Improved Synthesis and Crystal Structure of Dalcetrapib


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
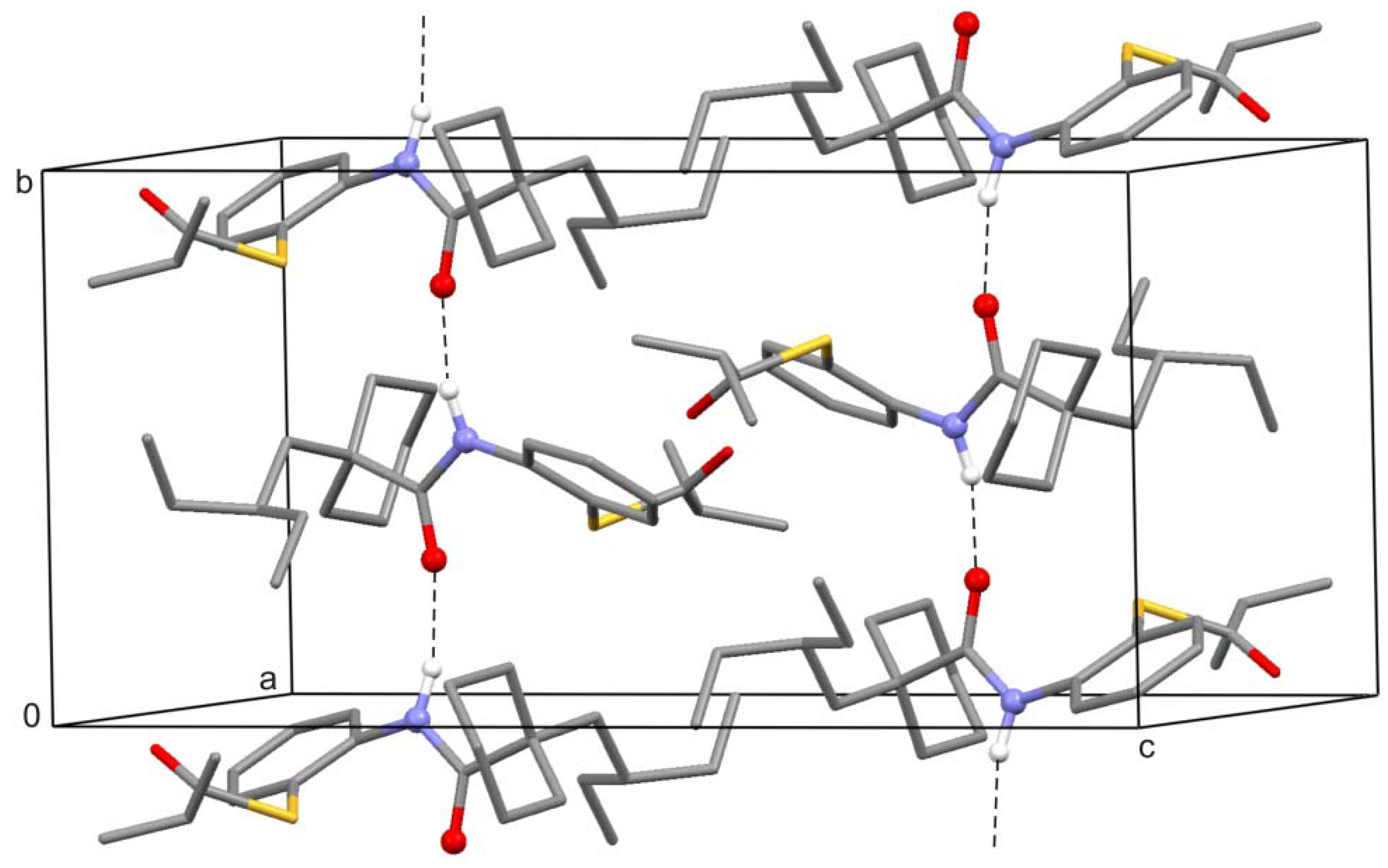
2. Results and Discussion

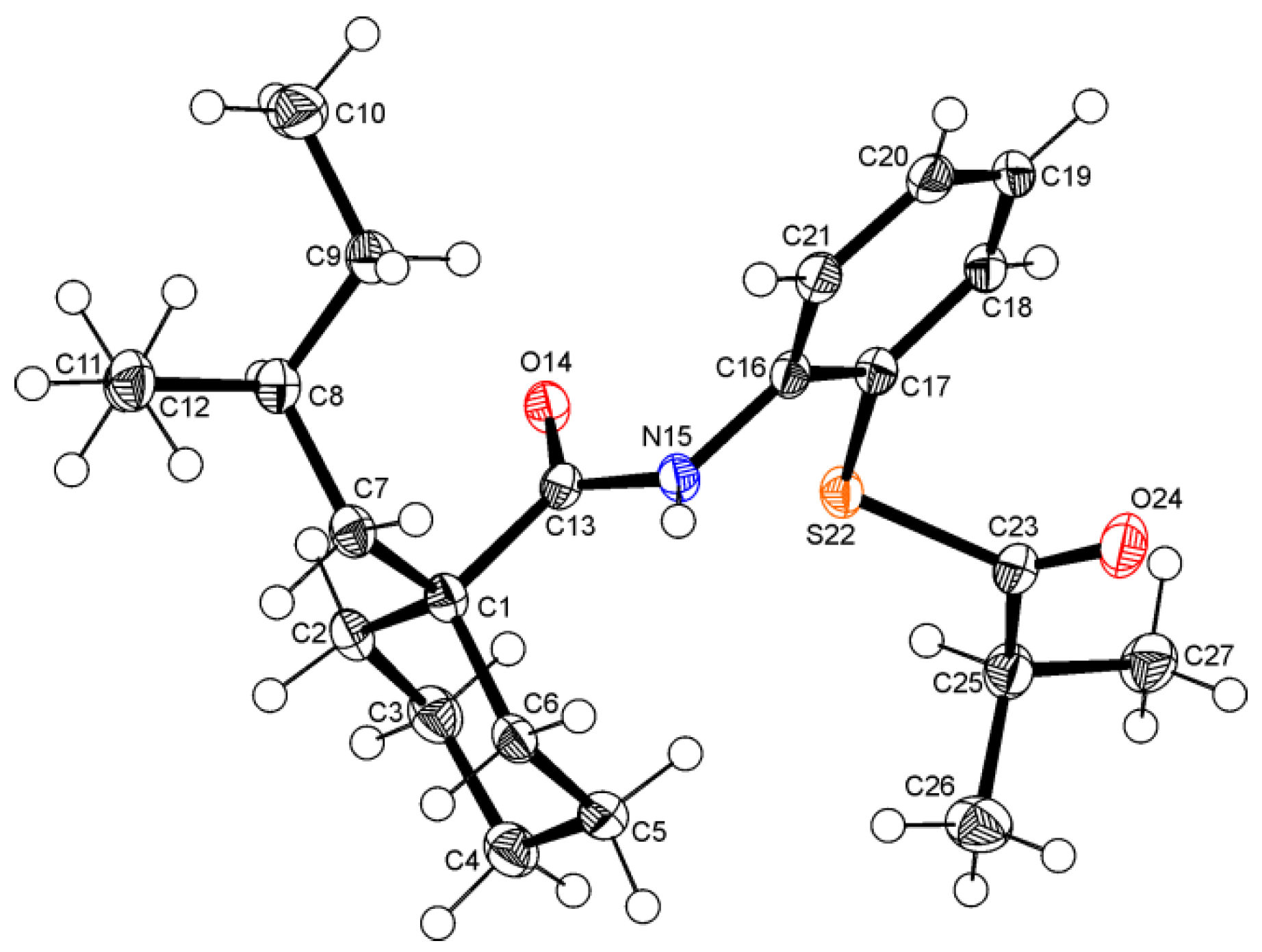
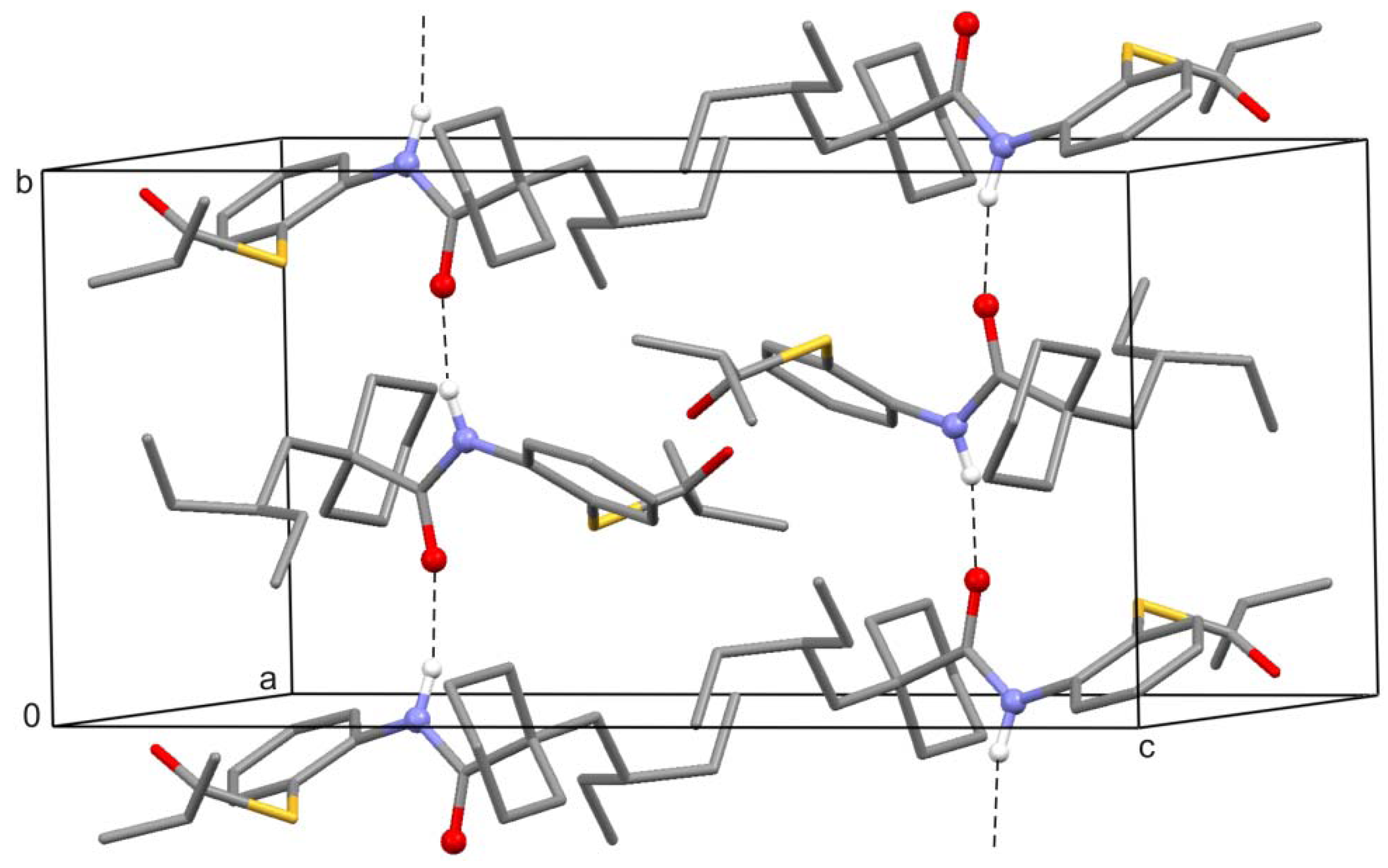
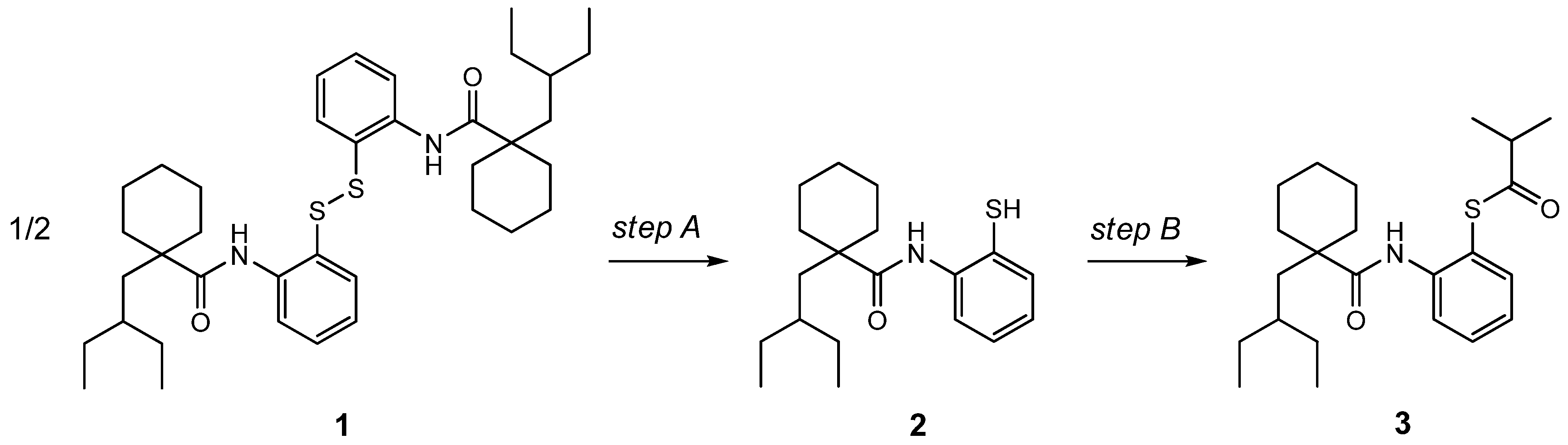
3. Experimental Section
3.1. Two-Step Synthesis of Dalcetrapib (3)
3.2. One-Pot Synthesis of Dalcetrapib (3)
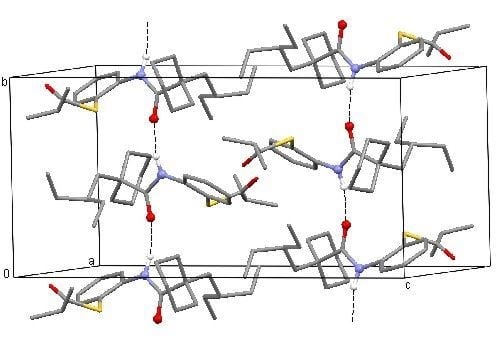
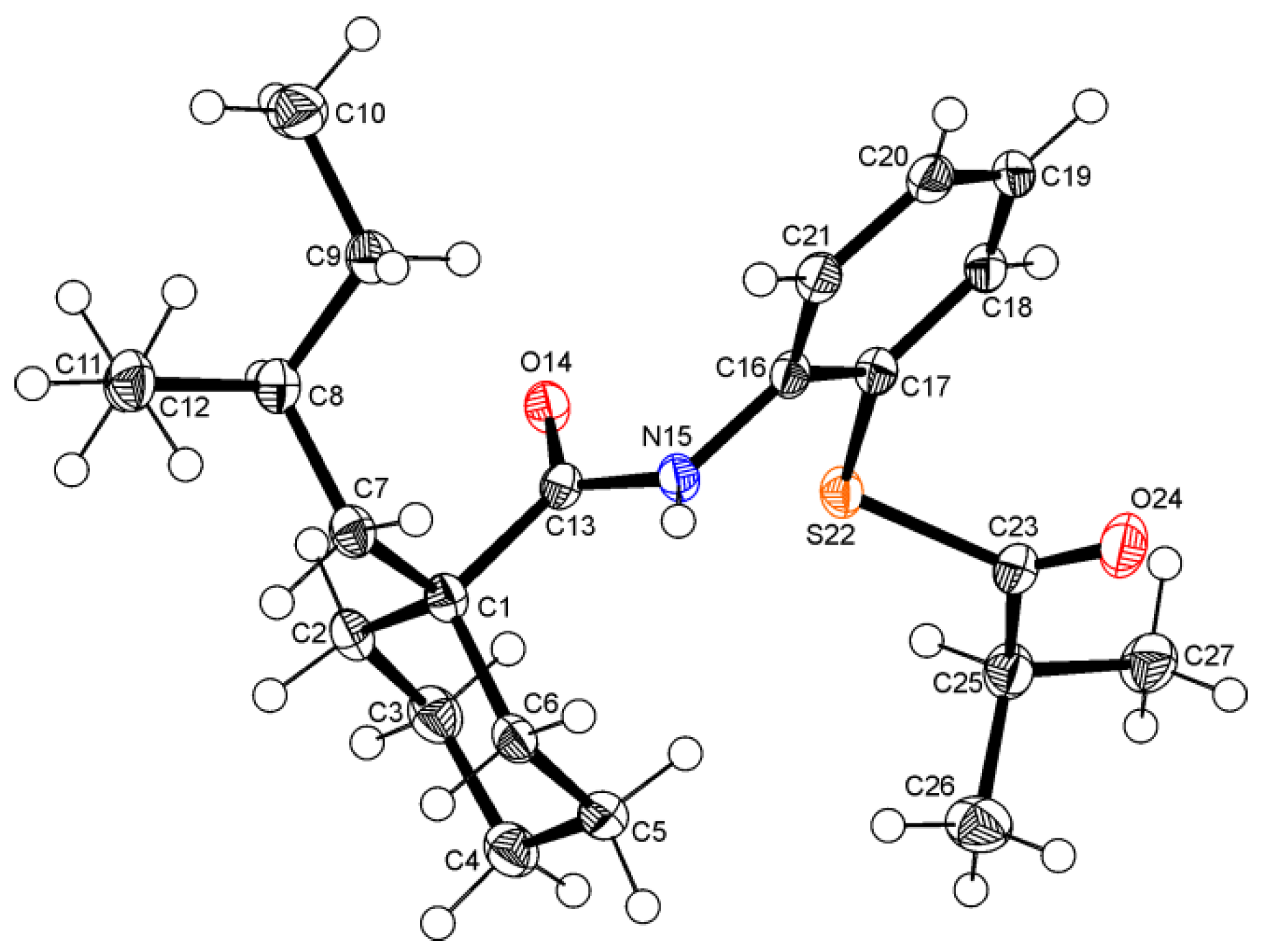
3.3. Spectroscopic and Crystal Structure Data
4. Conclusions
References
- Sinclair, P.J. Emerging topics in atherosclerosis: HDL raising therapies. Annu. Rep. Med. Chem. 2005, 40, 71–83. [Google Scholar] [CrossRef]
- Shinkai, H.; Maeda, K.; Yamasaki, T.; Okamoto, H.; Uchida, I. Bis(2-(Acylamino)phenyl) disulfides, 2-(acylamino)benzenethiols, and S-(2-(acylamino)phenyl) alkanethioates as novel inhibitors of cholesteryl ester transfer protein. J. Med. Chem. 2000, 43, 3566–3572. [Google Scholar] [CrossRef]
- Sunami, M.; Serigano, T. Pharmaceutical compositions of CETP inhibitors. Int. Patent WO 2004/082593 A2, 2004. [Google Scholar]
- Shinkai, H. Cholesteryl ester transfer-protein modulator and inhibitors and their potential for the treatment of cardiovascular diseases. Vasc. Health Risk Manag. 2012, 8, 323–331. [Google Scholar] [CrossRef]
- Luescher, T.F.; Taddei, S.; Kaski, J.-C.; Jukema, J.W.; Kallend, D.; Muenzel, T.; Kastelein, J.J.P.; Deanfield, J.E. Vascular effects and safety of dalcetrapib in patients with or at risk of coronary heart disease: The dal-VESSEL randomized clinical trial. Eur. Heart J. 2012, 33, 857–865. [Google Scholar] [CrossRef]
- Costello, D.; Harnett, G.J.; Hidber, P.; Hoffmann, U.; McCarthy, T.; Reents, R.; Smith, D.A.; Smyth, T. Process for the preparation of S-2-[1-(2-ethylbutyl)cyclohexylcarbonylamino]phenyl] 2-methylthiopropionate (dalcetrapib) using isobutyric anhydride and a reducing agent. Int. Patent WO 2011/000793 A2, 2011. [Google Scholar]
- Sridhar, M.; Vadivel, S.K.; Bhalerao, U.T. Reduction of symmetric disulfides to thiols using Mg in methanol. Synth. Commun. 1997, 27, 1347–1350. [Google Scholar] [CrossRef]
- Cleland, W.W. Dithiothreitol, a new protective reagent for SH groups. Biochemistry 1964, 3, 480–482. [Google Scholar] [CrossRef]
- Lakouraj, M.M.; Movassagh, B.; Fadaei, Z. Convenient synthesis of thiol esters from acyl chlorides and disulfides using Zn/AlCl3. Monatsh. Chem. 2002, 133, 1085–1088. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, F.; Liu, G. Dynamic covalent chemistry of disulfides offers a highly efficient synthesis of diverse benzofused nitrogen-sulfur heterocycles in one pot. J. Comb. Chem. 2010, 12, 531–540. [Google Scholar] [CrossRef]
- Anonymous. S-[2-({[2-Ethylbutyl)cyclohexyl]carbonyl}amino)phenyl] 2-methylpropanethioate. Available online: http://ip.com/IPCOM/000205232 (accessed on 15 October 2012).
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laus, G.; Kahlenberg, V.; Richter, F.; Nerdinger, S.; Schottenberger, H. Improved Synthesis and Crystal Structure of Dalcetrapib. Crystals 2012, 2, 1455-1459. https://doi.org/10.3390/cryst2041455
Laus G, Kahlenberg V, Richter F, Nerdinger S, Schottenberger H. Improved Synthesis and Crystal Structure of Dalcetrapib. Crystals. 2012; 2(4):1455-1459. https://doi.org/10.3390/cryst2041455
Chicago/Turabian StyleLaus, Gerhard, Volker Kahlenberg, Frank Richter, Sven Nerdinger, and Herwig Schottenberger. 2012. "Improved Synthesis and Crystal Structure of Dalcetrapib" Crystals 2, no. 4: 1455-1459. https://doi.org/10.3390/cryst2041455
APA StyleLaus, G., Kahlenberg, V., Richter, F., Nerdinger, S., & Schottenberger, H. (2012). Improved Synthesis and Crystal Structure of Dalcetrapib. Crystals, 2(4), 1455-1459. https://doi.org/10.3390/cryst2041455