σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures


Abstract
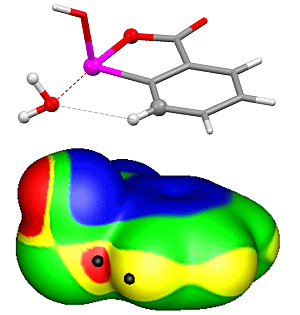
:
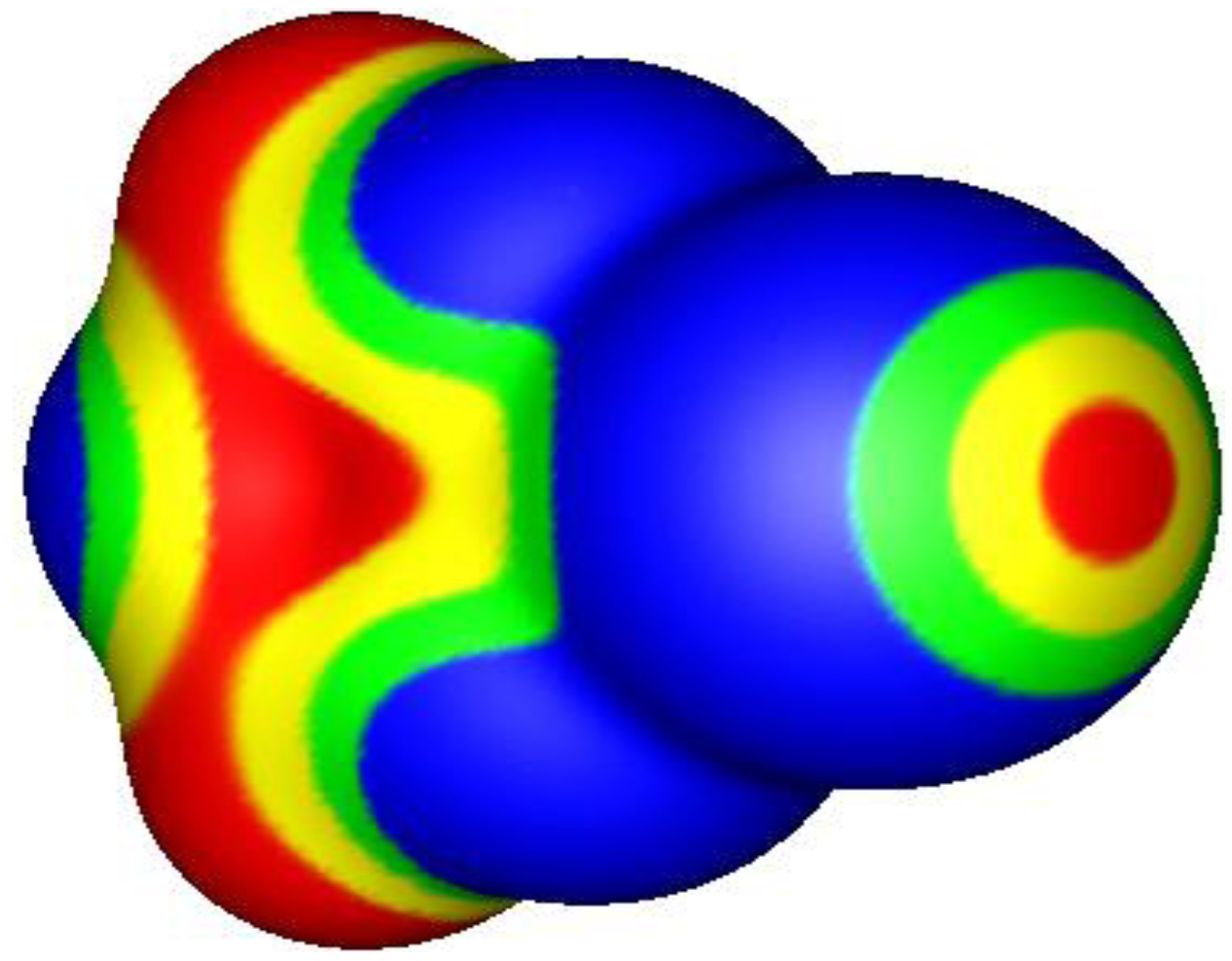
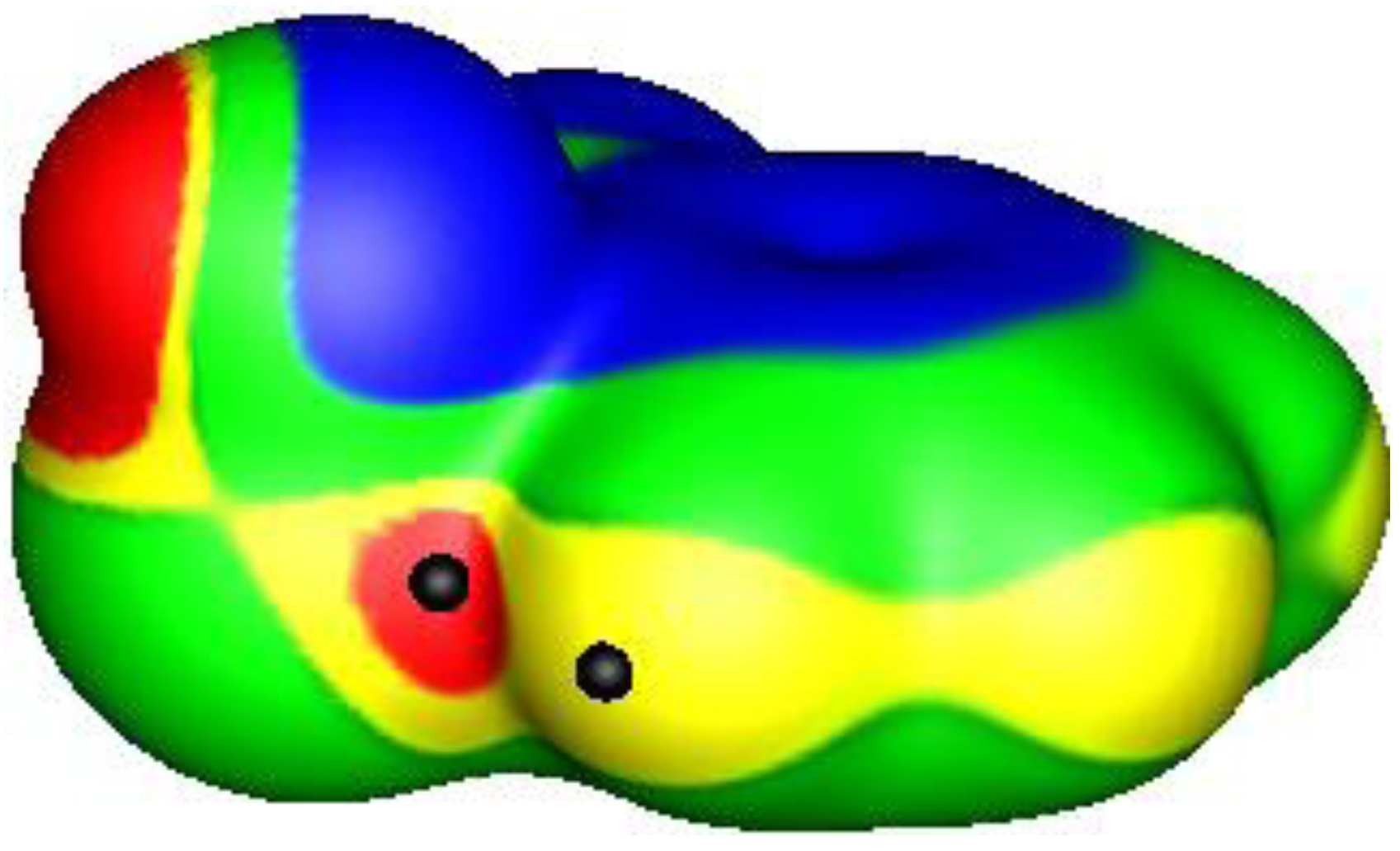
1. The σ-Hole and Halogen Bonding
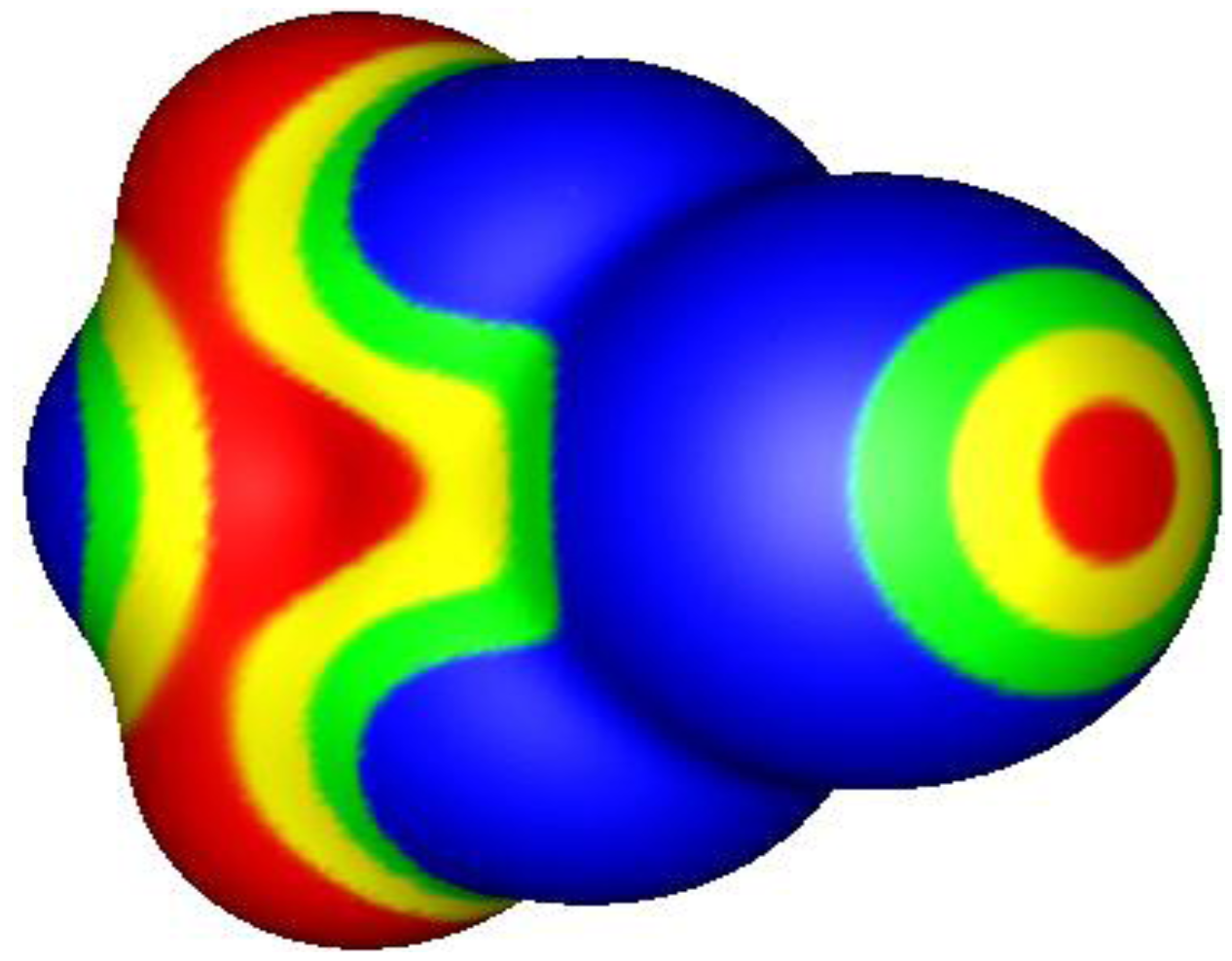

2. σ-Holes and Groups IV–VI Noncovalent Interactions
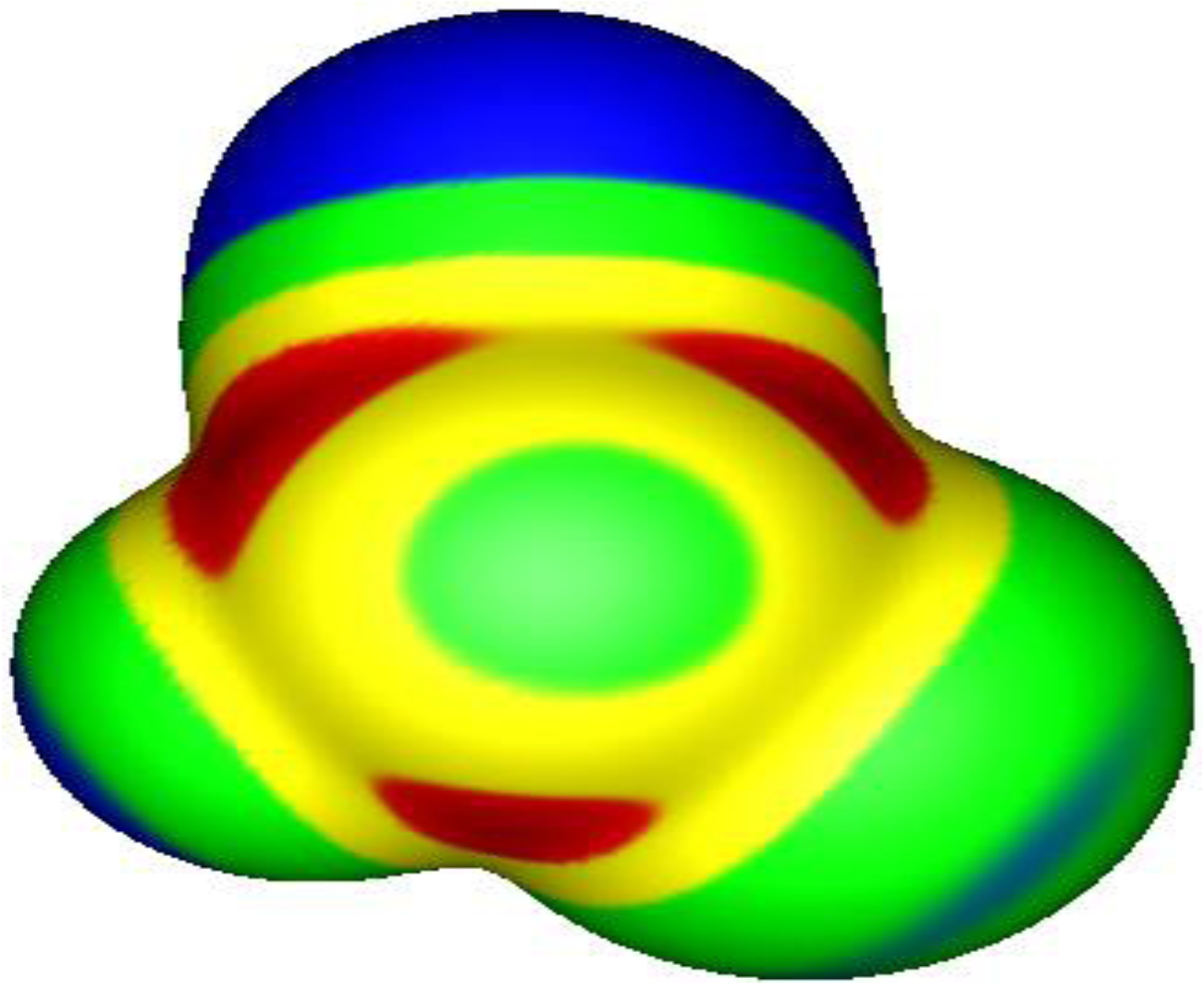
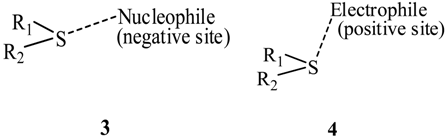
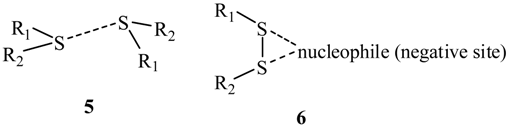
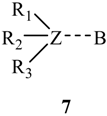
3. Present Study
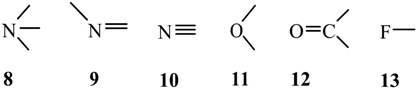
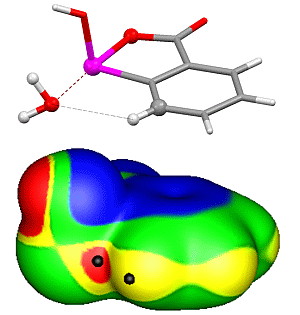
4. Survey of Crystal Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Z | B | |||||
 |  |  |  |  |  | |
| N | 0 | 0 | 0 | 0 | 0 | 13 |
| P | 16 | 0 | 10 | 43 | 30 | 61 |
| As | 6 | 0 | 6 | 69 | 5 | 26 |
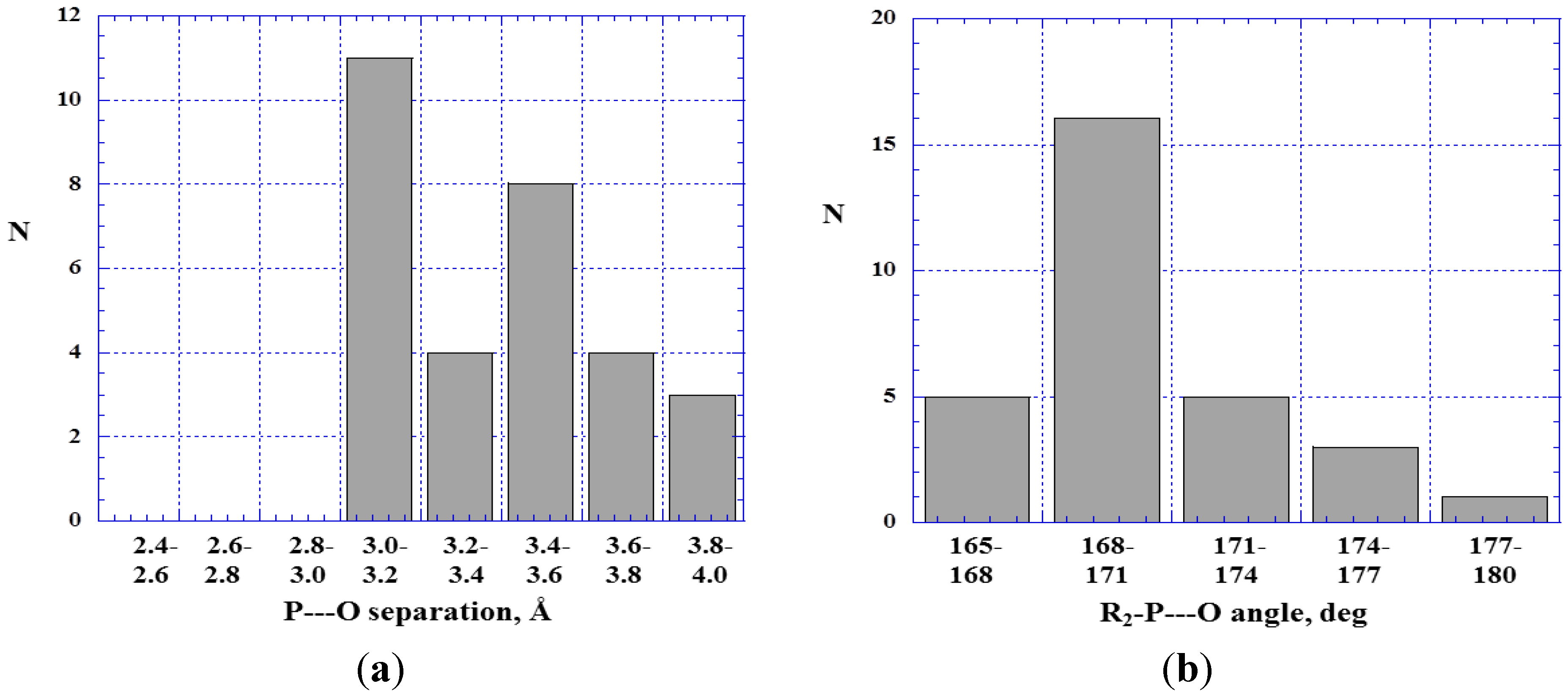
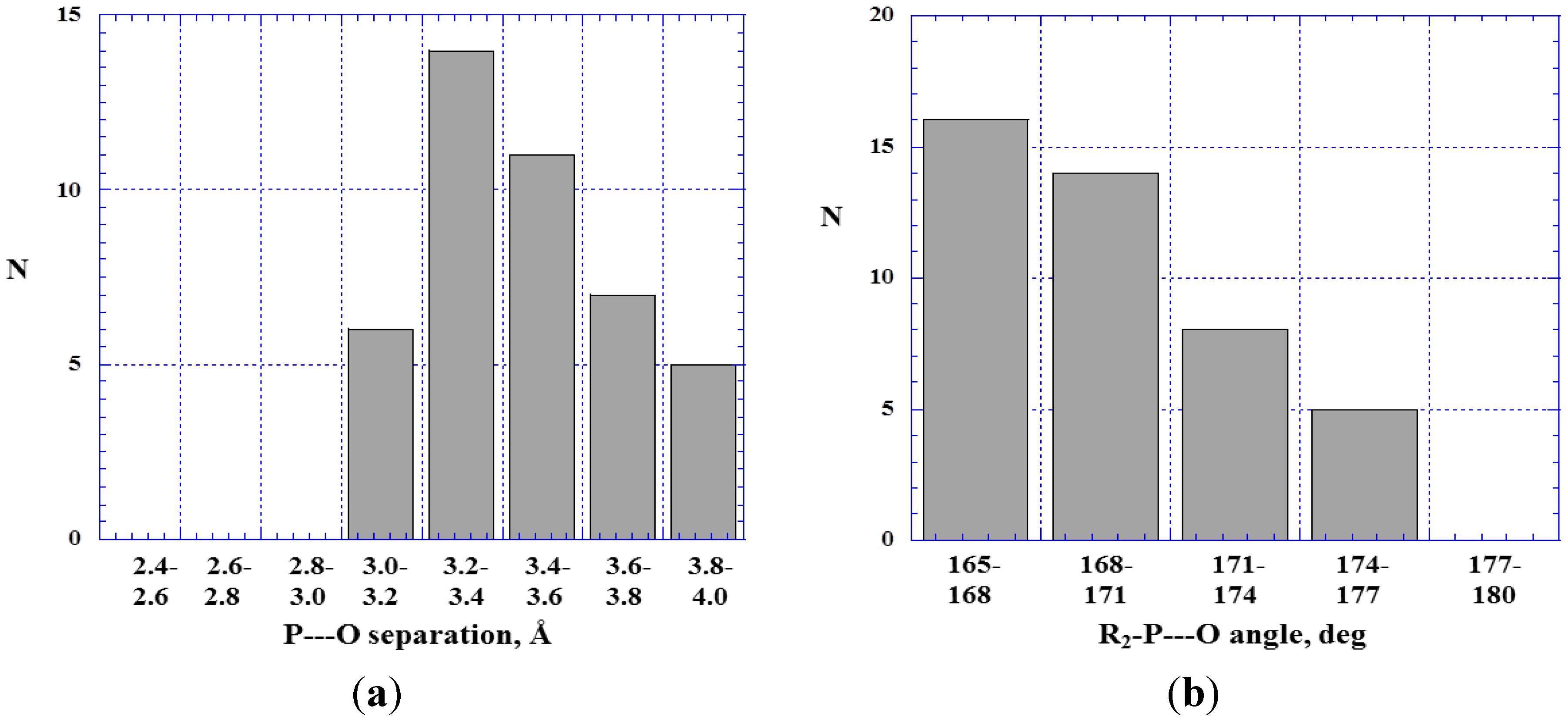
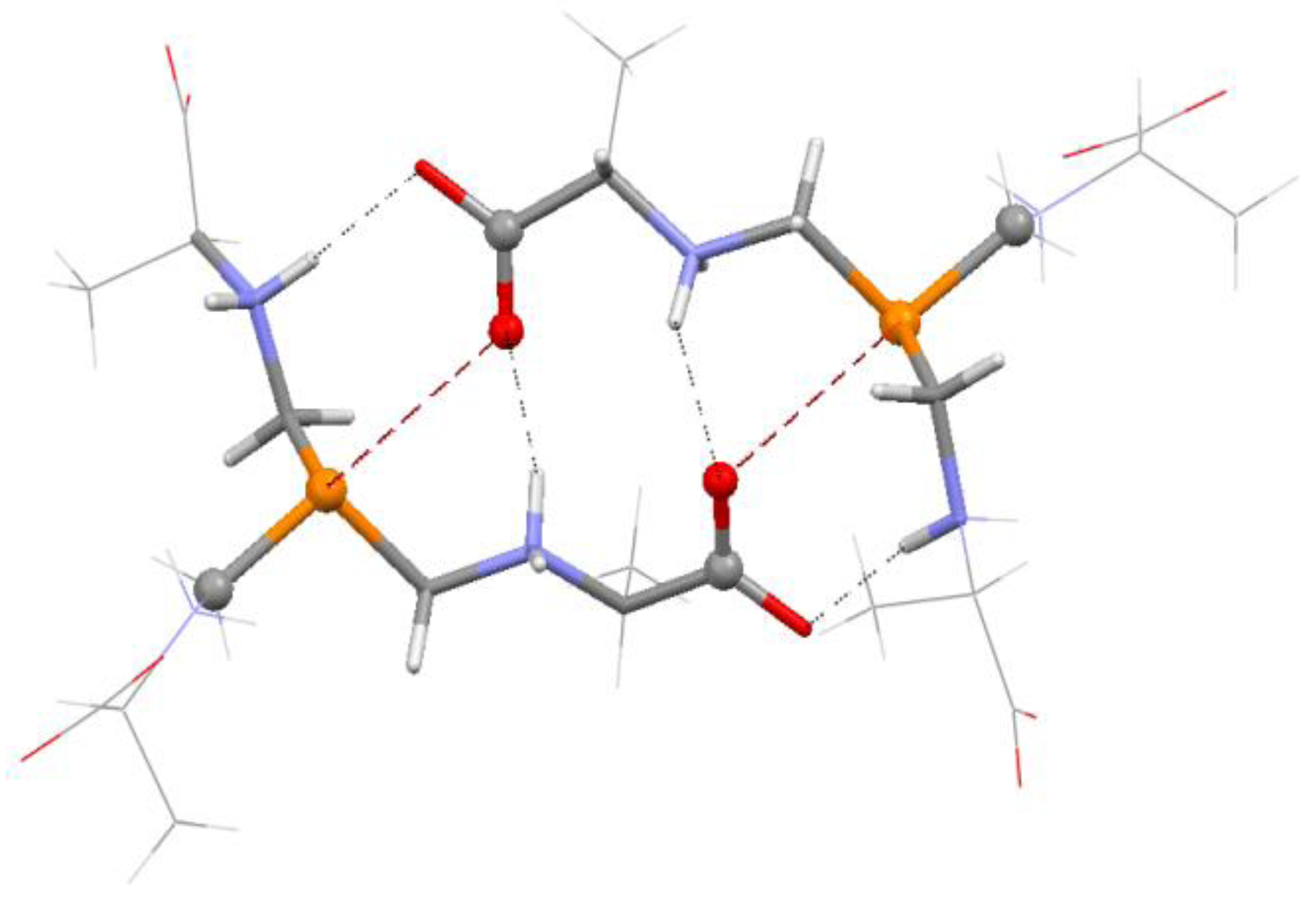
4.1. Close Contacts of Trivalent Phosphorus with Oxygens of Types 11 and 12
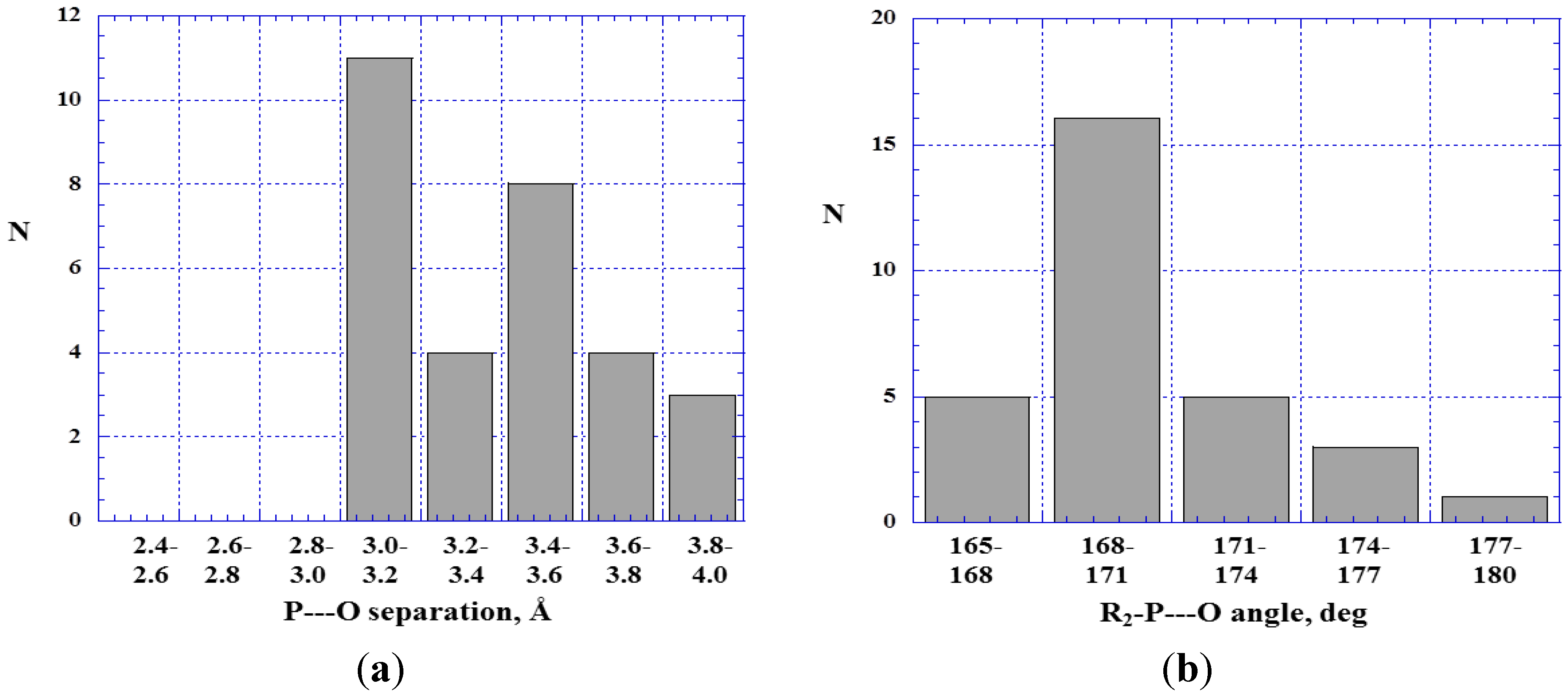
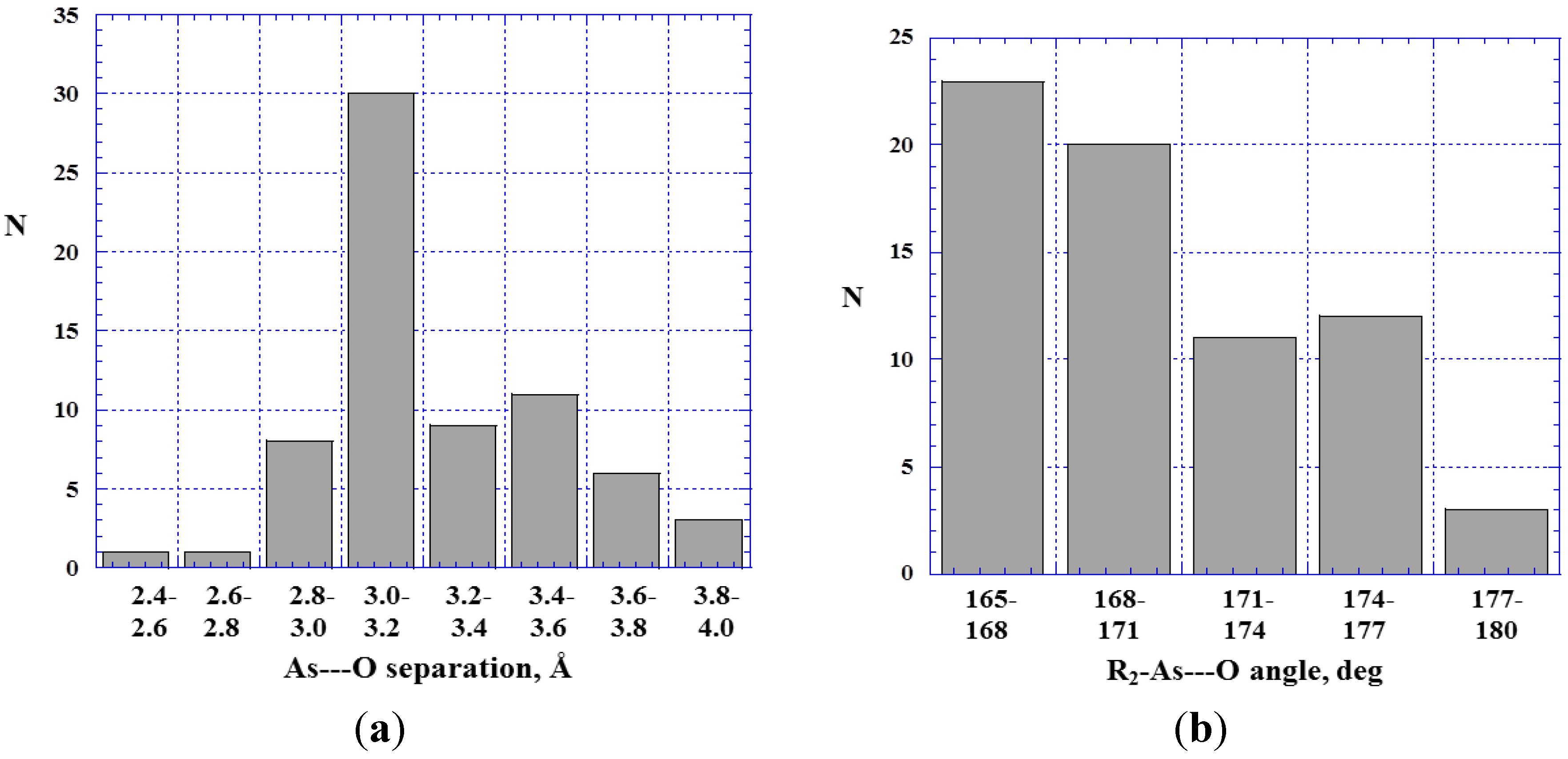
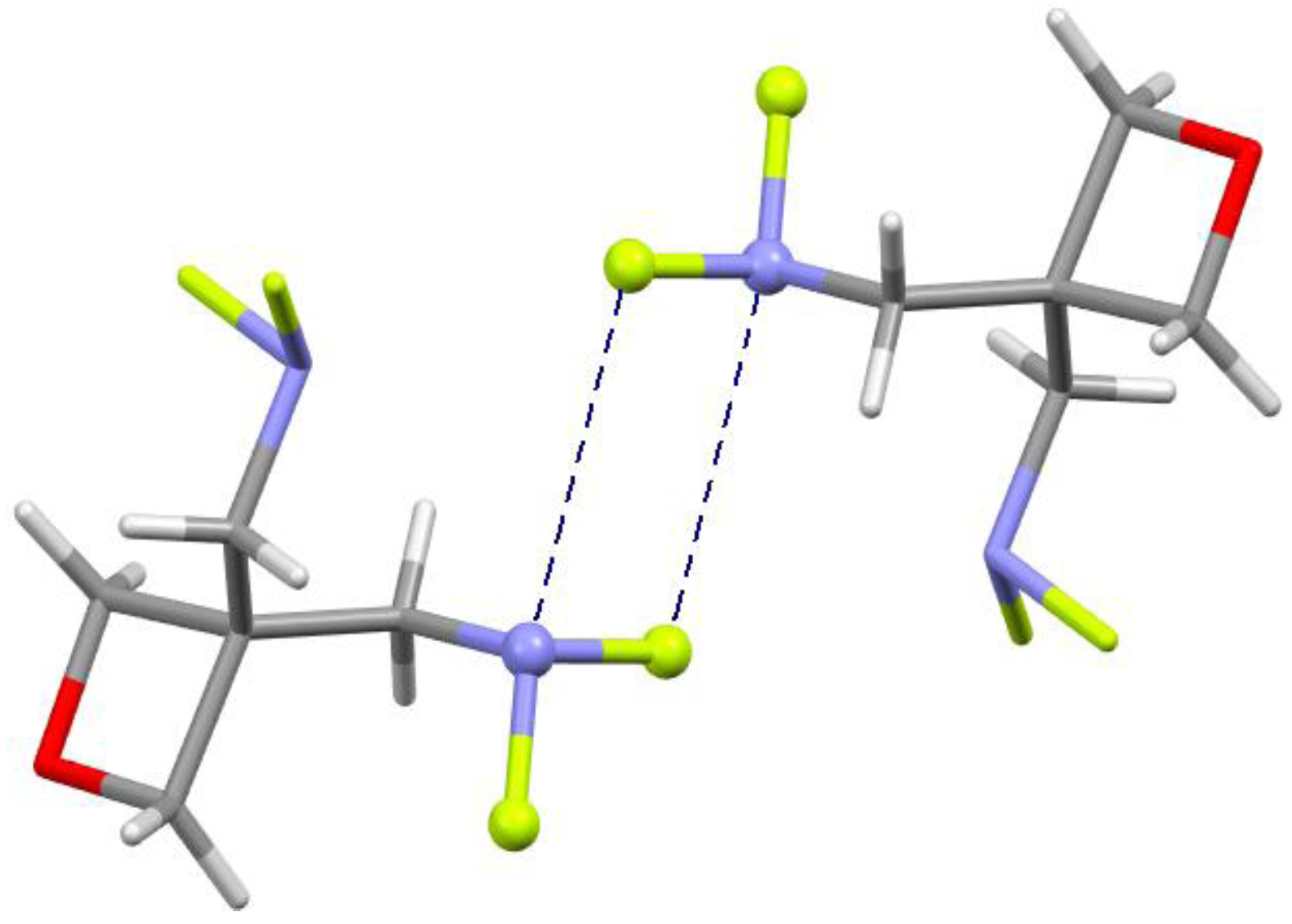
4.2. Close Contacts of Trivalent Arsenic with Oxygens of Type 11

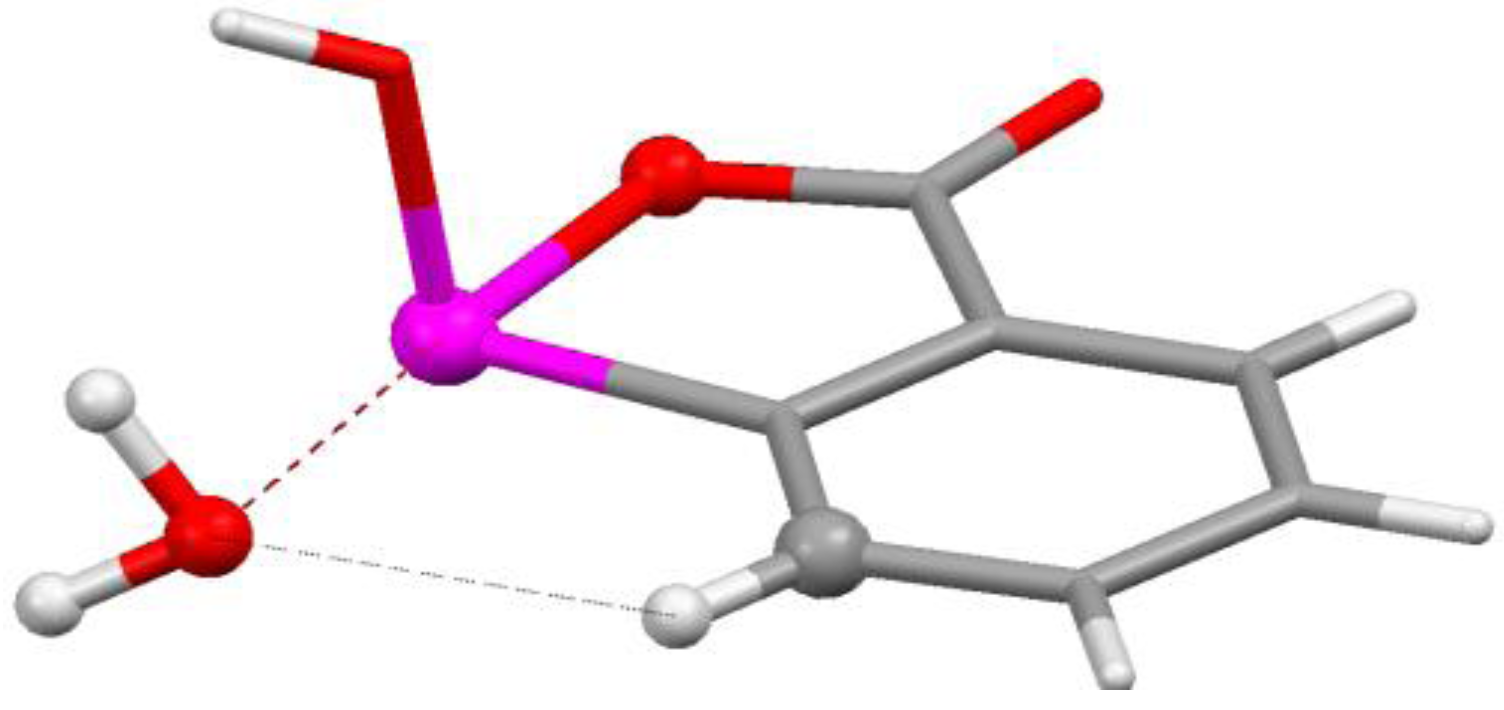

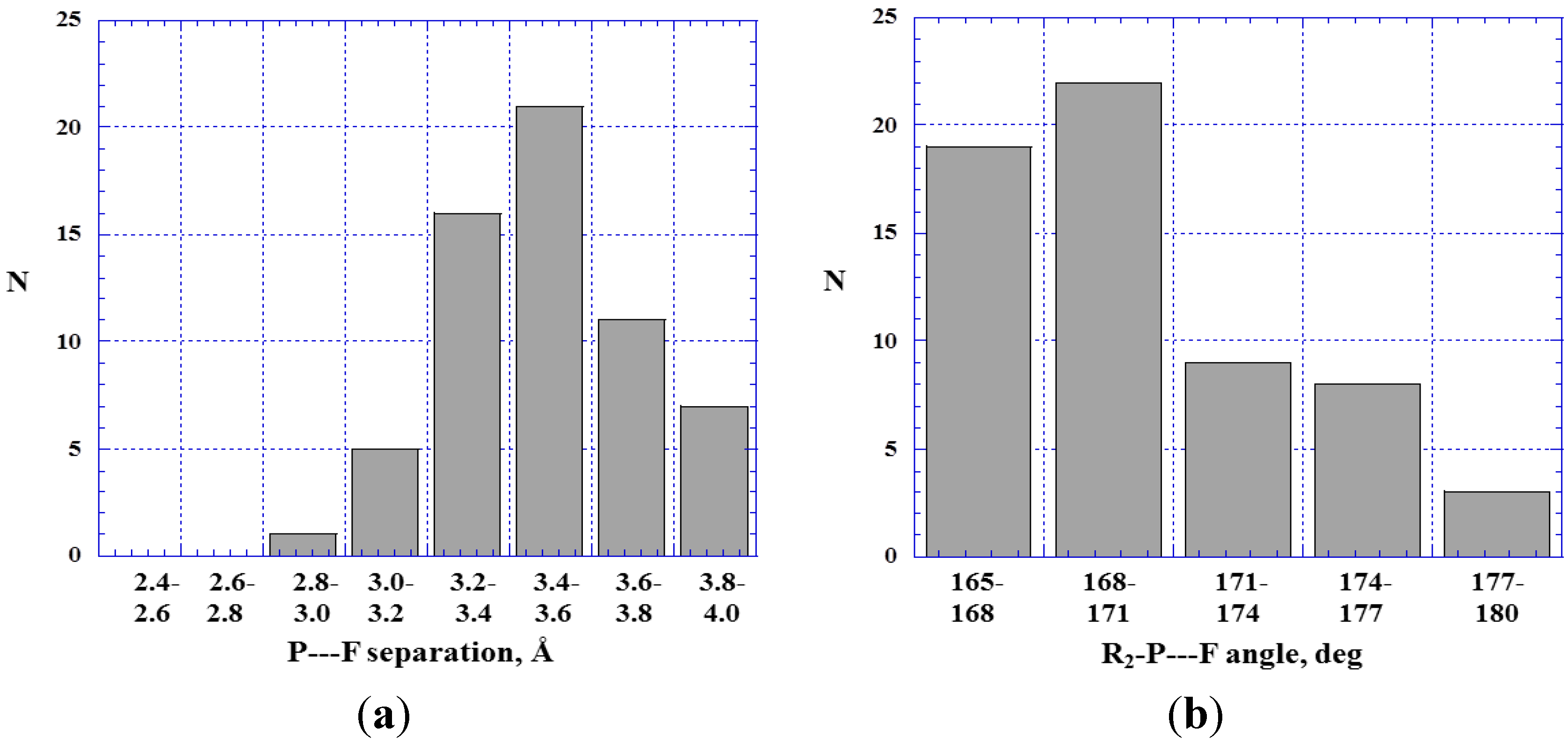
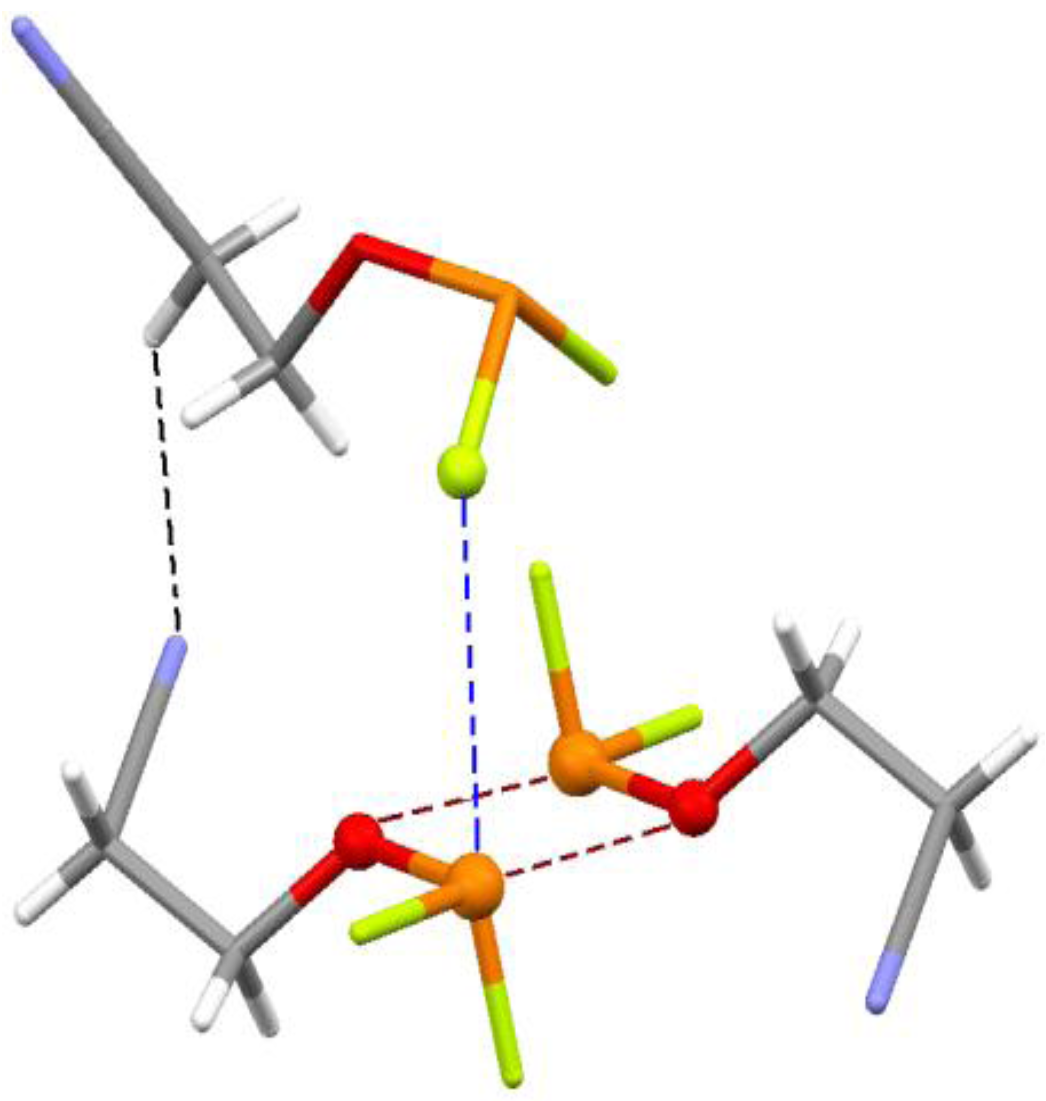
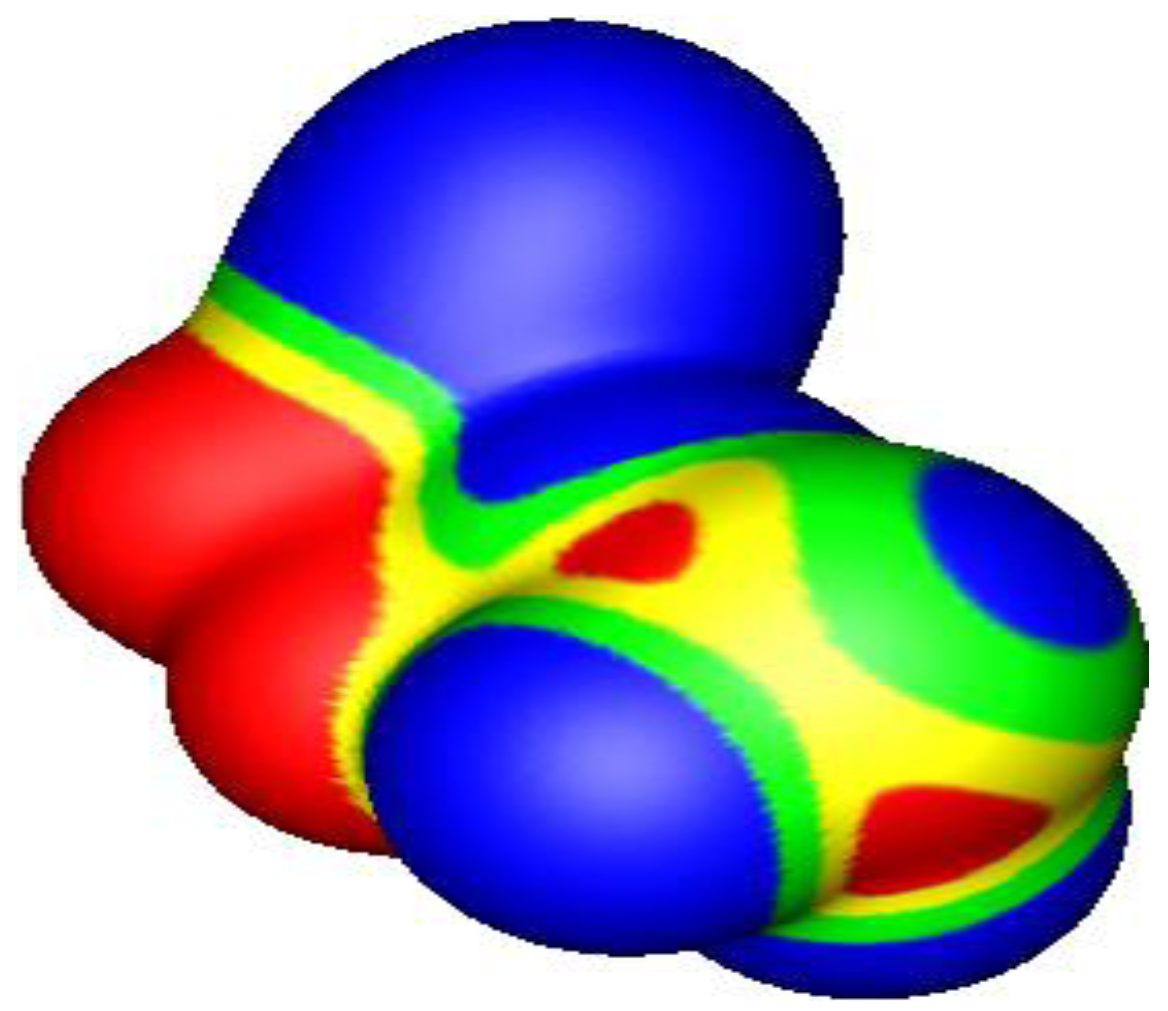
4.3. Close Contacts of Trivalent Phosphorus with Fluorines
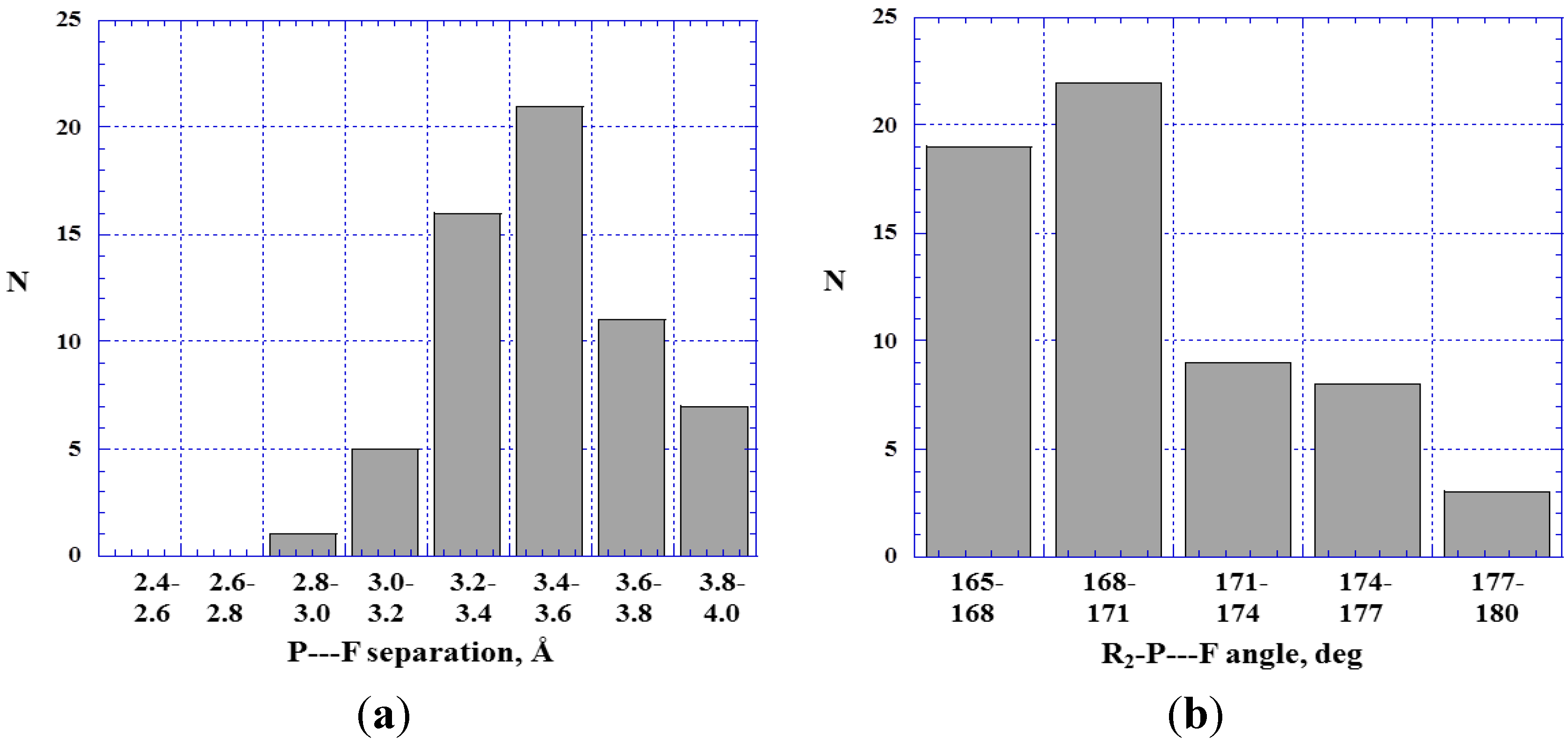

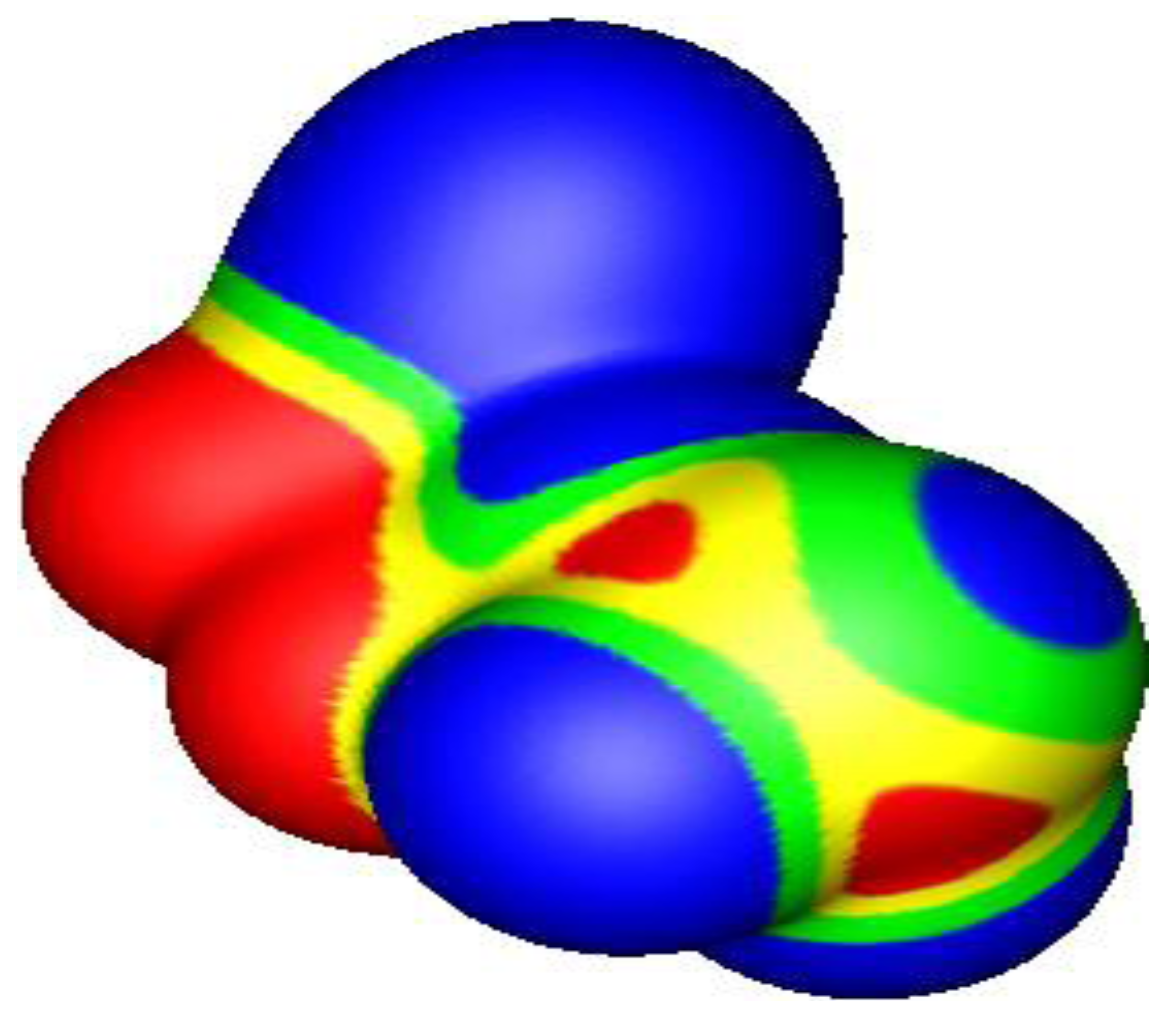
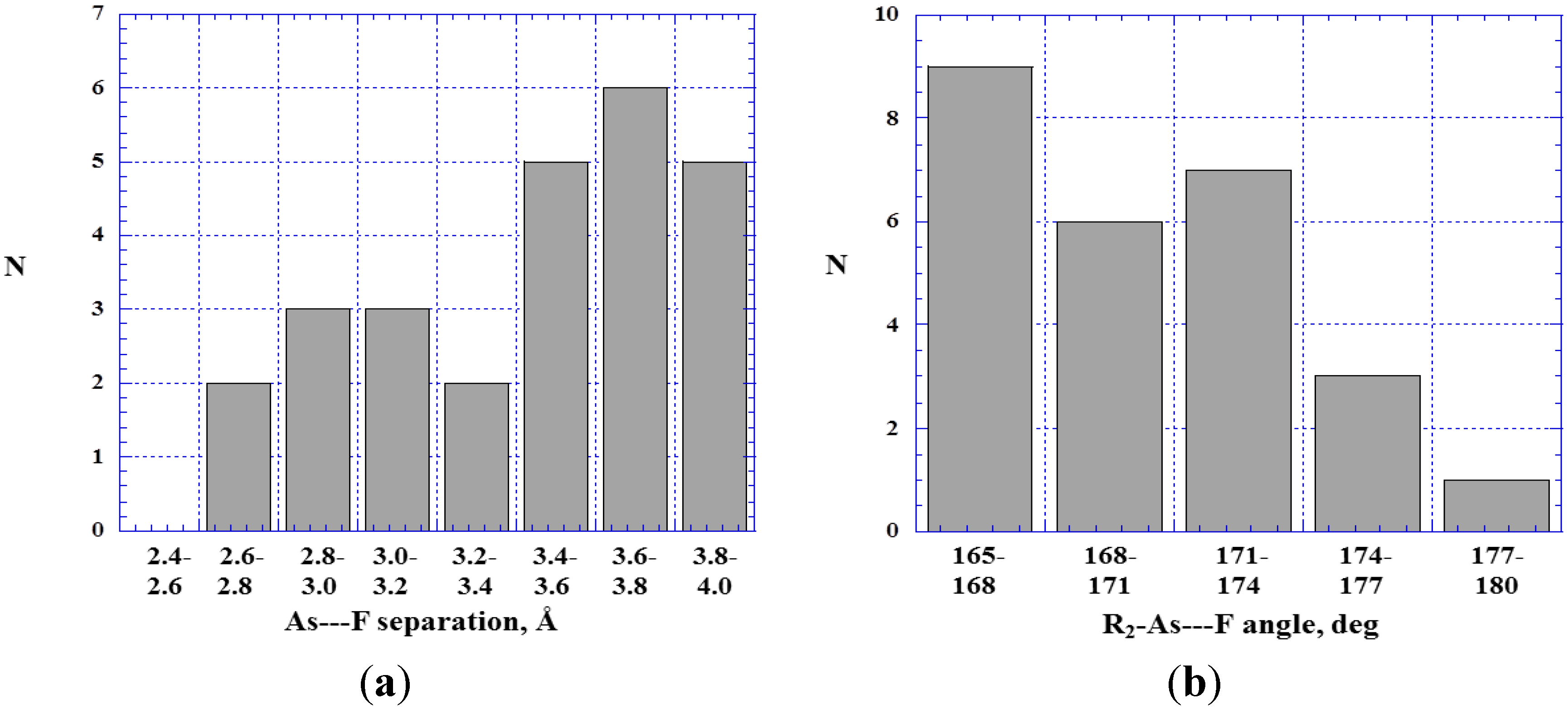
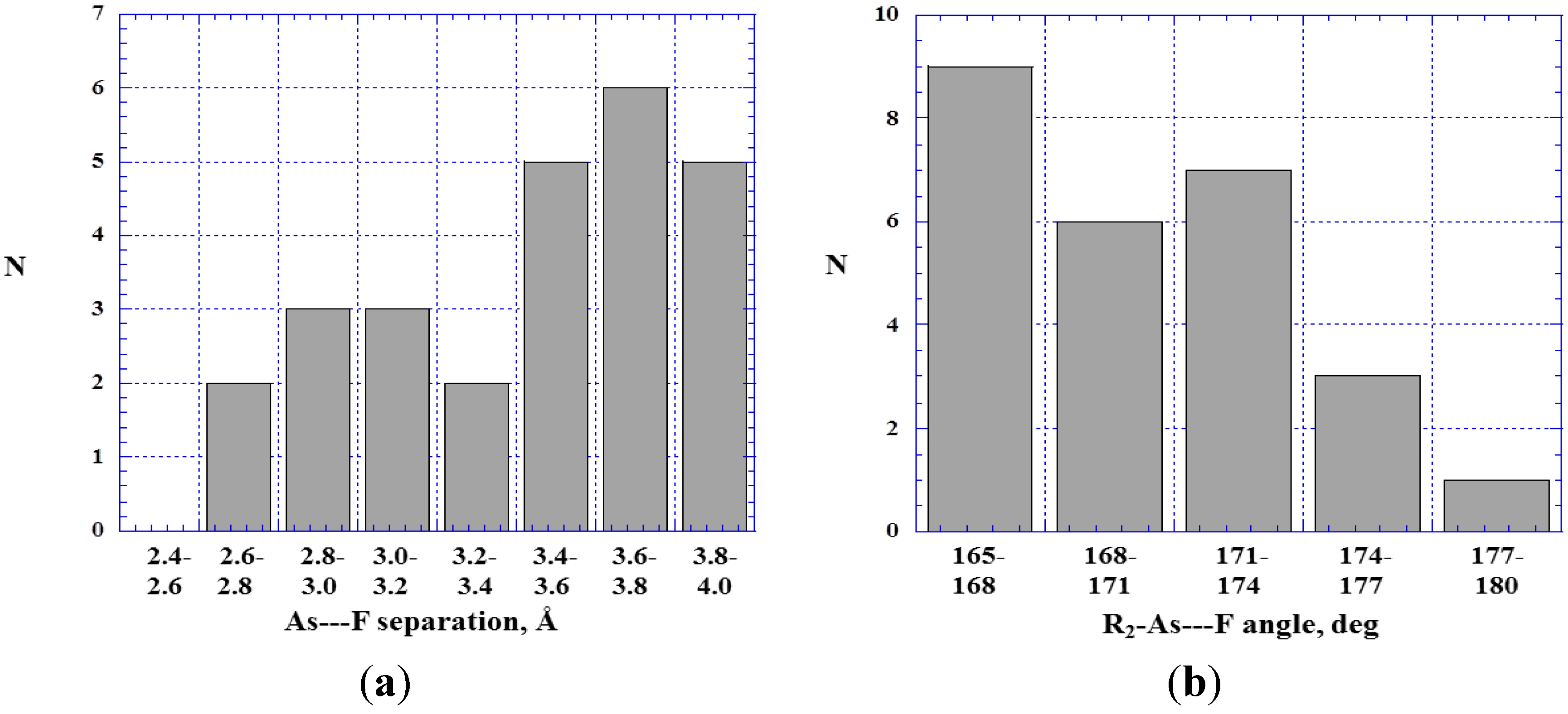
4.4. Close Contacts of Trivalent Arsenic with Fluorines
4.5. Close Contacts of Trivalent Nitrogens with Fluorines
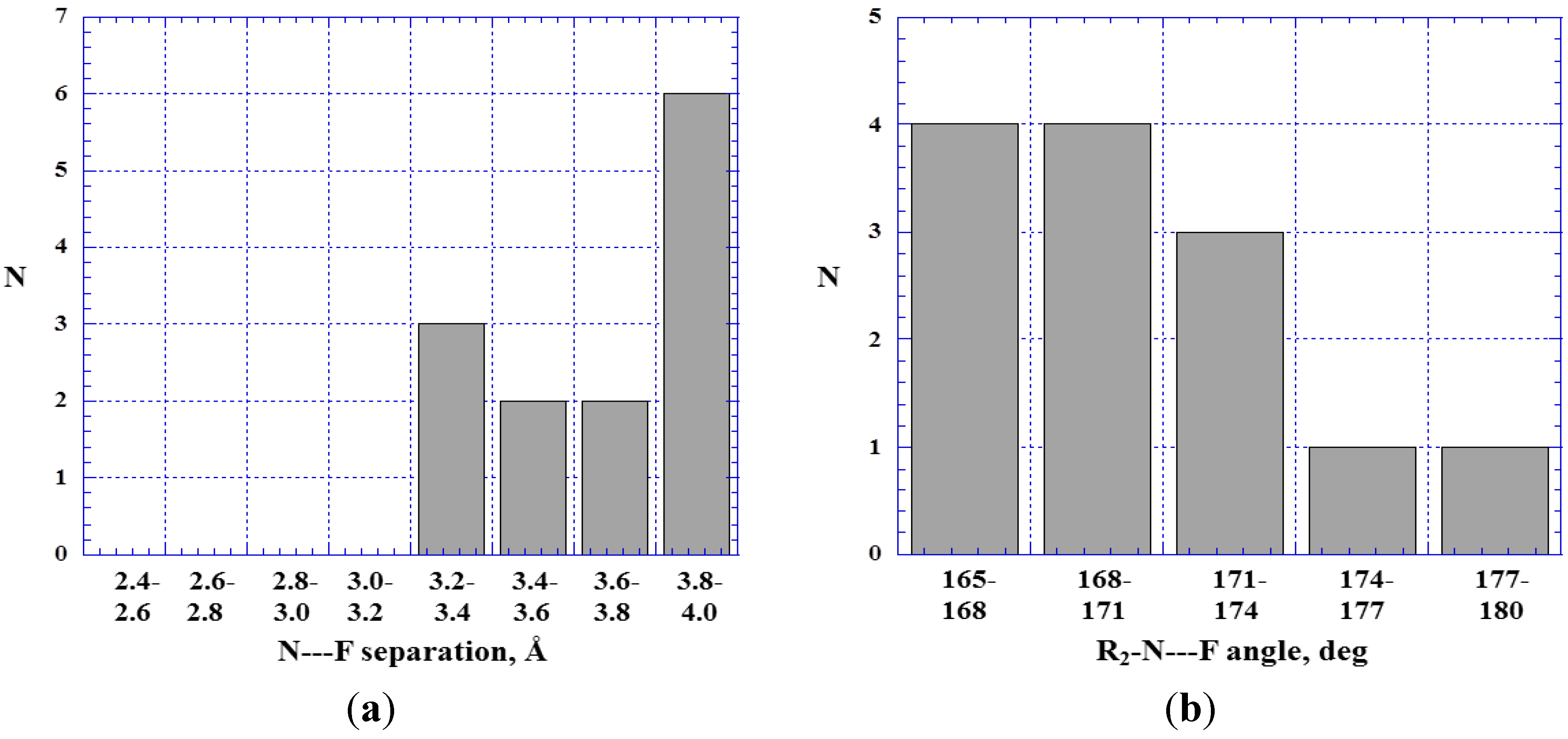
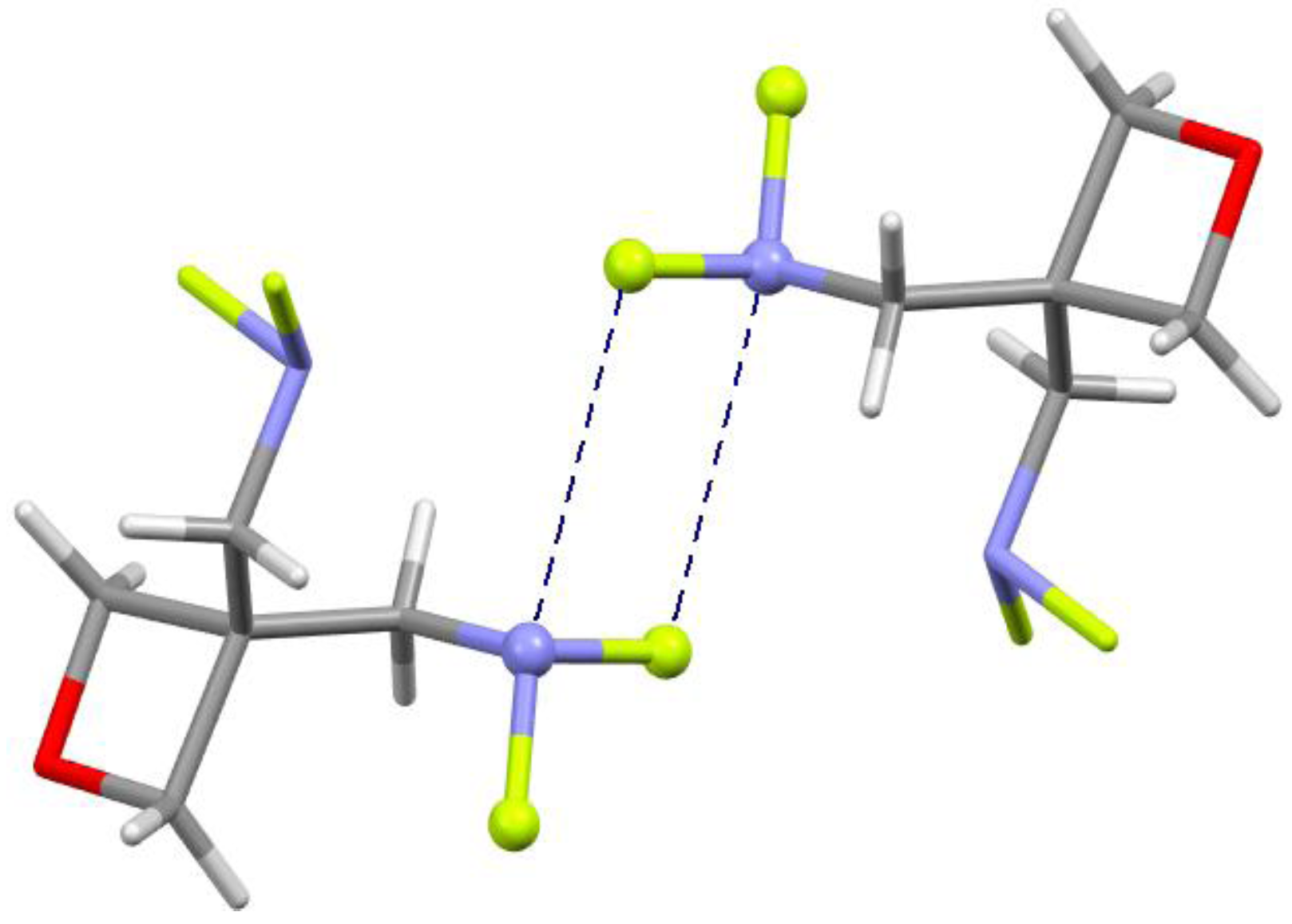
5. Discussion and Summary
Acknowledgments
Conflicts of Interest
References
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Molecular surface electrostatic potentials and local ionization energies of group V–VII hydrides and their anions: Relationships for aqueous and gas-phase acidities. Int. J. Quantum Chem. 1993, 48, 73–88. [Google Scholar] [CrossRef]
- Guthrie, F. XXVIII.—On the iodide of iodammonium. J. Chem. Soc. 1863, 16, 239–244. [Google Scholar] [CrossRef]
- Prescott, A.B. Notes on a few pyridine alkyl iodides. Am. Chem. J. 1896, 18, 91–96. [Google Scholar] [CrossRef]
- Hassel, O.; Rømming, C. Direct structural evidence for weak charge transfer bonds in solids containing chemically saturated molecules. Quart. Rev. Chem. Soc. 1962, 16, 1–18. [Google Scholar] [CrossRef]
- Bent, H.A. Structural chemistry of donor-acceptor interactions. Chem. Rev. 1968, 68, 587–648. [Google Scholar] [CrossRef]
- Bernard-Houplain, M.-C.; Sandorfy, C. A low temperature infrared study of hydrogen bonding in N-alkylacetamides. Can. J. Chem. 1973, 51, 3640–3647. [Google Scholar] [CrossRef]
- Di Paolo, T.; Sandorfy, C. On the hydrogen bond breaking ability of fluorocarbons containing higher halogens. Can. J. Chem. 1974, 52, 3612–3622. [Google Scholar] [CrossRef]
- Metrangolo, P.; Neukirch, H.; Pilati, T.; Resnati, G. Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res. 2005, 38, 386–395. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef]
- Murray, J.S.; Paulsen, K.; Politzer, P. Molecular surface electrostatic potentials in the analysis of non-hydrogen-bonding noncovalent interactions. Proc. Indian Acad. Sci. Chem. Sci. 1994, 106, 267–275. [Google Scholar]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. 2004, 101, 16789–16794. [Google Scholar] [CrossRef]
- Awwadi, F.F.; Willett, R.D.; Peterson, K.A.; Twamley, B. The nature of halogen---halogen synthons: Crystallographic and theoretical studies. Chem. Eur. J. 2006, 12, 8952–8960. [Google Scholar] [CrossRef]
- Stevens, E.D. Experimental electron density distribution of molecular chlorine. Mol. Phys. 1979, 37, 27–45. [Google Scholar] [CrossRef]
- Nyburg, S.C.; Faerman, C.H. A revision of van der Waals atomic radii for molecular crystals: N, O, F, S, Cl, Se, Br and I bonded to carbon. Acta Cryst. 1985, B41, 274–279. [Google Scholar] [CrossRef]
- Tsirelson, V.G.; Zou, P.F.; Tang, T.-H.; Bader, R.F.W. Topological definition of crystal structure: Determination of the bonded interactions in solid molecular chlorine. Acta Cryst. A 1995, 51, 143–153. [Google Scholar] [CrossRef]
- Bilewicz, E.; Rybarczyk-Pirek, A.J.; Dubis, A.T.; Grabowski, S.J. Halogen bonding in crystal structure of 1-methylpyrrol-2-yl trichloromethyl ketone. J. Mol. Struct. 2007, 829, 208–211. [Google Scholar] [CrossRef]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: lex parsimoniae (Occam’s Razor). Comput. Theoret. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. Halogen bonding and the design of new materials: Organic bromides, chlorides and even fluorides as donors. J. Mol. Model. 2007, 13, 643–650. [Google Scholar] [CrossRef]
- Chopra, D.; Guru Row, T.N. Role of organic fluorine in crystal engineering. CrystEngComm 2011, 13, 2175–2186. [Google Scholar] [CrossRef]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-centered halogen bonding: A factor in recognition phenomena and reactivity. Cryst. Growth Des. 2011, 11, 4238–4246. [Google Scholar] [CrossRef]
- Murray-Rust, P.; Motherwell, W.D.S. Computer retrieval and analysis of molecular geometry. 4. Intermolecular interactions. J. Am. Chem. Soc. 1979, 101, 4374–4376. [Google Scholar] [CrossRef]
- Murray-Rust, P.; Stallings, W.C.; Monti, C.T.; Preston, R.K.; Glusker, J.P. Intermolecular interactions of the carbon-fluorine bond: The crystallographic environment of fluorinated carboxylic acids and related structures. J. Am. Chem. Soc. 1983, 105, 3206–3214. [Google Scholar] [CrossRef]
- Ramasubbu, N.; Parthasarathy, R.; Murray-Rust, P. Angular preferences of intermolecular forces around halogen centers: Preferred directions of approach of electrophiles and nucleophiles around carbon-halogen bonds. J. Am. Chem. Soc. 1986, 108, 4308–4314. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Politzer, P. σ-hole bonding: Molecules containing group VI atoms. J. Mol. Model. 2007, 13, 1033–1038. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. A predicted new type of directional interaction. Int. J. Quant. Chem. 2007, 107, 2286–2292. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Lane, P.; Politzer, P. Why are dimethyl sulfoxide and dimethyl sulfone such good solvents? J. Mol. Model. 2008, 14, 689–697. [Google Scholar] [CrossRef]
- Guru Row, T.N.; Parthasarathy, R. Directional preferences of nonbonded atomic contacts with divalent sulfur in terms of its orbital orientations. 2. S---S interactions and nonspherical shape of sulfur in crystals. J. Am. Chem. Soc. 1981, 103, 477–479. [Google Scholar] [CrossRef]
- Ramasubbu, N.; Parthasarathy, R. Stereochemistry of incipient electrophilic and nucleophilic reactions at divalent selenium center: Electrophilic-Nucleophilic pairing and anisotropic shape of Se in Se---Se interactions. Phosphorus Sulfur 1987, 31, 221–229. [Google Scholar] [CrossRef]
- Murray, J.S.; Macaveiu, L.; Politzer, P. Factors affecting the strengths of σ-hole electrostatic potentials. J. Comput. Sci. 2014. [Google Scholar] [CrossRef]
- Iwaoka, M.; Komatsu, H.; Katsuda, T.; Tomoda, S. Quantitative evaluation of weak nonbonded Se---F interactions and their remarkable nature as orbital interactions. J. Am. Chem. Soc. 2002, 124, 1902–1909. [Google Scholar]
- Cozzolino, A.F.; Vargas-Baca, I.; Mansour, S.; Mahmoudkhani, A.H. The nature of the supramolecular association of 1,2,5-chalcogenadiazoles. J. Am. Chem. Soc. 2005, 127, 3184–3190. [Google Scholar] [CrossRef]
- Bleiholder, C.; Werz, D.B.; Köppel, H.; Gleiter, R. Theoretical investigations on chalcogen-chalcogen interactions: What makes these nonbonded interactions bonding? J. Am. Chem. Soc. 2006, 128, 2666–2674. [Google Scholar] [CrossRef]
- Mohajeri, A.; Pakiari, A.H.; Bagheri, N. Theoretical studies on the nature of bonding in σ-hole complexes. Chem. Phys. Lett. 2009, 467, 393–397. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, D.; Sun, Y.; Hao, J.; Cai, Z. Theoretical investigations on the weak nonbonded C=S---CH2 interactions: Chalcogen-based complexes with singlet carbene as an electron donor. Int. J. Quantum Chem. 2011, 111, 3881–3887. [Google Scholar]
- Jing, B.; Li, Q.; Gong, B.; Li, R.; Liu, Z.; Li, W.; Cheng, J.; Sun, J. Hydrogen bond and σ-hole interaction in M2C=S---HCN (M = H, F, Cl, Br, HO, H3C, H2N) complex: Dual roles of C=S group and substitution effects. Int. J. Quantum Chem. 2012, 112, 1491–1498. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Li, R.; Guo, P.; Li, H.; Li, W.-Z.; Cheng, J.-B. Competition of chalcogen bond, halogen bond and hydrogen bond in SCS---HOX and SeCSe---HOX (X = Cl and Br) complexes. Comput. Theor. Chem. 2012, 980, 56–61. [Google Scholar] [CrossRef]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef]
- Rosenfield, R.E., Jr.; Parthasarathy, R.; Dunitz, J.D. Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J. Am. Chem. Soc. 1977, 99, 4860–4862. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Concha, M.C. σ-Hole bonding between like atoms: A fallacy of atomic charges. J. Mol. Model. 2008, 14, 659–665. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Molecular Electrostatic Potentials: Some Observations. In Concepts and Methods in Modern Theoretical Chemistry, Vol. 1: Electronic Structure and Reactivity; Ghosh, K., Chattaraj, P., Eds.; Taylor & Francis: New York, NY, 2013; Chapter 9; pp. 181–199. \. [Google Scholar]
- Widhalm, M.; Kratky, C. Synthesis and X-ray structure of binaphthyl-based macrocyclic diphosphanes and their Ni(II) and Pd(II) complexes. Chem. Ber. 1992, 125, 679–689. [Google Scholar] [CrossRef]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1,2-Dicarba-closo-dodecaboranes with short P-P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, W. The bifurcate chalcogen bond: Some theoretical observations. J. Mol. Struct. Theochem. 2009, 916, 135–138. [Google Scholar] [CrossRef]
- Mammi, M.; Bardi, R.; Traverso, G.; Bezzi, S. Crystal structure of 2,5-dimethyl-dithio-furophthene, a homologue of thio-thiophthene. Nature 1961, 192, 1282–1283. [Google Scholar] [CrossRef]
- Lynch, T.R.; Mellor, I.P.; Nyburg, S.C.; Yates, P. The structure and stereochemistry of the desaurin from acetophenone. Tetrahedron Lett. 1967, 373–377. [Google Scholar]
- Shishkin, O.V.; Omelchenko, I.V.; Kalyuzhny, A.L.; Paponov, B.V. Intramolecular S---O chalcogen bond in thioindirubin. Struct. Chem. 2010, 21, 1005–1011. [Google Scholar] [CrossRef]
- Junming, L.; Yunxiang, L.; Subin, Y.; Weiliang, Z. Theoretical and crystallographic data investigations of noncovalent S---O interactions. Struct. Chem. 2011, 22, 757–763. [Google Scholar] [CrossRef]
- Cohen-Addad, C.; Lehmann, M.S.; Becker, P.; Párkányi, L.; Kálmán, A. Nature of S---O interaction in short X–S---O contacts: Charge density experimental studies and theoretical interpretation. J. Chem. Soc. Perkin Trans. II 1984, 191–196. [Google Scholar]
- Burling, F.T.; Goldstein, B.M. Computational studies of nonbonded sulfur-oxygen and selenium-oxygen interactions in the thiazole and selenazole nucleosides. J. Am. Chem. Soc. 1992, 114, 2313–2320. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Simultaneous σ-hole and hydrogen bonding by sulfur- and selenium-containing heterocycles. Int. J. Quantum Chem. 2008, 108, 2770–2781. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Krumm, B.; Scherr, M. Homoleptic selenium cyanides: Attempted preparation of Se(CN)4 and redetermination of the crystal structure of Se(CN)2. Inorg. Chem. 2008, 47, 7025–7028. [Google Scholar] [CrossRef]
- Viguera, A.R.; Serrano, L. Side-chain interactions between sulfur-containing amino acids and phenylalanine in α-helixes. Biochemistry 1995, 34, 8771–8779. [Google Scholar] [CrossRef]
- Tatko, C.D.; Waters, M.L. Investigation of the nature of the methionine-π interaction in β-hairpin peptide model systems. Protein Sci. 2004, 13, 2515–2522. [Google Scholar] [CrossRef]
- Carré, F.; Chuit, C.; Corriu, R.J.P.; Monforte, P.; Nayyar, N.K.; Reyé, C. Intramolecular coordination at phosphorus: Donor acceptor interaction in three- and four-coordinated phosphorus compounds. J. Organometall. Chem. 1995, 499, 147–154. [Google Scholar] [CrossRef]
- Tschirschwitz, S.; Lönnecke, P.; Hey-Hawkins, E. Aminoalkylferrocenyldichloro-phosphanes: Facile synthesis of versatile chiral starting materials. Dalton Trans. 2007, 14, 1377–1382. [Google Scholar] [CrossRef]
- Drago, R.S.; Wong, N.; Ferris, D.C. The E, C, T interpretation of bond dissociation energies and anion-neutral molecule interactions. J. Am. Chem. Soc. 1991, 113, 1970–1977. [Google Scholar] [CrossRef]
- Hill, N.J.; Levason, W.; Reid, G. Arsenic (III) halide complexes with phosphine and arsine Co-ligands: Synthesis, spectroscopic and structural properties. J. Chem. Soc. Dalton Trans. 2002, 1188–1192. [Google Scholar] [CrossRef]
- Starbuck, J.; Norman, N.C.; Orpen, A.G. Secondary bonding as a potential design element for crystal engineering. New J. Chem. 1999, 23, 969–972. [Google Scholar] [CrossRef]
- Brellère, C.; Carré, F.; Corriu, R.J.P.; Poirier, M.; Royo, G.; Zwecker, J. Hexacoordinated silicon species: A possible model for reaction intermediates. 1. X-ray determination of the geometry in the solid state. Organometallics 1989, 8, 1831–1833. [Google Scholar] [CrossRef]
- Carré, F.; Chuit, C.; Corriu, R.J.P.; Mehdi, A.; Reye, C. Synthesis, structure and fluxional behavior of a dihydrosilane bearing an aryldiamine pincer ligand. Organometallics 1995, 14, 2754–2759. [Google Scholar] [CrossRef]
- Belzner, J.; Schär, D.; Herbst-Irmer, R.; Kneisel, B.O.; Noltemeyer, M. Synthesis and structure of silicon compounds intramolecularly coordinated to hydrazino groups. Tetrahedron 1998, 54, 8481–8500. [Google Scholar] [CrossRef]
- Ahdab, A.A.-E.; Rima, G.; Gornitzka, H.; Barrau, J. Synthesis and Characterization of 2,4,6-tris((dimethylamino)methyl)phenoxysilicon compounds. J. Organomet. Chem. 2001, 636, 96–107. [Google Scholar] [CrossRef]
- Berceanc, V.; Crainic, C.; Haiduc, I.; Mahon, M.F.; Molloy, K.C.; Venter, M.M.; Wilson, P.J. The structural chemistry of organotin derivatives of 5-Mercapto-3-Phenyl-1,3,4-thiadiazoline-2-thione: Supramolecular structures involving intermolecular Sn---S, N-H---S or S---S interactions. J. Chem. Soc. Dalton Trans. 2002, 6, 1036–1045. [Google Scholar]
- Kost, D.; Gostevskii, B.; Kalikhman, I. Silicon rehybridization and molecular rearrangements in hypercoordinate silicon dichelates. Pure Appl. Chem. 2007, 79, 1125–1134. [Google Scholar] [CrossRef]
- Cheng, F.; Hector, A.L.; Levason, W.; Reid, G.; Webster, M.; Zhang, W. Preparation and structure of the unique silicon (IV) cation [SiF3(Me3tacn)]+. Chem. Commun. 2009, 1334–1336. [Google Scholar]
- Shainyan, B.A.; Lazareva, N.F. P=O → Si intramolecular coordination in the derivatives of 1,4-phosphasilacyclohexane 1-oxides. Int. J. Quantum Chem. 2009, 109, 301–307. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel-bond σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Banzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Mitzel, N.W.; Blake, A.J.; Rankin, D.W.H. β-donor bonds in SiON units: An inherent structure determining property leading to (4 + 4)-coordination in tetrakis-(N,N-dimethylhydroxylamido)silane. J. Am. Chem. Soc. 1997, 119, 4143–4148. [Google Scholar] [CrossRef]
- Losehand, U.; Mitzel, N.W.; Rankin, D.W.H. Synthesis and molecular structures of N,N-dimethylhydroxylamino-trichlorosilane and -germane. J. Chem. Soc. Dalton Trans. 1999, 4291–4297. [Google Scholar] [CrossRef]
- Mitzel, N.W.; Vojinovic, K.; Froehlich, R.; Foerster, T.; Robertson, H.E.; Borisenko, K.B.; Rankin, D.W.H. Three-membered ring or open chain molecule—(F3C)F2SiONMe2 a model for the α-effect in silicon chemistry. J. Am. Chem. Soc. 2005, 127, 13705–13713. [Google Scholar] [CrossRef]
- Murray, J.S.; Concha, M.C.; Politzer, P. Molecular surface electrostatic potentials as guides to Si–O–N angle contraction: Tunable σ-holes. J. Mol. Model. 2011, 17, 2151–2157. [Google Scholar] [CrossRef]
- Allen, F.H. The cambridge structural database: A quarter of a million crystal structures and rising. Acta Cryst. 2002, B58, 380–388. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; Cabe, P.M.; Pearson, J.; Taylor, R. New software for searching the cambridge structural database and visualizing crystal structures. Acta Cryst. 2002, B58, 389–397. [Google Scholar]
- Riley, K.E.; Murray, J.S.; Fanfrlík, J.; Řezáč, J.; Solá, R.J.; Concha, M.C.; Ramos, F.M.; Politzer, P. Halogen bond tunability I: The effects of aromatic fluorine substitution on the strengths of halogen-bonding interactions involving chlorine, bromine and iodine. J. Mol. Model. 2011, 17, 3309–3318. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular nonbonded contact distances in organic crystal structures: Comparison with distances expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Raghuraman, K.; Katti, K.K.; Barbour, L.J.; Pillarsetty, N.; Barnes, C.L.; Katti, K.V. Characterization of supramolecular (H2O)18 water morphology and water-methanol (H2O)15(CH3OH)3 clusters in a novel phosphorus functionalized trimeric amino acid host. J. Am. Chem. Soc. 2003, 125, 6955–6961. [Google Scholar]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1689–1691. [Google Scholar]
- Murray, J.S.; Politzer, P. The electrostatic potential: An overview. WIREs Comp. Mol. Sci. 2011, 1, 153–163. [Google Scholar] [CrossRef]
- Hundal, M.S.; Hundal, G.; Parmar, S.S. 1-Hydroxy-2,1λ5-benzoxarsol-3-one monohydrate. Acta Cryst. 1996, C52, 2726–2728. [Google Scholar]
- Politzer, P.; Murray, J.S.; Concha, M.C. The complementary roles of molecular surface electrostatic potentials and average local ionization energies with respect to electrophilic processes. Int. J. Quantum Chem. 2002, 88, 19–27. [Google Scholar] [CrossRef]
- Blake, A.J.; Davis, M.J.; Rankin, D.W.H. Instant ligands: Part 5. The molecular structures of (PF2)OCH2C CCH2O(PF2) in the gas phase and PF2OCH2CH2CN in gaseous and crystalline phases. J. Mol. Struct. 1990, 221, 25–44. [Google Scholar] [CrossRef]
- Gilardi, R.; Evans, R.N.; Manser, G.E. 3,3-bis(difluoroaminomethyl)oxetane, a promising new energetic material. Acta Cryst. 2003, E59, o2032–o2034. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Politzer, P.; Murray, J.S.; Janjić, G.V.; Zarić, S.D. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals 2014, 4, 12-31. https://doi.org/10.3390/cryst4010012
Politzer P, Murray JS, Janjić GV, Zarić SD. σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures. Crystals. 2014; 4(1):12-31. https://doi.org/10.3390/cryst4010012
Chicago/Turabian StylePolitzer, Peter, Jane S. Murray, Goran V. Janjić, and Snežana D. Zarić. 2014. "σ-Hole Interactions of Covalently-Bonded Nitrogen, Phosphorus and Arsenic: A Survey of Crystal Structures" Crystals 4, no. 1: 12-31. https://doi.org/10.3390/cryst4010012