Abstract
We have prepared polycrystalline samples of the trimer Ir oxide BaIrO3 with face-shared Ir3O12 trimers, and have investigated the origin of the phase transition at 182 K by measuring resistivity, thermopower, magnetization and synchrotron X-ray diffraction. We propose a possible electronic model and transition mechanism, starting from a localized electron picture on the basis of the Rietveld refinement. Within this model, BaIrO3 can be basically regarded as a Mott insulator, when the Ir3O12 trimer is identified to one pseudo-atom or one lattice site. The transition can be viewed as a transition from the Mott insulator phase to a kind of charge ordered insulator phase.
1. Introduction
Strongly correlated electrons are seen in a certain class of solids in which the Coulomb repulsion is too strong to hold a simple one-electron picture based on the band theory. They have occupied a central position in condensed matter sciences for many decades, and have attracted keen interests from vast numbers of researchers since the discovery of high-temperature superconducting copper oxides in 1986. They store a macroscopic number of degeneracy and entropy based on spin and charge degrees of freedom on each lattice site at high temperature [1,2], and release them through various phase transitions towards 0 K. Various forms of phase transitions and ordered states have been discovered one after another with the progress in the studies of strongly correlated electrons. In this context, understanding of new phase transitions and ordered states is a major purpose for strong-correlation physics.
BaIrO3 ( phase) is an interesting oxide as a playground for a new phase transition. The crystal structure is schematically shown in the inset of Figure 1b, in which the three IrO6 octahedra are connected with one another in a face-sharing network, and form an Ir3O12 trimer structure, as indicated in light brown and dark yellow. The trimers are connected with each other in a corner-sharing network and construct zig-zag chains along the c axis and corrugated honeycomb lattices in the plane. Owing to the low crystal symmetry of , two trimers are inequivalent in the unit cell, as painted in different colors in the inset of Figure 1b.
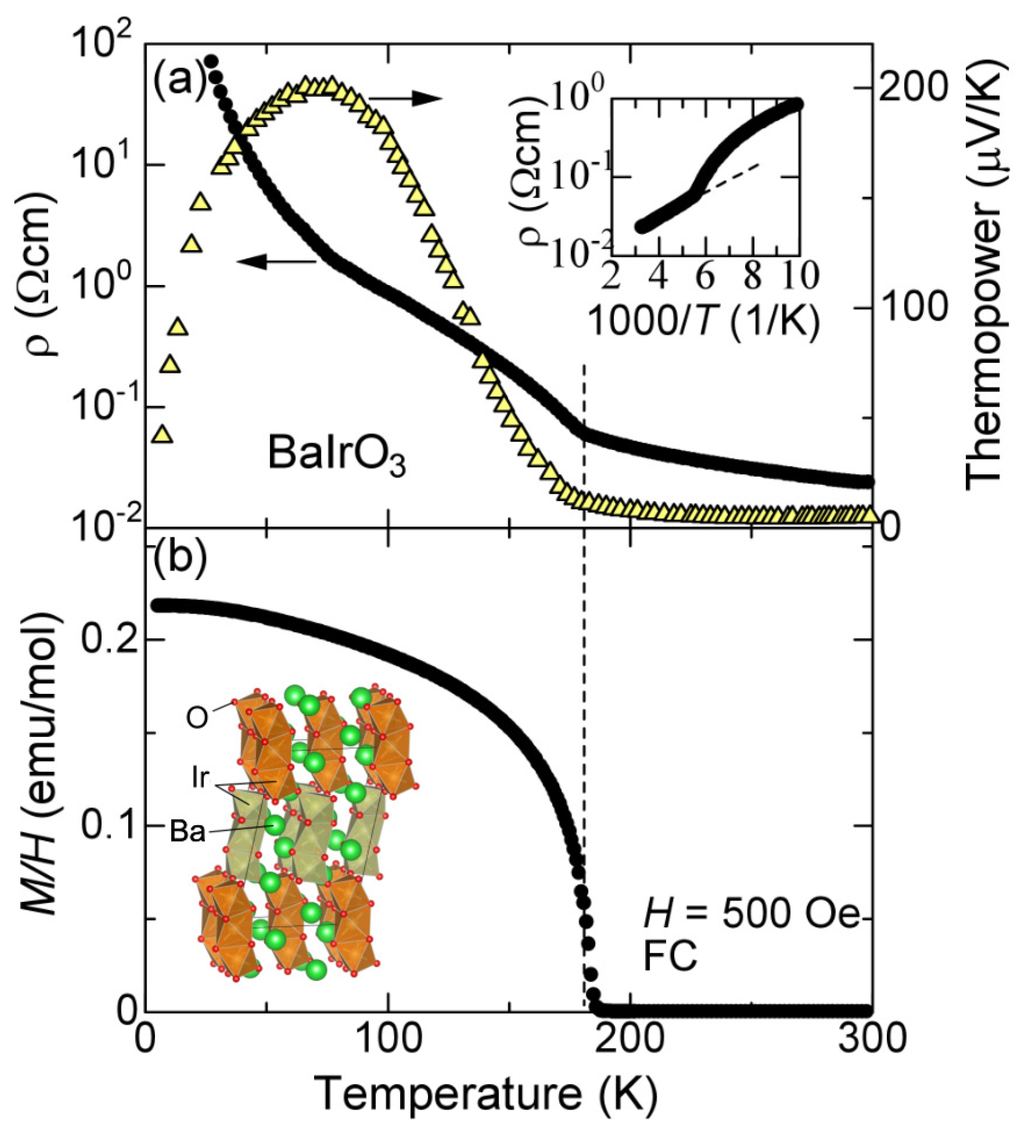
Figure 1.
(a) resistivity and thermopower of a polycrystalline sample of BaIrO3. The inset shows the Arrhenius plot of the resistivity, in which the dotted line indicates the activation transport above the transition temperature ; (b) magnetization M divided by an external field H of 500 Oe of the same sample. The dotted line represents 182 K. The inset shows the schematic picture of the crystal structure of BaIrO3.
The study of this oxide has a rather long history; Donohue et al. first reported the synthesis and structure of this material in the 60s, [3], and later Gai et al. [4] studied the poly-types of 5H and 6H. Chamberland [5] extensively studied the relationship of the poly-types to the synthetic routes, and further found a magnetic phase transition near 200 K. The physical and chemical properties of the poly-types have been measured rather recently because of difficulty in the sample synthesis [6,7]. Siegrist and Chamberland [8] prepared single crystals and determined the crystal symmetry of , which can be viewed as pseudo-rhombohedral R-centered. Later Powell and Battle [9] identified that this magnetic order is ferromagnetic, by measuring the temperature hysteresis and the magnetization-field curve. Linsay et al. [10] measured the remnant magnetization to demonstrate the ferromagnetic ground state. Cao et al. [11] measured the transport and optical properties of a single-crystal sample of BaIrO3, and found that the ferromagnetic order accompanies rapid increase in the resistivity below 180 K. In addition, they observed a gap-like structure in the optical conductivity below around 1000 cm−1, and attributed this transition to charge-density-wave (CDW) formation with ferromagnetic order. They further found the additional two temperature anomalies near 80 and 30 K. Although the 80-K anomaly was ill-defined, the 30-K anomaly was detected by other groups in a muon-spin-relaxation experiment [12] and in a nonlinear conduction measurement [13]. Kini et al. [14] measured the specific heat of BaIrO3 and observed a small jump of 2 J/mol K around 180 K, not around 30 and 80 K. They also found that the thermopower rapidly increases below , and pointed out the existence of a charge gap below . Maiti et al. [15] discussed the CDW state through photoemission spectroscopy.
In this paper, we try to address the nature and mechanism of the 180-K phase transition in BaIrO3 on the basis of the electronic states constructed from a localized electron picture. A concern in the CDW scenario is that there is no evidence for the lattice distortion at . In conventional CDW materials, a lattice modulation of emerges below through the electron-phonon interaction, where is the Fermi wavenumber [16]. To the best of our knowledge, no superlattice reflections below have been reported in BaIrO3, and this implies that the charge gap opens without translational-symmetry breaking. We have investigated the structure-property relationship through synchrotron X-ray diffraction, and propose a possible mechanism of the transition.
2. Experimental Section
Polycrystalline samples of ( 0.1) were prepared using a conventional solid-state reaction. A stoichiometric mixture of powder sources of BaCO3 (99.9%), RuO2 (99.9%) and Ir (99%) was thoroughly ground in an agate mortar. The mixture was then pre-sintered in an alumina crucible at 900 °C for 12 h in air, and was pressed into pellets after re-grinding. The pellets were sintered at 1000 °C for 36 h in air.
The synchrotron X-ray diffraction was taken at BL8A & 8B (Photon-Factory, KEK, Tsukuba, Japan). The X-ray energy was adjusted to be 18 keV, which was carefully calibrated using a standard powder sample of CeO2. Powder samples were sealed in a silica-glass capillary of 0.1-mm diameter, and the capillary was rotated by an angle of 30° from the sample-stage axis during measurement. The sample temperature was controlled using a cool helium gas or nitrogen gas. The diffraction patterns were analyzed using the Rietveld refinement with Rietan-FP code [17].
The resistivity was measured with a four-probe technique using a home-made measurement system in a liquid-helium cryostat. The thermopower was measured with a two-probe method using a home-made measurement system in a liquid-helium cryostat, and the thermopower of the measurement leads was carefully subtracted. The temperature difference was typically 0.5 K which was monitored with a copper-constantan differential thermocouple. The magnetization was measured with a commercial product of Quantum Design Magnetic-Property-Measurement-System.
3. Results
Figure 2 shows a synchrotron X-ray diffraction pattern of BaIrO3 at room temperature. Since the scattering intensity of oxygen is 100 times weaker than that of Ba and Ir, we gave up determining the atomic position of oxygen atoms, but instead employed the values reported in the literature [8]. Then, we optimized the atomic positions of Ba and Ir atoms above 100 K by iterating the calculation until the and values reached less than 3 % and 4%, respectively. We accepted worse values of = 3% − 4.5% for the data below 100 K. The experimental data are compared with the refinement at 300, 150 and 34 K in Figure 2b–d, respectively. As shown in Figure 2, the refinement is satisfactory; there are neither detectable impurity phases nor poly-type phases, and the deviation from the refinement seems small enough to discuss the atomic positions of the Ir atoms. We measured the diffraction patterns around carefully, and found no trace of superlattice reflections. This consolidates that the phase transition does not accompany the violation of translational symmetry, also indicating that this is unlikely to be a CDW transition.

Figure 2.
(a) synchrotron X-ray diffraction pattern of BaIrO3 at room temperature. For the details of the refinement, see text. Comparison between the observed data and the refinement at (b) 300 K; (c) 150 K; and (d) 34 K.
Figure 1a shows the transport properties of the polycrystalline sample of BaIrO3 obtained from the same batch as used in the X-ray measurement. The resistivity is non-metallic at all temperatures with an anomalous increase near = 182 K. We should note that the resistivity above is of activation type as indicated by the dotted line shown in the inset of Figure 1a. The slope of the dotted line gives a rough estimate of the activation gap around 240 K, which is small in comparison with a band gap of conventional semiconductors. This is comparable to or even smaller than , and it is nontrivial whether or not we may regard this non-metallic behavior as activation-type transport. According to the band calculation of BaIrO3 [18,19,20], the electronic states above are metallic with no gap in the density of states.
The thermopower also shows an anomaly near , below which it rapidly enhances with decreasing temperatures. As Kini et al. pointed out [14], this is a clear indication of the charge gap in the density of states [21,22], strongly indicating an insulating nature below . The thermopower takes a broad maximum near 100 K, and goes towards zero with decreasing temperature. We think this behavior is a crossover between the activation-type transport from 100 to 180 K and the hopping conduction in the impurity states below 100 K [23]. The thermopower above is difficult to interpret; the magnitude is as small as that of metals, but the temperature dependence seems of activation type just above . We evaluate an activation energy to be 90 K by assuming . Again, this activation energy is comparable or smaller than . We will come back to this point later.
Figure 1b shows the magnetization divided by an external field of 500 Oe measured in the field-cooling process. Around the same temperature of , the magnetization suddenly shows up with decreasing temperature. This ferromagnetic behavior is consistent with the previous reports [10,11,24,25]. From this figure, the magnetization at 4 K is evaluated to be around 100 emu/mol, which corresponds to a saturation magnetization of 0.016 /Ir. This unusually small moment is consistent with the previous works [10,11,24,25], and is associated with weak ferromagnetism of itinerant magnets. However, the electronic ground state of this oxide is an insulator with a finite energy gap in the density of states, and the origin of the ferromagnetism is yet to be explored.
Figure 3a–d show the lattice parameters plotted as a function of temperature obtained from the Rietveld refinement of the X-ray diffraction patterns. The relative uncertainty is less than 0.01%, and the size of the error bars are smaller than the size of the closed circles. All the parameters smoothly change with temperature across , indicating a nature of second-order transition. By having a closer look at the data in Figure 3a, one finds a slight cusp in the a-axis length near as a piece of evidence for finite electron-lattice coupling.
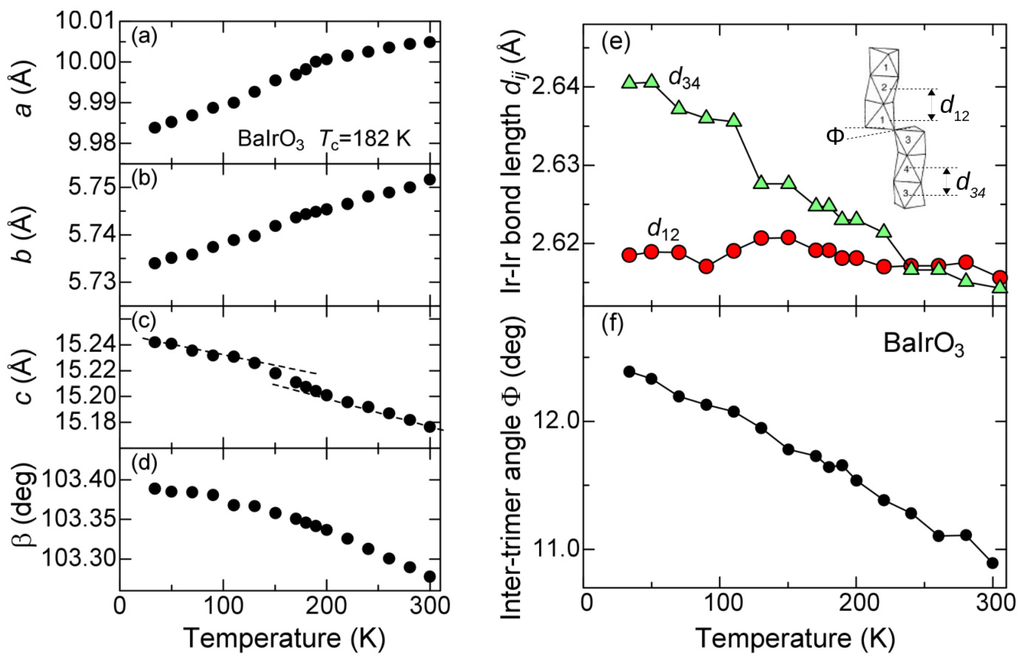
Figure 3.
(a–d) lattice parameters of BaIrO3. (a) a-; (b) b-; and (c) c-axis length; (d) monoclinic angle β; (e) The intra-timer Ir-Ir bond length and ; and (f) the inter-timer angle Φ. The inset schematically designates , and Φ.
The c-axis length parallel to the zig-zag chain of the trimers exhibits remarkable and nontrivial temperature dependence; it increases with decreasing temperature as shown in Figure 3c. Such negative temperature dependence is difficult to understand from solely lattice effects [26] except for ZrW2O8 [27]. The negative expansion is not observed in CDW materials [16,28], and can be another piece of evidence that this transition is not CDW-like, but, rather, the negative lattice expansion comes from various electronic origins [29]. Prime examples are Invar alloy [30], BaNiO3 [31] and Mn3AN (A = Cu, Zn, Ga) [32], in which electronic or magnetic instability tightly couples with the lattice. The slopes of the negative expansion in the c-axis length significantly change across as shown by the dotted lines, clearly indicating that the negative expansion characterizes the phase transition of the title compound. One may notice that the angle β also increases with decreasing temperature. Since the increase in β reduces the lattice volume, the c axis tends to expand in spite of the volume shrinkage at low temperature.
In order to clarify the structural origin of the negative expansion in the c-axis length, we examined what kind of atomic displacement is responsible, and have finally arrived at the intra-trimer Ir-Ir bond length. As schematically shown in the inset of Figure 3e, there are four crystallographically inequivalent Ir sites labeled 1, 2, 3 and 4. We define the bond length between Ir1 and Ir2 as and that between Ir3 and Ir4 as . As shown in Figure 3e, increases with decreasing temperature, whereas remains constant. This clearly indicates that the two timers are more inequivalent at lower temperatures. The data are somewhat scattered in comparison with the lattice parameters, possibly because the latter is determined only by the values of the diffraction peaks whereas the former needs accuracy in the relative intensity. Nevertheless, we emphasize that and were independently determined at each temperature within an uncertainty of 10−4, and that increasing inequivalence at low temperatures is inherent in this oxide. In addition, we have ascribed the structural origin for the temperature dependence of β to the inter-timer angle Φ as indicated at the inset of Figure 3e. Figure 3f shows Φ plotted as a function of temperature, where Φ increases with decreasing temperature. We can summarize the above results as follows: One trimer of Ir3–Ir4–Ir3 elongates with decreasing temperature, and concomitantly the lattice distorts to accept the elongation.
As mentioned above, it is difficult to determine the atomic position of the oxygen atoms from the synchrotron X-ray diffraction used in the present experiment, and this constraint prevents us from evaluating the formal valence of Ir ions using bond-valence-sum calculation. In the next section, we try to estimate the formal Ir valence by using the atomic position data for oxygen from [8]. Nevertheless, we may infer from and that the average number of d electrons in the Ir3–Ir4–Ir3 timer is larger than that in the Ir1–Ir2–Ir1 trimer, for d electrons feel the static Coulomb repulsion with one another to stay as far as possible. We further speculate that a finite fraction of charge rapidly transfers at between the two trimers, and also expect that this charge transfer is a driving force for the phase transition. In this respect, this charge transfer should be associated with the CDW scenario by Cao et al. [11]. One essential difference from the conventional CDW is that the two trimers are already inequivalent above , and this charge transfer causes no superlattice reflections with holding the same unit cell at all temperature.
4. Discussion
Having the above results in mind, let us propose here a possible electronic model for BaIrO3 starting from the localized picture. As schematically drawn in Figure 4a, one Ir ion has the electronic configuration of in the triply degenerate level, for the formal valence of Ir is . Since the oxygen octahedron is trigonally distorted, the level lifts the degeneracy to have the and doubly-degenerate levels. In the present case, the octahedron is compressed along the (111) direction of the local coordinate, and thus the level is the higher level with one electron occupied.

Figure 4.
A model for the electronic states of BaIrO3 starting from a localized electron picture. (a) the electronic configuration of Ir4+ in a regular and trigonally distorted IrO6 (left). The highest occupied orbitals of Ir3O12 trimer (center) and the molecular orbital formation (right). B, NB and AB denote bonding, non-bonding and anti-bonding orbitals, respectively; (b) the electronic configuration of Ir4+ in the strong spin-orbit coupling limit (taken from [33]); (c) the electronic states of BaIrO3 above . 1–2–1 and 3–4–3 correspond to the inequivalent Ir3O12 trimers; The case for strong spin-orbit coupling is drawn in the right side. (d) the electronic states of BaIrO3 below .
There is a strong hybridization between the levels within a trimer owing to the short intra-trimer Ir-Ir distance, and a kind of molecular orbital is expected to be formed within the trimer as shown in the right side of Figure 4a, where we denote the bonding, non-bonding and anti-bonding orbitals made of the three orbitals as B, NB and AB, respectively. By putting the three electrons from the bottom, we find one electron in the non-bonding orbital. Thus, we may regard this state as half-filling, when we identify one trimer to one pseudo-atom. In this sense, we propose that BaIrO3 is a half-filled trimer solid.
Very recently, Ir compounds have attracted extensive interest as a strong spin-orbit interaction system, in which the total angular momentum of j dominates the band structure [34]. A recent X-ray absorption study has revealed the strong spin-orbit nature in BaIrO3, and the highest occupied level should be regarded as the level rather than the level from the levels as shown in Figure 4b [33]. Even in such a case, we can make similar molecular orbitals. The highest occupied state then corresponds to the NB instead of the NB , and is unlikely to alter the picture of the half-filled trimer solid (compare the left and right sides of Figure 4c,d. As is widely accepted, half-filled electron systems is unstable against Mott insulator phase in the presence of electron-electron correlation. We propose the normal state of BaIrO3 is essentially in the Mott insulator phase, and name this “trimer Mott insulator”.
Let us discuss here about the charge transport above . According to the Landau theory of metal-insulator transition [35], there is a critical end point above which metals and insulators cannot be distinguished. In fact, the transport properties above the critical end point can be viewed as either bad metals or bad insulators, as reported in V2O3 [36] and κ-(BEDT-TTF)Cu[N(CN)2]Cl [37]. We propose that the normal state of BaIrO3 should be understood basically in terms of trimer Mott insulator, with the critical end point below . If so, the charge transport seems to show intermediate properties between metal and insulator, which are exemplified by the small activation energies of and . Zhao et al. [38] have found that external pressure increases the resistivity to enhance the insulating nature of the title compound. They further found that the transition temperature decreases with pressure. Kida et al. [25] have also found the large pressure dependence of in the magnetization measurement. Their results indicate that the pressure stabilizes the insulating normal state, being consistent with our trimer-Mott-insulator picture above . Cao et al. [39] have also stated that BaIrO3 is in the verge of a metallic state.
Figure 4c schematically shows the trimer Mott insulator phase above . The Ir1–Ir2–Ir1 trimer is in the higher level because of the shorter intra-trimer Ir–Ir bond length. All the timers have one electron in the NB or level, and the energy difference is too small to cause charge transfer. As the intra-trimer Ir–Ir bond length of the Ir3–Ir4–Ir3 trimer increases with decreasing temperature, is expected to increase. At a certain temperature, a rapid increase of and a charge transfer simultaneously happen, and the electronic states schematically drawn in Figure 4d realize below , where the NB orbital of the Ir1–Ir2–Ir1 trimer is empty and that of the Ir3–Ir4–Ir3 timer is fully occupied. From the view point of valence change, the Ir4+ state changes into a disproportionate state of Ir3.67+ and Ir4.33+ in the neighboring trimers. We regard this state as a kind of charge ordered state rather than CDW state, because this has no relation to the Fermi surface information such as nesting or [16]. We therefore propose that the transition at 180 K in BaIrO3 is a phase transition from the trimer Mott insulator phase to the trimer charge ordered phase.
We will try a rough estimate for the formal Ir valence ν using the bond valence sum calculation [40] given by , where is the Ir-O bond length in a IrO6 octahedron in units of angstrom, and 1.87 Å for Ir4+ [41]. Since the positions of oxygen atoms are not well determined in the present refinement, we have instead employed the atomic position data at 300 K from [8] and have calculated ν as listed in Table 1. The 300-K data indicate that the two trimers already have different charges; the Ir1–Ir2–Ir1 trimer has a net charge of 12.23, whereas the Ir3–Ir4–Ir3 trimer has a 0.45-shorter value of 11.78. Unlike the related ruthenate Ba4Ru3O10 [42], the charge is not disproportionated within trimer significantly. A slight difference (0.16 for Ir1 and Ir2; 0.07 for Ir3 and Ir4) may correspond to the bonding orbital widely spread within trimer, as is pointed out in [43]. For the calculation of the 16-K values, we used our data of and shown in Figure 3e, and assume that the trimers uniformly expand or shrink with temperature. Namely, we assume that the Ir-O bond lengths change with temperature in the same ratio as the Ir-Ir bond lengths. For the Ir3–Ir4–Ir3 trimer, increases from 2.615 to 2.640 Å from 300 down to 16 K, and thus the Ir-O bond lengths at 16 K equals the 300-K value multiplied by a factor of 2.640/2.615 = 1.0096, while they remain unchanged for the Ir1–Ir2–Ir1 trimer. With this assumption, ν for Ir3 and Ir4 is further reduced from the values at 300 K, as was discussed in the last paragraph of Section 3. At 16 K, the Ir1–Ir2–Ir1 trimer has a net charge of 12.23, whereas the Ir3–Ir4–Ir3 trimer has a 1.05-shorter value of 11.18. This is in a reasonable agreement with the picture in Figure 4d in spite of our rough and bold assumption.

Table 1.
Estimation of the formal Ir valence. The atomic positions for 300 K are employed from [8]. For the data at 16 K, we assume that Ir-O bond length changes with the same ratio as and . Namely, we use the same Ir-O bond lengths as at 300 K for Ir1 and Ir2, and values 0.96%-larger than at 300 K for Ir3 and Ir4.
Here, let us compare our model with band calculations reported previously [18,19,20]. Whangbo and Koo [18] have pointed out that the density of states of Ir2 and Ir4 (the central Ir ion in the trimer) show pseudo gap at the Fermi energy. This is explained in terms of molecular orbital. The highest occupied states belong to the non-bonding orbital of the or state of the Ir ion, which has a node at the center of the trimer. Note that the non-bonding orbital is expressed by , where and are the or state at the left and right edges of the trimer. The hybridization effects are also seen in other calculations [19,20], and in particular, the calculation with strong spin-orbit interaction [20] gives a small Mott gap which is consistent with the observed small activation energies of and . Since the band calculation was based on the structure at room temperature, the calculated insulating states should be those above .
Our proposed model can explain various experimental results at least semi-quantitatively. It explains bad metal/bad insulator behavior above , a charge gap below without translational-symmetry breaking, and the transition susceptible to external pressure. Since one electron in the NB or state per trimer is responsible for magnetism, we can understand the small effective magnetic moment of 0.16 per Ir above [11,24]. The transition is basically an insulator-insulator transition, which can give anomalous critical exponents [24]. This transition looks similar to the neutral-ionic transition in the charge transfer organic complexes showing non-linear conduction [44,45]. BaIrO3 also shows giant non linear conduction at low temperature [13], the origin of which may be compared with the organic complexes. Within our model, however, the weak ferromagnetism is difficult to understand, for all the trimers are nonmagnetic below . The ideal charge transfer of e in Figure 4c,d may not occur in real materials, but only a fraction of e may move between the trimers. In such a case, all the trimers can be slightly magnetic and may show ferromagnetism with the much smaller moment of 0.016 below .
It would be fair to point out controversial points and limitations of our model. First, Figure 4 is an oversimplified picture where transfer energies except for the intra-timer case are neglected. In a real material, the complicated band structure may invalidate our model, although the band calculations reported so far do not always explain the experimental results satisfactorily. Secondly, the strong correlation, i.e., the on-site Coulomb repulsion forces the electron density to be as homogeneous as possible and disfavors the charge transfer from one trimer to another. The proposed charge order may come from a delicate balance among the on-site Coulomb repulsion, the energy difference between the two timers, and the transfer energies. A more complete theoretical modeling is needed to advance further. Third, IrO6 octahedra are distorted so that there are no pure and orbitals in a strict sense. Accordingly the molecular orbital such as NB may be misleading. Even in such a case, one hole on Ir4+ of occupies the non-degenerate, highly occupied orbital (say for Iri). Then, we can make linear combinations from and for one Ir trimer, from and for the other trimer. Again we can construct a one electron in a corresponding non-bonding-like state for one trimer, and find the essence of our model left unchanged.
To examine the validity of our model, we have investigated the impurity effects on the 182-K transition for a set of polycrystalline samples of BaIr1−xRuxO3. Figure 5 shows the magnetization and the resistivity of the samples. The phase transition is systematically and drastically suppressed with the Ru concentration x, and essentially vanishes at only 10% substitution; For , the resistivity is non-metallic without any trace of anomalies at all temperatures measured, whereas a tiny trace of the weak ferromagnetic phase of is visible in the magnetization measurement. The resistivity for indicates the electronic states above is essentially insulating, being consistent with the picture of the trimer Mott insulator. The inset of Figure 5a shows the intra-trimer Ir-Ir bond lengths for the sample, where the two lengths of and are essentially temperature-independent, and identical. Concomitantly the negative expansion of the c-axis is severely suppressed (not shown). The Ru substitution simultaneously suppresses the 182 K-transition and the electron transfer from Ir1–Ir2–Ir1 to Ir3–Ir4–Ir3, and therefore these two phenomena are two sides of the same coin. We further note that the charge order is known to be vulnerable to impurities [46], while the charge density wave is more or less robust against impurities [47].
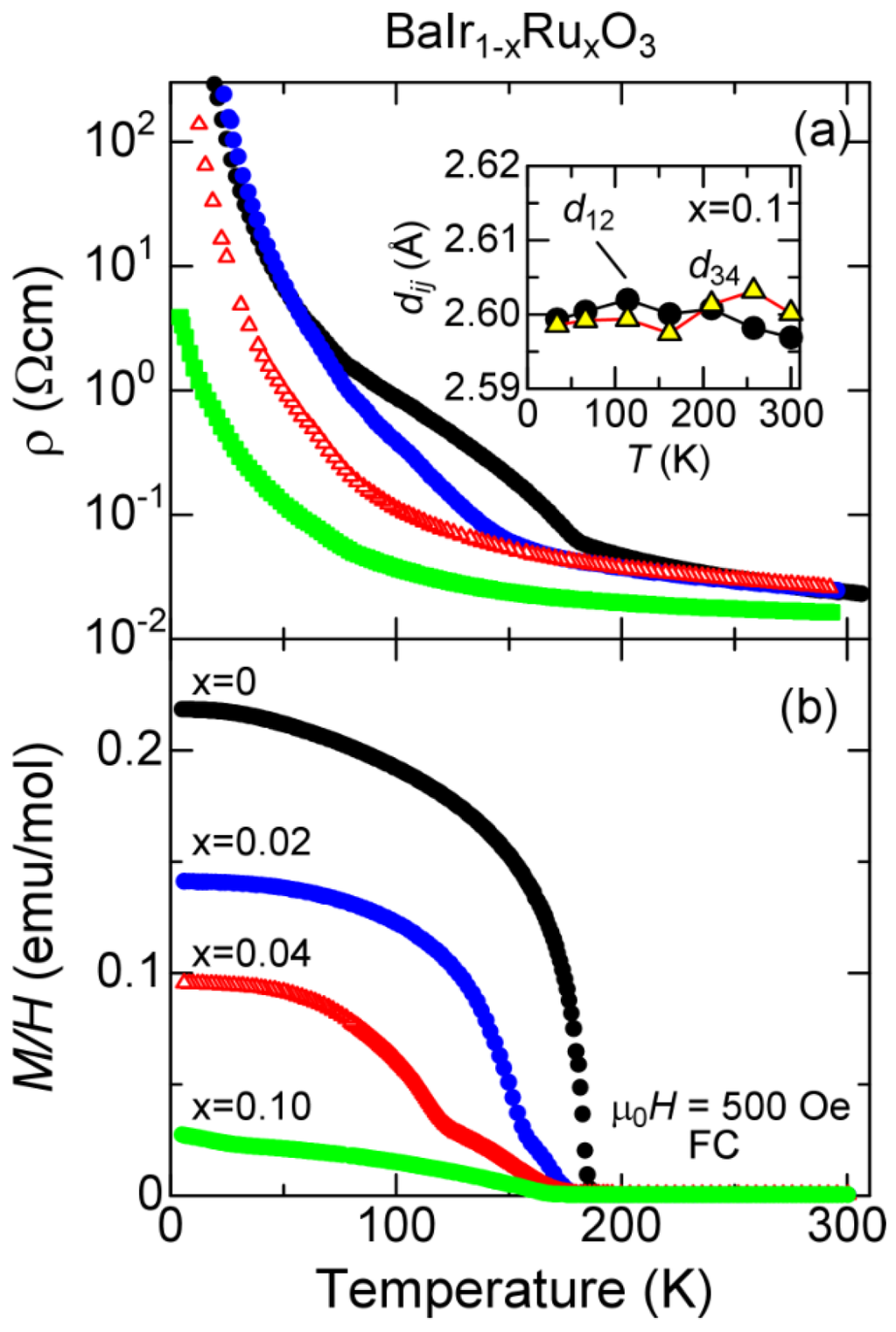
Figure 5.
The impurity effect on the 182-K transition. (a) resistivity ρ of polycrystalline samples of BaIr1−xRuxO3. The inset shows the intra-trimer Ir-Ir bond lengths for the sample. The two trimers have the identical, temperature-independent bond length; (b) magnetization M divided by an external field H of 500 Oe of the same samples.
A similar type of the phase transition occurs in the organic salt β-(meso-DMBEDT-TTF)2PF6 [48]. Many conductive organic salts with a chemical formula of consists of a dimer molecule of as a composition unit. In the present case, two molecules of meso-DMBEDT-TTF form a dimer structure, and share one hole per dimer to behave as dimer Mott insulators [49]. Okazaki et al. [50] identified the phase transition in this compound at 70 K to a transition from dimer Mott insulator phase to charge ordered insulator phase. This results from a competition between the intra- and inter-dimer Coulomb interactions, which causes intrinsic inhomogeneous state detected through infrared microscope measurement. As is similar to BaIrO3, this organic salt also shows non-linear conduction below [51].
Finally, we briefly comment that we have applied a similar molecular orbital concept to the related timer Ru oxide Ba4Ru3O10 in which the three face-shared RuO6 octahedra form a Ru3O12 trimer [22,42,52]. This related oxide shows a phase transition at 105 K, where a metallic, paramagnetic state changes into an insulating, antiferromagnetic state at low temperature. However, the electronic states are different; In this related oxide, each trimer is three electrons short because Ru4+ has four electrons. On the basis of a similar molecular orbital approach, we have to consider partially occupied orbitals. In this case, the static electronic potential is deeper at the center of the trimer, and a finite fraction of charge is expected to transfer from the edge. Namely, intra-timer charge transfer drives the phase transition in Ba4Ru3O10. This makes a stark contrast to the inter-trimer charge transfer in BaIrO3. The intra-trimer charge disproportionation has been also discussed from the local-density-approximation calculations [43,53].
5. Conclusions
In summary, we have prepared polycrystalline samples of the trimer oxide BaIrO3 with the face-shared Ir3O12 trimers, and have measured the resistivity, thermopower, magnetization and synchrotron X-ray diffraction from room temperature down to 4 K. On the basis of the structure analysis, we have made a model for the electronic states of this oxide and have pointed out that this oxide can be regarded as a kind of Mott insulator, when the Ir3O12 trimer is identified to one lattice site. Within the same framework, the 182-K transition can be viewed as a transition from the trimer Mott insulator phase to the trimer charge-ordered phase, which is essentially different from conventional charge density wave as was used to be a candidate for the transition. We have further succeeded in explaining various properties above such as the non-metallic resistivity and thermopower, the large pressure effect, and the unusually small effective magnetic moment. We expect that the proposed concept of trimer solid will be applied to other face-shared transition metal oxides.
Acknowledgments
The authors would like to thank Mike Whangbo for showing unpublished data for the electronic states of BaIrO3. They are indebted to T. Nakano, T. Kida, M. Hagiwara, Y. Nogami, S. Mori for collaboration at an early stage of this work. They also wish to appreciate J. Akimitsu, T. Arima, S.-W. Cheong, M. Tsuchiizu for fruitful discussion and useful advice. This work was partially supported by Grant-in-Aid for Scientific Research, Japan Society for the Promotion of Science, Japan (Kakenhi No. 25610091, 26247060), and by the Program for Leading Graduate Schools “Integrative Graduate Education and Research in Green Natural Sciences”, MEXT, Japan. The synchrotron X-ray diffraction was performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2012G718 and 2012S2-005).
Author Contributions
Ichiro Terasaki conducted the whole research project and considered the electronic states of the title compound. Shun Ito made samples, and measured and analyzed all the quantities including the Rietveld refinement. Taichi Igarashi, Shinichiro Asai, Kensuke Kobayashi, Reiji Kumai, Hironori Nakao and Youichi Murakami supported the measurements of synchrotron X-ray diffraction at KEK. Hiroki Taniguchi, Ryuji Okazaki and Yukio Yasui joined the research project by mainly discussing the results and by supervising Shun Ito as a group staff member.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Furukawa, N.; Imada, M. Charge Mass Singularity in Two-Dimensional Hubbard Model. J. Phys. Soc. Jpn. 1993, 62, 2557–2560. [Google Scholar] [CrossRef]
- Terasaki, I.; Tsukada, I.; Iguchi, Y. Impurity-induced transition and impurity-enhanced thermopower in the thermoelectric oxide NaCo2−xCuxO4. Phys. Rev. B 2002, 65, 195106. [Google Scholar] [CrossRef]
- Donohue, P.C.; Katz, L.; Ward, R. The Crystal Structure of Barium Ruthenium Oxide and Related Compounds. Inorg. Chem. 1965, 4, 306–310. [Google Scholar] [CrossRef]
- Gai, P.L.; Jacobson, A.J.; Rao, C.N.R. Investigation of the structure of barium iridate (BaIrO3) by high-resolution electron microscopy. Inorg. Chem. 1976, 15, 480–483. [Google Scholar] [CrossRef]
- Chamberland, B. A study on the BaIrO3 system. J. Less Common Met. 1991, 171, 377–394. [Google Scholar] [CrossRef]
- Cheng, J.G.; Zhou, J.S.; Alonso, J.A.; Goodenough, J.B.; Sui, Y.; Matsubayashi, K.; Uwatoko, Y. Transition from a weak ferromagnetic insulator to an exchange-enhanced paramagnetic metal in the BaIrO3 polytypes. Phys. Rev. B 2009, 80, 104430. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, L.; Yu, Y.; Li, F.; Yu, R.; Jin, C. Structural and physical properties evolution of BaIr1−xMnxO3 solid solutions synthesized by high-pressure sintering. J. Solid State Chem. 2010, 183, 720–726. [Google Scholar] [CrossRef]
- Siegrist, T.; Chamberland, B. The crystal structure of BaIrO3. J. Less Common Met. 1991, 170, 93–99. [Google Scholar] [CrossRef]
- Powell, A.V.; Battle, P.D. The electronic properties of non-stoichiometric barium iridate, BaIrO3−δ. J. Alloys Comp. 1993, 191, 313–318. [Google Scholar] [CrossRef]
- Lindsay, R.; Strange, W.; Chamberland, B.; Moyer, R. Weak ferromagnetism in BaIrO3. Solid State Commun. 1993, 86, 759–763. [Google Scholar] [CrossRef]
- Cao, G.; Crow, J.; Guertin, R.; Henning, P.; Homes, C.; Strongin, M.; Basov, D.; Lochner, E. Charge density wave formation accompanying ferromagnetic ordering in quasi-one-dimensional BaIrO3. Solid State Commun. 2000, 113, 657–662. [Google Scholar] [CrossRef]
- Brooks, M.L.; Blundell, S.J.; Lancaster, T.; Hayes, W.; Pratt, F.L.; Frampton, P.P.C.; Battle, P.D. Unconventional magnetic properties of the weakly ferromagnetic metal BaIrO3. Phys. Rev. B 2005, 71, 220411. [Google Scholar] [CrossRef]
- Nakano, T.; Terasaki, I. Giant nonlinear conduction and thyristor-like negative differential resistance in BaIrO3 single crystals. Phys. Rev. B 2006, 73, 195106. [Google Scholar] [CrossRef]
- Kini, N.; Bentien, A.; Ramakrishnan, S.; Geibel, C. Specific heat and transport study of the co-existence of charge-density-wave and weak ferromagnetism in BaIrO3. Physica B 2005, 359–361, 1264–1266. [Google Scholar] [CrossRef]
- Maiti, K.; Singh, R.S.; Medicherla, V.R.R.; Rayaprol, S.; Sampathkumaran, E.V. Origin of Charge Density Wave Formation in Insulators from a High Resolution Photoemission Study of BaIrO3. Phys. Rev. Lett. 2005, 95, 016404. [Google Scholar] [CrossRef] [PubMed]
- Grüner, G. The dynamics of charge-density waves. Rev. Mod. Phys. 1988, 60, 1129–1181. [Google Scholar] [CrossRef]
- Izumi, F.; Momma, F. Three-Dimensional Visualization in Powder Diffraction. Solid State Phenom. 2007, 130, 15–20. [Google Scholar] [CrossRef]
- Whangbo, M.H.; Koo, H.J. Structural and electronic features of BaIrO3 causing the simultaneous occurrence of weak ferromagnetism and charge density wave formation. Solid State Commun. 2001, 118, 491–495. [Google Scholar] [CrossRef]
- Maiti, K. Electronic structure of BaIrO3: A first-principles study using the local spin density approximation. Phys. Rev. B 2006, 73, 115119. [Google Scholar] [CrossRef]
- Ju, W.; Liu, G.Q.; Yang, Z. Exotic spin-orbital Mott insulating states in BaIrO3. Phys. Rev. B 2013, 87, 075112. [Google Scholar] [CrossRef]
- Sharath Chandra, L.; Lakhani, A.; Gangrade, M.; Ganesan, V. Impurity band conduction in FeSi1−xAlx. J. Phys. Condens. Matter 2008, 20, 325238. [Google Scholar] [CrossRef]
- Klein, Y.; Rousse, G.; Damay, F.; Porcher, F.; André, G.; Terasaki, I. Antiferromagnetic order and consequences on the transport properties of Ba4Ru3O10. Phys. Rev. B 2011, 84, 054439. [Google Scholar] [CrossRef]
- Takahashi, H.; Okazaki, R.; Yasui, Y.; Terasaki, I. Low-temperature magnetotransport of the narrow-gap semiconductor FeSb2. Phys. Rev. B 2011, 84, 205215. [Google Scholar] [CrossRef]
- Kida, T.; Senda, A.; Yoshii, S.; Hagiwara, M.; Takeuchi, T.; Nakano, T.; Terasaki, I. Unconventional critical behavior in the weak ferromagnet BaIrO3. EPL 2008, 84, 27004. [Google Scholar] [CrossRef]
- Kida, T.; Senda, A.; Yoshii, S.; Hagiwara, M.; Nakano, T.; Terasaki, I. Pressure effect on magnetic properties of a weak ferromagnet BaIrO3. J. Phys. Conf. Ser. 2010, 200, 012084. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 7th ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Mary, T.A.; Evans, J.S.O.; Vogt, T.; Sleight, A.W. Negative Thermal Expansion from 0.3 to 1050 Kelvin in ZrW2O8. Science 1996, 272, 90–92. [Google Scholar] [CrossRef]
- Tian, M.; Chen, L.; Zhang, Y. Temperature dependence of the structural parameters of the host lattice in blue bronze K0.3MoO3. Phys. Rev. B 2000, 62, 1504–1507. [Google Scholar] [CrossRef]
- Takenaka, K. Negative thermal expansion materials: Technological key for control of thermal expansion. Sci. Technol. Adv. Mater. 2012, 13, 013001. [Google Scholar] [CrossRef]
- Colling, D.A.; Carr, W.J. Invar Anomaly. J. Appl. Phys. 1970, 41, 5125–5129. [Google Scholar] [CrossRef]
- Ishiwata, S.; Azuma, M.; Hanawa, M.; Moritomo, Y.; Ohishi, Y.; Kato, K.; Takata, M.; Nishibori, E.; Sakata, M.; Terasaki, I.; et al. Pressure/temperature/substitution-induced melting of A-site charge disproportionation in Bi1−xLaxNiO3 (0 ≤ x ≤ 0.5). Phys. Rev. B 2005, 72, 045104. [Google Scholar] [CrossRef]
- Takenaka, K.; Takagi, H. Giant negative thermal expansion in Ge-doped anti-perovskite manganese nitrides. Appl. Phys. Lett. 2005, 87, 261902. [Google Scholar] [CrossRef]
- Laguna-Marco, M.A.; Haskel, D.; Souza-Neto, N.; Lang, J.C.; Krishnamurthy, V.V.; Chikara, S.; Cao, G.; van Veenendaal, M. Orbital Magnetism and Spin-Orbit Effects in the Electronic Structure of BaIrO3. Phys. Rev. Lett. 2010, 105, 216407. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Jin, H.; Moon, S.J.; Kim, J.Y.; Park, B.G.; Leem, C.S.; Yu, J.; Noh, T.W.; Kim, C.; Oh, S.J.; et al. Novel Jeff = 1/2 Mott State Induced by Relativistic Spin-Orbit Coupling in Sr2IrO4. Phys. Rev. Lett. 2008, 101, 076402. [Google Scholar] [CrossRef] [PubMed]
- Kotliar, G.; Lange, E.; Rozenberg, M.J. Landau Theory of the Finite Temperature Mott Transition. Phys. Rev. Lett. 2000, 84, 5180–5183. [Google Scholar] [CrossRef] [PubMed]
- Limelette, P.; Georges, A.; Jérome, D.; Wzietek, P.; Metcalf, P.; Honig, J.M. Universality and Critical Behavior at the Mott Transition. Science 2003, 302, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, F.; Itou, T.; Miyagawa, K.; Kanoda, K. Transport criticality of the first-order Mott transition in the quasi-two-dimensional organic conductor κ-BEDT-TTF2Cu[N(CN)2]Cl. Phys. Rev. B 2004, 69, 064511. [Google Scholar] [CrossRef]
- Zhao, J.; Yang, L.; Mydeen, K.; Li, F.; Yu, R.; Jin, C. Effects of pressure on electrical property of BaIrO3. Solid State Commun. 2008, 148, 361–364. [Google Scholar] [CrossRef]
- Cao, G.; Lin, X.N.; Chikara, S.; Durairaj, V.; Elhami, E. High-temperature weak ferromagnetism on the verge of a metallic state: Impact of dilute Sr doping on BaIrO3. Phys. Rev. B 2004, 69, 174418. [Google Scholar] [CrossRef]
- Brown, I.D. Recent Developments in the Methods and Applications of the Bond Valence Model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed]
- Bond valence parameters. Available online: http://www.iucr.org/resources/data/datasets/bond-valence-parameters (accessed on 16 January 2016).
- Igarashi, T.; Nogami, Y.; Klein, Y.; Rousse, G.; Okazaki, R.; Taniguchi, H.; Yasui, Y.; Terasaki, I. X-ray Crystal Structure Analysis and Ru Valence of Ba4Ru3O10 Single Crystals. J. Phys. Soc. Jpn. 2013, 82, 104603. [Google Scholar] [CrossRef]
- Streltsov, S.V.; Khomskii, D.I. Unconventional magnetism as a consequence of the charge disproportionation and the molecular orbital formation in Ba4Ru3O10. Phys. Rev. B 2012, 86, 064429. [Google Scholar] [CrossRef]
- Torrance, J.B.; Vazquez, J.E.; Mayerle, J.J.; Lee, V.Y. Discovery of a Neutral-to-Ionic Phase Transition in Organic Materials. Phys. Rev. Lett. 1981, 46, 253–257. [Google Scholar] [CrossRef]
- Tokura, Y.; Okamoto, H.; Koda, T.; Mitani, T.; Saito, G. Nonlinear electric transport and switching phenomenon in the mixed-stack charge-transfer crystal tetrathiafulvalene-p-chloranil. Phys. Rev. B 1988, 38, 2215–2218. [Google Scholar] [CrossRef]
- Tatami, N.; Ando, Y.; Niioka, S.; Kira, H.; Onodera, M.; Nakao, H.; Iwasa, K.; Murakami, Y.; Kakiuchi, T.; Wakabayashi, Y.; et al. Orbital ordering and the dilute effect in copper fluoride. J. Mag. Mag. Mater. 2007, 310, 787–789. [Google Scholar] [CrossRef]
- Kawabata, K. Impurity Effects on Superconductivity and Charge Density Waves in NbSe3. J. Phys. Soc. Jpn. 1985, 54, 762–770. [Google Scholar] [CrossRef]
- Kimura, S.; Suzuki, H.; Maejima, T.; Mori, H.; Yamaura, J.I.; Kakiuchi, T.; Sawa, H.; Moriyama, H. Checkerboard-Type Charge-Ordered State of a Pressure-Induced Superconductor, β-(meso-DMBEDT-TTF)2PF6. J. Am. Chem. Soc. 2006, 128, 1456–1457. [Google Scholar]
- Kino, H.; Fukuyama, H. Electronic States of Conducting Organic κ-(BEDT-TTF)2X. J. Phys. Soc. Jpn. 1995, 64, 2726–2729. [Google Scholar] [CrossRef]
- Okazaki, R.; Ikemoto, Y.; Moriwaki, T.; Shikama, T.; Takahashi, K.; Mori, H.; Nakaya, H.; Sasaki, T.; Yasui, Y.; Terasaki, I. Optical Conductivity Measurement of a Dimer Mott-Insulator to Charge-Order Phase Transition in a Two-Dimensional Quarter-Filled Organic Salt Compound. Phys. Rev. Lett. 2013, 111, 217801. [Google Scholar] [CrossRef] [PubMed]
- Niizeki, S.; Yoshikane, F.; Kohno, K.; Takahashi, K.; Mori, H.; Bando, Y.; Kawamoto, T.; Mori, T. Dielectric Response and Electric-Field-Induced Metastable State in an Organic Conductor β-(meso-DMBEDT-TTF)2PF6. J. Phys. Soc. Jpn. 2008, 77, 073710. [Google Scholar] [CrossRef]
- Igarashi, T.; Okazaki, R.; Taniguchi, H.; Yasui, Y.; Terasaki, I. Effects of the Ir Impurity on the Thermodynamic and Transport Properties of Ba4Ru3O10. J. Phys. Soc. Jpn. 2015, 84, 094601. [Google Scholar] [CrossRef]
- Radtke, G.; Saúl, A.; Klein, Y.; Rousse, G. Magnetism of Ba4Ru3O10 revealed by density functional calculations: Structural trimers behaving as coupled magnetic dimers. Phys. Rev. B 2013, 87, 054436. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).