Polyoxotungstates in Molecular Boxes of Purine Bases


Abstract
:
1. Introduction
2. Results and Discussion
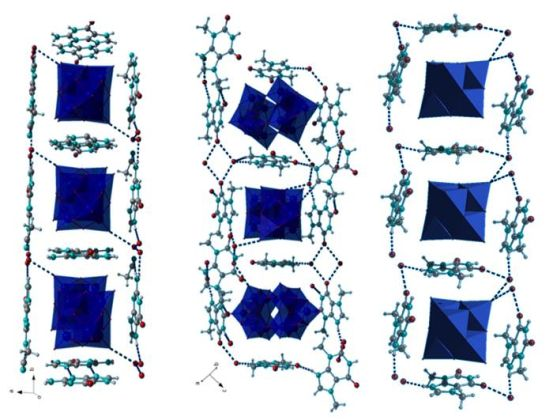
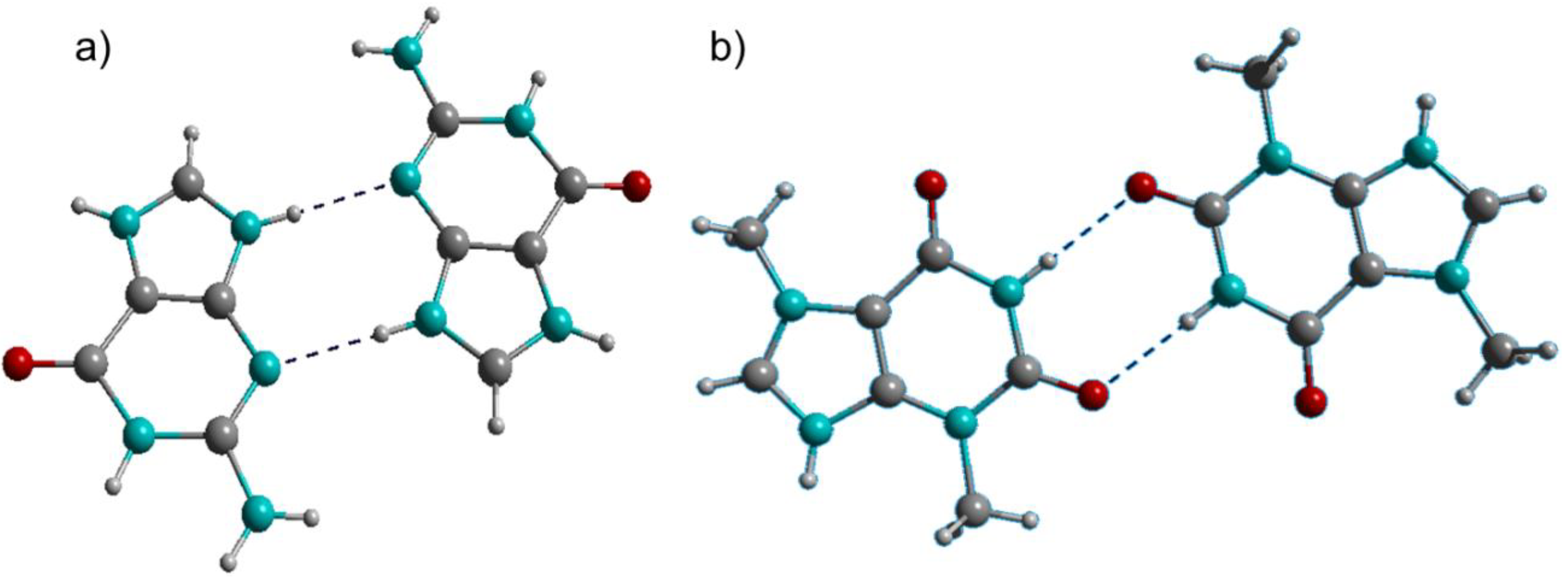
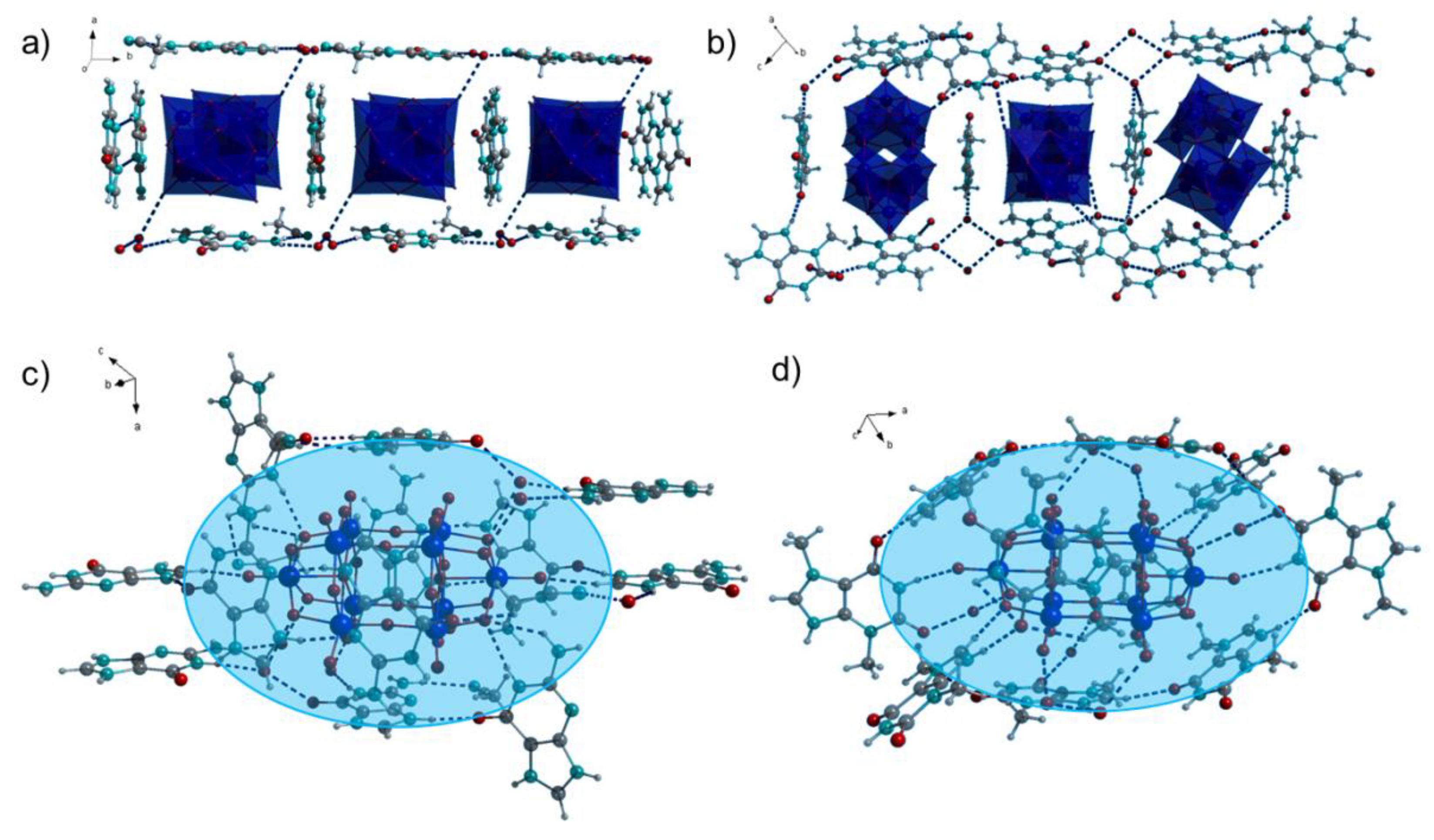
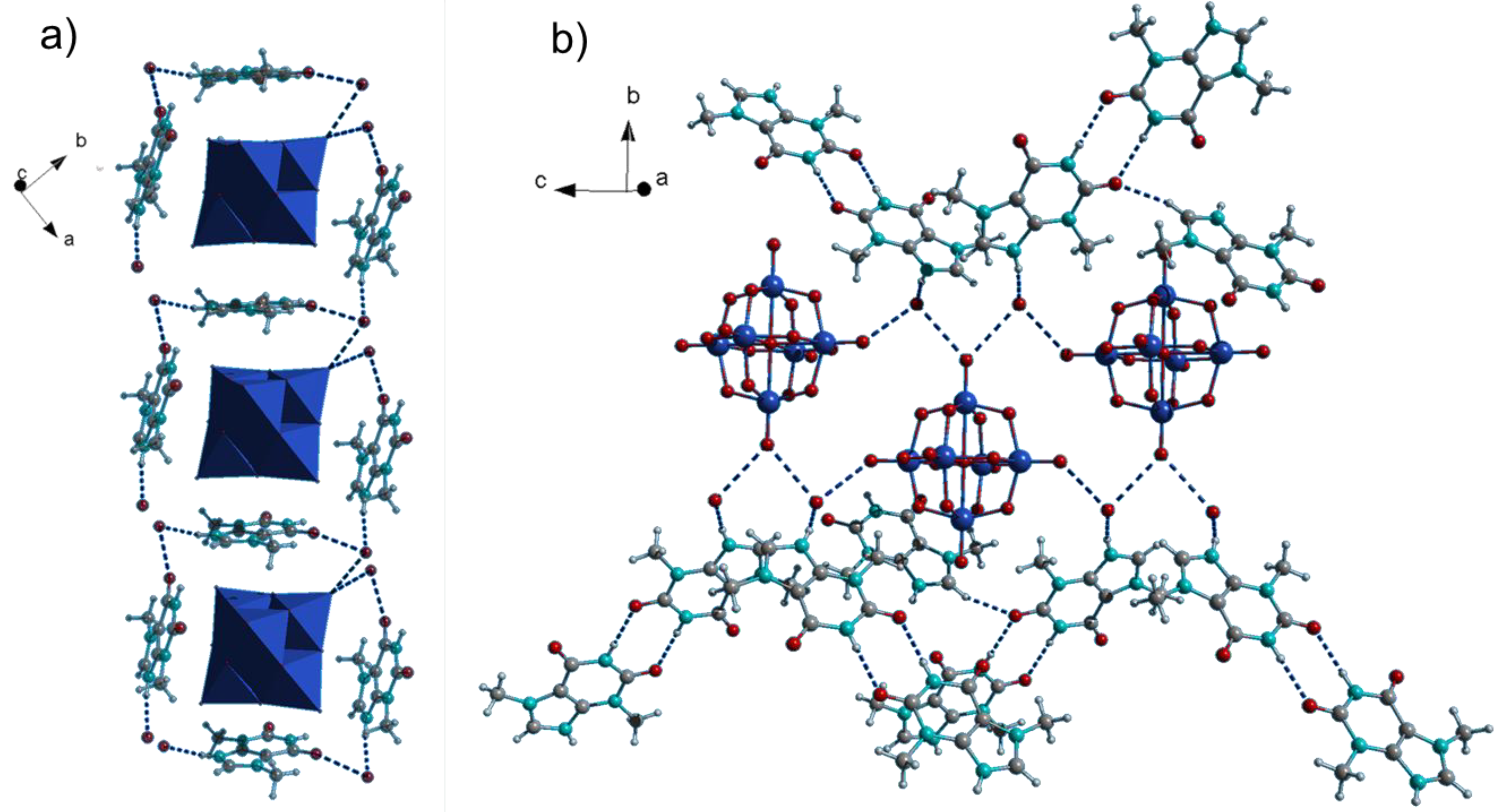
2.1. Crystal Structures
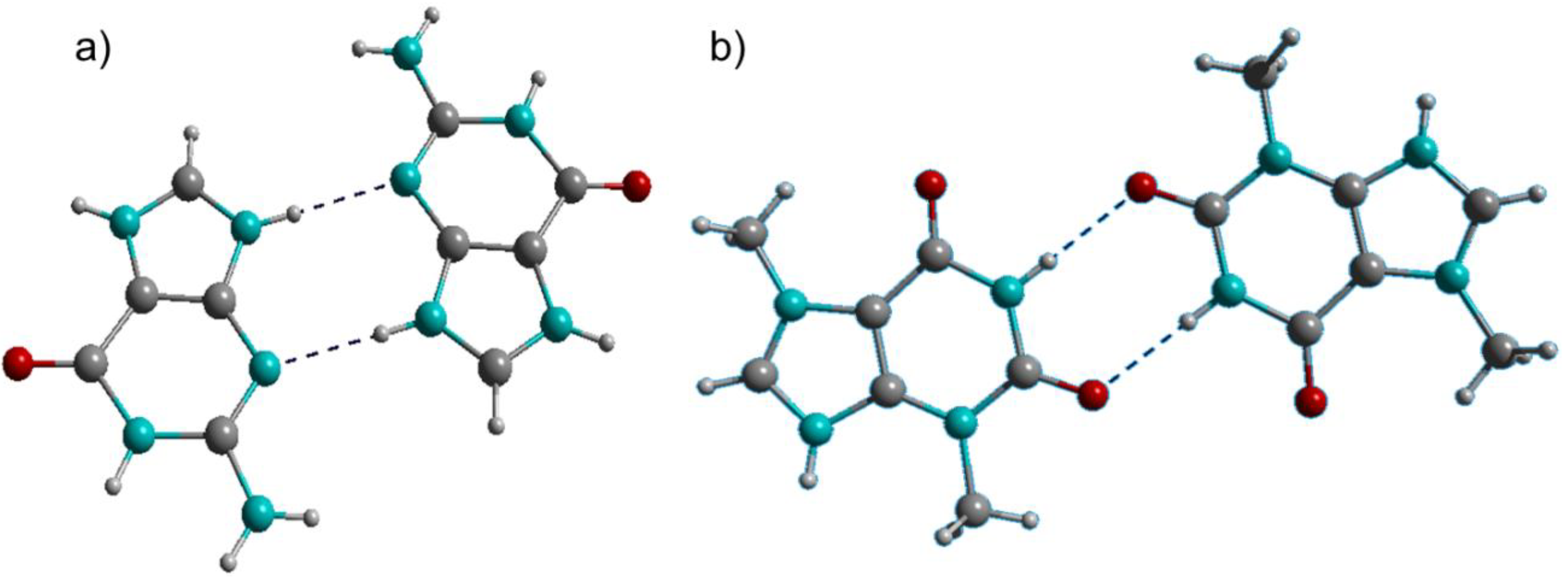
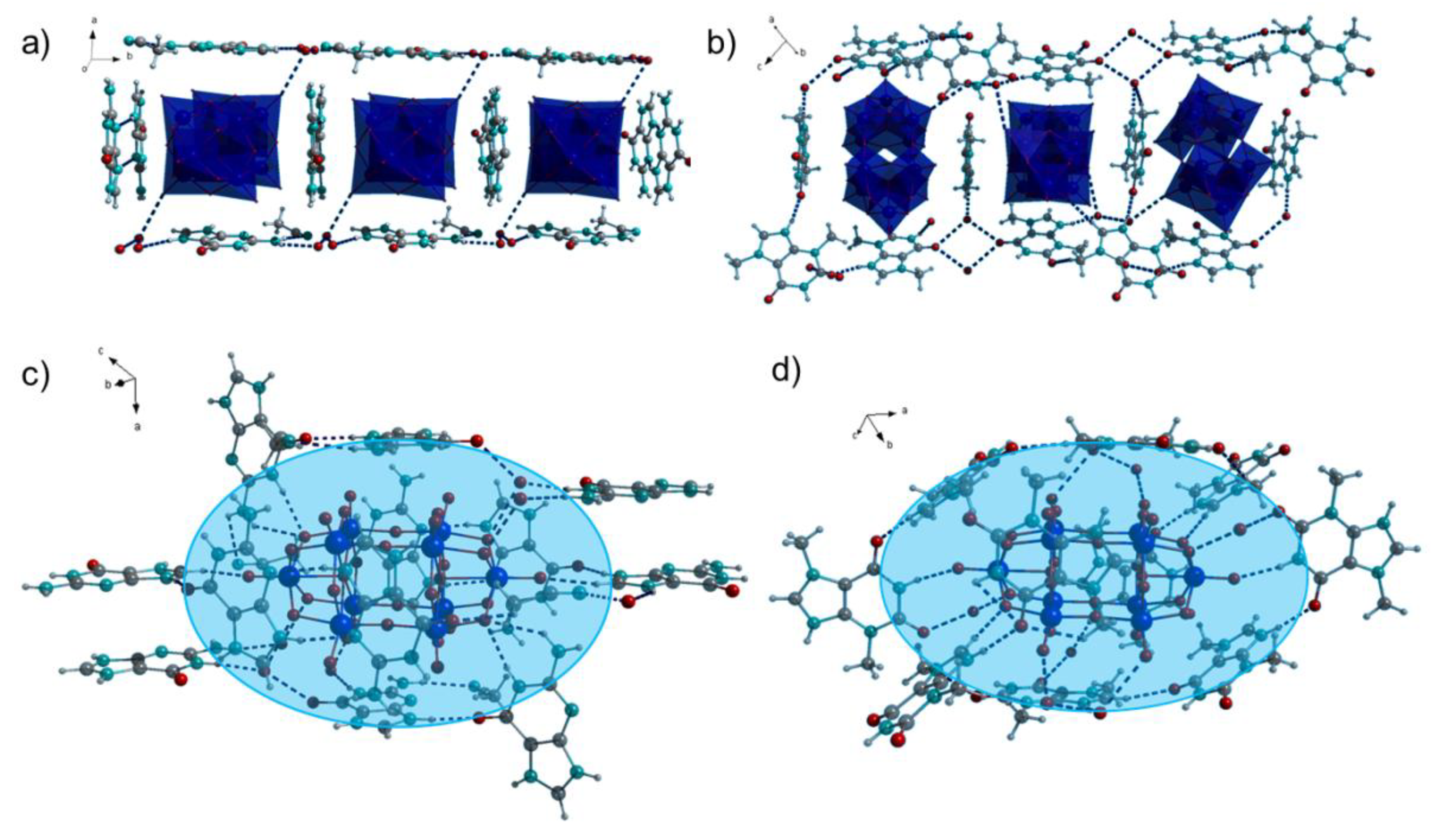
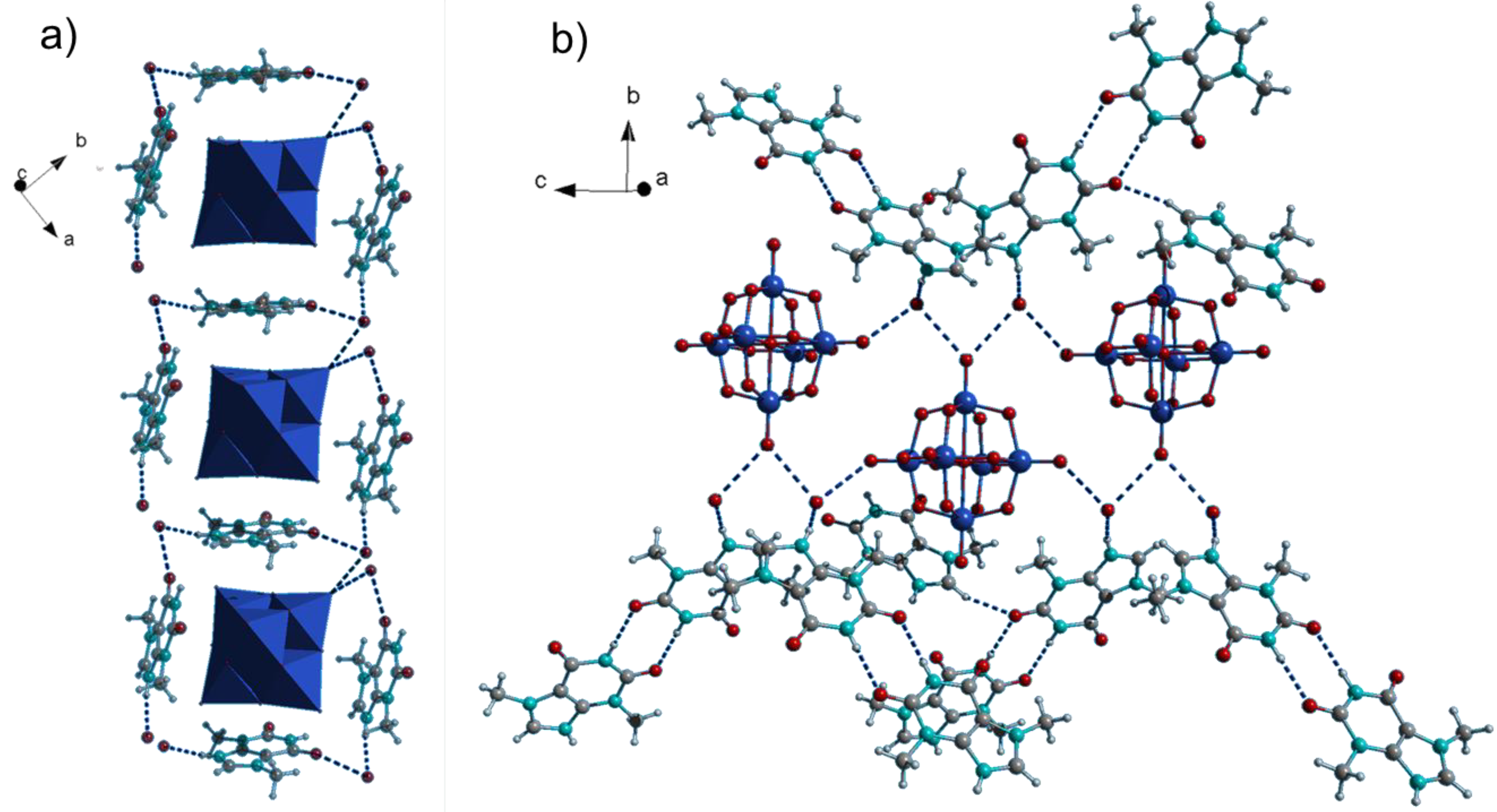
2.2. IR Spectroscopy
2.3. Thermal Properties
3. Experimental Section

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| empirical formula | C22H37N20O41W10 | C21H45N12O46.5W10 | C14H22N8O25W6 |
| M (g/mol) | 3076.2 | 3048.2 | 1805.47 |
| crystal system | monoclinic | triclinic | monoclinic |
| space group | P 21/c | P 1 | C 2/c |
| a (Å) | 14.477 (1) | 13.491 (1) | 16.810 (3) |
| b (Å) | 10.8678 (9) | 15.171 (1) | 13.051 (2) |
| c (Å) | 21.627 (2) | 17.069 (2) | 15.676 (3) |
| α (°) | 90 | 79.594 (8) | 90 |
| β (°) | 122.780 (6) | 89.386 (8) | 109.73 (1) |
| γ (°) | 90 | 70.001 (7) | 90 |
| ρcalc. (g/cm3) | 3.526 | 3.057 | 3.6960 |
| V (Å3) | 2860.8 (4) | 3223.8 (6) | 3237.2 (9) |
| Z | 2 | 2 | 4 |
| μ(MoKα) (mm−1) | 20.131 | 17.86 | 21.35 |
| T (K) | 170 (2) | 293 (2) | 293 (2) |
| reflns measured | 33411 | 41410 | 21383 |
| independent reflns | 6202 | 14242 | 3590 |
| parameters | 443 | 767 | 242 |
| R1 (I > 4σ) | 0.0434 | 0.0347 | 0.0361 |
| R1 (all data) | 0.0612 | 0.0590 | 0.1037 |
| wR2 (all data) | 0.1316 | 0.0950 | 0.0745 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pope, M.; Müller, A. Polyoxometalate Chemistry from Topology via Self-Assembly to Applications, 1st ed.; Springer: Dordrecht, The Netherlands, 2001. [Google Scholar]
- Gouzerh, P.; Che, M. From Scheele and Berzelius to Müller: Polyoxometalates (POMs) revisited and the “missing link” between the bottom up and top down approaches. L’Actualité Chim. 2006, 298, 9–21. [Google Scholar]
- Long, D.-L.; Tsunashima, R.; Cronin, L. Polyoxometalates: Building blocks for functional nanoscale systems. Angew. Chem. Int. Ed. 2010, 49, 1736–1758. [Google Scholar] [CrossRef]
- Yamase, T.; Pope, M.T. Polyoxometalate Chemistry for Nano-Composite Design (Nanostructure Science and Technology); Kluwer Academic/Plenum Publishers: New York, NY, USA, 2002. [Google Scholar]
- Symes, M.D.; Cronin, L. Decoupling hydrogen and oxygen evolution during electrolytic water splitting using an electron-coupled-proton buffer. Nat. Chem. 2013, 5, 403–409. [Google Scholar] [CrossRef]
- Long, D.-L.; Abbas, H.; Kögerler, P.; Cronin, L. A high-nuclearity “celtic-ring” isopolyoxotungstate, [H12W36O120]12−, that captures trace potassium ions. J. Am. Chem. Soc. 2004, 126, 13880–13881. [Google Scholar] [CrossRef]
- Miras, H.N.; Yan, J.; Long, D.-L.; Cronin, L. Structural evolution of “s”-shaped [H4W22O74]12− and “§”-shaped [H12W34O116]18− isopolyoxotungstate clusters. Angew. Chem. Int. Ed. 2008, 47, 8420–8423. [Google Scholar] [CrossRef]
- Ito, T.; Fujimoto, N.; Uchida, S.; Iijima, J.; Naruke, H.; Mizuno, N. Polyoxotungstate-surfactant layered crystal toward conductive inorganic-organic hybrid. Crystals 2012, 2, 362–373. [Google Scholar] [CrossRef]
- Rhule, J.T.; Hill, C.L.; Judd, D.A.; Schinazi, R.F. Polyoxometalates in medicine. Chem. Rev. 1998, 98, 327–358. [Google Scholar] [CrossRef]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; John Wiley & Sons, Inc.: San Francisco, CA, USA, 2011. [Google Scholar]
- Kortz, U.; Savelieff, M.G.; Ghali, F.Y.A.; Khalil, L.M.; Maalouf, S.A.; Sinno, D.I. Heteropolymolybdates of AsIII, SbIII, BiIII, SeIV, and TeIV functionalized by amino acids. Angew. Chem. Int. Ed. 2002, 41, 4070–4073. [Google Scholar] [CrossRef]
- Biradha, K.; Samai, S.; Maity, A.C.; Goswami, S. Supramolecular assembly of protonated xanthine alkaloids in their perchlorate salts. Cryst. Growth Des. 2010, 10, 937–942. [Google Scholar] [CrossRef]
- Zhai, H.; Liu, S.; Peng, J.; Hu, N.; Jia, H. Synthesis, crystal structure, and thermal property of a novel supramolecular assembly: (NH4)2(C8H10N4O2)4[H4V10O28]·2H2O, constructed from decavanadate and caffeine. J. Chem. Crystallogr. 2004, 34, 541–548. [Google Scholar] [CrossRef]
- Ng, S.W. Interpretation of Diammonium Decavanadate(V)-Tetra(caffeine) Dihydrate as Tetra(caffeinium) Decavanadate(V) Tetrahydrate. J. Chem. Crystallogr. 2008, 38, 483. [Google Scholar] [CrossRef]
- Inoue, M.; Yamase, T. Crystal structure of the pentamolybdate complex coordinated by adenosine-5'-monophosphoric acid. Bull. Chem. Soc. Jpn. 1996, 69, 2863–2868. [Google Scholar] [CrossRef]
- Kulikov, V.; Meyer, G. A new strategy for the synthetic assembly of inorganic–organic silver(I)-polyoxometalate hybrid structures employing noncovalent interactions between theobromine ligands. Cryst. Growth Des. 2013, 13, 2916–2927. [Google Scholar] [CrossRef]
- Kulikov, V.; Meyer, G. Hexa- to octamolybdate rearrangement leads to the new coordination polymer [Ag(PhCN)(thb)]4[β-Mo8O26](PhCN)2. Z. Anorg. Allg. Chem. 2014, 640, 19–22. [Google Scholar] [CrossRef]
- Hoogsten, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Cryst. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Egli, M.; Sarkhel, S. Lone pair-aromatic interactions: To stabilize or not to stabilize. Acc. Chem. Res. 2007, 40, 197–205. [Google Scholar] [CrossRef]
- Fuchs, J.; Hartl, H.; Schiller, W.; Gerlach, U. Die Kristallstruktur des Tributylammoniumdekawolframats [(C4H9)2NH]4W10O23. Acta Cryst. B 1976, 32, 740–749. [Google Scholar] [CrossRef]
- Poblet, J.M.; Lopez, X.; Bo, C. Ab initio and DFT modelling of complex materials: Towards the understanding of electronic and magnetic properties of polyoxometalates. Chem. Soc. Rev. 2003, 32, 297–308. [Google Scholar] [CrossRef]
- Fernández, J.A.; López, X.; Poblet, J.M. A DFT study on the effect of metal, anion charge, heteroatom and structure upon the relative basicities of polyoxoanions. J. Mol. Catal. A Chem. 2007, 262, 236–242. [Google Scholar] [CrossRef]
- Wills, A.S. VaList-Program. Available online: https://dl.dropboxusercontent.com/u/8933134/Website/Site/Software/Software.html (accessed on 15 February 2014).
- Mattes, R.; Bierbüsse, H.; Fuchs, J. Schwingungsspektren und kraftkonstanten von polyanionen mit M6O19-gruppen. Z. Anorg. Allg. Chem. 1971, 385, 230–242. [Google Scholar] [CrossRef]
- Gunasekaran, S.; Sankari, G.; Ponnusamy, S. Vibrational spectral investigation on xanthine and its derivatives—Theophylline, caffeine and theobromine. Spectrochim. Acta Part A 2005, 61, 117–127. [Google Scholar] [CrossRef]
- Fournier, M. Tetrabutylammonium Isopolyoxometallates. In Inorganic Syntheses; Ginsberg, A.P., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1990; Volume 27, pp. 74–85. [Google Scholar]
- Farrugia, L. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Cryst. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Stoe; Cie. X-SHAPE; Stoe & Cie GmbH: Darmstadt, Germany, 1999. [Google Scholar]
- Stoe; Cie. X-RED; Stoe & Cie GmbH: Darmstadt, Germany, 2001. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kulikov, V.; Meyer, G. Polyoxotungstates in Molecular Boxes of Purine Bases. Crystals 2014, 4, 64-73. https://doi.org/10.3390/cryst4010064
Kulikov V, Meyer G. Polyoxotungstates in Molecular Boxes of Purine Bases. Crystals. 2014; 4(1):64-73. https://doi.org/10.3390/cryst4010064
Chicago/Turabian StyleKulikov, Vladislav, and Gerd Meyer. 2014. "Polyoxotungstates in Molecular Boxes of Purine Bases" Crystals 4, no. 1: 64-73. https://doi.org/10.3390/cryst4010064
APA StyleKulikov, V., & Meyer, G. (2014). Polyoxotungstates in Molecular Boxes of Purine Bases. Crystals, 4(1), 64-73. https://doi.org/10.3390/cryst4010064