Temperature-Dependent Ordering of the Methyl Group in the Crystal Structure of 5-(2-Chlorophenyl)-7-ethyl-1H-thieno [2,3-E][1,4]diazepin-2(3H)-one

 ,
,
Abstract
:1. Introduction
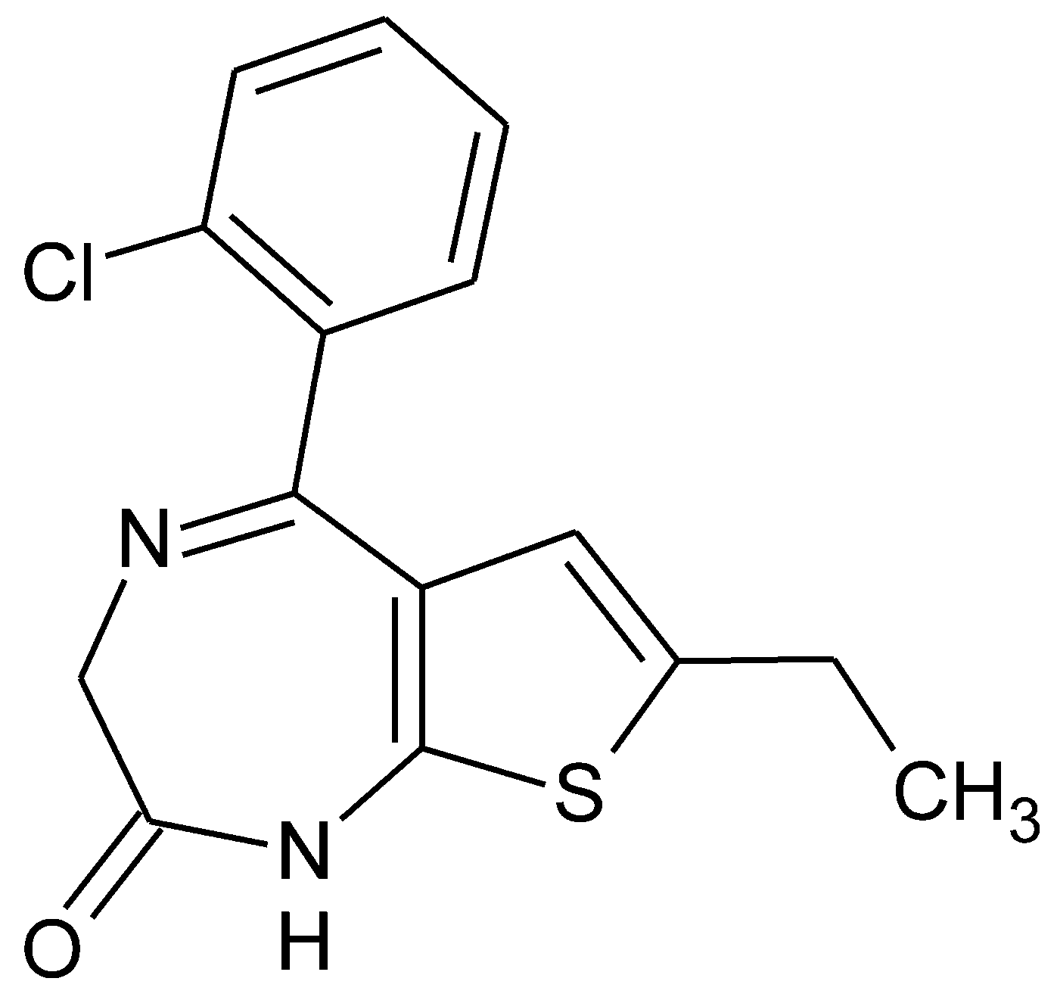
2. Results and Discussion
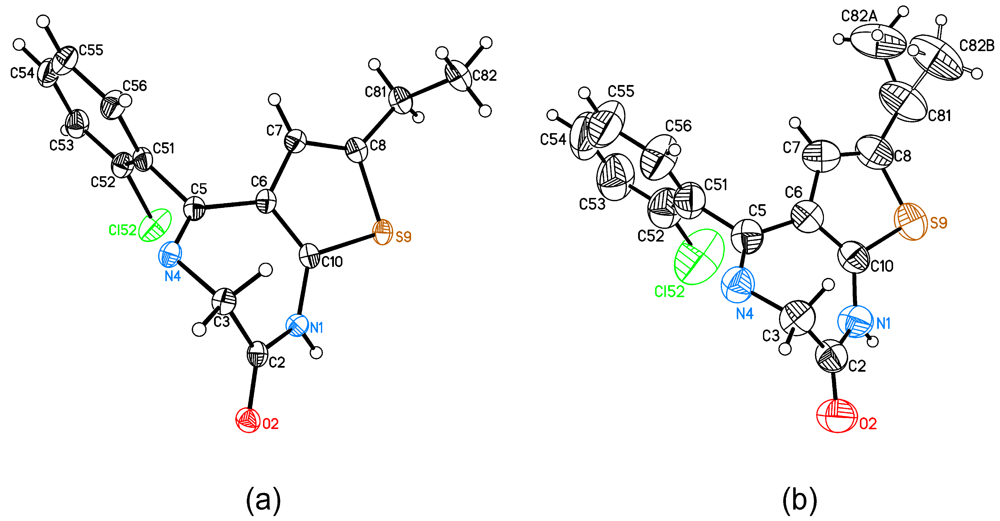
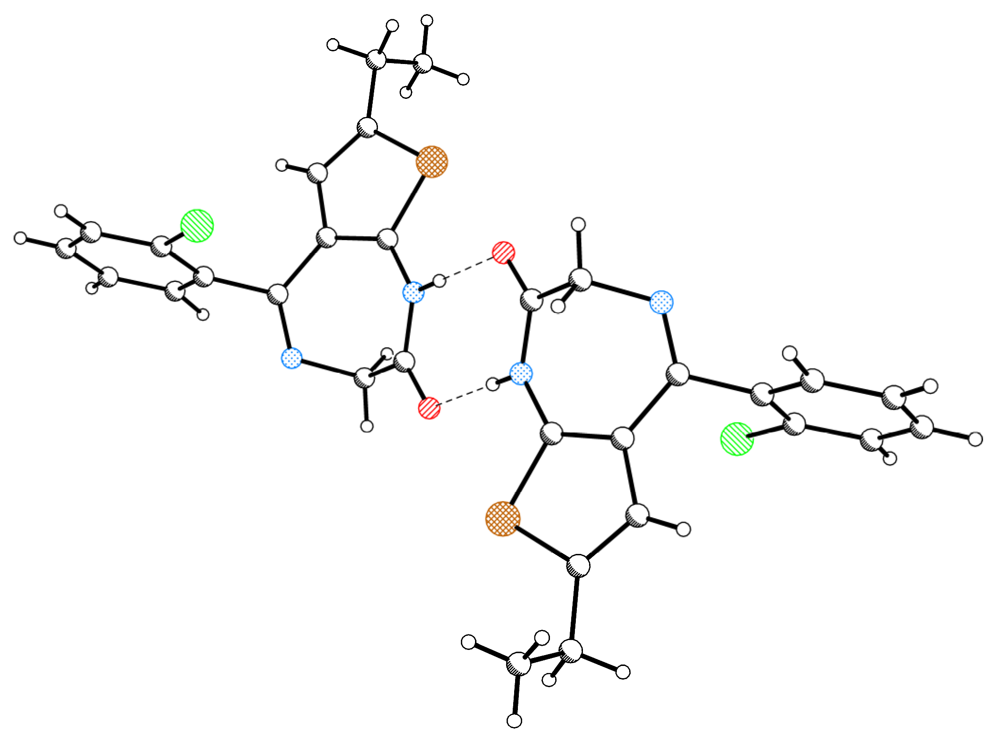
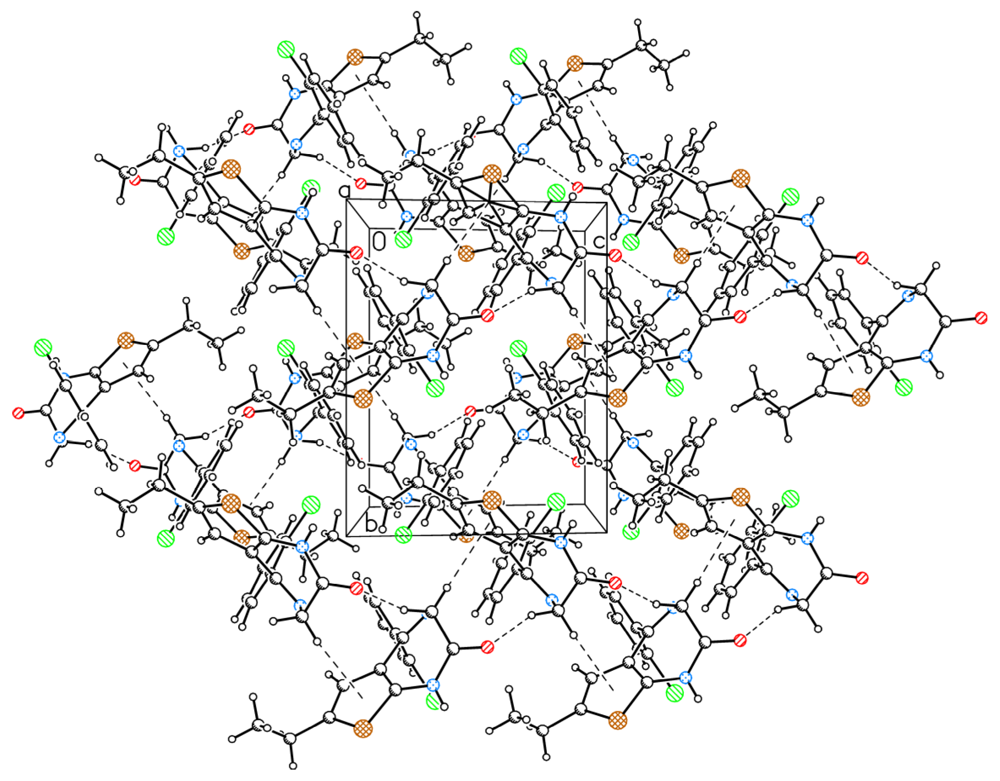
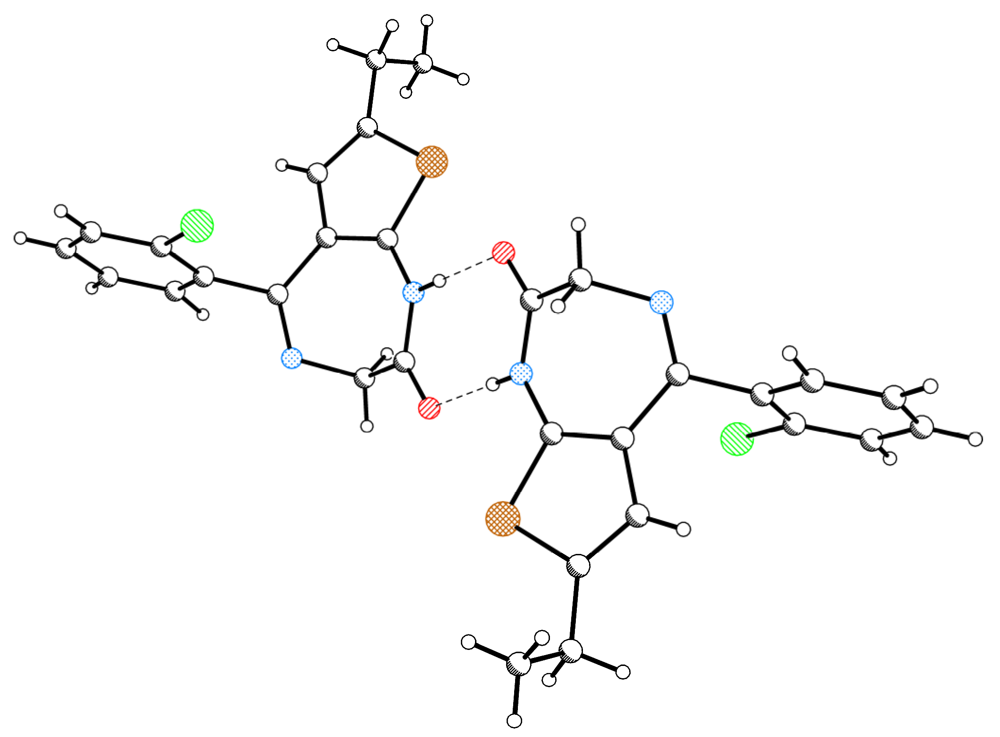
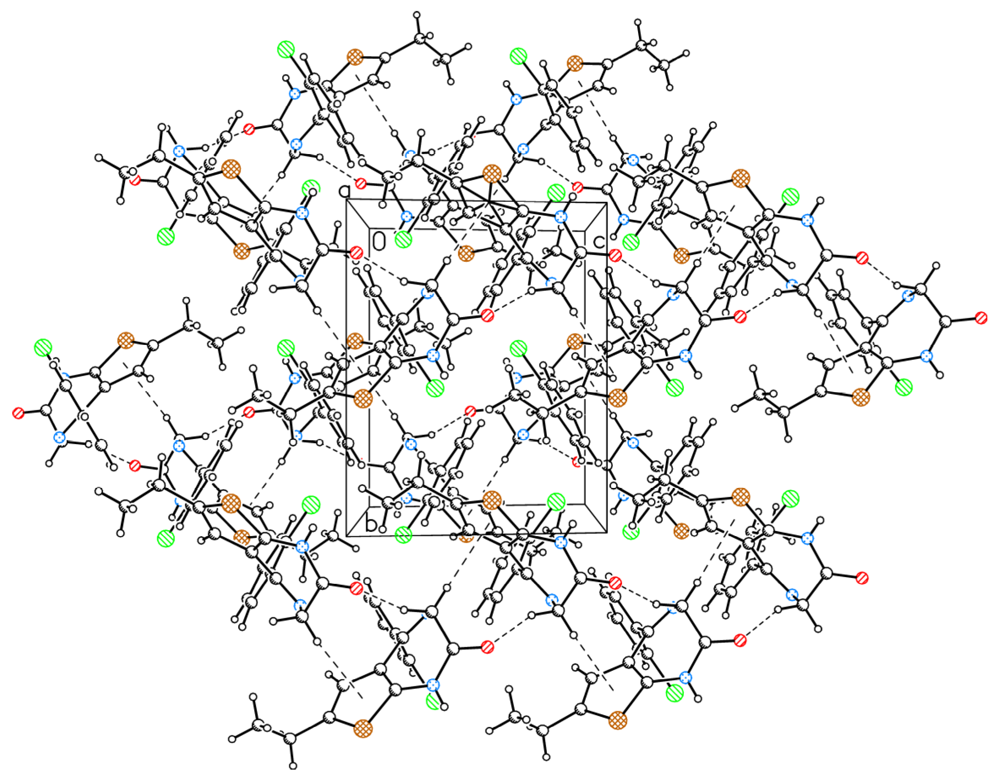
2.1. Molecular Structure and Crystal Packing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 125 K | 380 K | |
|---|---|---|
| N1-C2 | 1.3541(19) | 1.342(4) |
| N1-C10 | 1.3905(19) | 1.385(4) |
| C2-O2 | 1.2360(18) | 1.234(4) |
| C2-C3 | 1.507(2) | 1.487(5) |
| C3-N4 | 1.4716(19) | 1.463(4) |
| N4-C5 | 1.279(2) | 1.282(4) |
| C5-C6 | 1.468(2) | 1.457(5) |
| C6-C10 | 1.375(2) | 1.365(5) |
| C6-C7 | 1.428(2) | 1.421(5) |
| C7-C8 | 1.353(2) | 1.344(5) |
| C8-S9 | 1.744(2) | 1.730(4) |
| S9-C10 | 1.726(2) | 1.714(3) |
| C2-N1-C10 | 125.0(1) | 125.0(3) |
| C3-N4-C5 | 117.7(1) | 116.8(3) |
| N4-C5-C6 | 126.7(1) | 126.9(3) |
| C8-S9-C10 | 91.60(7) | 91.9(2) |
| N1-C2-C3-N4 | 67.4(2) | 65.7(4) |
| C2-C3-N4-C5 | −72.2(2) | −72.4(4) |
| C3-N4-C5-C6 | 7.9(2) | 8.6(5) |
| N4-C5-C6-C10 | 32.2(2) | 33.1(5) |
| C5-C6-C10-N1 | 0.6(2) | −1.6(6) |
| C6-C10-N1-C2 | −35.3(2) | −35.3(6) |
| C10-N1-C2-C3 | −1.2(2) | 1.4(5) |
| N4-C5-C51-C52 | −107.9(2) | −105.3(4) |
| N4-C5-C51-C56 | 71.0(2) | 74.3(5) |
| C6-C7-C8-C81 | −174.6(2) | 178.6(4) |
| C10-S9-C8-C81 | 175.3(1) | −178.8(4) |
| C7-C8-C81-C82 | 97.8(2) | 78.7(9) |
| 18(1) | ||
| S9-C8-C81-C82 | −75.8(2) | −103.0(8) |
| −164(1) |
| 125K | ||||||
| D | H | A | D–H(Å) | H···A(Å) | D···A(Å) | D–H···A(°) |
| N1 | H1 | O2 i | 0.86(2) | 1.94(2) | 2.793(2) | 178(2) |
| C3 | H3B | O2 ii | 1.01(2) | 2.38(2) | 3.316(2) | 154(1) |
| C3 | H3A | CgA iii | 0.96(2) | 2.83(2) | 3.637(2) | 143(1) |
| 1LT | ||||||
| D | H | A | D–H(Å) | H···A(Å) | D···A(Å) | D–H···A(°) |
| N1 | H1 | O2 i | 0.86 | 1.99 | 2.836(4) | 167 |
| C3 | H3B | O2 ii | 0.97 | 2.48 | 3.389(5) | 155 |
| C3 | H3A | CgA iii | 0.97 | 3.01 | 3.866(5) | 148 |
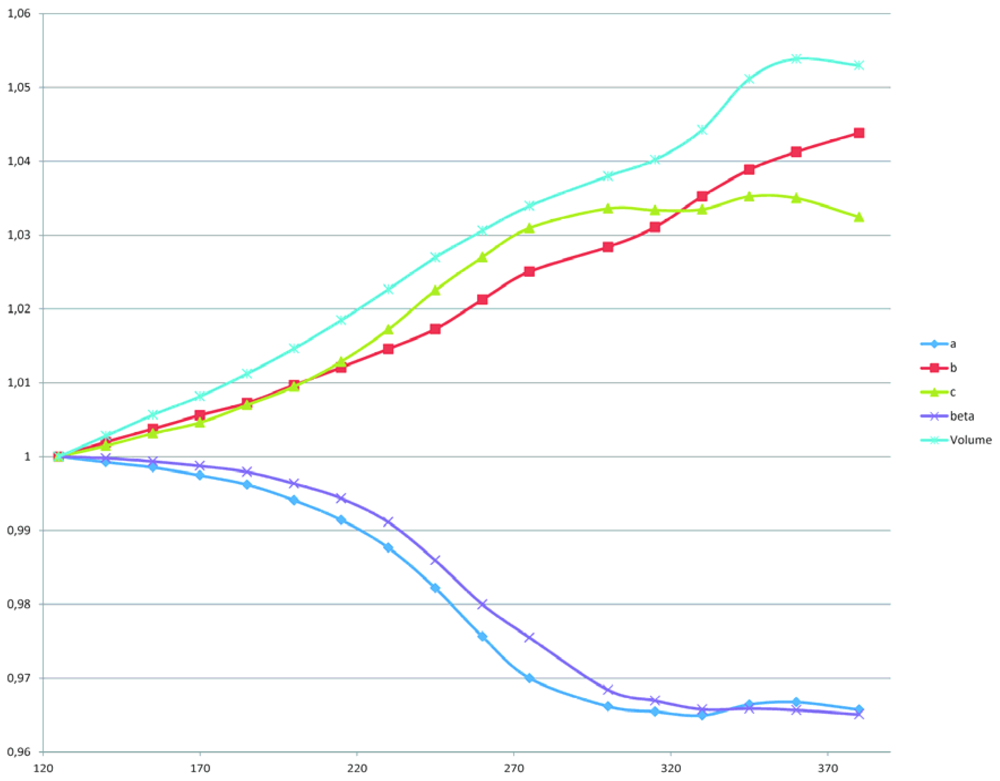
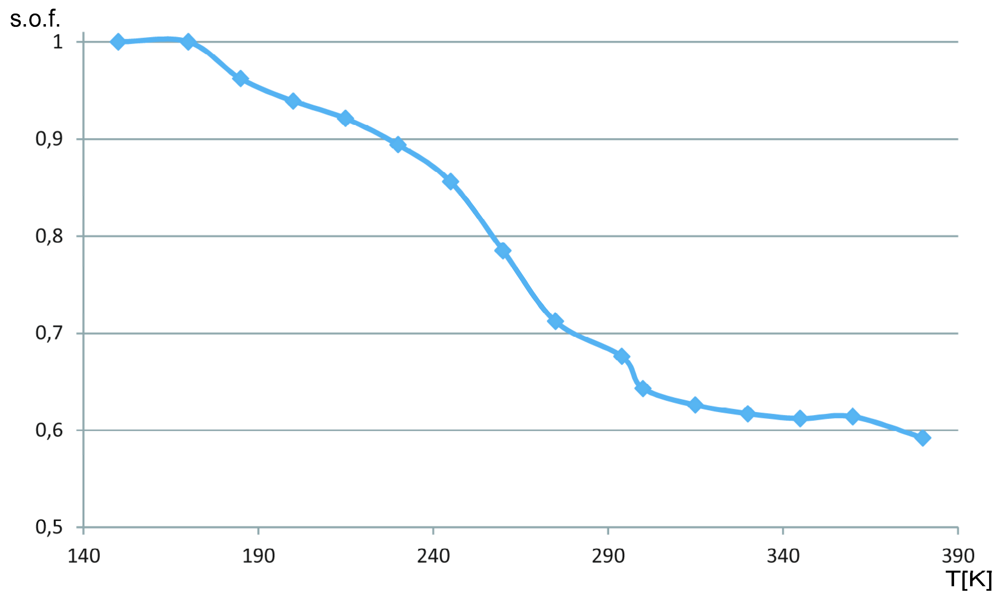
2.2. The Structural Changes
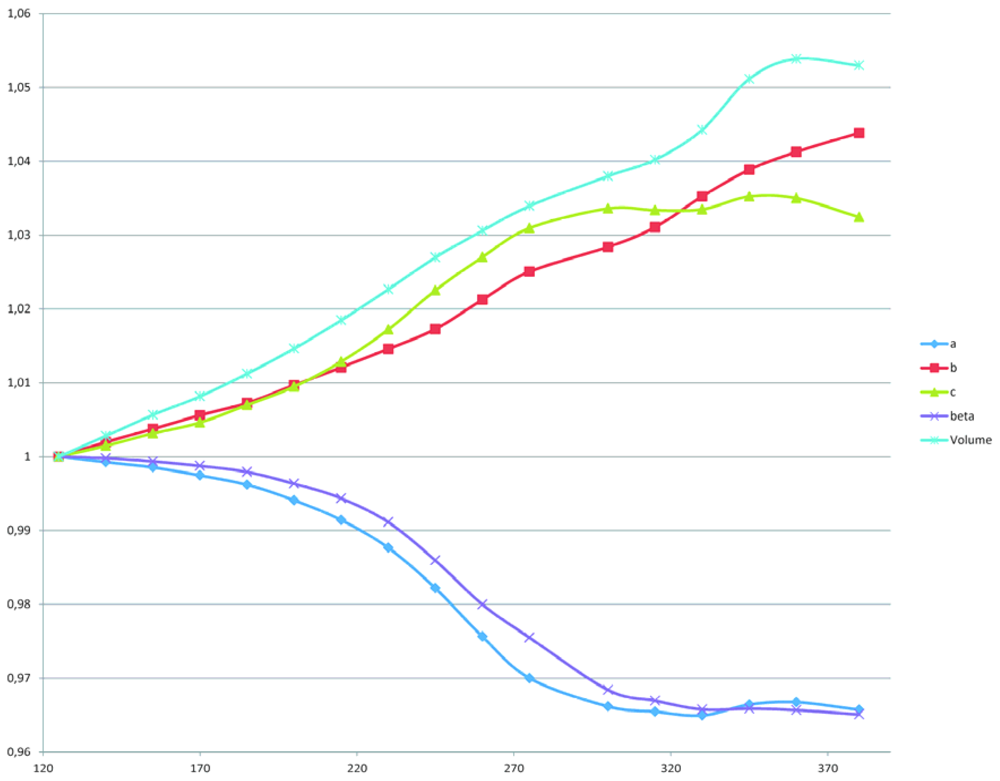
3. Experimental Section
| Compound | 125 K | 230 K (D) | 230 K (No_D) | 380 K | |||
|---|---|---|---|---|---|---|---|
| Formula | C15H13ClN2OS | ||||||
| Formula weight | 304.78 | ||||||
| Crystal system | monoclinic | ||||||
| Space group | P21/c | ||||||
| a (Å) | 15.6941(6) | 15.5187(8) | 15.177(2) | ||||
| b (Å) | 10.7909(4) | 10.9358(5) | 11.281(1) | ||||
| c (Å) | 8.6586(3) | 8.8141(4) | 8.933(1) | ||||
| β (°) | 102.184(4) | 101.245(5) | 98.73(1) | ||||
| V (Å3) | 1433.33(9) | 1467.1(1) | 1511.7(4) | ||||
| Z | 4 | ||||||
| Dx (g·cm−3) | 1.41 | 1.38 | 1.34 | ||||
| F(000) | 632 | ||||||
| μ (mm−1) | 0.41 | 0.40 | 0.39 | ||||
| Crystal size (mm) | 0.25 × 0.15 × 0.1 | ||||||
| No. of reflections used for unit cell determination | 2249 | 1661 | |||||
| Θ range (°) | 3.06–28.89 | 3.00–28.91 | 2.93–25.99 | ||||
| hkl range | −20 ≤ h ≤ 18 | −19 ≤ h ≤ 18 | −17 ≤ h ≤ 18 | ||||
| −14 ≤ k ≤ 9 | −14 ≤ k ≤ 9 | −12 ≤ k ≤ 13 | |||||
| −11 ≤ l ≤ 11 | −11 ≤ l ≤ 11 | −11 ≤ l ≤ 9 | |||||
| Reflections: | |||||||
| collected | 6034 | 6177 | 6031 | ||||
| unique (Rint) | 3218 (0.024) | 3286 (0.024) | 2942 (0.023) | ||||
| with I > 2σ(I) | 2723 | 2393 | 1625 | ||||
| Number of parameters | 233 | 222 | 222 | 192 | |||
| Weighting scheme: | |||||||
| A | 0.0405 | 0.0483 | 0.0483 | 0.0976 | |||
| B | 0.3247 | 0.3847 | 0.3847 | 0.3814 | |||
| R(F) [I > 2σ(I)] | 0.034 | 0.045 | 0.046 | 0.065 | |||
| wR(F2) [I > 2σ(I)] | 0.083 | 0.105 | 0.105 | 0.176 | |||
| R(F) [all data] | 0.043 | 0.068 | 0.069 | 0.116 | |||
| wR(F2) [all data] | 0.088 | 0.119 | 0.120 | 0.207 | |||
| Goodness of fit | 1.032 | 1.030 | 1.018 | 0.993 | |||
| max/min ∆ρ (e Å−3) | 0.32/−0.33 | 0.37/−0.26 | 0.34/−0.29 | 0.32/−0.30 | |||
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Block, M.G.; DiPardo, R.M.; Evans, B.E.; Rittle, K.E.; Witter, W.L.; Veber, D.F.; Anderson, P.S.; Freidinger, R.M. Benzodiazepine gastrin and brain cholecystokinin receptor ligands: L-365, 260. J. Med. Chem. 1989, 32, 13–16. [Google Scholar] [CrossRef]
- Di Braccio, M.; Grossi, G.; Roma, G.; Vargiu, L.; Mura, M.; Marongiu, M.E. 1,5-Benzodiazepines. Part XII. Synthesis and biological evaluation of tricyclic and tetracyclic 1,5-benzodiazepine derivatives as nevirapine analogues. Eur. J. Med. Chem. 2001, 36, 935–949. [Google Scholar] [CrossRef]
- Hollister, L.E. Principles of therapeutic applications of the benzodiazepines. J. Psychoactive Drugs 1983, 15, 41–44. [Google Scholar] [CrossRef]
- Moroz, G. High-Potency benzodiazepines: recent clinical results. J. Clin. Psychiatry 2004, 65, 13–18. [Google Scholar]
- Robol, J.A.; Cimarusti, M.P.; Simpkins, L.M.; Brown, B.; Ryono, D.E.; Bird, M.M.; Asad, T.R.; Schaeffer, N.C.; Trippodo, N.C. Dual metalloprotease inhibitors. 6. Incorporation of bicyclic and substituted monocyclic azepinones as dipeptide surrogates in angiotensin-converting enzyme/neutral endopeptidase inhibitors. J. Med. Chem. 1996, 39, 494–502. [Google Scholar] [CrossRef]
- Wang, T.; Lui, A.S.; Cloudsdale, I.S. A novel route to pyrrolo[2,1-c][1,4]benzodiazepin-5-ones. Formal total synthesis of (±)-DC-81. Org. Lett. 1999, 1, 1835–1837. [Google Scholar] [CrossRef]
- Novelli, F.; Sparatore, F.A.; Tassso, B.; Sparatore, F. Quinolizidinyl derivatives of 5,11-dihydro-6H-pyrido[2,3-b][1,4]benzodiazepin-6-one as ligands for muscarinic receptors. Bioorg. Med. Chem. Lett. 1999, 9, 3031–3034. [Google Scholar] [CrossRef]
- Evans, B.; Pipe, A.; Clarke, L.; Banks, M. Identification of a potent and selective oxytocin antagonist, from screening a fully encoded differential release combinatorial chemical library. Bioorg. Med. Chem. Lett. 2001, 11, 1297–1300. [Google Scholar] [CrossRef]
- Scammells, P.J.; Tranberg, C.E.; Tiekink, E.R.T. 6,7-Dimethyl-5-phenyl-1H-thieno[2,3-e][1,4]diazepin-2(3H)-one. Acta Cryst. 2001, E57, o344–o345. [Google Scholar]
- Bruno, G.; Nicolo, F.; Gitto, R.; Micale, N.; Rosace, G. 5-Phenyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-8(7H)-one. Acta Cryst. 2003, C59, o117–o119. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Betz, R.; Gerber, T.; Hosten, E.; Dayananda, A.S.; Yathirajan, H.S.; Ramesha, A.R. (E)-5-(2-chlorophenyl)-7-ethyl-2-oxo-2,3-dihydro-1H-thieno[2,3-e][1,4]-diazepin-4-ium 2,4,6-trinitrophenola-te. Acta Cryst. 2012, E68, o516–o517. [Google Scholar]
- Duax, W.L.; Norton, D.A. Atlas of Steroid Structure; Plenum: New York, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar]
- Agilent Technologies Ltd. CrysAlis PRO, Version 1.171.35.15, Agilent Technologies Ltd.: Yarnton, UK, 2010.
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Cryst. 1993, 26, 343–350. [Google Scholar] [CrossRef]
- Kubicki, M.; Dutkiewicz, G.; Jasinski, J.P.; Butcher, R.J.; Siddegowda, M.S.; Yathirajan, H.S.; Narayana, B. Ordering of water molecule in the crystal structure of 2-[3-methyl-4-(2,2,2-trifluoroethoxy)-2-pyridinyl]methylthio-1H-benzimidazole hydrate (Lansprazole sulphide hydrate). J. Chem. Cryst. 2012, 42, 245–250. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dutkiewicz, G.; Kubicki, M.; Dayananda, A.S.; Yathirajan, H.S.; Ramesha, A.R. Temperature-Dependent Ordering of the Methyl Group in the Crystal Structure of 5-(2-Chlorophenyl)-7-ethyl-1H-thieno [2,3-E][1,4]diazepin-2(3H)-one. Crystals 2012, 2, 1347-1356. https://doi.org/10.3390/cryst2031347
Dutkiewicz G, Kubicki M, Dayananda AS, Yathirajan HS, Ramesha AR. Temperature-Dependent Ordering of the Methyl Group in the Crystal Structure of 5-(2-Chlorophenyl)-7-ethyl-1H-thieno [2,3-E][1,4]diazepin-2(3H)-one. Crystals. 2012; 2(3):1347-1356. https://doi.org/10.3390/cryst2031347
Chicago/Turabian StyleDutkiewicz, Grzegorz, Maciej Kubicki, Alaloor S. Dayananda, Hemmige S. Yathirajan, and Andagar R. Ramesha. 2012. "Temperature-Dependent Ordering of the Methyl Group in the Crystal Structure of 5-(2-Chlorophenyl)-7-ethyl-1H-thieno [2,3-E][1,4]diazepin-2(3H)-one" Crystals 2, no. 3: 1347-1356. https://doi.org/10.3390/cryst2031347