl)pyridines. Aust. J. Chem. 1993, 46, 1269–1290. [Google Scholar] [CrossRef]
- Baker, A.T.; Goodwin, H.A. Iron(II) and nickel(II) complexes of 2,6-di(thiazol-4-yl)pyridine and related ligands: Magnetic, spectral and structural studies. Aust. J. Chem. 1986, 39, 209–220. [Google Scholar] [CrossRef]
- Mohammed, R.; Chastanet, G.; Tuna, F.; Malkin, T.L.; Barrett, S.A.; Kilner, C.A.; Létard, J.-F.; Halcrow, M.A. Synthesis of 2,6-di(pyrazol-1-yl)pyrazine derivatives and the spin-state behavior of their iron(II) complexes. Eur. J. Inorg. Chem. 2013, 2013, 819–831. [Google Scholar] [CrossRef]
- Childs, B.J.; Craig, D.C.; Ross, K.A.; Scudder, M.L.; Goodwin, H.A. Structural and electronic properties of bis[2-(pyrazin-2-ylamino)-4-(pyridin-2-yl)thiazolato]iron(II) and its solvated derivatives. Aust. J. Chem. 1994, 47, 891–902. [Google Scholar] [CrossRef]
- Tweedle, M.F.; Wilson, L.J. Variable spin iron(III) chelates with hexadentate ligands derived from triethylenetetramine and various salicylaldehydes. Synthesis, characterization, and solution state studies of a new 2T ⇌ 6A spin equilibrium system. J. Am. Chem. Soc. 1976, 98, 4824–4834. [Google Scholar] [CrossRef]
- Masárová, P.; Zoufalý, P.; Moncol, J.; Nemec, I.; Pavlik, J.; Gembický, M.; Trávníček, Z.; Boča, R.; Šalitroš, I. Spin crossover and high spin electroneutral mononuclear iron(III) Schiff base complexes involving terminal pseudohalido ligands. New J. Chem. 2015, 39, 508–519. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Nakano, K.; Suemura, N.; Yoneda, K.; Kawata, S.; Kaizaki, S. Substituent effect of the coordinated pyridine in a series of pyrazolato bridged dinuclear diiron(II) complexes on the spin-crossover behavior. Dalton Trans. 2005, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Létard, J.-F.; Carbonera, C.; Real, J.A.; Kawata, S.; Kaizaki, S. Photomagnetism of a series of dinuclear iron(II) complexes. Chem. Eur. J. 2009, 15, 4146–4155. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.; Deeth, R.J. Spin-state energetics of FeII complexes—The continuing voyage through the density functional minefield. Eur. J. Inorg. Chem. 2014, 4573–4580. [Google Scholar] [CrossRef]
- Takahashi, K.; Hasegawa, Y.; Sakamoto, R.; Nishikawa, M.; Kume, S.; Nishibori, E.; Nishihara, H. Solid-state ligand-driven light-induced spin change at ambient temperatures in bis(dipyrazolylstyryl-pyridine)iron(II) complexes. Inorg. Chem. 2012, 51, 5188–5198. [Google Scholar] [CrossRef] [PubMed]
- Creutz, S.E.; Peters, J.C. Spin-state tuning at pseudo-tetrahedral d6 ions: Spin crossover in [BP3]FeII−X complexes. Inorg. Chem. 2016, 55, 3894–3906. [Google Scholar] [CrossRef] [PubMed]
- Park, J.G.; Jeon, I.-R.; Harris, T.D. Electronic effects of ligand substitution on spin crossover in a series of diiminoquinonoid-bridged FeII2 complexes. Inorg. Chem. 2015, 54, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Enamullah, M.; Hasegawa, M.; Fukuda, Y.; Linert, W.; Hoshi, T. Synthesis, characterization and spin-crossover behaviors of [Co(hydroxycarboxylato)(triphos)] complexes. Bull. Chem. Soc. Jpn. 2002, 75, 2449–2453. [Google Scholar] [CrossRef]
- Kershaw Cook, L.J.; Kulmaczewski, R.; Mohammed, R.; Dudley, S.; Barrett, S.A.; Little, M.A.; Deeth, R.J.; Halcrow, M.A. A unified treatment of the relationship between ligand substituents and spin state in a family of iron(II) complexes. Angew. Chem. Int. Ed. 2016, 55, 4327–4331. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.D.; Little, M.A.; Kershaw Cook, L.J.; Halcrow, M.A. Iron(II) complexes of 2,6-di(1H-pyrazol- 3-yl)pyridine derivatives with hydrogen bonding and sterically bulky substituents. Dalton Trans. 2014, 43, 7577–7588. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.W.; Schultz, F.A. Solution characterization of the iron(II) bis(1,4,7-triazacyclononane) spin-equilibrium reaction. Inorg. Chem. 2001, 40, 5296–5298. [Google Scholar] [CrossRef] [PubMed]
- Chum, H.L.; Vanin, J.A.; Holanda, M.I.D. Tris(2-(aminomethyl)pyridine)iron(II): A new spin-state equilibrium in solution. Inorg. Chem. 1982, 21, 1146–1152. [Google Scholar] [CrossRef]
- Linert, W.; Konecny, M.; Renz, F. Spin-state equilibria in non-aqueous solution and quantum mechanical investigations of iron(II) and nickel(II) complexes with 4-substituted 2,6-bis(benzimidazol-2-yl)pyridines. J. Chem. Soc. Dalton Trans. 1994, 1523–1531. [Google Scholar] [CrossRef]
- Ni, Z.; Shores, M.P. Magnetic observation of anion binding in iron coordination complexes: Toward spin-switching chemosensors. J. Am. Chem. Soc. 2009, 131, 32–33. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; McDaniel, A.M.; Shores, M.P. Ambient temperature anion-dependent spin state switching observed in “mostly low spin” heteroleptic iron(II) diimine complexes. Chem. Sci. 2010, 1, 615–621. [Google Scholar] [CrossRef]
- Ni, Z.; Shores, M.P. Supramolecular effects on anion-dependent spin-state switching properties in heteroleptic iron(II) complexes. Inorg. Chem. 2010, 49, 10727–10735. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Fiedler, S.R.; Shores, M.P. Investigation of anion-dependence in the spin-state switching properties of [(H2bip)2Fe(6-Mebpy)]X2. Dalton Trans. 2011, 40, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.A.; Halcrow, M.A. Anion-dependent spin crossover in solution for an iron(II) complex of a 1H-pyrazolyl ligand. RSC Adv. 2014, 4, 11240–11243. [Google Scholar] [CrossRef]
- Kubik, S. Anion recognition in water. Chem. Soc. Rev. 2010, 39, 3648–3663. [Google Scholar] [CrossRef] [PubMed]
- Young, M.C.; Liew, E.; Ashby, J.; McCoy, K.E.; Hooley, R.J. Spin state modulation of iron spin crossover complexes via hydrogen-bonding self-assembly. Chem. Commun. 2013, 49, 6331–6333. [Google Scholar] [CrossRef] [PubMed]
- Young, M.C.; Liew, E.; Hooley, R.J. Colorimetric barbiturate sensing with hybrid spin crossover assemblies. Chem. Commun. 2014, 50, 5043–5045. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.J. X-seed—A software tool for supramolecular crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- SIGMAPLOT; v. 8.02; SPSS Scientific Inc.: Chicago, IL, USA, 2002.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halcrow, M.A. The Effect of Ligand Design on Metal Ion Spin State—Lessons from Spin Crossover Complexes. Crystals 2016, 6, 58. https://doi.org/10.3390/cryst6050058
Halcrow MA. The Effect of Ligand Design on Metal Ion Spin State—Lessons from Spin Crossover Complexes. Crystals. 2016; 6(5):58. https://doi.org/10.3390/cryst6050058
Chicago/Turabian StyleHalcrow, Malcolm A. 2016. "The Effect of Ligand Design on Metal Ion Spin State—Lessons from Spin Crossover Complexes" Crystals 6, no. 5: 58. https://doi.org/10.3390/cryst6050058