
PRINCEPS: A Computer-Based Approach to the Structural Description and Recognition of Trends within Structural Databases, and Its Application to the Ce-Ni-Si System

Abstract
:1. Introduction
2. The PRINCEPS Algorithm
2.1. Comparing Elemental CEs
- (1)
- First, the CE of the atom is defined, as is accomplished unambiguously (for an elemental structure) by a Voronoi–Dirichlet partitioning of the space, a common method for CE determination in intermetallic phases [15]. An atom is considered to be part of the CE if its Voronoi cell shares a face with the central atom’s Voronoi cell [55].
- (2)
- Next, the CE is processed by assigning a weight inversely related to its distance to the central atom, as atoms further from the central atom should play a smaller role in defining the CE. In the current algorithm, we follow the common practice of using the solid angle of the shared Voronoi face in this role: we weight each neighbor by the solid angle it takes up, normalized by the average over all atoms in the CE. Following this step, we obtain a function defined on the unit sphere around the central atom that has a (weighted) δ-peak for each coordinating atom and is zero everywhere else.
- (3)
- This function is then expanded into an orthonormal basis of real-valued functions on the unit sphere, the real spherical harmonics (SHs). The result of this expansion is a set of vectors of SH coefficients corresponding to each l value: , , , etc. This expansion is then truncated up to an arbitrary order. In the current algorithm, , i.e., only the s, p, d, f and g components are considered. Including higher order coefficients may enhance the level of detail treated in the algorithm at the expense of longer execution time. The program then proceeds to compute the total norm of SH coefficients corresponding to each l value, i.e., . Since these norms are isometry invariant, they collectively serve as a descriptor of the CE.
- (4)
- Finally, the distance between two CEs can be calculated as , where with are the descriptors of the two CEs. The factors are used to normalize all terms in the summation to the same scale, since has components.
2.2. Comparing Multi-Element CEs
- (1)
- The CE is determined through a power diagram partition of the space using atomic radii.
- (2)
- Each atom in the total CE is assigned a weight equal to the solid angle of the shared PD face, normalized by the average over all atoms in CE. We thus obtain a function defined on the unit sphere around the central atom for each elemental CE.
- (3)
- The function for each elemental CE is then expanded into real SHs and truncated up to , and the coefficients are collected in matrices .
- (4)
- cross-element matrices are calculated from the coefficients by . The resulting 3D-matrix is the descriptor of the total CE.
- (5)
- Determination of the distance between two CEs involves a loop over all possible permutations of the elements:
- (5a)
- For each permutation , the symmetric recombination matrix is calculated, and the recombination matrix is calculated by performing a Cholesky decomposition on .
- (5b)
- For each , the distance of the two CEs under permutation is calculated by , where is the l-th order cross-element matrix of the descriptor of CE , with rows and columns permuted according to . denotes the standard matrix norm.
- (5c)
- The distance between the two CEs under permutation is defined as , where is the normalization factor for .
- (6)
- The overall distance between the two CEs is then calculated by taking the average of over all permutations .
2.3. From CEs to Structures
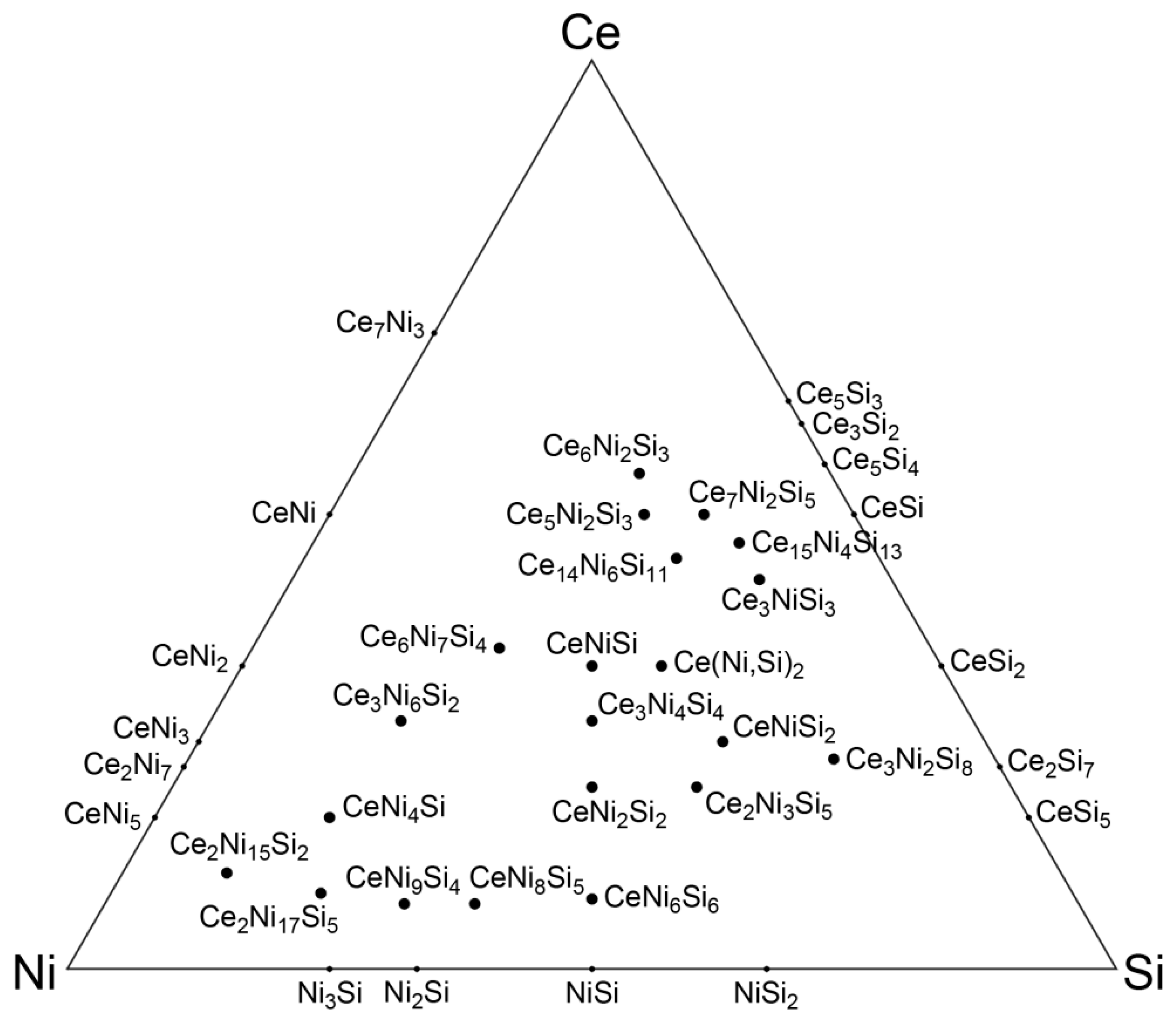
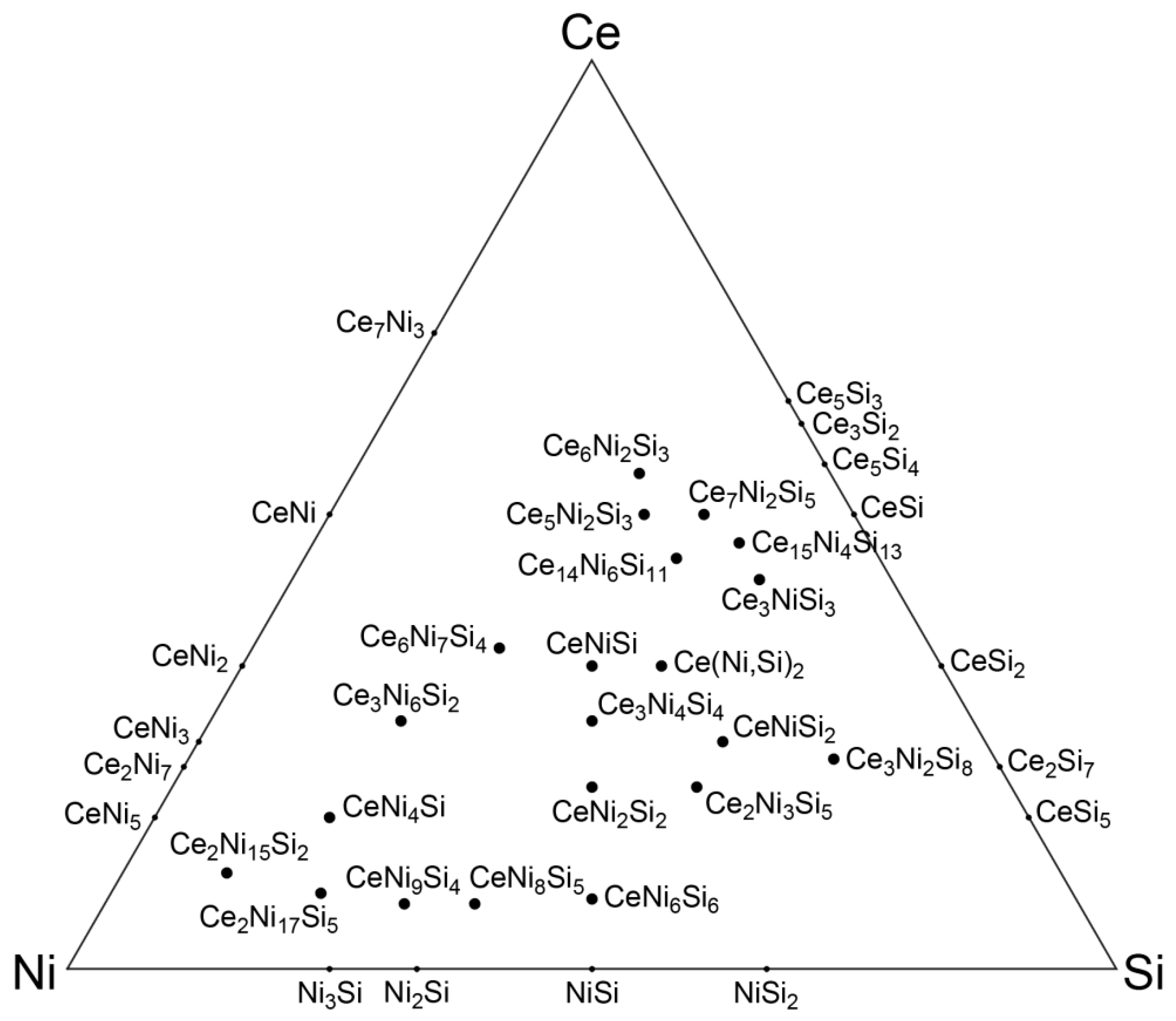
3. Analysis of Crystal Structures in the Ce-Ni-Si System
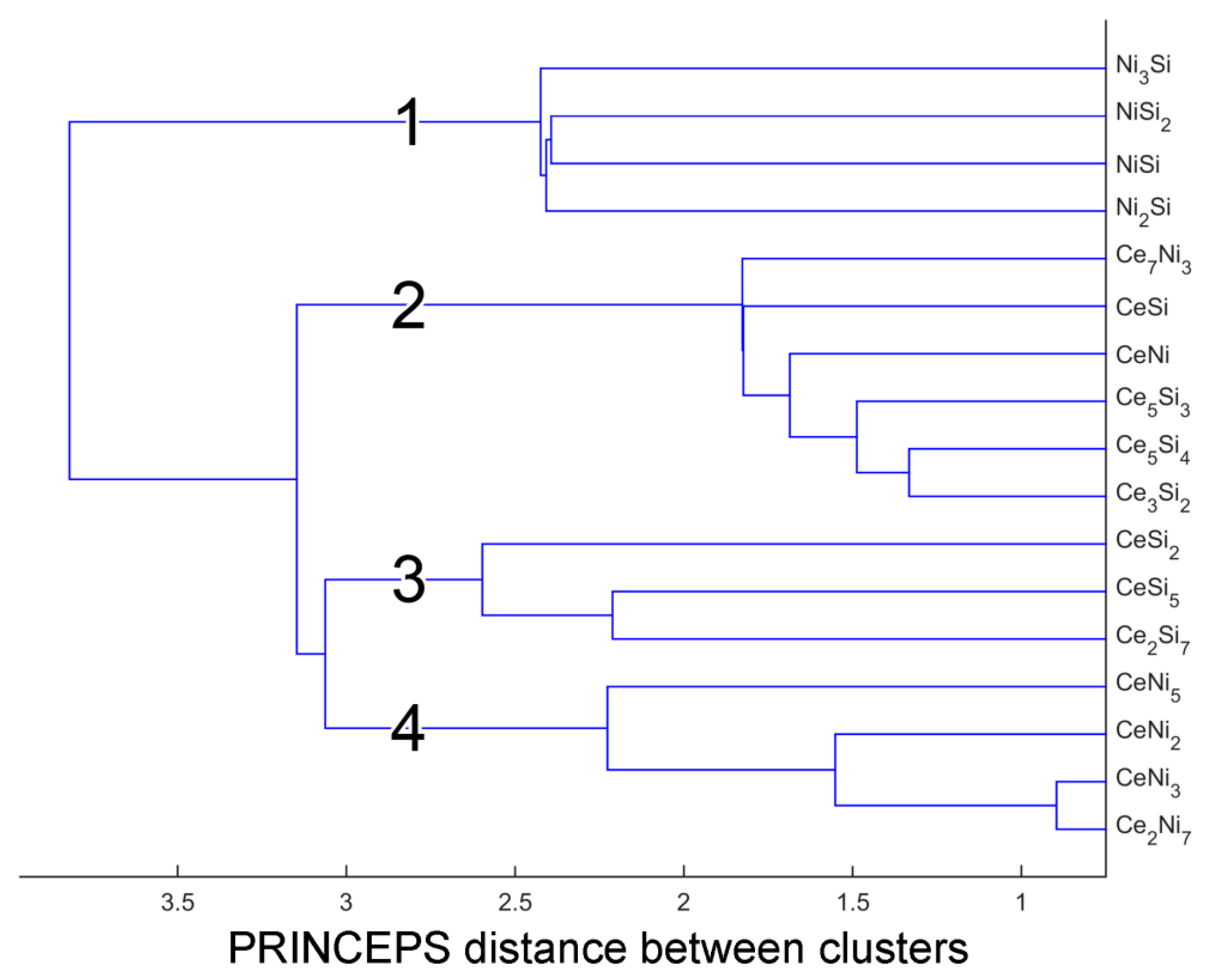
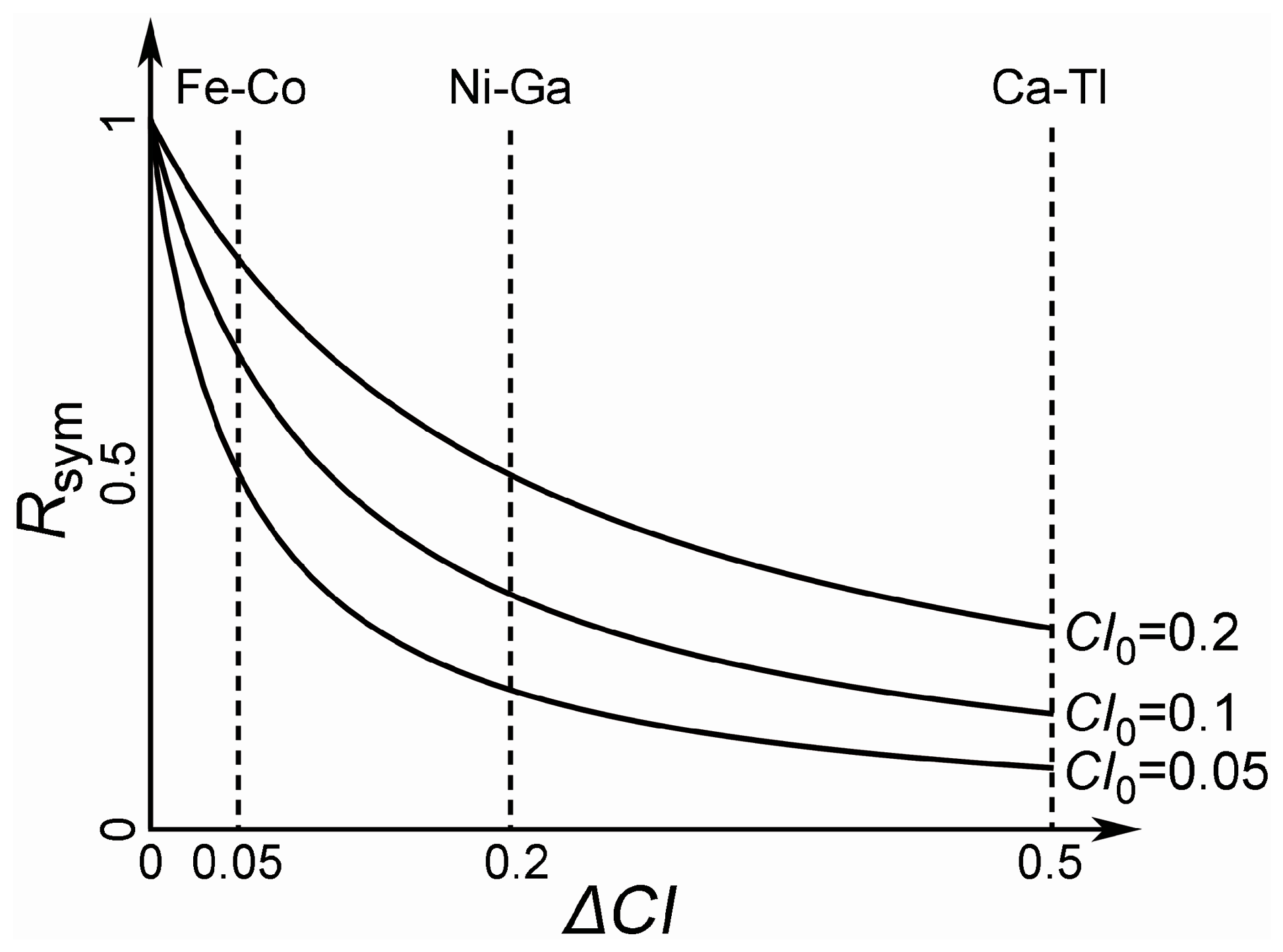
3.1. Defining the Reference Database: Elimination of Redundant Structures
| Cluster 1: | Ni2Si ( oP12, ), Ni3Si (cP4, ), NiSi (oP8, ), NiSi2 (cF12, ) |
| Cluster 2: | Ce3Si2 (tP10, ), Ce5Si3 (tI32, ), Ce5Si4 (tP36, ), Ce7Ni3 (hP20, ), CeNi (oS8, ), CeSi (oP8, ) |
| Cluster 3: | Ce2Si7 (oS18, ), CeSi2 (tI12, ) [81], CeSi5 (oI12, ) |
| Cluster 4: | Ce2Ni7 (hP36, ), CeNi2 (cF24, ), CeNi3 (hP24, ), CeNi5 (hP6, ) |
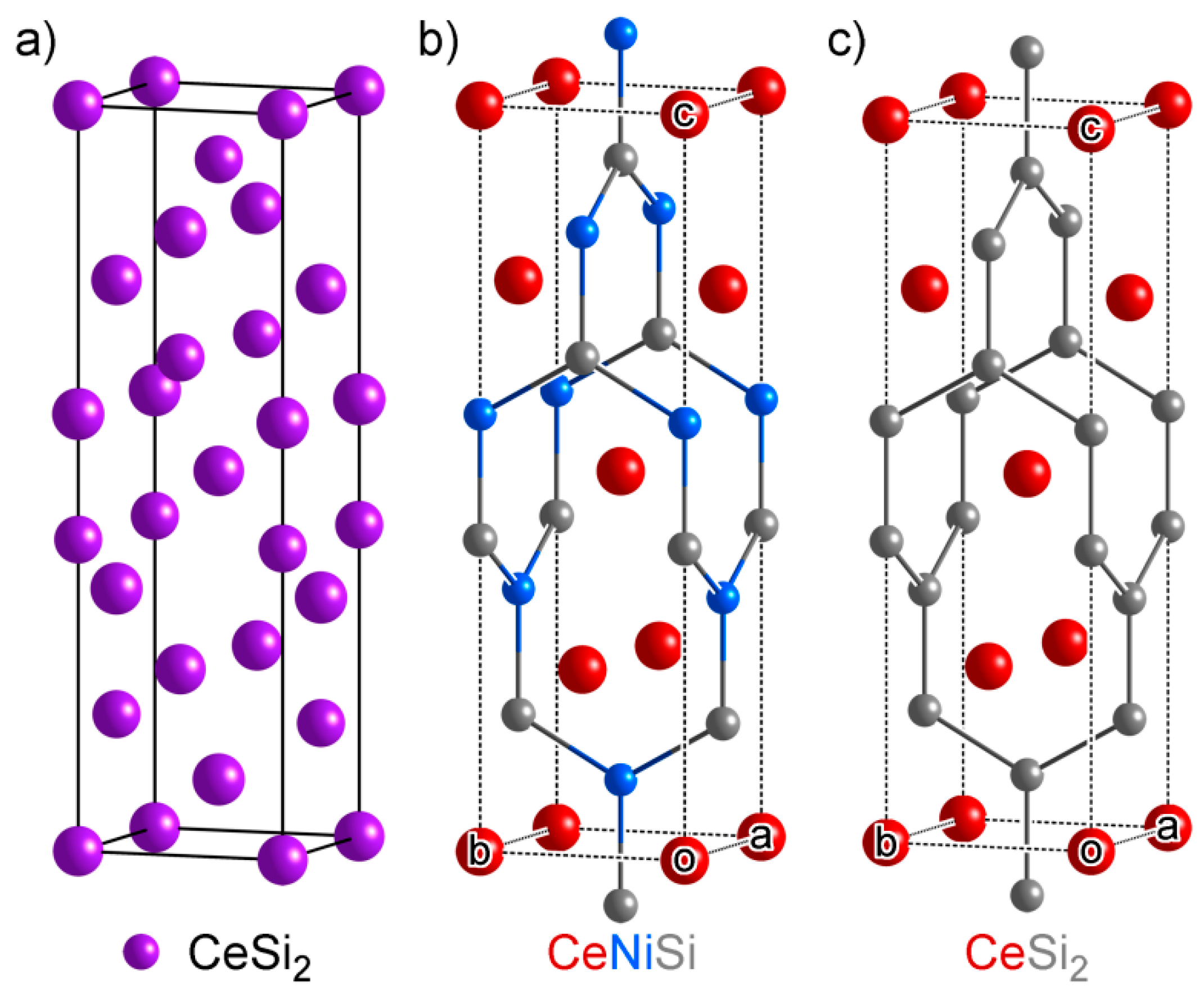
3.2. Projection of Ternary Phases onto Simple Binary Phases: Sample Output
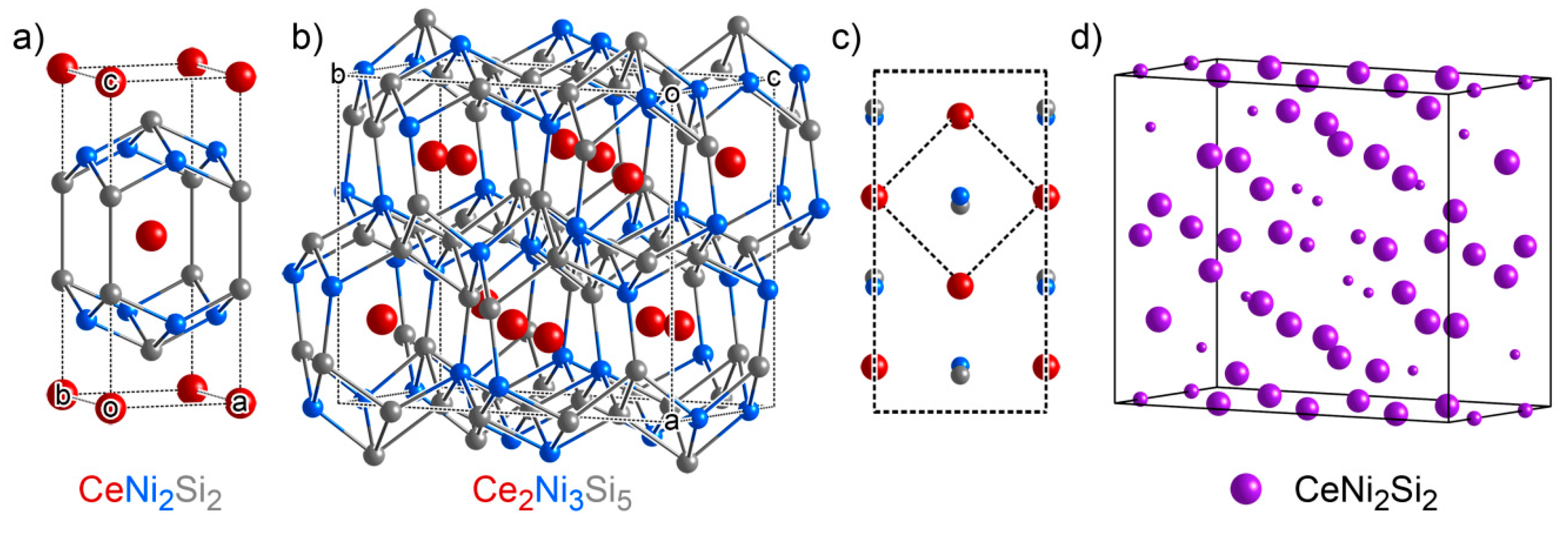
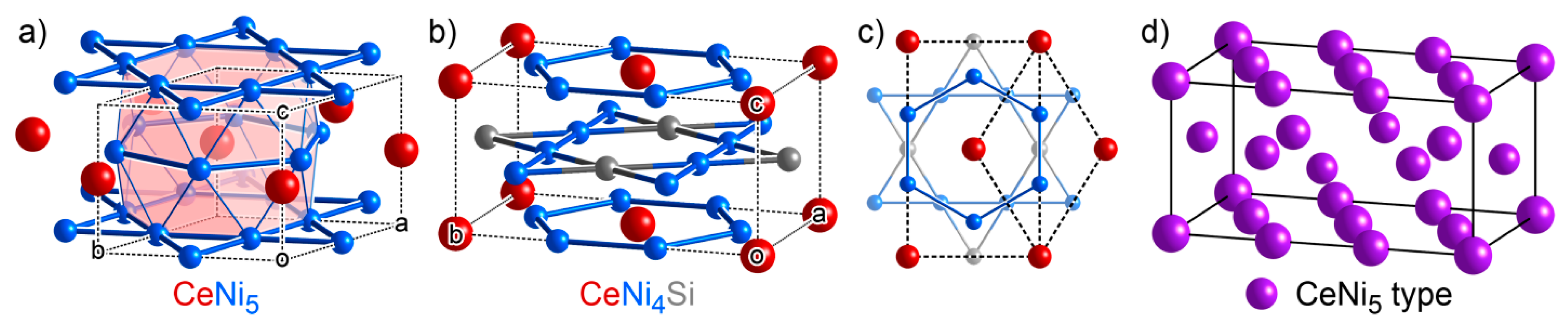
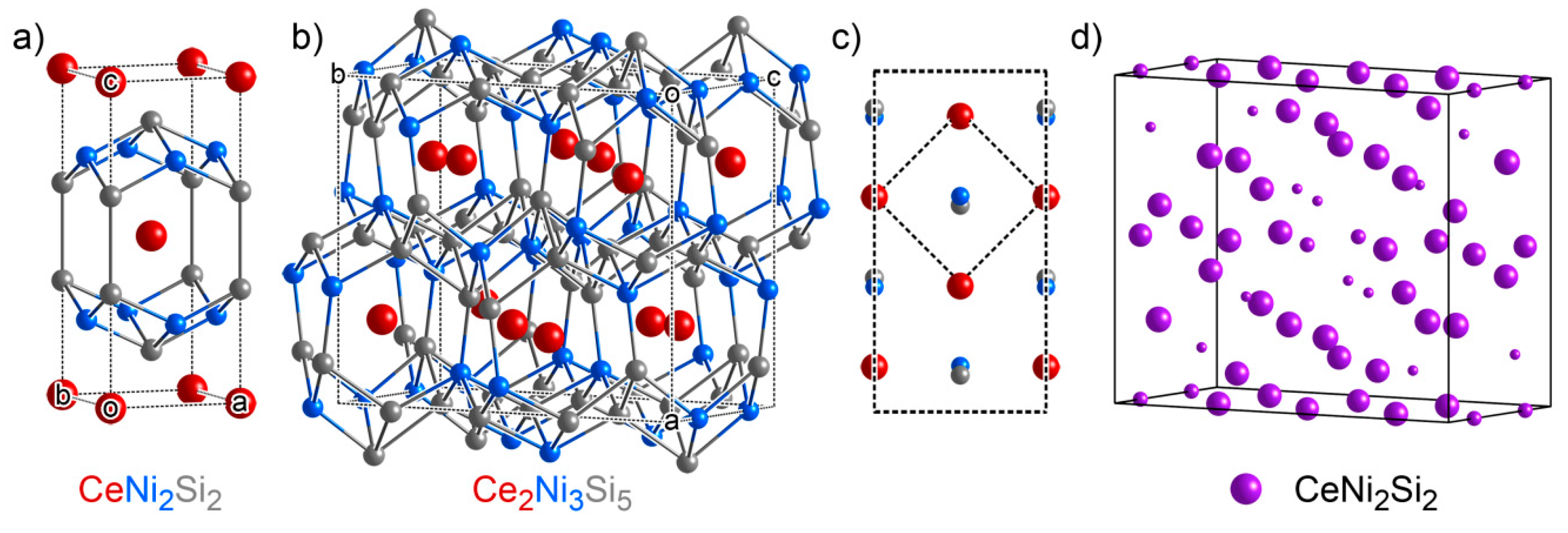
3.3. CeNi4Si: An Ordered Ternary Variant of a Binary Structure
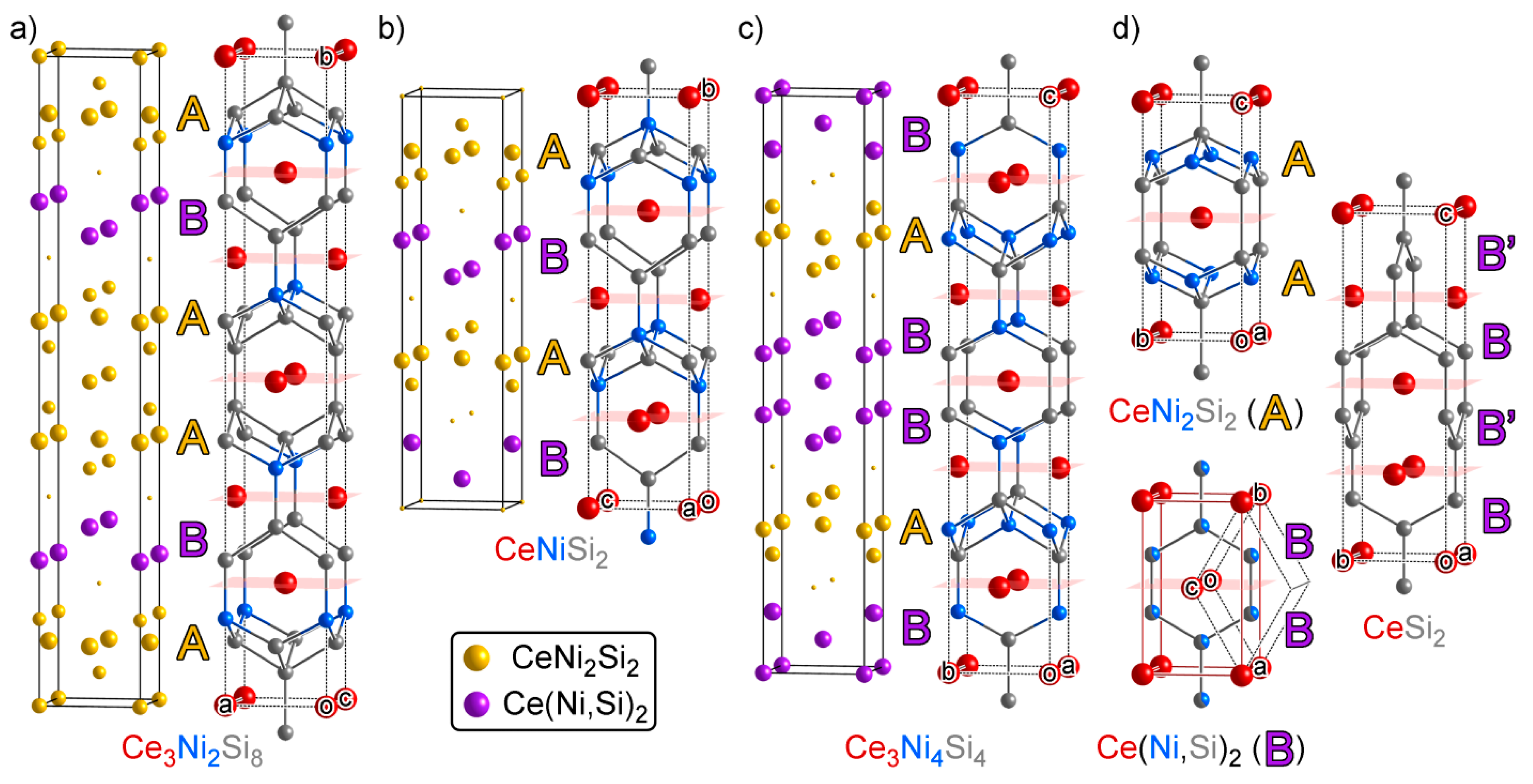
3.4. Ce2Ni3Si5: Another Ternary Variant Structure
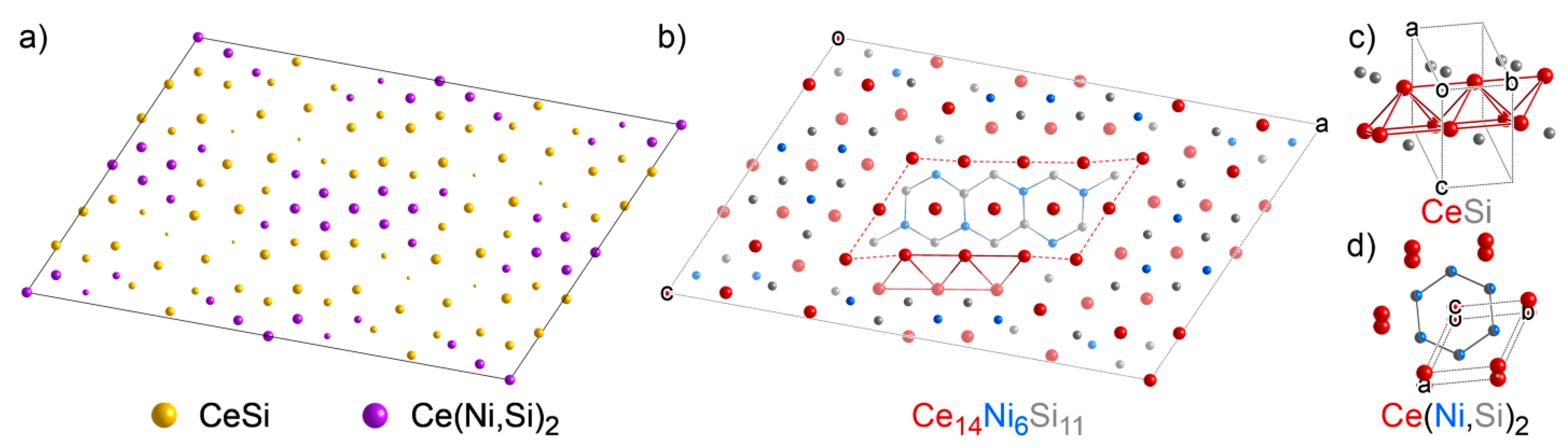
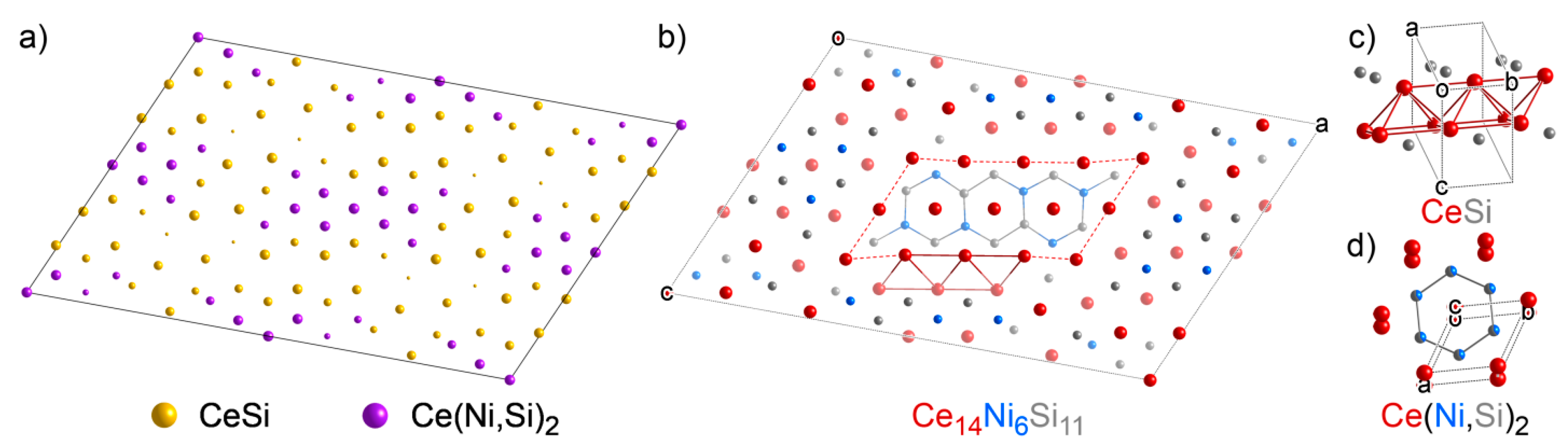
3.5. Ce14Ni6Si11: An Intergrowth of Reference Structures
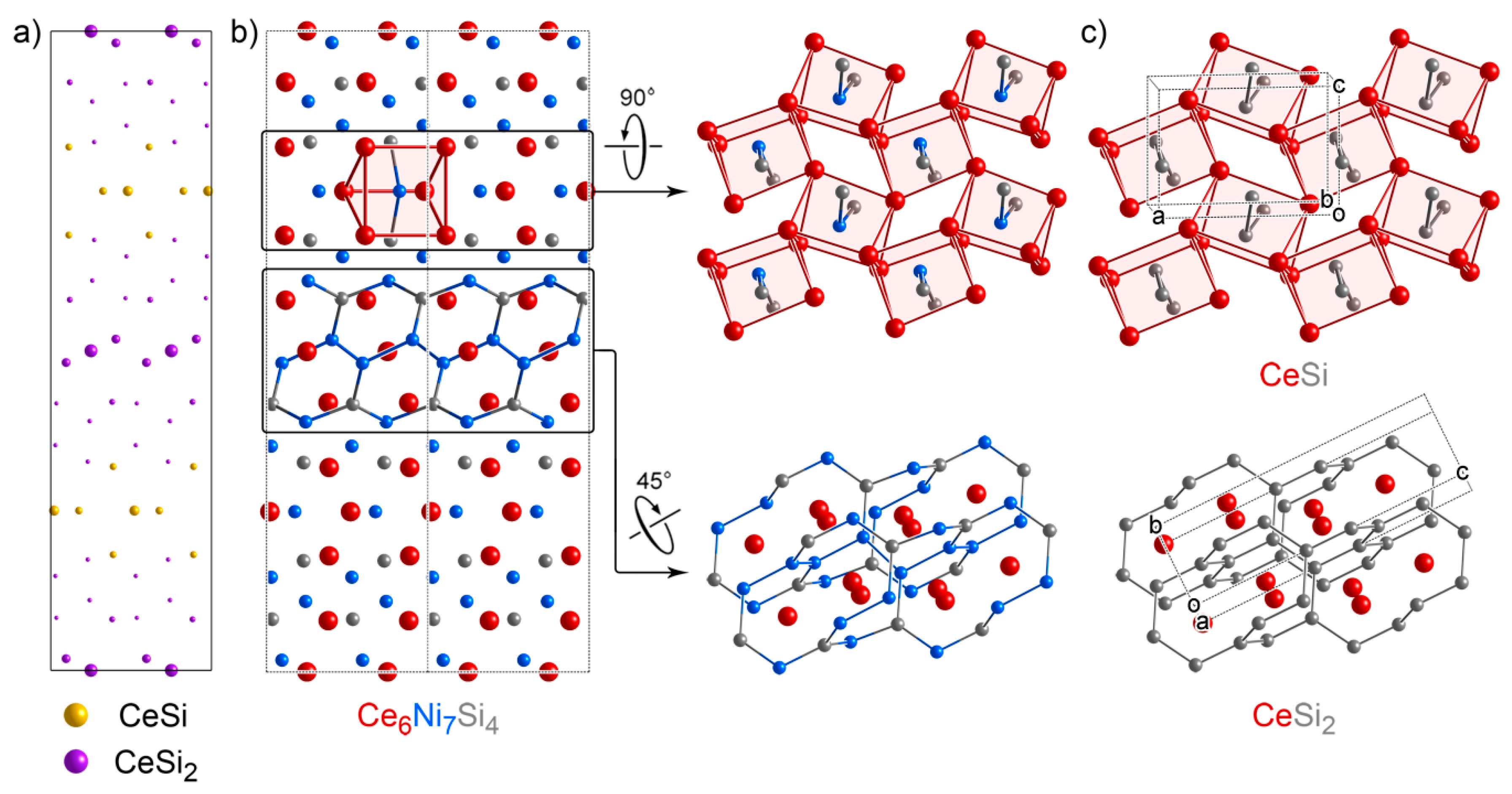
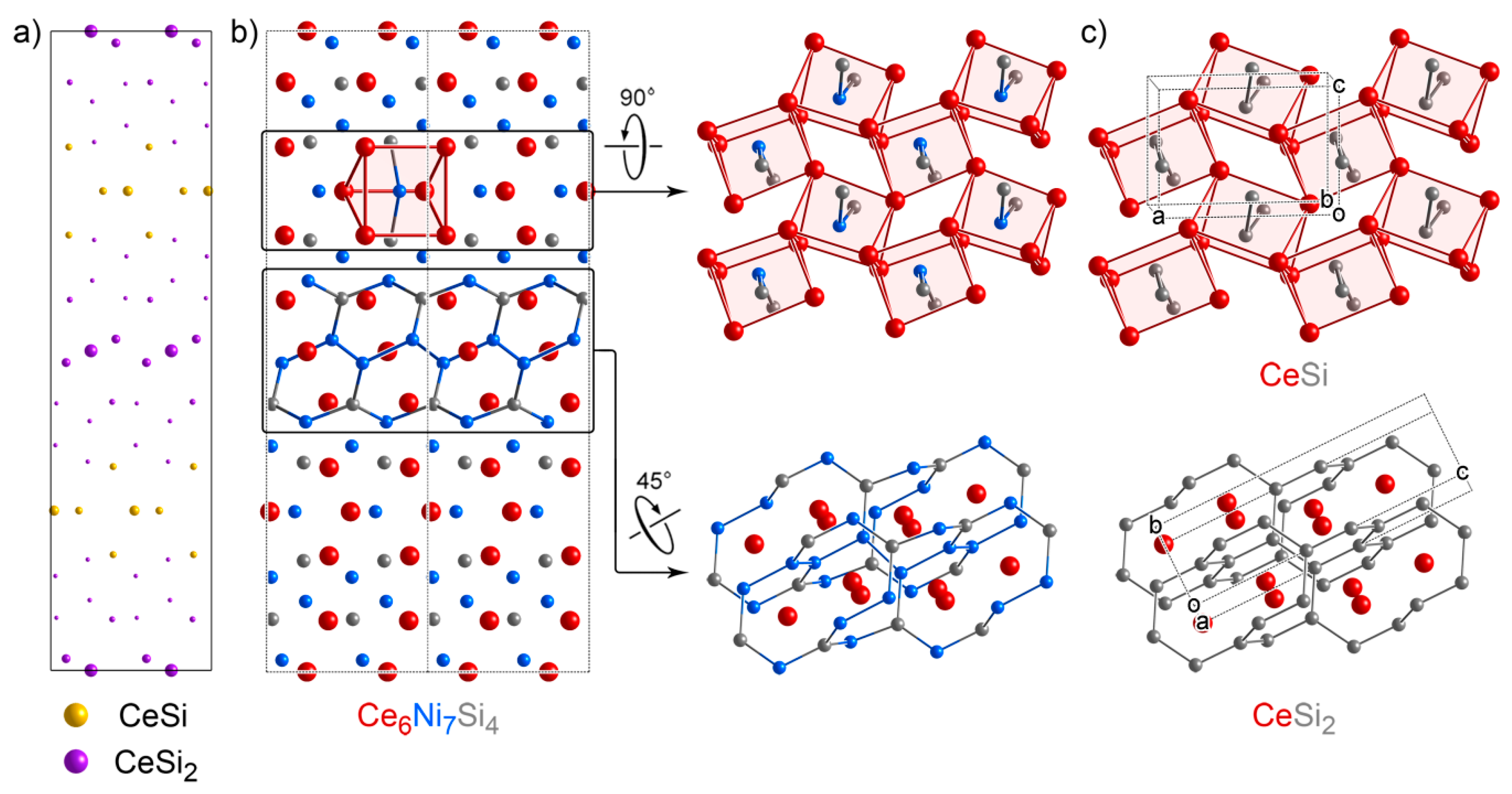
3.6. Ce6Ni7Si4: An Intergrowth with Substantial Relaxation at the Interface
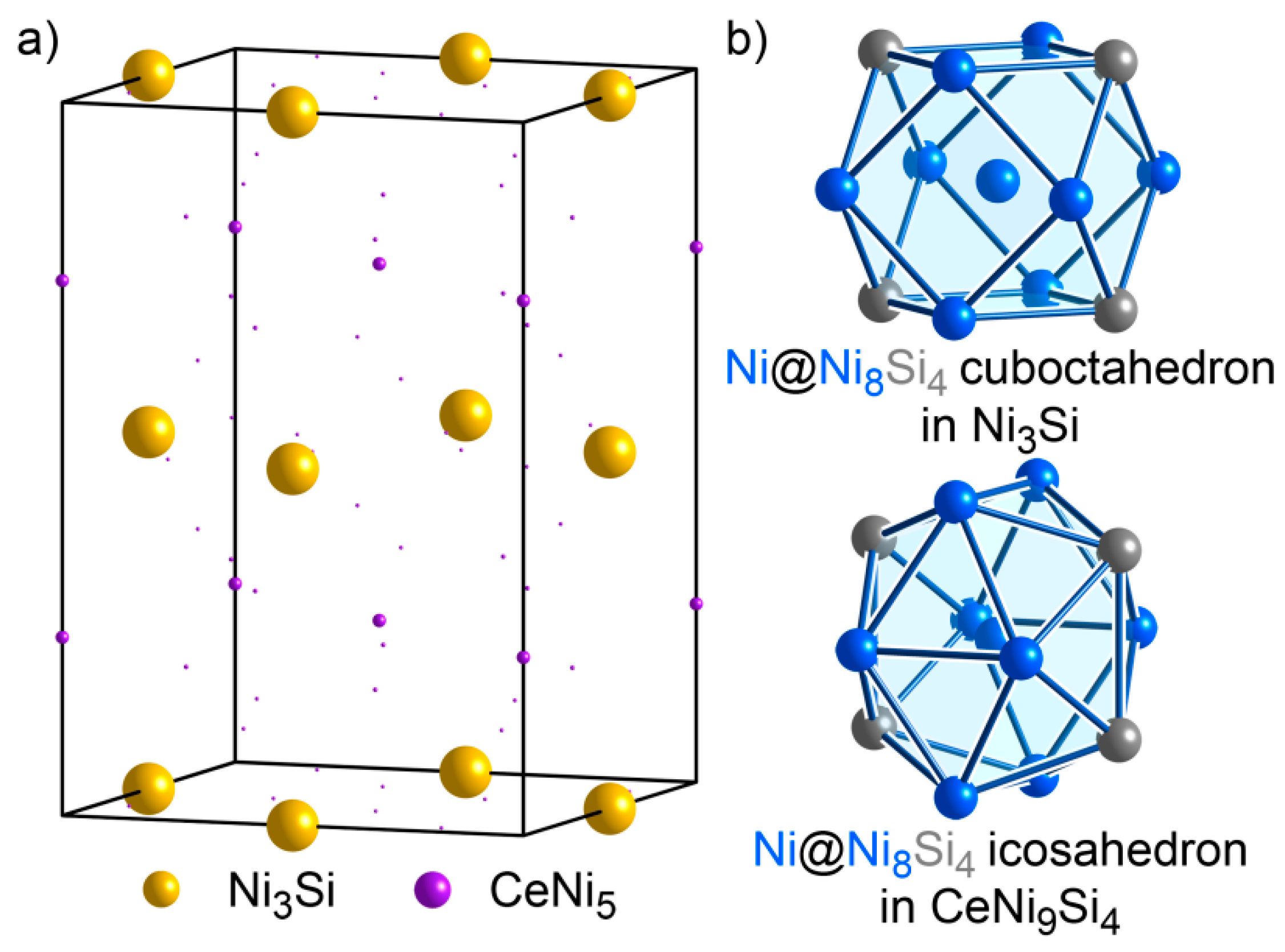
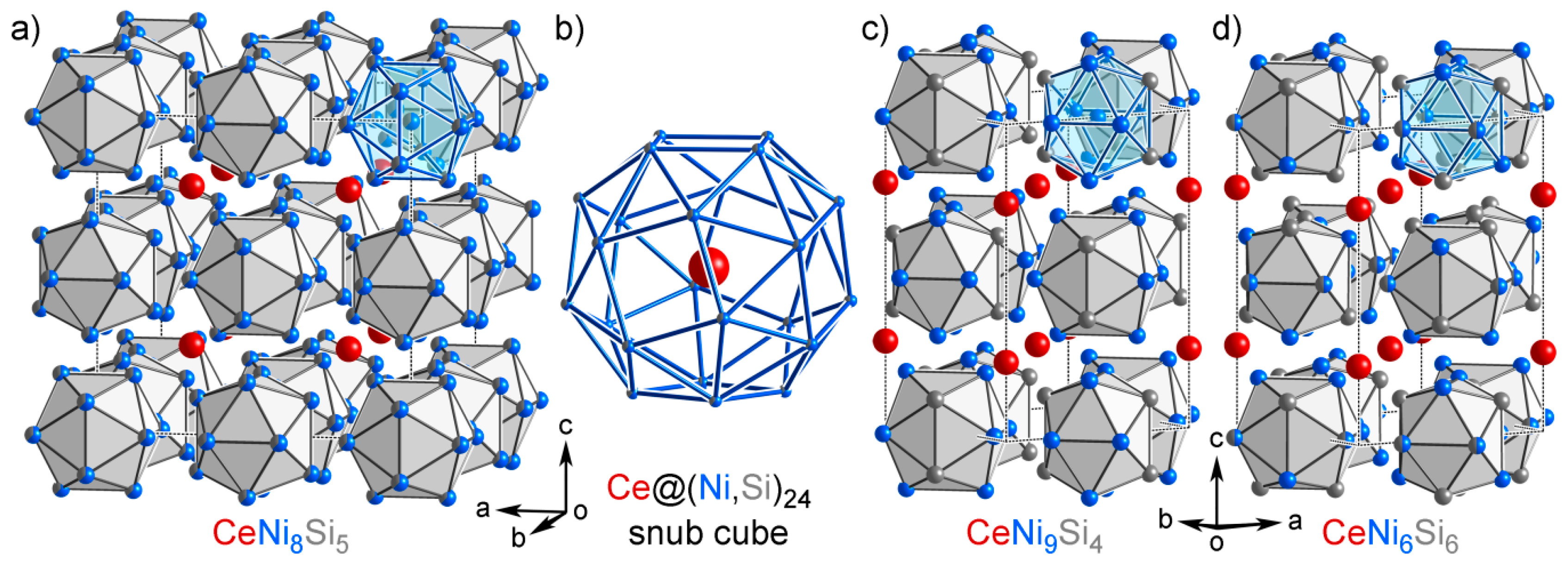
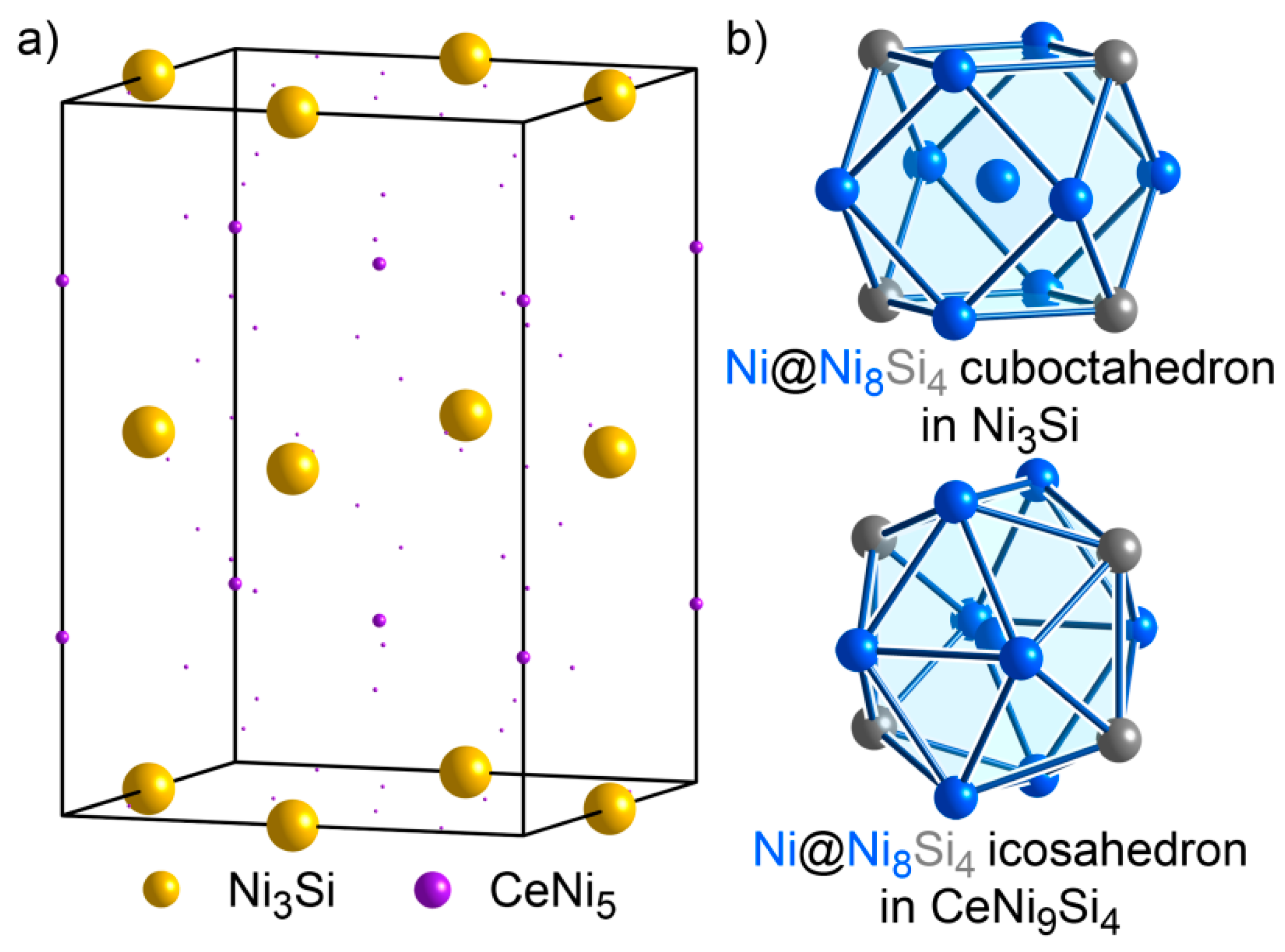
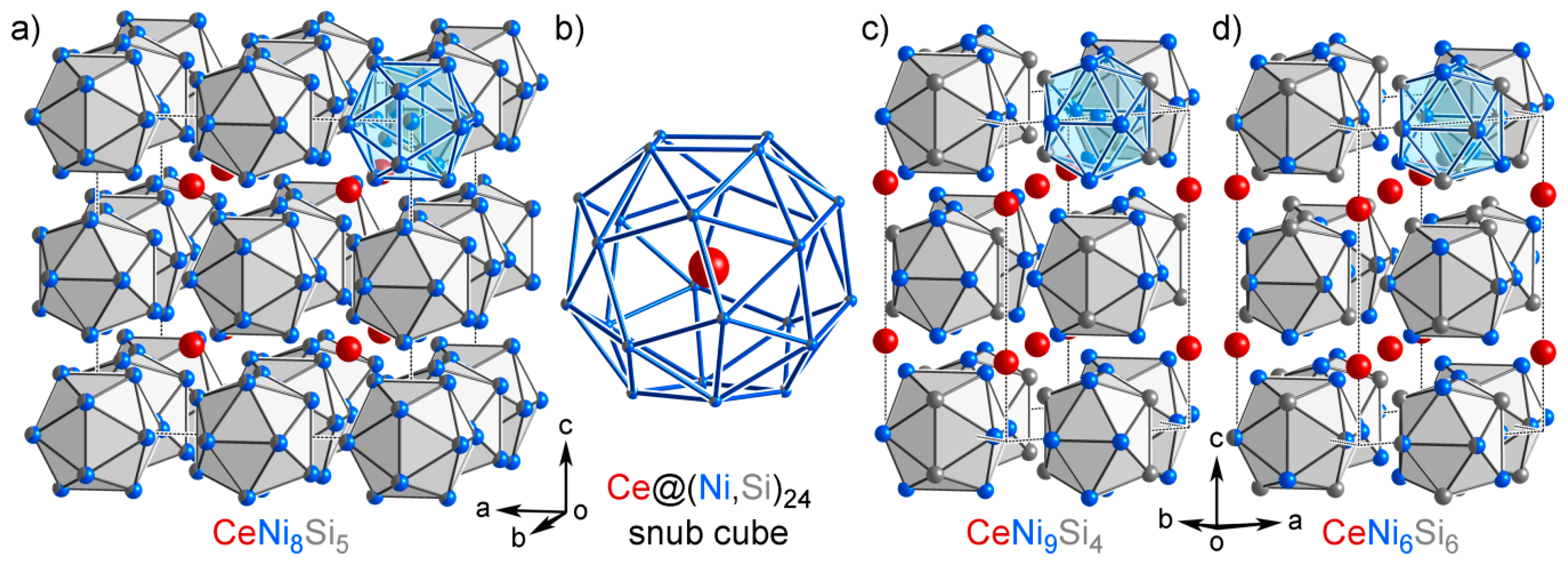
3.7. CeNi9Si4: An Isolated Structure
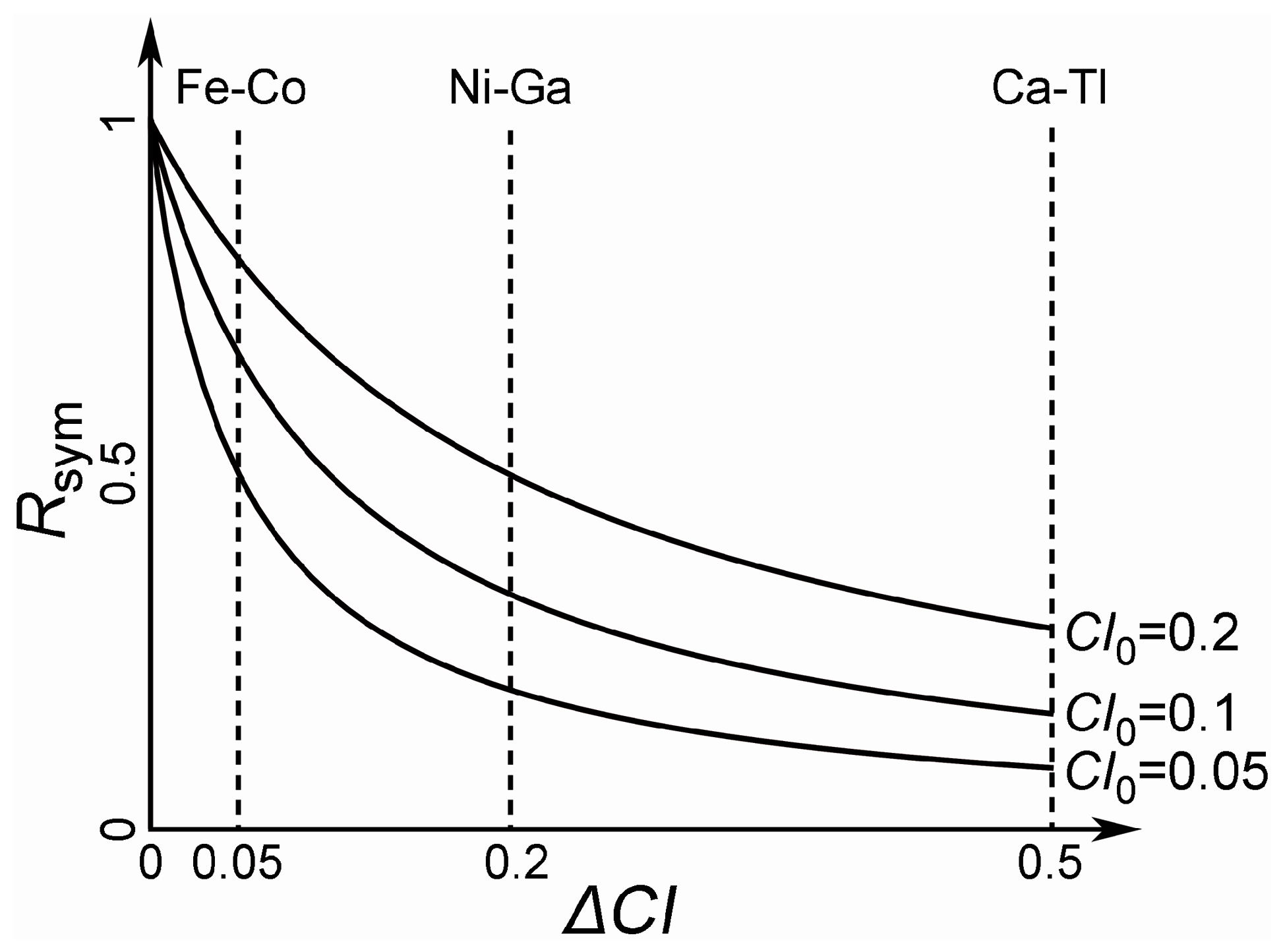
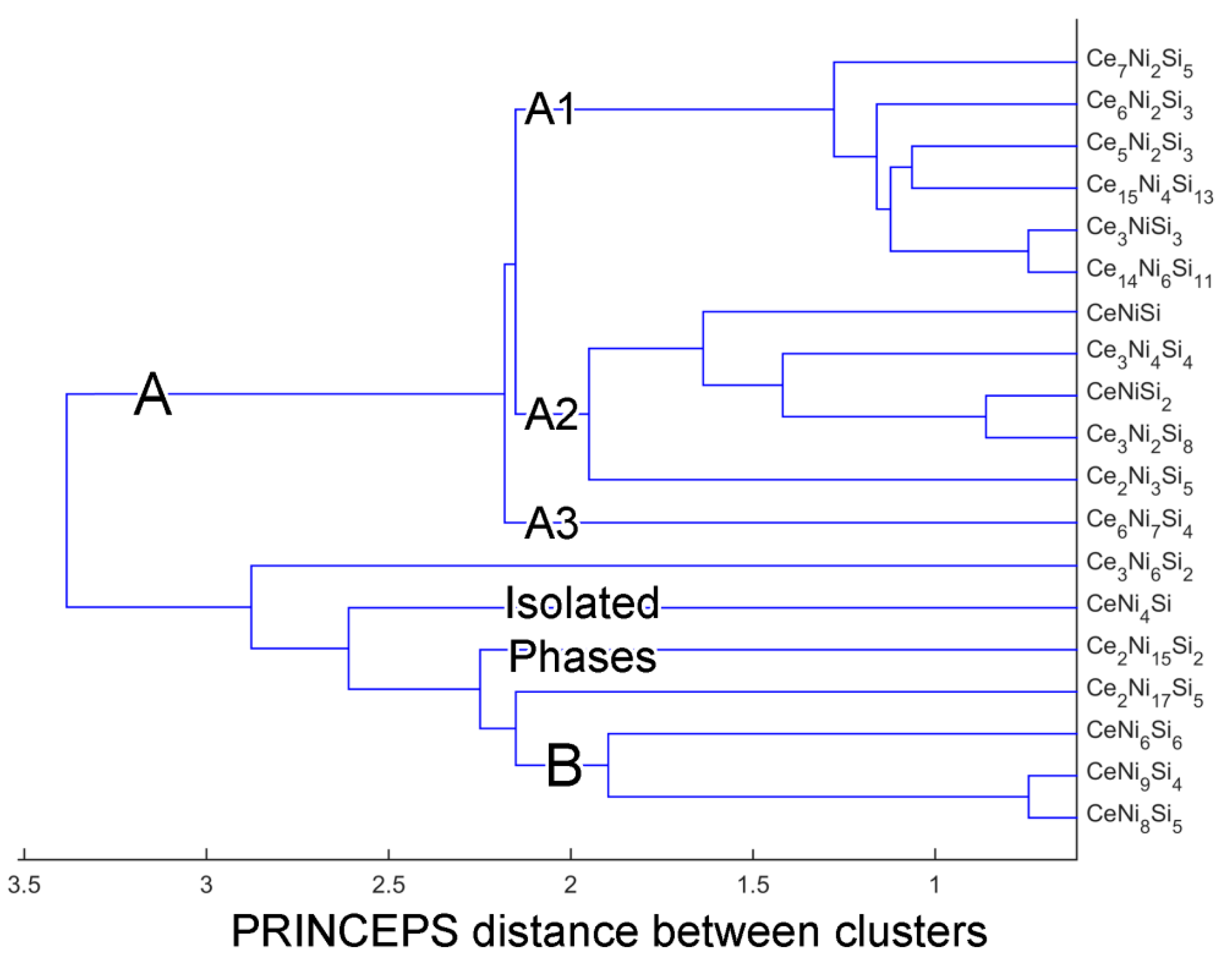
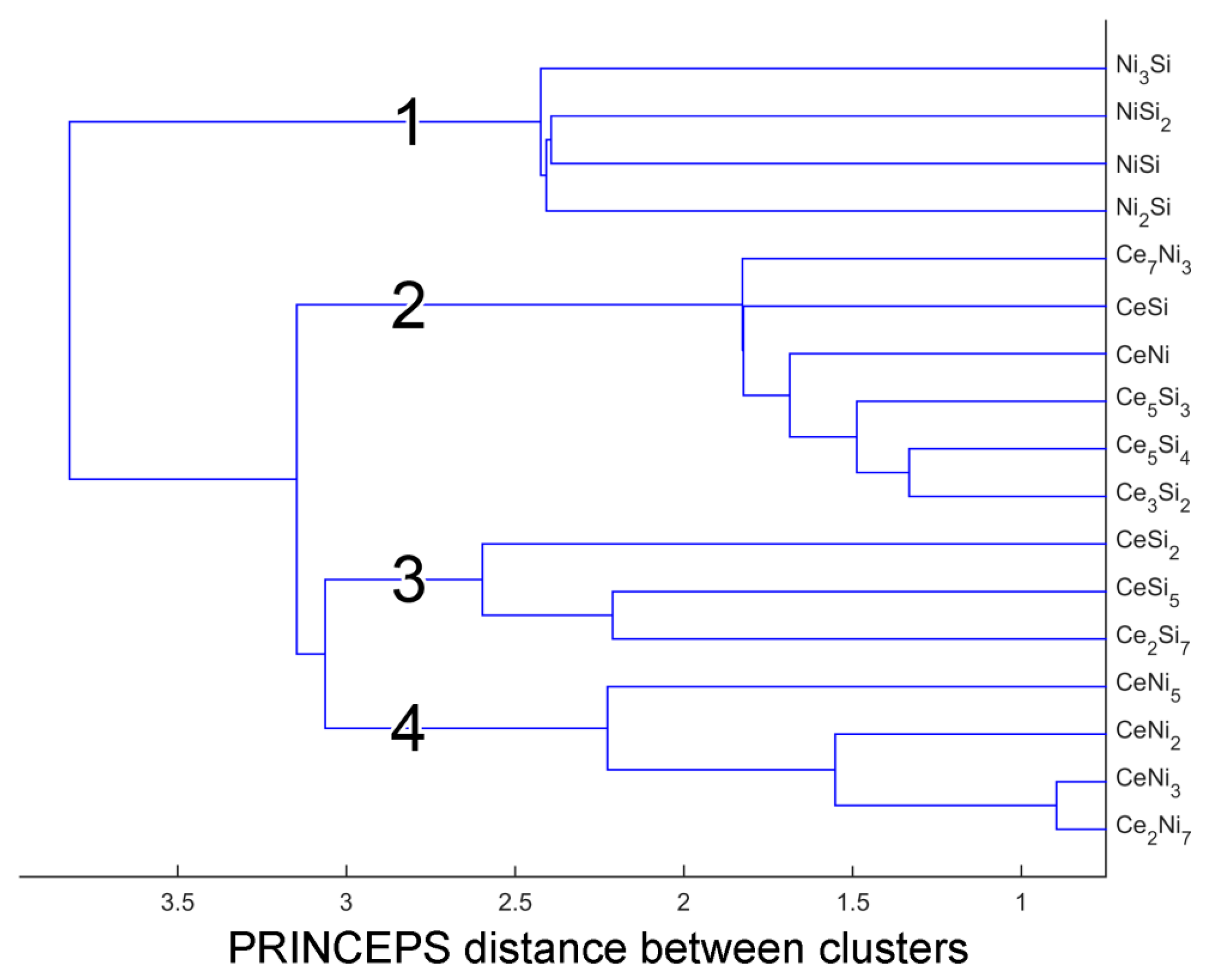
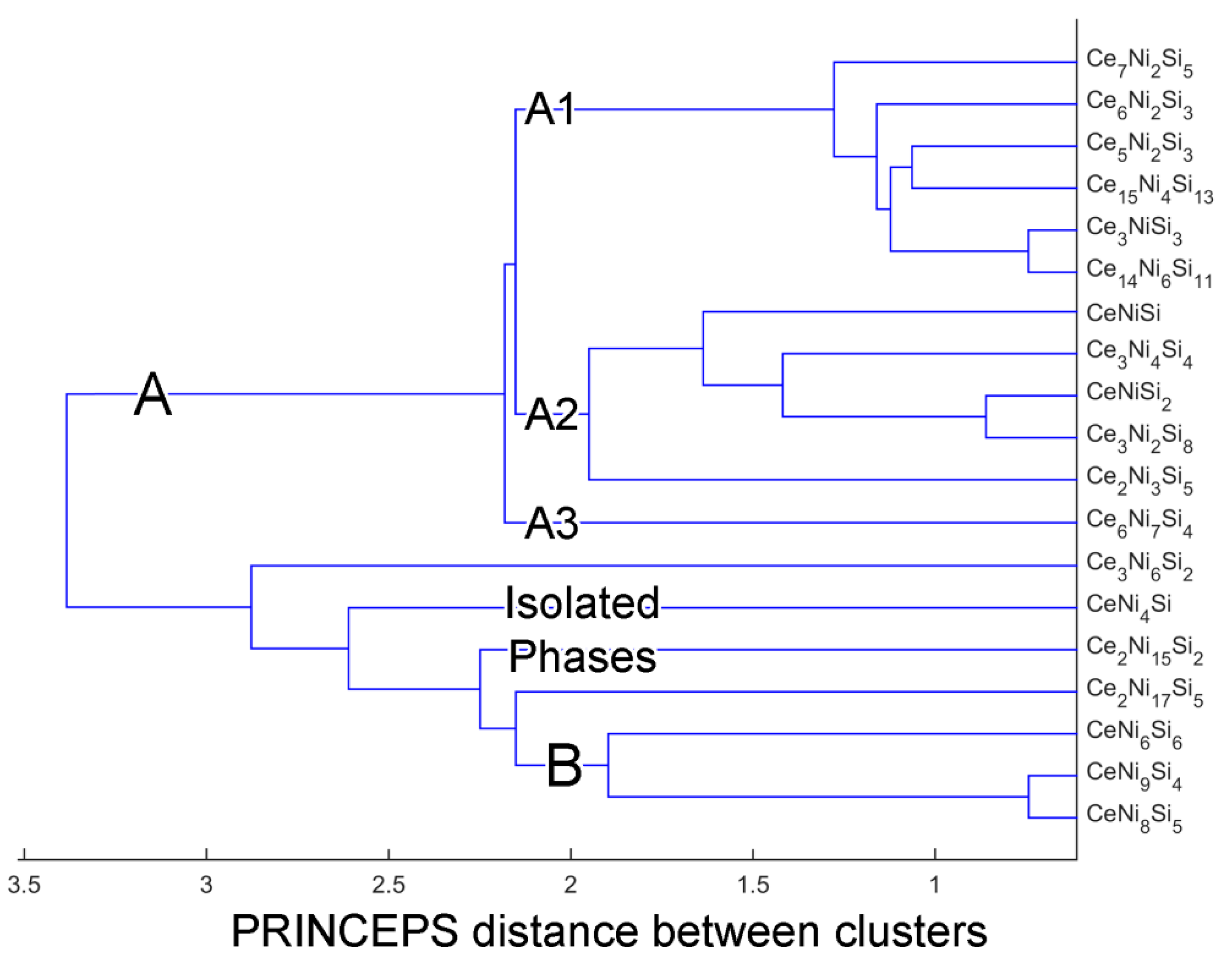
4. Clustering of Ternary Phases and Group-Wise Structural Analysis
4.1. Clustering of Ternary Phases in the Ce-Ni-Si System
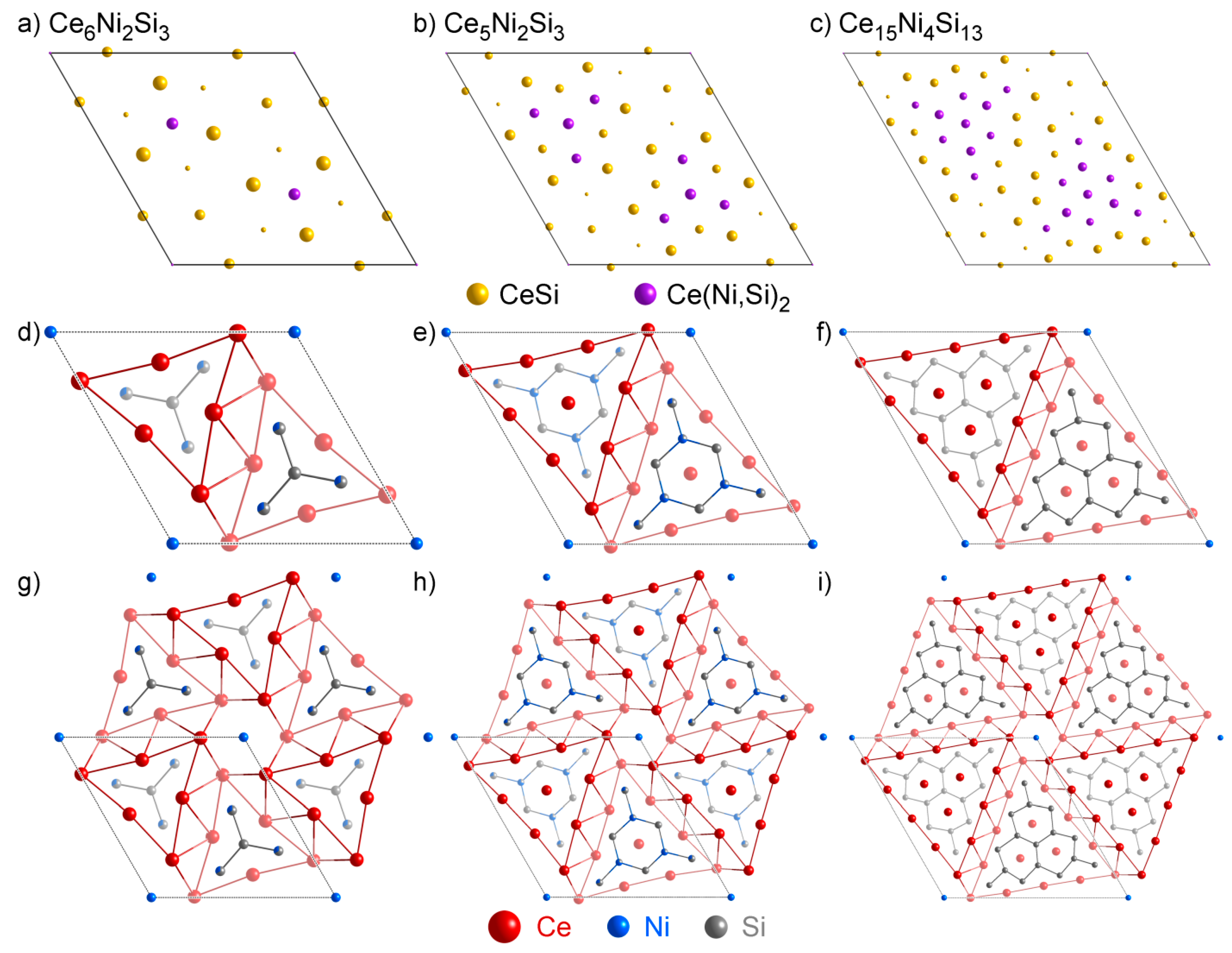
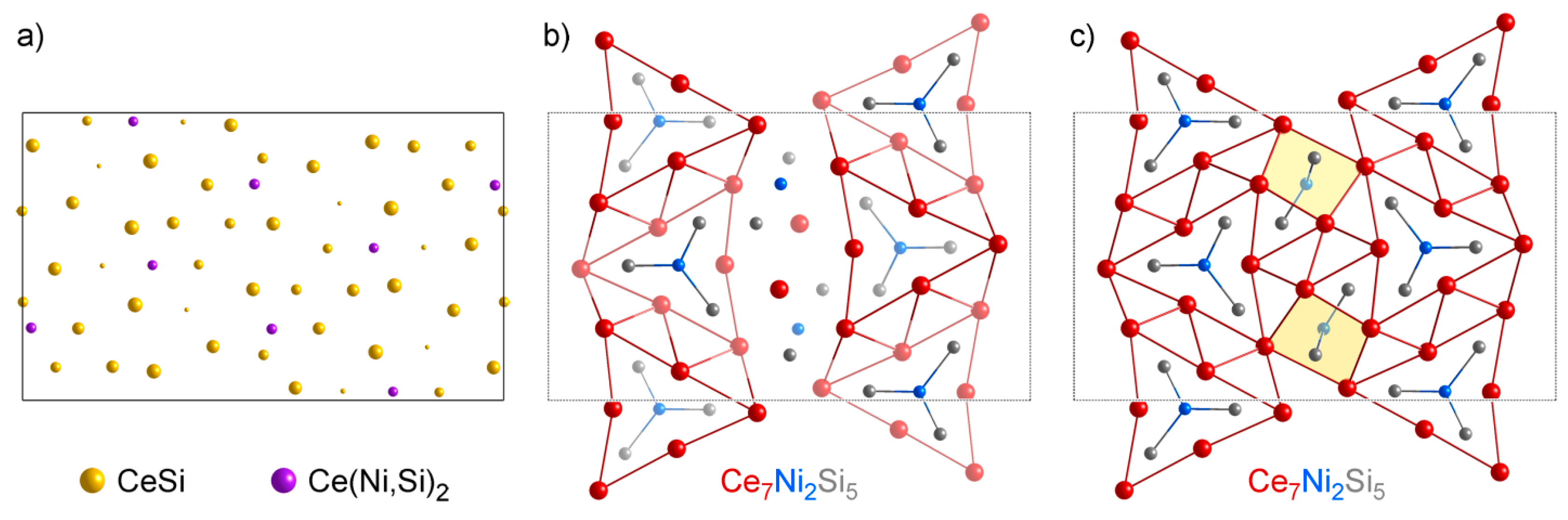
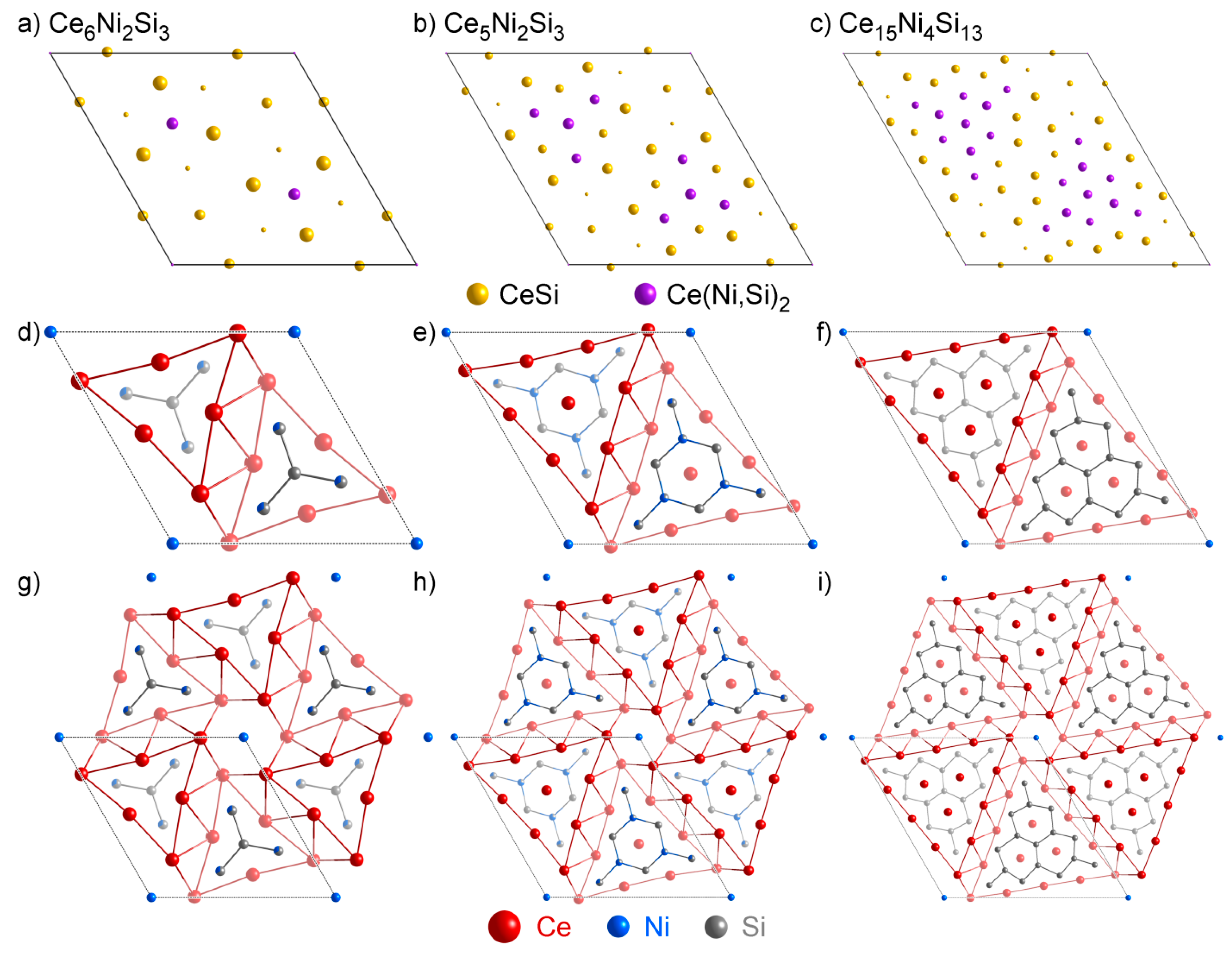
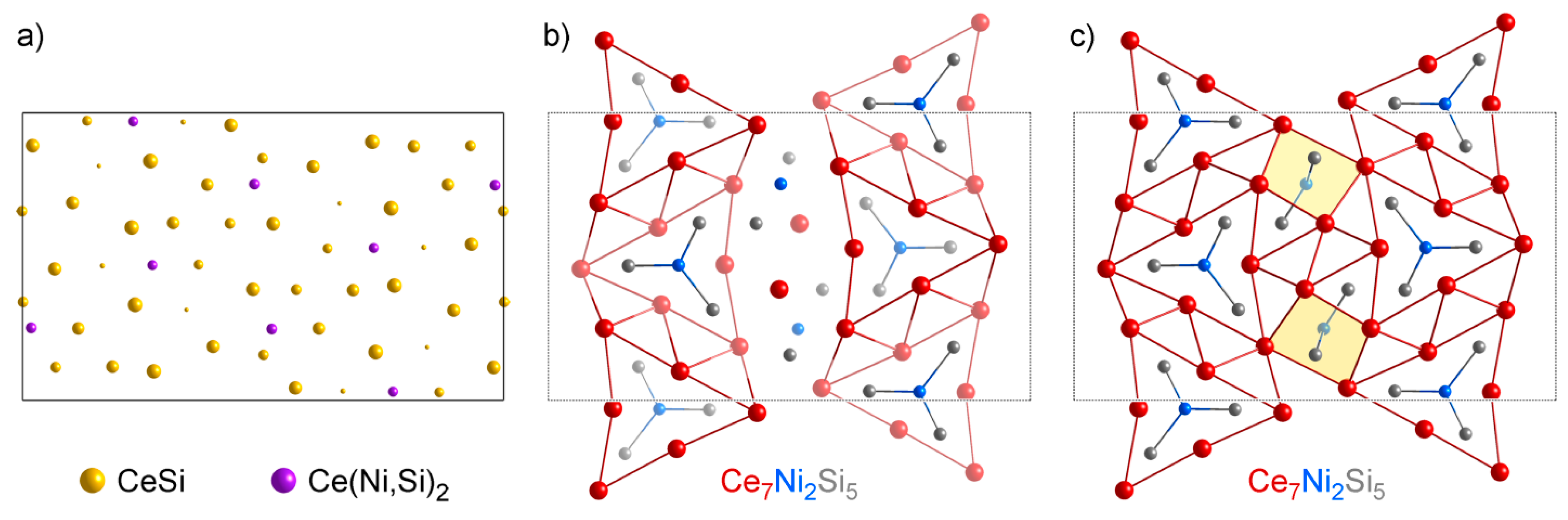
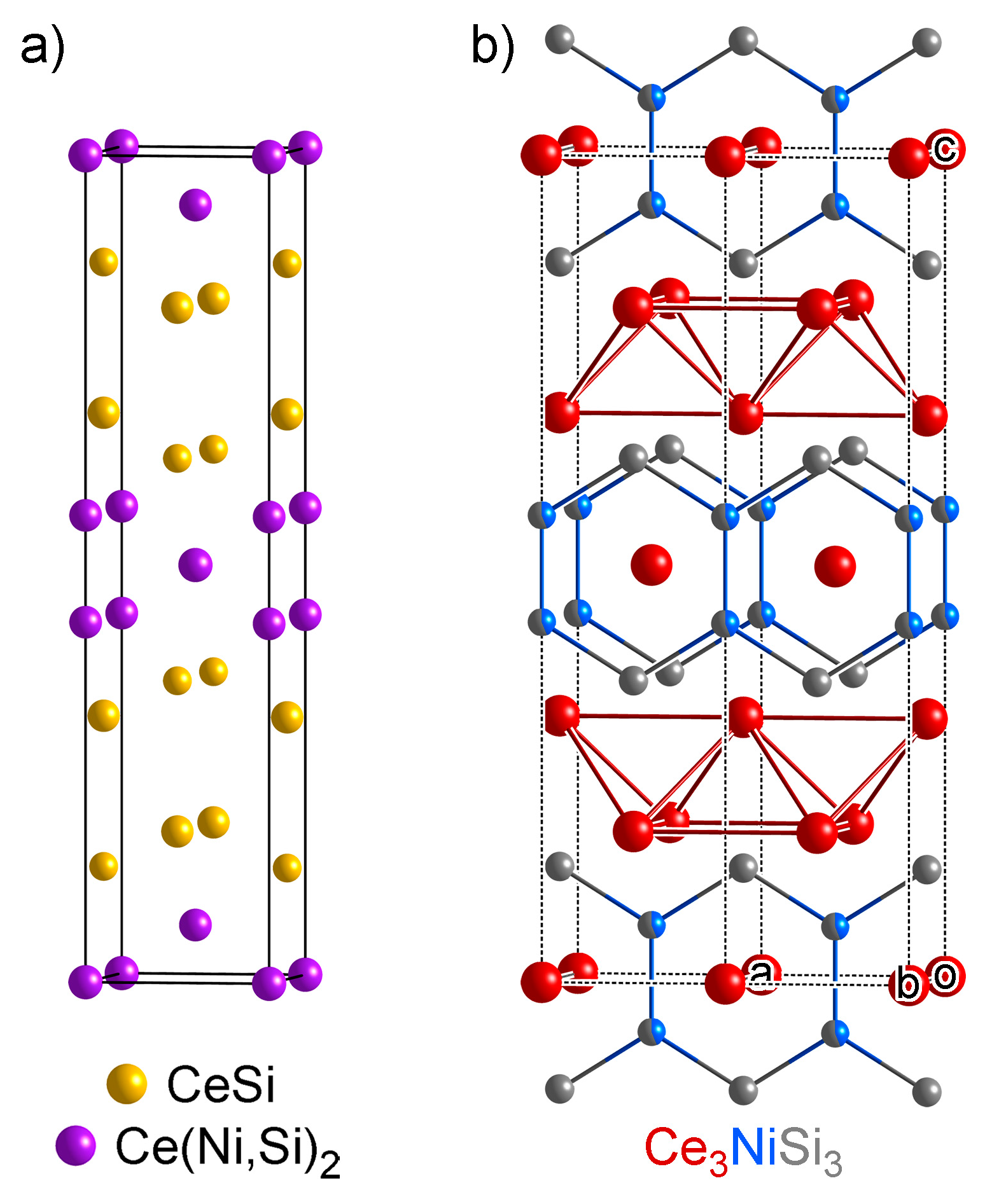
4.2. Analysis of Group A1
4.3. Analysis on Group A2
4.4. Group A3: The Ce6Ni7Si4 Structure
4.5. Analysis on Group B
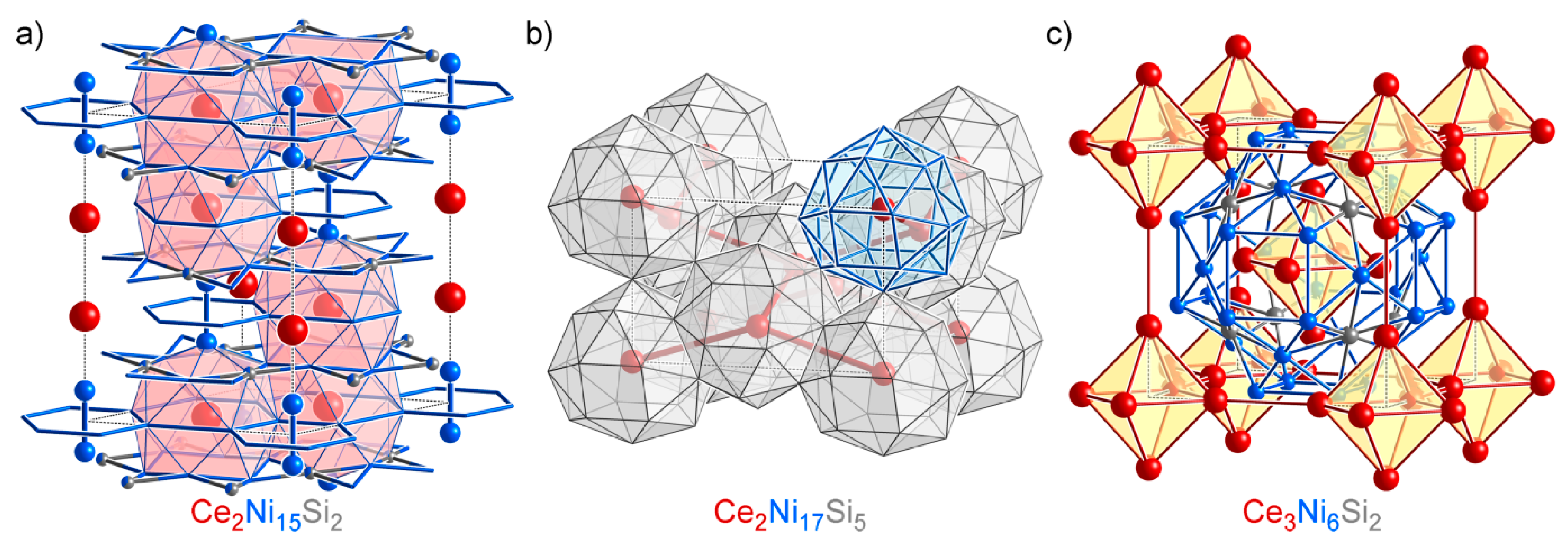
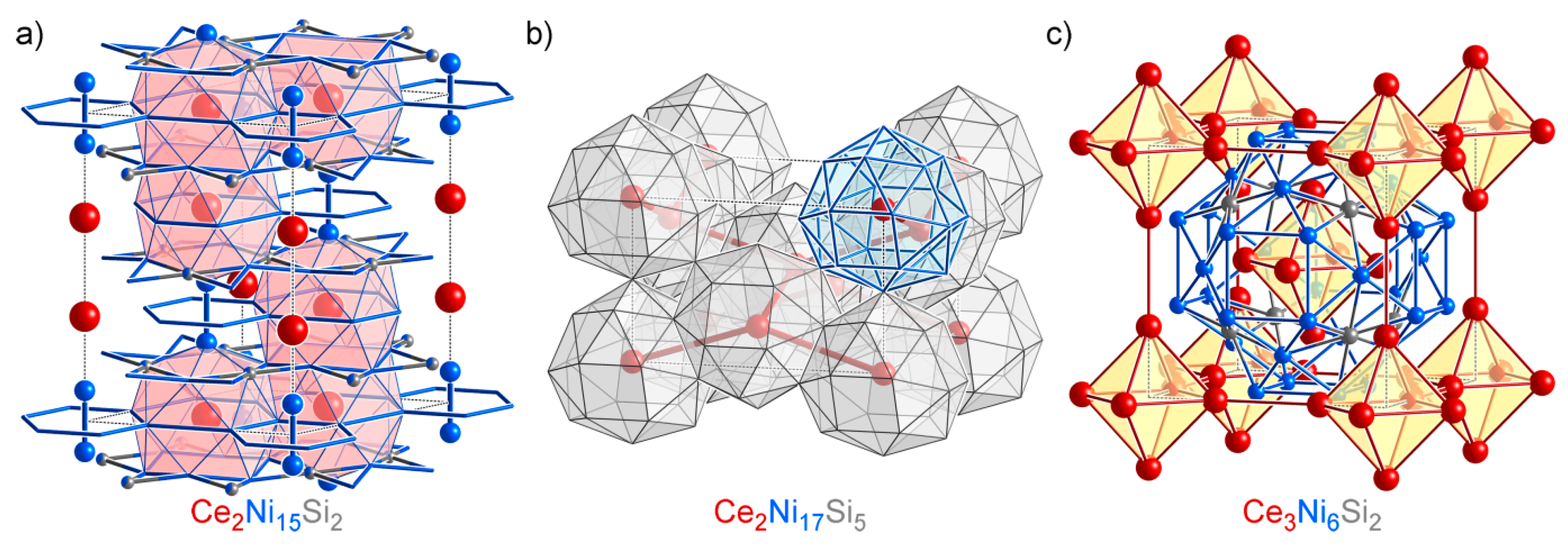
4.6. Overview of the Isolated Phases
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Andersson, S. On the Description of Complex Inorganic Crystal Structures. Angew. Chem. Int. Ed. 1983, 2, 69–81. [Google Scholar] [CrossRef]
- Andersson, S. The Description of Complex Alloy Structures. In Structure and Bonding in Crystals; Elsevier: Amsterdam, The Netherlands, 1981; pp. 233–258. [Google Scholar]
- Hyde, B.G.; Andersson, S. Inorganic Crystal Structures; Wiley: New York, NY, USA, 1989. [Google Scholar]
- Goodenough, J.B. Metallic Oxides. Prog. Solid State Chem. 1971, 5, 145–399. [Google Scholar] [CrossRef]
- O’Keeffe, M. Nets, Tiles, and Metal-Organic Frameworks. APL Mater. 2014, 12, 124106. [Google Scholar] [CrossRef]
- Corbett, J.D. Exploratory Synthesis: The Fascinating and Diverse Chemistry of Polar Intermetallic Phases. Inorg. Chem. 2009, 1, 13–28. [Google Scholar]
- Pearson, W.B. The Crystal Chemistry and Physics of Metals and Alloys; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Ferro, R.; Saccone, A. Structure of Intermetallic Compounds and Phases. Mater. Sci. Technol. 1996, 1, 123–215. [Google Scholar]
- Fredrickson, D.C.; Lee, S.; Hoffmann, R. Interpenetrating Polar and Nonpolar Sublattices in Intermetallics: The NaCd2 Structure. Angew. Chem. Int. Ed. 2007, 12, 1958–1976. [Google Scholar] [CrossRef] [PubMed]
- Gladyshevskii, E.I.; Krypiakevich, P.I. Homologous series including the new structure types of ternary silicides. Acta Crystallogr. Sect. A Found. Crystallogr. 1972, 28, S97. [Google Scholar]
- Andersson, S. Structures Related to the β-tungsten or Cr3Si Structure type. J. Solid State Chem. 1978, 1, 191–204. [Google Scholar] [CrossRef]
- Andersson, S. An Alternative Description of the Structure of Cu4Cd3. Acta Crystallogr. B 1980, 11, 2513–2516. [Google Scholar] [CrossRef]
- Parthé, E.; Chabot, B.A.; Cenzual, K. Complex structures of intermetallic compounds interpreted as intergrowth of segments of simple structures. Chimia 1985, 39, 164–174. [Google Scholar]
- Grin, Y. The intergrowth concept as a useful tool to interpret and understand complicated intermetallic structures. In Modern Perspectives in Inorganic Crystal Chemistry; Parthé, E., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992; pp. 77–96. [Google Scholar]
- Blatov, V. Nanocluster Analysis of Intermetallic Structures with the Program Package TOPOS. Struct. Chem. 2012, 4, 955–963. [Google Scholar] [CrossRef]
- Blatov, V.A.; Ilyushin, G.D.; Proserpio, D.M. Nanocluster Model of Intermetallic Compounds with Giant Unit Cells: β,β′-Mg2Al3 Polymorphs. Inorg. Chem. 2010, 4, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Ilyushin, G.; Blatov, V. Structures of the ZrZn22 Family: Suprapolyhedral Nanoclusters, Methods of Self-Assembly and Superstructural Ordering. Acta Crystallogr. B—Struct. Sci. 2009, 3, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, V.Y.; Blatov, V.; Ilyushin, G. Intermetallic Compounds of the NaCd2 Family Perceived as Assemblies of Nanoclusters. Struct. Chem. 2009, 6, 975–982. [Google Scholar] [CrossRef]
- Jana, P.P.; Pankova, A.A.; Lidin, S. Au10Mo4Zn89: A Fully Ordered Complex Intermetallic Compound Analyzed by TOPOS. Inorg. Chem. 2013, 19, 11110–11117. [Google Scholar] [CrossRef] [PubMed]
- Fässler, T.F.; Hoffmann, S.D. Endohedral Zintl Ions: Intermetalloid Clusters. Angew. Chem. Int. Ed. 2004, 46, 6242–6247. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Hoffman, S.D.; Fässler, T.F. Na29Zn24Sn32: A Zintl Phase Containing a Novel Type of Sn14 Enneahedra and Heteroatomic Zn8Sn4 Icosahedra. Angew. Chem. Int. Ed. 2007, 17, 3144–3148. [Google Scholar] [CrossRef] [PubMed]
- Kovnir, K.; Shatruk, M. Magnetism in Giant Unit Cells—Crystal Structure and Magnetic Properties of R117Co52+δSn112+γ (R = Sm, Tb, Dy). Eur. J. Inorg. Chem. 2011, 26, 3955–3962. [Google Scholar] [CrossRef]
- Steurer, W.; Deloudi, S. Fascinating Quasicrystals. Acta Crystallogr. A 2007, 1, 1–11. [Google Scholar]
- Steurer, W.; Feuerbacher, M.; Thomas, C.; Makongo, J.P.A.; Hoffmann, S.; Carrillo-Cabrera, W.; Cardoso, R.; Grin, Y.; Kreiner, G.; Joubert, J.-M.; et al. The Samson Phase, β-Mg2Al3, Revisited. Z. Kristallogr. 2007, 6, 259–288. [Google Scholar] [CrossRef]
- Lin, Q.; Smetana, V.; Miller, G.J.; Corbett, J.D. Conventional and Stuffed Bergman-Type Phases in the Na-Au-T (T = Ga, Ge, Sn) Systems: Syntheses, Structures, Coloring of Cluster Centers, and Fermi Sphere-Brillouin Zone Interactions. Inorg. Chem. 2012, 16, 8882–8889. [Google Scholar] [CrossRef] [PubMed]
- Dshemuchadse, J.; Steurer, W. More of the “Fullercages”. Z. Anorg. Allg. Chem. 2014, 5, 693–700. [Google Scholar] [CrossRef]
- Dshemuchadse, J.; Jung, D.Y.; Steurer, W. Structural building principles of complex face-centered cubic intermetallics. Acta Crystallogr. B 2011, 4, 269–292. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, M. Self-Dual Plane Nets in Crystal Chemistry. Aust. J. Chem. 1992, 9, 1489–1498. [Google Scholar] [CrossRef]
- Andersson, S. An Alternative Description of the Structures of Rh7Mg44 and Mg6Pd. Acta Crystallogr. A 1978, 6, 833–835. [Google Scholar] [CrossRef]
- Samson, S. The Crystal Structure of the Intermetallic Compound Cu4Cd3. Acta Crystallogr. 1967, 4, 586–600. [Google Scholar] [CrossRef]
- Samson, S.; Hansen, D. Complex Cubic A6B Compounds. I. The Crystal Structure of Na6Tl. Acta Crystallogr. B 1972, 3, 930–935. [Google Scholar] [CrossRef]
- Frank, F.T.; Kasper, J. Complex Alloy Structures Regarded as Sphere Packings. I. Definitions and Basic Principles. Acta Crystallogr. 1958, 3, 184–190. [Google Scholar] [CrossRef]
- Frank, F.T.; Kasper, J. Complex Alloy Structures Regarded as Sphere Packings. II. Analysis and Classification of Representative Structures. Acta Crystallogr. 1959, 7, 483–499. [Google Scholar] [CrossRef]
- Elser, V.; Henley, C.L. Crystal and Quasicrystal Structures in Al-Mn-Si Alloys. Phys. Rev. Lett. 1985, 26, 2883–2886. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.F.; Lee, S.; Johnson, J.; Nebgen, B.; Sha, F.; Xu, J. The Mystery of Perpendicular Fivefold Axes and the Fourth Dimension in Intermetallic Structures. Chem. Eur. J. 2008, 13, 3908–3930. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Henderson, R.; Kaminsky, C.; Nelson, Z.; Nguyen, J.; Settje, N.F.; Schmidt, J.T.; Feng, J. Pseudo-Fivefold Diffraction Symmetries in Tetrahedral Packing. Chem. Eur. J. 2013, 31, 10244–10270. [Google Scholar] [CrossRef] [PubMed]
- Elenius, M.; Zetterling, F.H.; Dzugutov, M.; Fredrickson, D.C.; Lidin, S. Structural Model for Octagonal Quasicrystals Derived from Octagonal Symmetry Elements Arising in β-Mn Crystallization of a Simple Monatomic Liquid. Phys. Rev. B 2009, 14. [Google Scholar] [CrossRef]
- Andersson, S.; Hyde, S.; Larsson, K.; Lidin, S. Minimal Surfaces and Structures: From Inorganic and Metal Crystals to Cell Membranes and Biopolymers. Chem. Rev. 1988, 1, 221–242. [Google Scholar] [CrossRef]
- Hyde, S.; Andersson, S. A Systematic Net Description of Saddle Polyhedra and Periodic Minimal Surfaces. Z. Kristallogr. 1984, 1–4, 221–254. [Google Scholar] [CrossRef]
- Grin, Y.; Wedig, U.; von Schnering, H.G. Hyperbolic Lone Pair Structure in RhBi4. Angew. Chem. Int. Ed. 1995, 11, 1204–1206. [Google Scholar] [CrossRef]
- Grin, Y.; Wedig, U.; Wagner, F.; von Schnering, H.G.; Savin, A. The Analysis of “Empty Space” in the PdGa5 Structure. J. Alloys Compd. 1997, 1, 203–208. [Google Scholar] [CrossRef]
- Fredrickson, D.C. DFT-Chemical Pressure Analysis: Visualizing the Role of Atomic Size in Shaping the Structures of Inorganic Materials. J. Am. Chem. Soc. 2012, 13, 5991–5999. [Google Scholar] [CrossRef] [PubMed]
- Yannello, V.J.; Kilduff, B.J.; Fredrickson, D.C. Isolobal Analogies in Intermetallics: The Reversed Approximation MO Approach and Applications to CrGa4- and Ir3Ge7-Type Phases. Inorg. Chem. 2014, 5, 2730–2741. [Google Scholar] [CrossRef] [PubMed]
- Stacey, T.E.; Fredrickson, D.C. The μ3 Model of Acids and Bases: Extending the Lewis Theory to Intermetallics. Inorg. Chem. 2012, 7, 4250–4264. [Google Scholar] [CrossRef] [PubMed]
- Berns, V.M.; Fredrickson, D.C. Structural Plasticity: How Intermetallics Deform Themselves in Response to Chemical Pressure, and the Complex Structures That Result. Inorg. Chem. 2014, 19, 10762–10771. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, R.T.; Fredrickson, D.C. The Modulated Structure of Co3Al4Si2: Incommensurability and Co-Co Interactions in Search of Filled Octadecets. Inorg. Chem. 2013, 6, 3178–3189. [Google Scholar] [CrossRef] [PubMed]
- Stacey, T.E.; Fredrickson, D.C. Structural Acid–Base Chemistry in the Metallic State: How μ3-Neutralization Drives Interfaces and Helices in Ti21Mn25. Inorg. Chem. 2013, 15, 8349–8359. [Google Scholar] [CrossRef] [PubMed]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 7, 3576–3586. [Google Scholar] [CrossRef]
- Blatov, V. Search for Isotypism in Crystal Structures by means of the Graph Theory. Acta Crystallogr. A 2000, 2, 178–188. [Google Scholar] [CrossRef]
- Blatov, V.A. Multipurpose Crystallochemical Analysis with the Program Package TOPOS. IUCr Crystallographic Computing Newsletter 2006, 7. [Google Scholar]
- Blatov, V.A. Topological Analysis of Ionic Packings in Crystal Structures of Inorganic Sulfides: The Method of Coordination Sequences. Z. Kristallogr. 2001, 216, 165–171. [Google Scholar] [CrossRef]
- Zakutkin, Y.A.; Blatov, V.A. A Comparative Analysis of Crystal Lattice Topology in Molybdates and Binary Compounds. J. Struct. Chem. 2001, 3, 436–445. [Google Scholar] [CrossRef]
- Kazhdan, M.; Funkhouser, T.; Rusinkiewicz, S. Rotation Invariant Spherical Harmonic Representation of 3D Shape Descriptors. In Proceedings of the Symposium on Geometry Processing, Aachen, Germany, 23–25 June 2003; pp. 156–164.
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-Orientational Order in Liquids and Glasses. Phys. Rev. B 1983, 2, 784–805. [Google Scholar] [CrossRef]
- In order to avoid ambiguity in degenerate cases (edge or vertex sharing, as can be found in some high-symmetry structures such as simple cubic or fcc), a threshold of 0.02 steradian is applied to the solid angle from the central atom to the shared face: Any coordinating atom with its corresponding solid angle higher than the threshold is considered part of the CE.
- Fischer, W.; Koch, E.; Hellner, E. Zur Berechnung von Wirkungsbereichen in Strukturen anorganischer verbindungen. Neues Jahrbuch Mineral. Monatsh. 1971, 5, 227–237. [Google Scholar]
- Pettifor, D.G. Bonding and Structure of Molecules and Solids; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
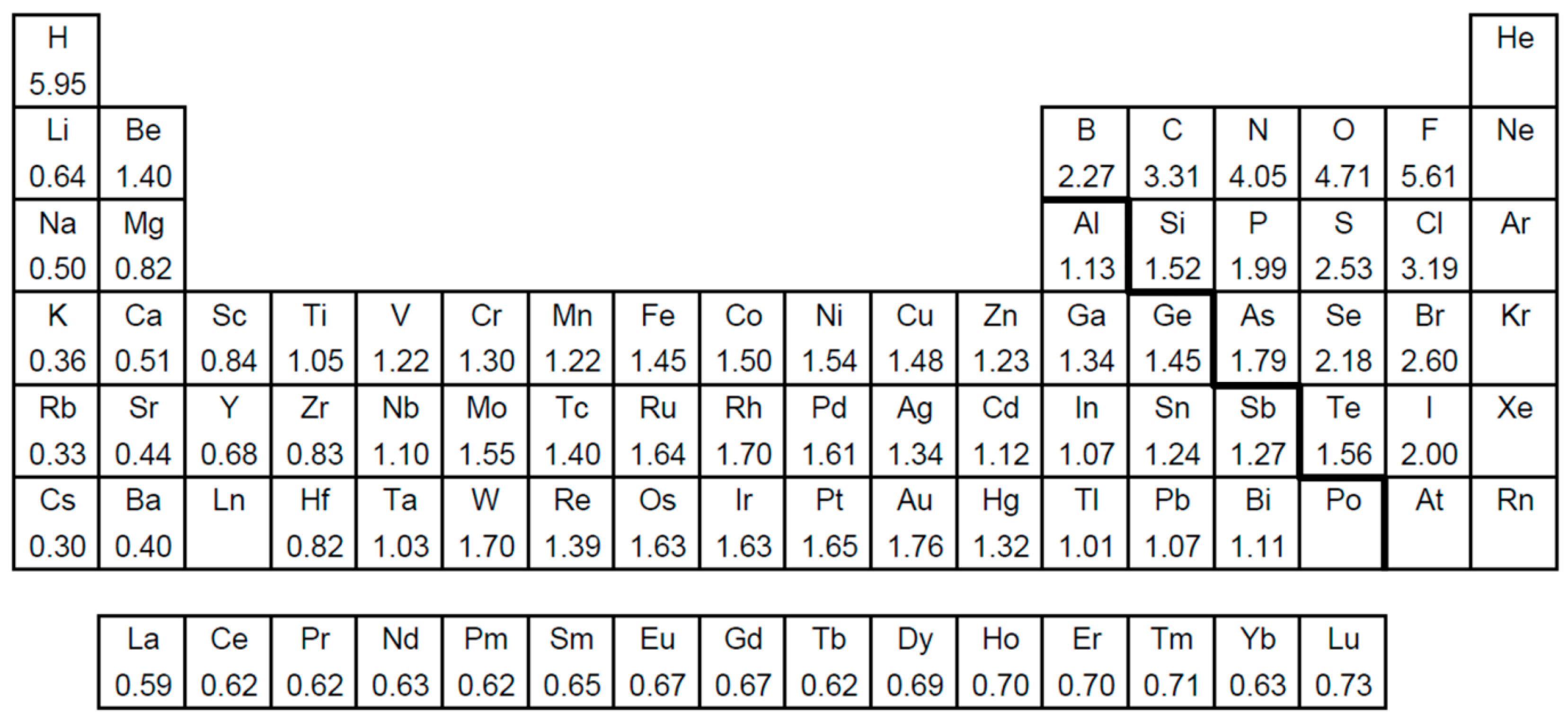
- Darken, L.S.; Gurry, R.W. Physical Chemistry of Metals; McGraw-Hill: New York, NY, USA, 1953. [Google Scholar]
- PRINCEPS also offers a user-defined weight applicable to different element types. This feature is added since it is common that researchers are more concerned about the CE of one specific element than about others in a multi-element system.
- Parthé, E.; Chabot, B. Crystal structures and crystal chemistry of ternary rare earth-transition metal borides, silicides and homologues. In Handbook on the Physics and Chemistry of Rare Earths; Gschneidner, K., Eyring, L., Eds.; Elsevier: Amsterdam, The Netherlands, 1984; Volume 6, pp. 113–334. [Google Scholar]
- Parthé, E.; Gelato, L.; Chabot, B.; Penzo, M.; Cenzual, K.; Gladyshevskii, R. TYPIX—Standardized Data and Crystal Chemical Characterization of Inorganic Structure Types, 8th ed.; Springer: Berlin, Germany, 1993; Volume 1. [Google Scholar]
- Bodak, O.; Mis’kiv, M.; Tyvanchuk, A.; Kharchenko, O.; Gladyshevskii, E. System Cerium-Nickel-Silicon in the Region 33.3 to 100 at·% Ce; Lvov State University: Lviv, Ukraine, 1973. [Google Scholar]
- Beck, U.; Neumann, H.G.; Becherer, G. Phasenbildung in Ni/Si-Schichten. Krist. Tech. 1973, 10, 1125–1129. [Google Scholar] [CrossRef]
- Bobet, J.-L.; Grigorova, E.; Chevalier, B.; Khrussanova, M.; Peshev, P. Hydrogenation of CeNi: Hydride Formation, Structure and Magnetic Properties. Intermetallics 2006, 2, 208–212. [Google Scholar] [CrossRef]
- Cromer, D.T.; Larson, A.C. The Crystal Structure of Ce2Ni7. Acta Crystallogr. 1959, 11, 855–859. [Google Scholar] [CrossRef]
- Cromer, D.T.; Olsen, C.E. The Crystal Structure of PuNi3 and CeNi3. Acta Crystallogr. 1959, 9, 689–694. [Google Scholar] [CrossRef]
- Ellner, M.; Heinrich, S.; Bhargava, M.; Schubert, K. Einige Strukturelle Untersuchungen in der Mischung NiSin. J. Less Common Met. 1979, 2, 163–173. [Google Scholar] [CrossRef]
- Pilström, G. The Crystal Structure of Ni3Si2 with some Notes on Ni5Si2. Acta Chem. Scand. 1961, 15, 893–902. [Google Scholar] [CrossRef]
- Gladyshevskii, E.T.; Kripyakevich, P. Monosilicides of Rare Earth Metals and their Crystal Structures. J. Struct. Chem. 1965, 6, 789–794. [Google Scholar] [CrossRef]
- Gout, D.; Benbow, E.; Miller, G.J. Structure and Bonding Consequences in the Pseudo-Binary System Ln5Si3−xMx (Ln = La, Ce or Nd; M = Ni or Co). J. Alloys Compd. 2002, 1, 153–164. [Google Scholar] [CrossRef]
- Kohgi, M.; Ito, M.; Satoh, T.; Asano, H.; Ishigaki, T.; Izumi, F. Crystal Structure Analysis of the Dense Kondo System CeSix. J. Magn. Magn. Mater. 1990, 90–91, 433–434. [Google Scholar] [CrossRef]
- Mishima, Y.; Ochiai, S.; Suzuki, T. Lattice Parameters of Ni (γ), Ni3Al (γ′) and Ni3Ga (γ′) Solid Solutions with Additions of Transition and B-Subgroup Elements. Acta Metall. 1985, 6, 1161–1169. [Google Scholar] [CrossRef]
- Pourarian, F.; Liu, M.; Lu, B.; Huang, M.; Wallace, W. Magnetic and Crystallographic Characteristics of CeNi5−xMx (M = Fe, Mn) Alloys and their Hydrides. J. Solid State Chem. 1986, 1, 111–117. [Google Scholar] [CrossRef]
- Roof, R.; Larson, A.; Cromer, D. The Crystal Structure of Ce7Ni3. Acta Crystallogr. 1961, 10, 1084–1087. [Google Scholar] [CrossRef]
- Shields, T.; Mayers, J.; Harris, I. Vacancy Induced Anomalies in the Laves Phase CeNi2. J. Magn. Magn. Mater. 1987, 587–590. [Google Scholar] [CrossRef]
- Toman, K. The Structure of NiSi. Acta Crystallogr. 1951, 4, 462–464. [Google Scholar] [CrossRef]
- Toman, K. The Structure of Ni2Si. Acta Crystallogr. 1952, 5, 329–331. [Google Scholar] [CrossRef]
- Weitzer, F.; Schuster, J.; Bauer, J.; Jounel, B. Phase Equilibria in Ternary RE-Si-N Systems (RE = Sc, Ce, Ho). J. Mater. Sci. 1991, 8, 2076–2080. [Google Scholar] [CrossRef]
- Zhang, H.; Mudryk, Y.; Zou, M.; Pecharsky, V.; Gschneidner, K.; Long, Y. Phase Relationships and Crystallography of Annealed Alloys in the Ce5Si4-Ce5Ge4 Pseudobinary System. J. Alloys Compd. 2009, 1, 98–102. [Google Scholar] [CrossRef]
- Leisegang, T.; Meyer, D.C.; Doert, T.; Zahn, G.; Weißbach, T.; Souptel, D.; Behr, G.; Paufler, P. Incommensurately modulated CeSi1.82. Z. Kristallogr. 2005, 220, 128–134. [Google Scholar] [CrossRef]
- CeSi2 adopts an incommensurately modulated structure based on the NdSi2−x type when there is a sufficient degree of Si-deficiency (see Ref 80). However, since the NdSi2−x is simply an orthorhombic variant of the ThSi2 type and the modulations would represent a superstructure normally removed from the reference list, it is unlikely that this transition will significantly affect the results of the PRINCEPS analysis.
- Mayer, I.; Tassa, M. Rare Earth-Iron (Cobalt, Nickel)-Silicon Compounds. J. Less Common Met. 1969, 3, 173–177. [Google Scholar] [CrossRef]
- Streletskii, A.; Morozkin, A.; Portnoy, V.; Berestetskaya, I.; Verbetskii, V. Possibilities of the CeNi2Si2 Hydrogenation under Mechanical Treatment. Mater. Res. Bull. 2000, 5, 719–726. [Google Scholar] [CrossRef]
- Bodak, O.; Gladyshevsky, E. Crystal Structure of CeNiSi2 and Kindred Compounds. Sov. Phys. Crystallogr. 1970, 6, 859–862. [Google Scholar]
- Kowalczyk, A.; Falkowski, M.; Tran, V.; Pugaczowa-Michalska, M. Electronic Structure and Thermoelectric Power of CeNi4Si. J. Alloys Compd. 2007, 1, 13–17. [Google Scholar] [CrossRef]
- Chabot, B.; Parthé, E. Ce2Co3Si5 and R2Ni3Si5 (R = Ce, Dy, Y) With the Orthorhombic U2Co3Si5-Type Structure and the Structural Relationship with the Tetragonal Sc2Fe3Si5-Type Structure. J. Less Common Met. 1984, 1, 285–290. [Google Scholar] [CrossRef]
- Hovestreydt, E. Crystal Data for Ce14Ni6Si11 Isotypic with Pr14Ni6Si11. J. Less Common Met. 1984, 1, L27–L29. [Google Scholar] [CrossRef]
- Michor, H.; El-Hagary, M.; Paul, C.; Bauer, E.; Hilscher, G.; Rogl, P.; Giester, G. Crystal Structure and Kondo Lattice Behavior of CeNi9Si4. Phys. Rev. B 2003, 22, 224428. [Google Scholar] [CrossRef]
- Bodak, O.; Gladyshevskij, E.; Kharchenko, O. Crystal Structure of Ce6Ni2Si3 and Related Compounds. Sov. Phys. Crystallogr. 1974, 19, 45–46. [Google Scholar]
- Bodak, O.; Gladyshevsky, E.; Miskiv, M. Crystal Structure of Ce2NiSi and Related Compounds. Sov. Phys. Crystallogr. 1972, 3, 439–441. [Google Scholar]
- Hladyschewskyj, E.; Krypiakewytsch, P.; Bodak, O. Die Kristallstruktur von Ce3Ni6Si2 und Verwandten Verbindungen. Z. Anorg. Allg. Chem. 1966, 1–2, 95–101. [Google Scholar] [CrossRef]
- Mys’kiv, M.G. The Crystal Structure of the Compound Ce7Ni2Si5. Visn. Lviv. Derzh. Univ. Ser. Khim. 1974, 17–21. [Google Scholar]
- Merlo, F.; Fornasini, M.; Pani, M. On the Existence and the Crystal Structure of Novel R3TSi3 Intermetallic Phases (R = Rare Earth; T = Fe, Co, Ni). J. Alloys Compd. 2005, 1, 165–171. [Google Scholar] [CrossRef]
- Myskiv, M.; Bodak, O.; Gladyshevsky, E. Crystal Structure of the Compound Ce15Ni4Si13. Krist. Graf. 1973, 4, 715–719. [Google Scholar]
- Bodak, O.I.; Gladyshevskii, E.I. Crystal Structure of the Compound CeNi8.6Si2.4 and Related Compounds. Dopov. Akad. Nauk Ukr. RSR, Ser. A 1969, 452–455. [Google Scholar]
- Pani, M.; Manfrinetti, P.; Provino, A.; Yuan, F.; Mozharivskyj, Y.; Morozkin, A.; Knotko, A.; Garshev, A.; Yapaskurt, V.; Isnard, O. New Tetragonal Derivatives of Cubic NaZn13-type Structure: RNi6Si6 Compounds, Crystal Structure and Magnetic Ordering (R = Y, La, Ce, Sm, Gd-Yb). J. Solid State Chem. 2014, 1, 45–52. [Google Scholar] [CrossRef]
- Pasturel, M.; Weill, F.; Bourée, F.; Bobet, J.-L.; Chevalier, B. Hydrogenation of the Ternary Silicides RENiSi (RE = Ce, Nd) Crystallizing in the Tetragonal LaPtSi-type Structure. J. Alloys Compd. 2005, 1, 17–22. [Google Scholar] [CrossRef]
- Stepien, J.; Lukaszewicz, K.; Hladyszewski, E.; Bodak, O. Crystalline Structure of the Intermetallic Compound Ce3Ni2Si8. Bull. Acad. Pol. Sci. Ser. Sci. Chim. 1972, 1029–1036. [Google Scholar]
- Gladyshevskii, E.I.; Kripyakevich, P.I.; Bodak, O.I. A New Example of Isotypism of the Intermetallic Compounds RCon and R(Ni,Si)n. Visn. Lviv. Derzh. Univ. Ser. Khim. 1967, 34–39. [Google Scholar]
- Prots, Y.M.; Jeitschko, W. Lanthanum Nickel Silicides with the General Formula La(n+1)(n+2)Nin(n-1)+2Sin(n+1) and Other Series of Hexagonal Structures with Metal:Metalloid Ratios Close to 2:1. Inorg. Chem. 1998, 21, 5431–5438. [Google Scholar] [CrossRef]
- Another difference between CeSi2 and Ce(Ni,Si)2 lies in their cell parameters: CeSi2 has tetragonal symmetry, so its a and b parameters are restricted to be equal (a = b = 4.156 Å), while Ce(Ni,Si)2 does not have such a symmetry constraint (a = 4.039 Å, c = 4.287 Å). For all three phases in the structural series, their two shorter cell axis lengths are almost equal, so the atomic CEs in CeSi2 is less distorted from the target atomic CEs compared to Ce(Ni,Si)2. This explains why CeSi2 takes up a higher weight than Ce(Ni,Si)2 in the decomposition of Ce3Ni2Si8 and CeNiSi2.
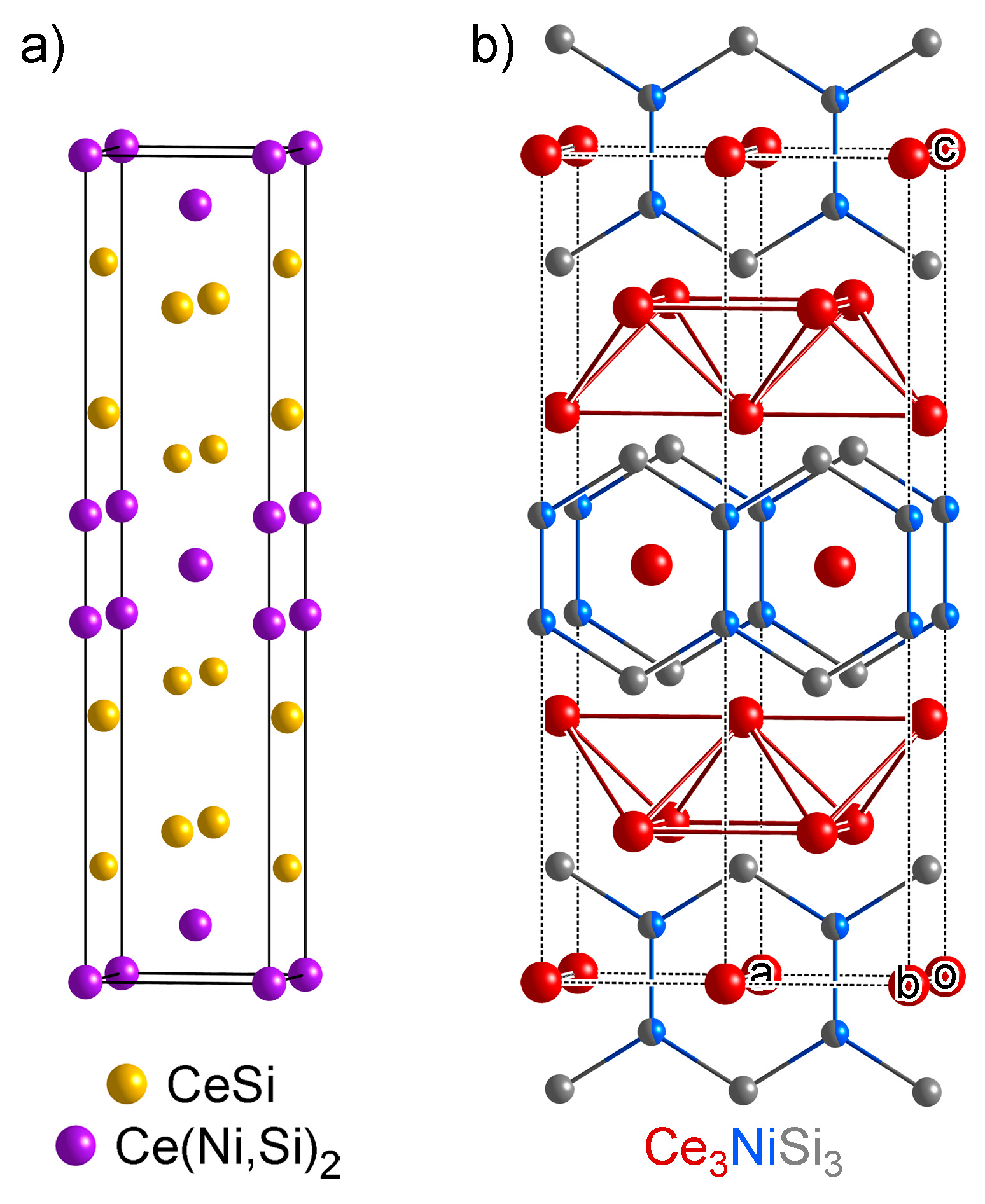
- One might notice that these three structures have very similar cell parameters to those of Ce3NiSi3 in Group A1: They all have tall and skinny unit cells with pseudo-tetragonal shapes. However, among these structures, Ce3NiSi3 is the only one that contains the CeSi type Ce double layers, and does not contain any of the CeNi2Si2-type fragments common in the Group A2 phases. Therefore, Ce3NiSi3 should be categorized as a member of Group A1, as is correctly pointed out by PRINCEPS.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
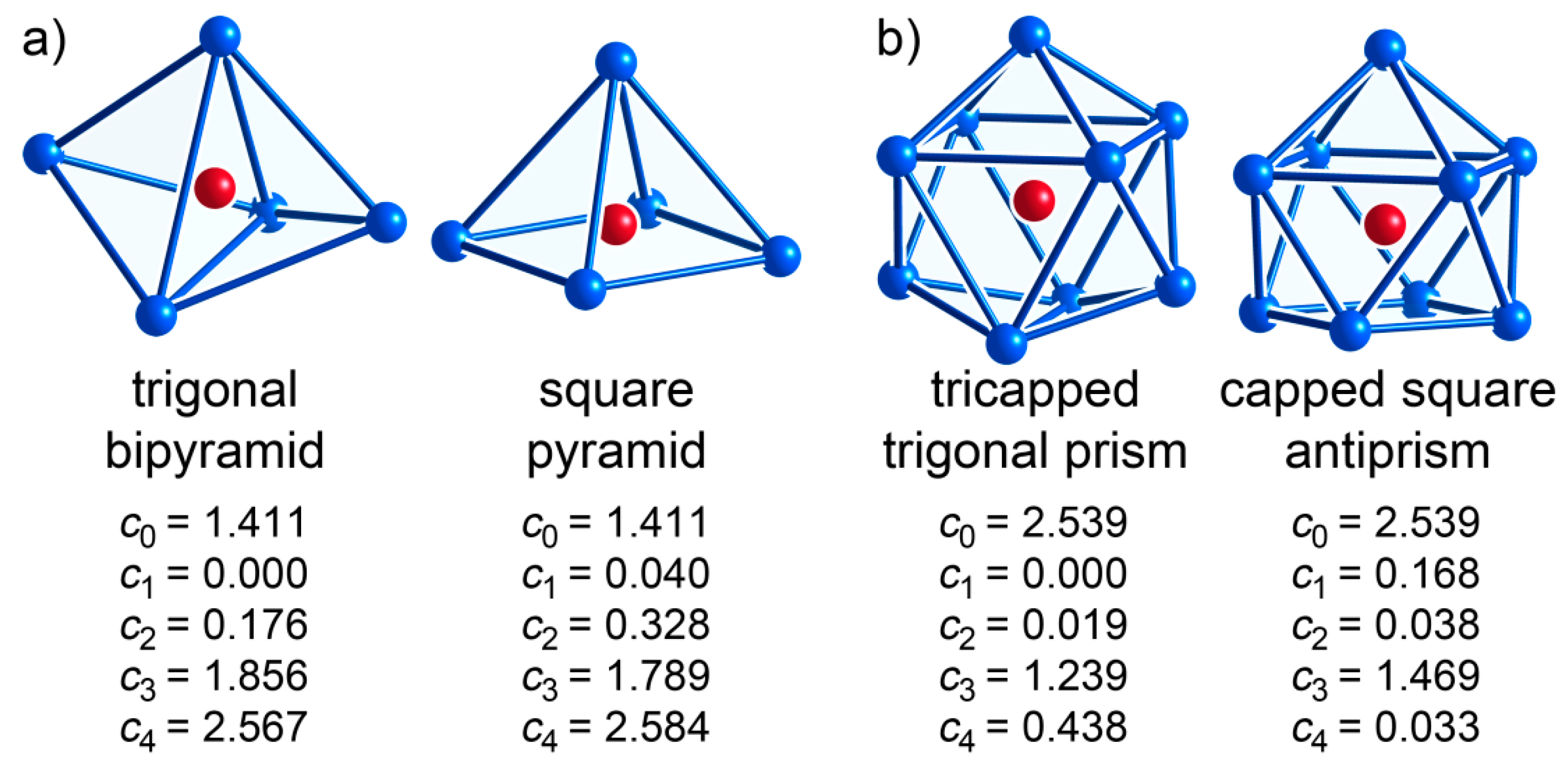
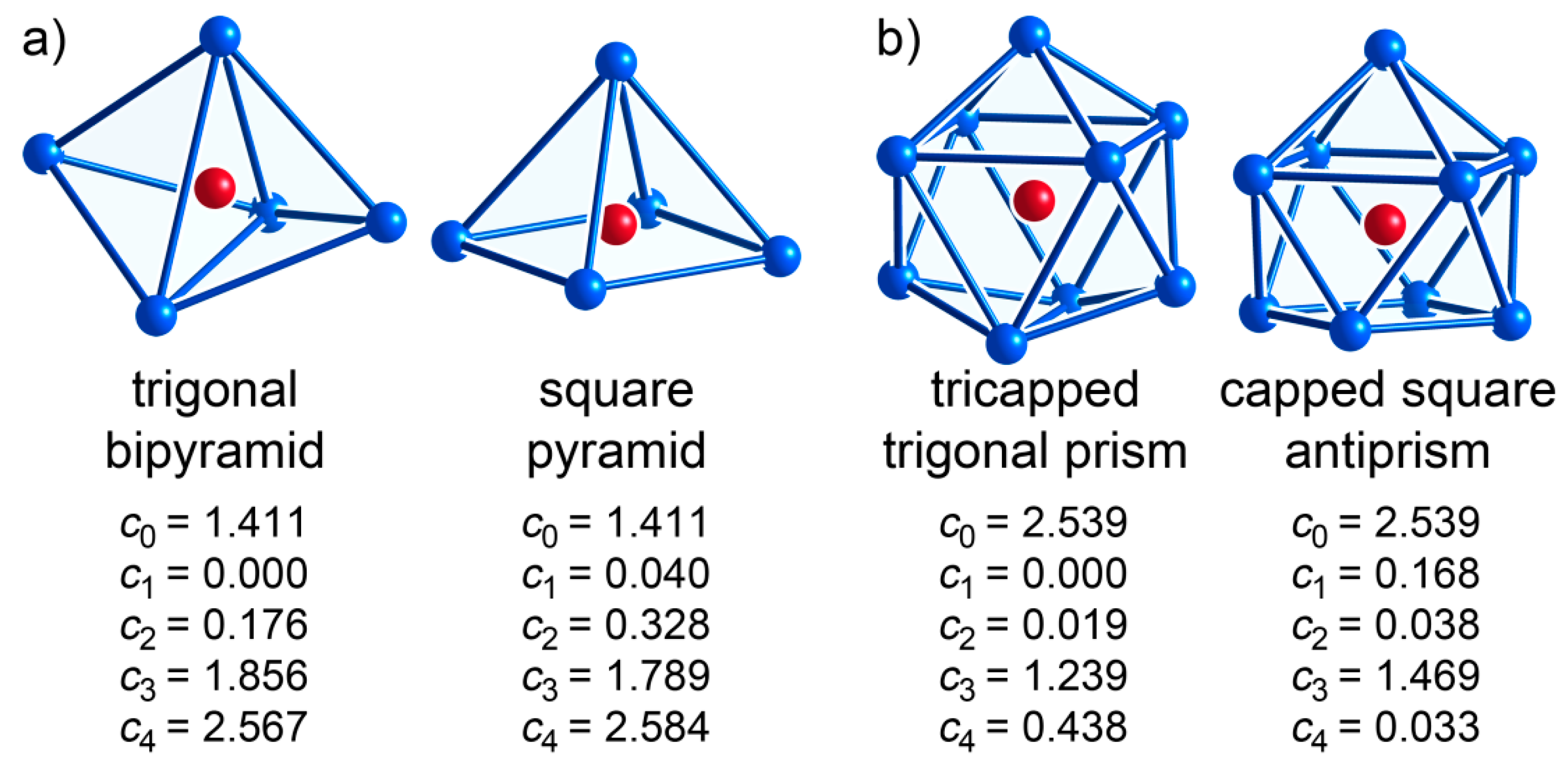
| CE Polyhedron Type | CN | (s) | (p) | (d) | (f) | (g) |
|---|---|---|---|---|---|---|
| tetrahedron | 4 | 1.128 | 0 | 0 | 2.225 | 1.723 |
| square planar | 4 | 1.128 | 0 | 1.262 | 0 | 2.807 |
| trigonal bipyramid | 5 | 1.411 | 0 | 0.176 | 1.856 | 2.567 |
| square pyramid | 5 | 1.411 | 0.04 | 0.328 | 1.789 | 2.584 |
| octahedron | 6 | 1.693 | 0 | 0 | 0 | 3.878 |
| trigonal prism | 6 | 1.693 | 0 | 0.541 | 1.529 | 2.176 |
| pentagonal bipyramid | 7 | 1.974 | 0 | 0.103 | 0 | 3.398 |
| capped trigonal prism | 7 | 1.974 | 0.274 | 0.972 | 0.916 | 2.972 |
| cube | 8 | 2.257 | 0 | 0 | 0 | 3.447 |
| square antiprism | 8 | 2.257 | 0 | 0.514 | 0 | 2.113 |
| bisdisphenoid | 8 | 2.257 | 0 | 0.051 | 0.196 | 2.913 |
| tricapped trigonal prism | 9 | 2.539 | 0 | 0.019 | 1.239 | 0.438 |
| capped square antiprism | 9 | 2.539 | 0.168 | 0.038 | 1.469 | 0.033 |
| bicapped cube | 10 | 2.821 | 0 | 0.844 | 0 | 2.91 |
| bicapped square antiprism | 10 | 2.821 | 0 | 0.519 | 0 | 1.276 |
| icosahedron | 12 | 3.385 | 0 | 0 | 0 | 0 |
| cuboctahedron | 12 | 3.385 | 0 | 0 | 0 | 1.939 |
| anticuboctahedron | 12 | 3.385 | 0 | 0 | 0.681 | 0.987 |
| bicapped pentagonal prism | 12 | 3.385 | 0 | 0.282 | 0 | 0.379 |
| Reference Phase | Structure Type | Pearson Symbol | Space Group |
|---|---|---|---|
| Ni3Si | AuCu3 | cP4 | |
| CeSi | FeB | oP8 | |
| CeSi2 | ThSi2 | tI12 | |
| CeNi5 | CaCu5 | hP6 | |
| Ce(Ni,Si)2 | AlB2 | hP3 | |
| CeNi2Si2 | ThCr2Si2 | tI10 |
| Distance | Ni3Si | CeSi | CeSi2 | CeNi5 | Ce(Ni,Si)2 | CeNi2Si2 |
|---|---|---|---|---|---|---|
| Ni3Si | 0 | 2.693 | 2.737 | 2.711 | 2.696 | 2.964 |
| CeSi | 2.693 | 0 | 1.925 | 2.412 | 2.088 | 2.154 |
| CeSi2 | 2.737 | 1.925 | 0 | 2.227 | 1.014 | 1.906 |
| CeNi5 | 2.711 | 2.412 | 2.227 | 0 | 2.019 | 1.746 |
| Ce(Ni,Si)2 | 2.696 | 2.088 | 1.014 | 2.019 | 0 | 1.754 |
| CeNi2Si2 | 2.964 | 2.154 | 1.906 | 1.746 | 1.754 | 0 |
| Site | CN Vector | Quality | Reference Phase | Reference Site | CN Vector | Distance |
|---|---|---|---|---|---|---|
| Ce1 | (2.00, 14.00, 4.00) | 99.01% | CeNi5 | Ce1 | (2.00, 18.00, 0.00) | 0.9775 |
| CeNi2Si2 | Ce1 | (4.00, 8.00, 10.00) | 3.6373 | |||
| Ce(Ni,Si)2 | Ce1 | (8.00, 4.80, 7.20) | 5.4135 | |||
| Ni1 | (3.00, 7.00, 2.00) | 94.22% | CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 0.8292 |
| CeNi2Si2 | Ni1 | (4.00, 4.00, 4.00) | 2.3017 | |||
| CeNi5 | Ni2 | (4.00, 8.00, 0.00) | 3.2811 | |||
| Ni2 | (4.00, 6.00, 2.00) | 90.27% | CeNi5 | Ni2 | (4.00, 8.00, 0.00) | 1.0008 |
| CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 3.2016 | |||
| CeNi2Si2 | Ni1 | (4.00, 4.00, 4.00) | 3.7154 | |||
| Si1 | (4.00, 8.00, 0.00) | 81.88% | CeNi5 | Ni2 | (4.00, 8.00, 0.00) | 1.2924 |
| CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 3.2767 | |||
| CeNi2Si2 | Ni1 | (4.00, 4.00, 4.00) | 3.8280 | |||
| overall decomposition | 91.64% | CeNi5 | weight: | 99.89% | ||
| CeNi2Si2 | weight: | 0.11% | ||||
| Ce(Ni,Si)2 | weight: | 0.00% | ||||
| Site | CN Vector | Quality | Reference Phase | Reference Site | CN Vector | Distance |
|---|---|---|---|---|---|---|
| Ce1 | (4.00, 7.00, 10.00) | 76.39% | CeNi2Si2 | Ce1 | (4.00, 8.00, 10.00) | 3.0627 |
| CeNi5 | Ce1 | (2.00, 18.00, 0.00) | 3.6569 | |||
| CeSi2 | Ce1 | (8.00, 0.00, 12.00) | 5.3249 | |||
| Ni2 | (5.00, 0.00, 5.00) | 72.01% | CeNi2Si2 | Si1 | (5.00, 4.00, 1.00) | 1.5344 |
| CeSi2 | Si1 | (6.00, 0.00, 3.00) | 3.1804 | |||
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 3.1999 | |||
| Si1 | (4.00, 4.00, 4.00) | 69.17% | CeNi2Si2 | Ni1 | (4.00, 4.00, 4.00) | 1.6321 |
| CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 2.5067 | |||
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 3.7712 | |||
| Si2 | (4.00, 3.00, 5.00) | 72.35% | CeNi2Si2 | Ni1 | (4.00, 4.00, 4.00) | 1.5506 |
| CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 2.1857 | |||
| CeNi5 | Ni2 | (4.00, 8.00, 0.00) | 3.8041 | |||
| overall decomposition | 63.12% | CeNi2Si2 | weight: | 80.69% | ||
| CeNi5 | weight: | 10.79% | ||||
| Ce(Ni,Si)2 | weight: | 4.97% | ||||
| Site | CN Vector | Quality | Reference Phase | Reference Site | CN Vector | Distance |
|---|---|---|---|---|---|---|
| Ce1 | (10.00, 2.00, 5.00) | 87.38% | CeSi | Ce1 | (10.00, 0.00, 7.00) | 3.2659 |
| CeSi2 | Ce1 | (8.00, 0.00, 12.00) | 5.2884 | |||
| CeNi2Si2 | Si1 | (5.00, 4.00, 1.00) | 6.3912 | |||
| Ce8 | (8.00, 6.00, 6.00) | 93.58% | Ce(Ni,Si)2 | Ce1 | (8.00, 4.80, 7.20) | 1.8547 |
| CeSi2 | Ce1 | (8.00, 0.00, 12.00) | 3.7543 | |||
| CeNi2Si2 | Ce1 | (4.00, 8.00, 10.00) | 5.6059 | |||
| Ni1 | (6.00, 0.00, 3.00) | 95.07% | CeSi2 | Si1 | (6.00, 0.00, 3.00) | 0.7839 |
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 0.8749 | |||
| CeSi | Si1 | (7.00, 0.00, 2.00) | 2.8443 | |||
| Si1 | (7.00, 1.00, 1.00) | 85.46% | CeSi | Si1 | (7.00, 0.00, 2.00) | 1.1839 |
| CeSi2 | Si1 | (6.00, 0.00, 3.00) | 2.6832 | |||
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 2.7863 | |||
| overall decomposition | 81.33% | CeSi | weight: | 65.34% | ||
| Ce(Ni,Si)2 | weight: | 18.70% | ||||
| CeSi2 | weight: | 15.93% | ||||
| Site | CN Vector | Quality | Reference Phase | Reference Site | CN Vector | Distance |
|---|---|---|---|---|---|---|
| Ce3 | (8.00, 5.00, 4.00) | 72.74% | CeSi | Ce1 | (10.00, 0.00, 7.00) | 3.2659 |
| CeSi2 | Ce1 | (8.00, 0.00, 12.00) | 5.2884 | |||
| Ce(Ni,Si)2 | Ce1 | (8.00, 4.80, 7.20) | 5.5187 | |||
| Ce4 | (8.00, 8.00, 4.00) | 85.61% | CeSi2 | Ce1 | (8.00, 0.00, 12.00) | 2.4999 |
| Ce(Ni,Si)2 | Ce1 | (8.00, 4.80, 7.20) | 3.7168 | |||
| CeSi | Ce1 | (10.00, 0.00, 7.00) | 5.6100 | |||
| Ni1 | (6.00, 2.00, 1.00) | 65.24% | CeSi2 | Si1 | (6.00, 0.00, 3.00) | 1.7046 |
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 1.8492 | |||
| CeSi | Si1 | (7.00, 0.00, 2.00) | 2.5276 | |||
| Ni4 | (7.00, 0.00, 2.00) | 55.83% | CeSi | Si1 | (7.00, 0.00, 2.00) | 1.9447 |
| CeSi2 | Si1 | (6.00, 0.00, 3.00) | 2.7660 | |||
| Ce(Ni,Si)2 | Mx1 | (6.00, 1.20, 1.80) | 2.8333 | |||
| overall projection | 51.91% | CeSi2 | weight: | 39.05% | ||
| CeSi | weight: | 37.59% | ||||
| Ce(Ni,Si)2 | weight: | 13.32% | ||||
| Site | CN Vector | Quality | Reference Phase | Reference Site | CN Vector | Distance |
|---|---|---|---|---|---|---|
| Ce1 | (0.00, 16.00, 8.00) | 31.31% | CeNi5 | Ce1 | (2.00, 18.00, 0.00) | 5.8858 |
| CeNi2Si2 | Ce1 | (4.00, 8.00, 10.00) | 6.6348 | |||
| Ce(Ni,Si)2 | Ce1 | (8.00, 4.80, 7.20) | 8.1575 | |||
| Ni1 | (2.00, 6.00, 4.00) | 12.07% | CeNi5 | Ni1 | (3.00, 9.00, 0.00) | 4.0759 |
| Ni3Si | Ni1 | (0.00, 8.00, 4.00) | 4.0763 | |||
| Ni3Si | Si1 | (0.00, 12.00, 0.00) | 4.0996 | |||
| Ni3 | (0.00, 8.00, 4.00) | 92.19% | Ni3Si | Ni1 | (0.00, 8.00, 4.00) | 0.9236 |
| Ni3Si | Si1 | (0.00, 12.00, 0.00) | 1.2999 | |||
| CeNi5 | Ni2 | (4.00, 8.00, 0.00) | 4.4683 | |||
| overall projection | 19.79% | Ni3Si | weight: | 44.77% | ||
| CeNi5 | weight: | 41.66% | ||||
| CeNi2Si2 | weight: | 9.01% | ||||
| Group A | Group A1 | Ce14Ni6Si11 (mC124, ), Ce6Ni2Si3 (hP22, ), Ce5Ni2Si3 (hP40, ), Ce15Ni4Si13 (hP64, ), Ce7Ni2Si5 (oP56, ), Ce3NiSi3 (oI14, ) |
| Group A2 | CeNiSi (tI12, ), Ce2Ni3Si5 (oI40, ), Ce3Ni2Si8 (oS26, ), CeNiSi2 (oS16, ), Ce3Ni4Si4 (oI22, ) | |
| Group A3 | Ce6Ni7Si4 (oP68, ) | |
| Group B | CeNi9Si4 (tI56, ), CeNi8Si5 (cF112, ), CeNi6Si6 (tI52, ) | |
| Isolated Phases | CeNi4Si (oS12, ), Ce2Ni15Si2 (hR57, ), Ce2Ni17Si5 (tI48, ), Ce3Ni6Si2 (cI44, ) | |
| Phase | Quality | Component 1 | Weight 1 | Component 2 | Weight 2 | Component 3 | Weight 3 |
|---|---|---|---|---|---|---|---|
| Ce14Ni6Si11 | 81.33% | CeSi | 65.34% | Ce(Ni,Si)2 | 18.70% | CeSi2 | 15.93% |
| Ce6Ni2Si3 | 64.63% | CeSi | 91.01% | Ce(Ni,Si)2 | 5.00% | CeSi2 | 3.79% |
| Ce5Ni2Si3 | 74.45% | CeSi | 79.43% | Ce(Ni,Si)2 | 13.93% | CeSi2 | 6.53% |
| Ce15Ni4Si13 | 80.09% | CeSi | 68.43% | Ce(Ni,Si)2 | 24.59% | CeSi2 | 6.90% |
| Ce7Ni2Si5 | 73.17% | CeSi | 89.82% | CeSi2 | 6.66% | Ce(Ni,Si)2 | 3.48% |
| Ce3NiSi3 | 90.80% | CeSi | 57.89% | Ce(Ni,Si)2 | 31.82% | CeSi2 | 10.28% |
| Phase | Quality | Component 1 | Weight 1 | Component 2 | Weight 2 | Component 3 | Weight 3 |
|---|---|---|---|---|---|---|---|
| Ce2Ni3Si5 | 63.12% | CeNi2Si2 | 80.69% | CeNi5 | 10.79% | Ce(Ni,Si)2 | 4.97% |
| CeNiSi | 94.12% | CeSi2 | 85.54% | Ce(Ni,Si)2 | 14.44% | CeSi | 0.01% |
| Ce3Ni2Si8 | 76.58% | CeNi2Si2 | 67.46% | CeSi2 | 24.66% | Ce(Ni,Si)2 | 6.54% |
| CeNiSi2 | 72.80% | CeNi2Si2 | 46.32% | CeSi2 | 42.52% | Ce(Ni,Si)2 | 9.89% |
| Ce3Ni4Si4 | 82.01% | CeSi2 | 42.73% | CeNi2Si2 | 31.58% | Ce(Ni,Si)2 | 24.87% |
| Phase | Quality | Component 1 | Weight 1 | Component 2 | Weight 2 | Component 3 | Weight 3 |
|---|---|---|---|---|---|---|---|
| CeNi9Si4 | 19.79% | Ni3Si | 44.77% | CeNi5 | 41.66% | CeNi2Si2 | 9.01% |
| CeNi8Si5 | 20.36% | CeNi5 | 48.97% | Ni3Si | 37.85% | CeNi2Si2 | 9.96% |
| CeNi6Si6 | 18.70% | CeNi5 | 64.12% | CeNi2Si2 | 21.61% | Ce(Ni,Si)2 | 7.01% |
| Distance | CeNi8Si5 | CeNi9Si4 | CeNi6Si6 |
|---|---|---|---|
| CeNi8Si5 | 0 | 0.477 | 1.147 |
| CeNi9Si4 | 0.477 | 0 | 1.139 |
| CeNi6Si6 | 1.147 | 1.139 | 0 |
| Phase | Quality | Component 1 | Weight 1 | Component 2 | Weight 2 | Component 3 | Weight 3 |
|---|---|---|---|---|---|---|---|
| CeNi4Si | 91.64% | CeNi5 | 99.89% | CeNi2Si2 | 0.11% | Ce(Ni,Si)2 | 0.00% |
| Ce2Ni15Si2 | 25.30% | CeNi5 | 84.20% | Ni3Si | 11.07% | CeNi2Si2 | 4.17% |
| Ce2Ni17Si5 | 19.88% | CeNi5 | 63.59% | Ni3Si | 24.48% | CeNi2Si2 | 9.43% |
| Ce3Ni6Si2 | 48.37% | CeNi5 | 60.12% | CeSi | 14.97% | CeNi2Si2 | 11.02% |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Fredrickson, D.C. PRINCEPS: A Computer-Based Approach to the Structural Description and Recognition of Trends within Structural Databases, and Its Application to the Ce-Ni-Si System. Crystals 2016, 6, 35. https://doi.org/10.3390/cryst6040035
Guo Y, Fredrickson DC. PRINCEPS: A Computer-Based Approach to the Structural Description and Recognition of Trends within Structural Databases, and Its Application to the Ce-Ni-Si System. Crystals. 2016; 6(4):35. https://doi.org/10.3390/cryst6040035
Chicago/Turabian StyleGuo, Yiming, and Daniel C. Fredrickson. 2016. "PRINCEPS: A Computer-Based Approach to the Structural Description and Recognition of Trends within Structural Databases, and Its Application to the Ce-Ni-Si System" Crystals 6, no. 4: 35. https://doi.org/10.3390/cryst6040035
APA StyleGuo, Y., & Fredrickson, D. C. (2016). PRINCEPS: A Computer-Based Approach to the Structural Description and Recognition of Trends within Structural Databases, and Its Application to the Ce-Ni-Si System. Crystals, 6(4), 35. https://doi.org/10.3390/cryst6040035