Crystal and Molecular Structures of Two 2-Aminothiophene Derivatives

 ,
, 
Abstract
:1. Introduction
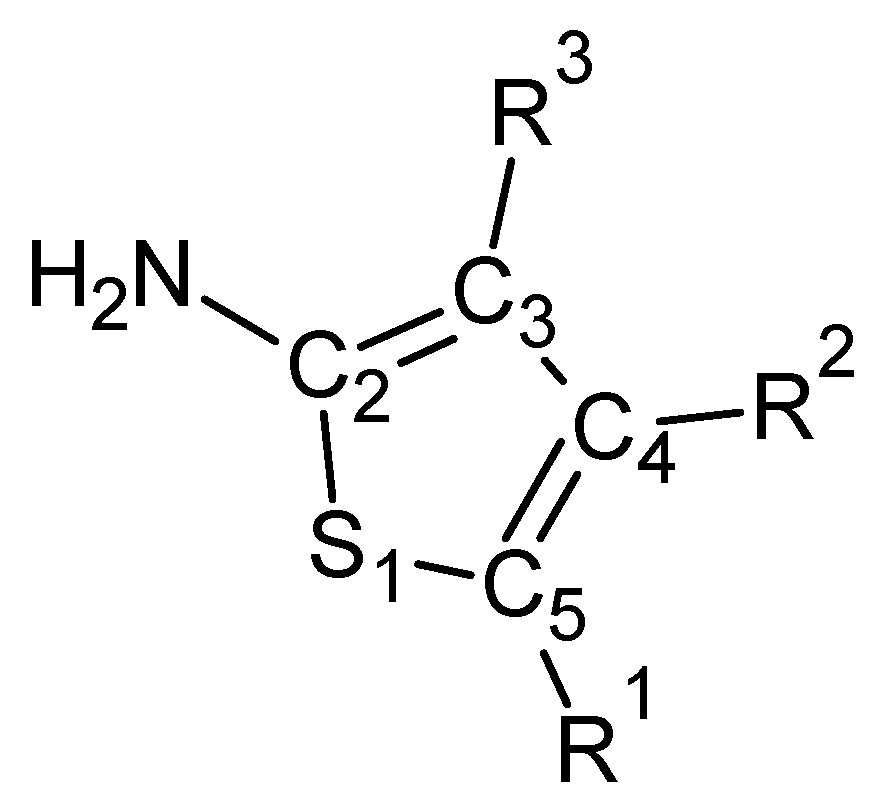
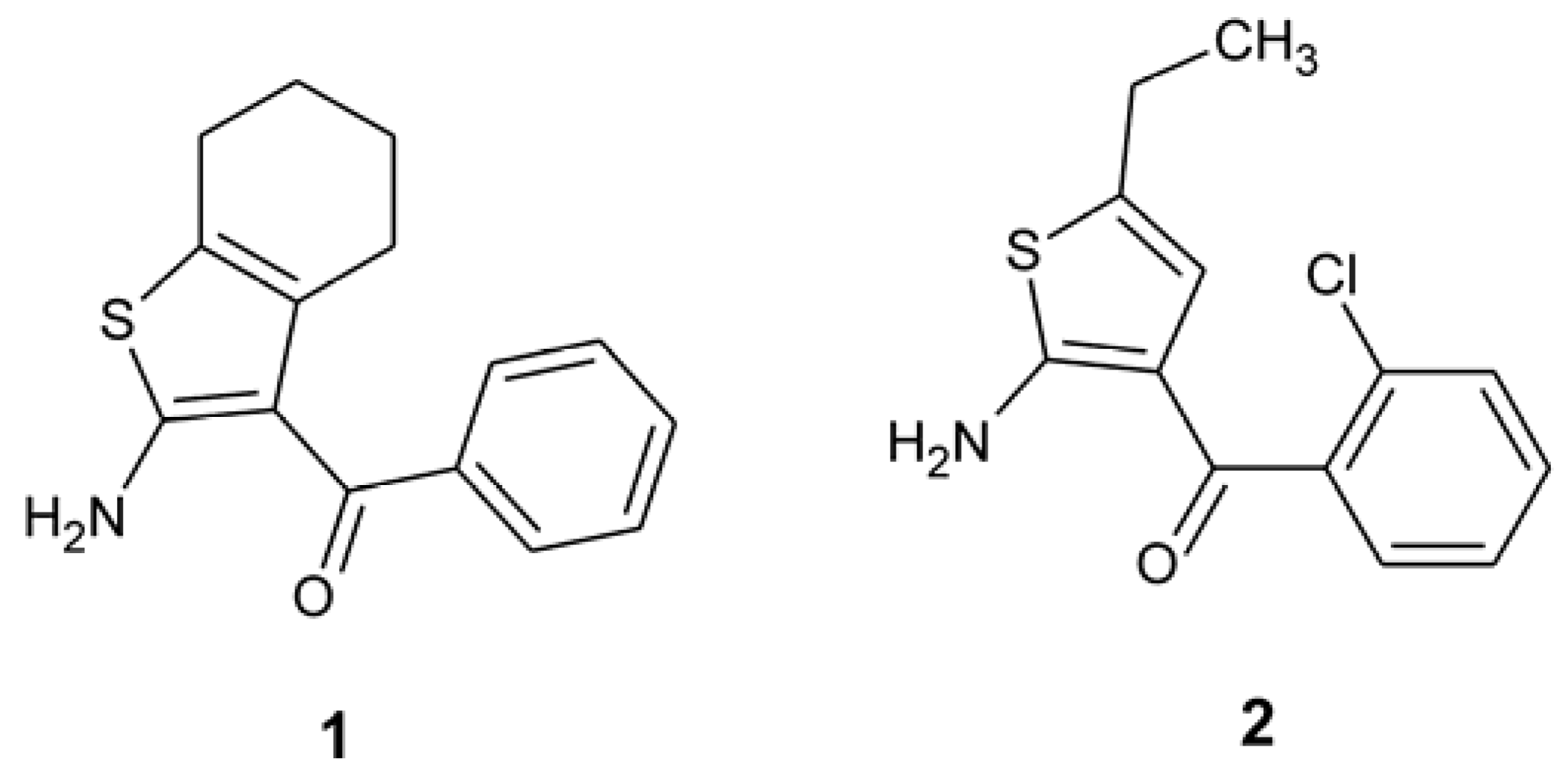
2. Results and Discussion

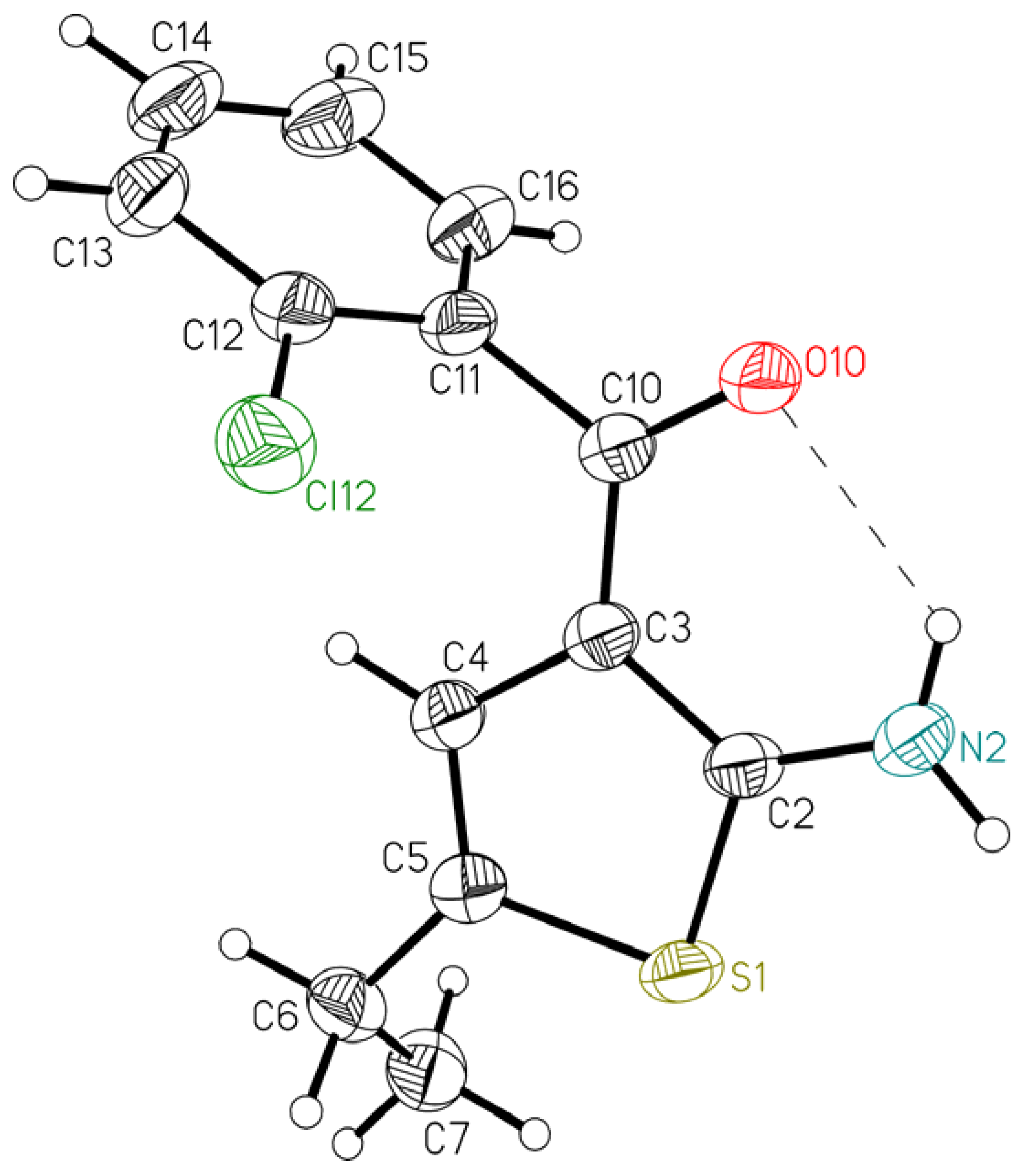

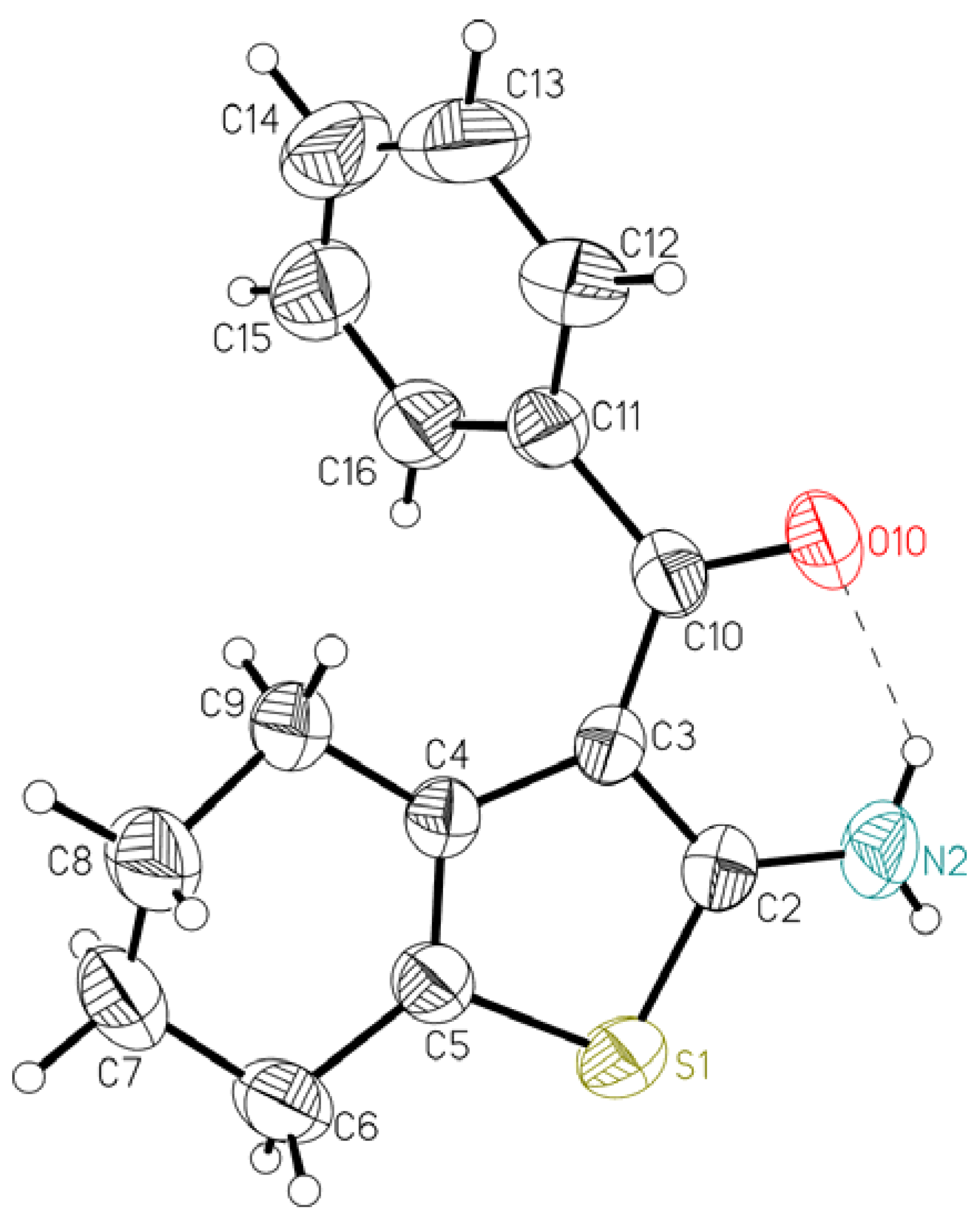{kind=link}
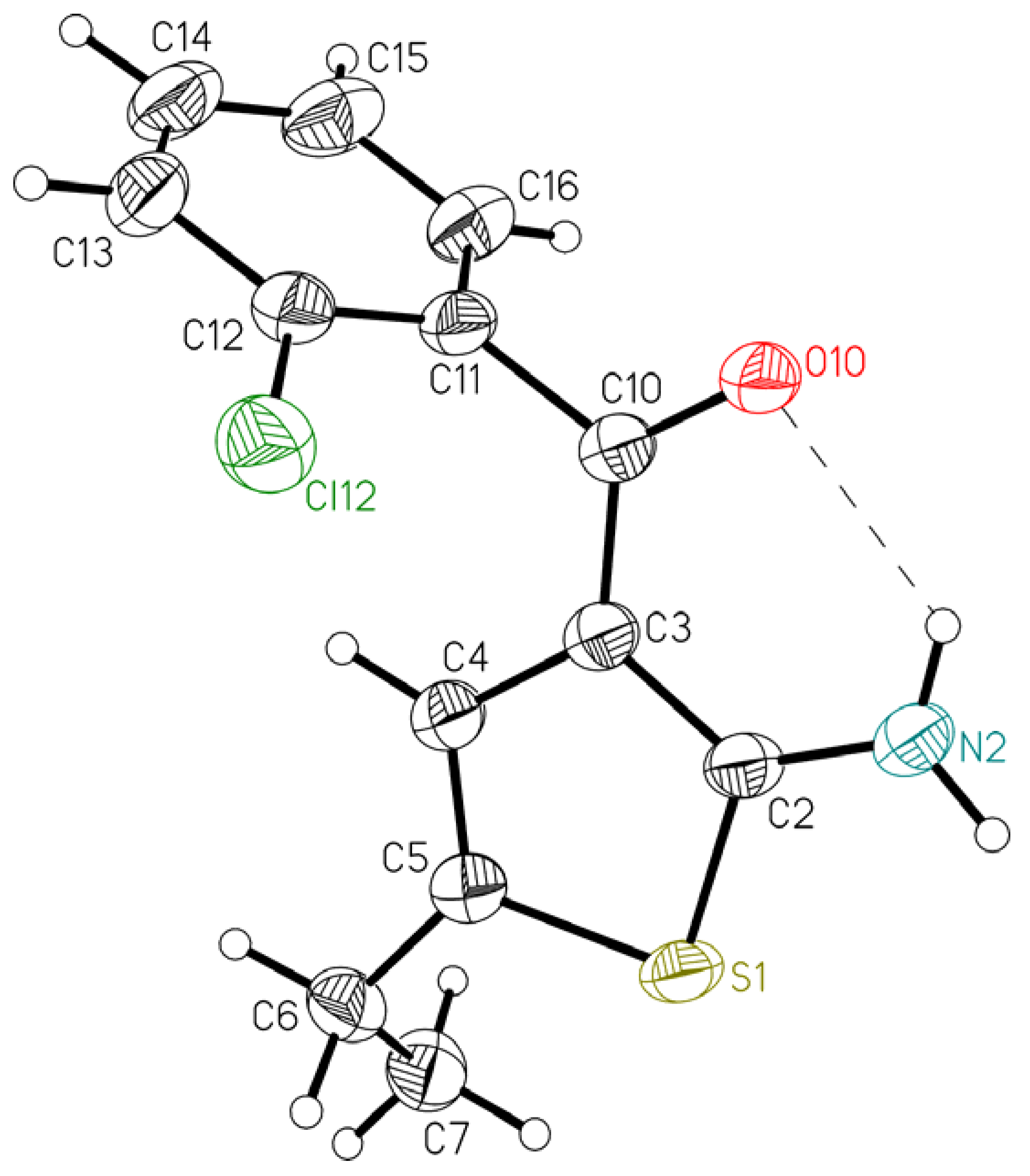{kind=link}
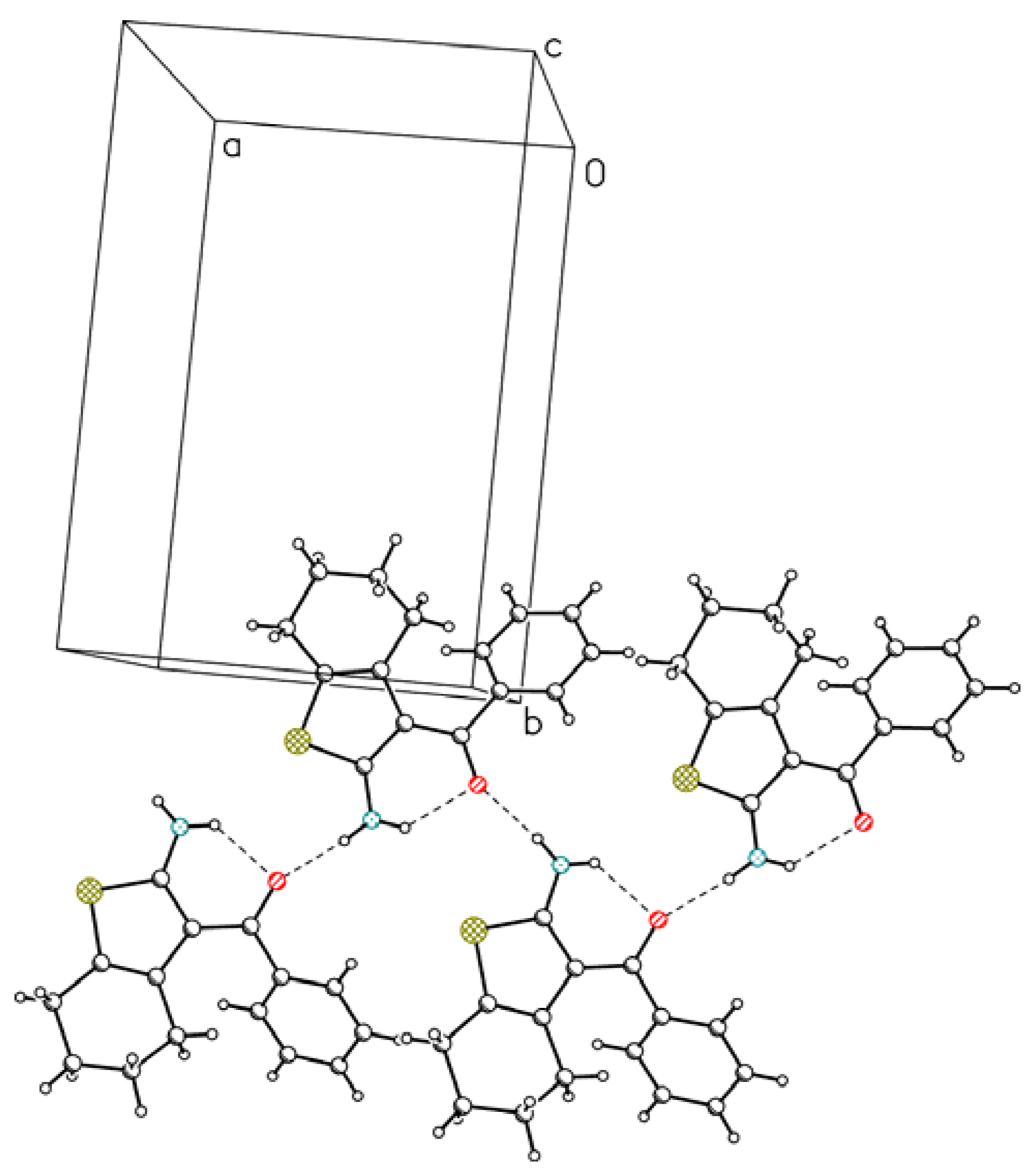{kind=link}
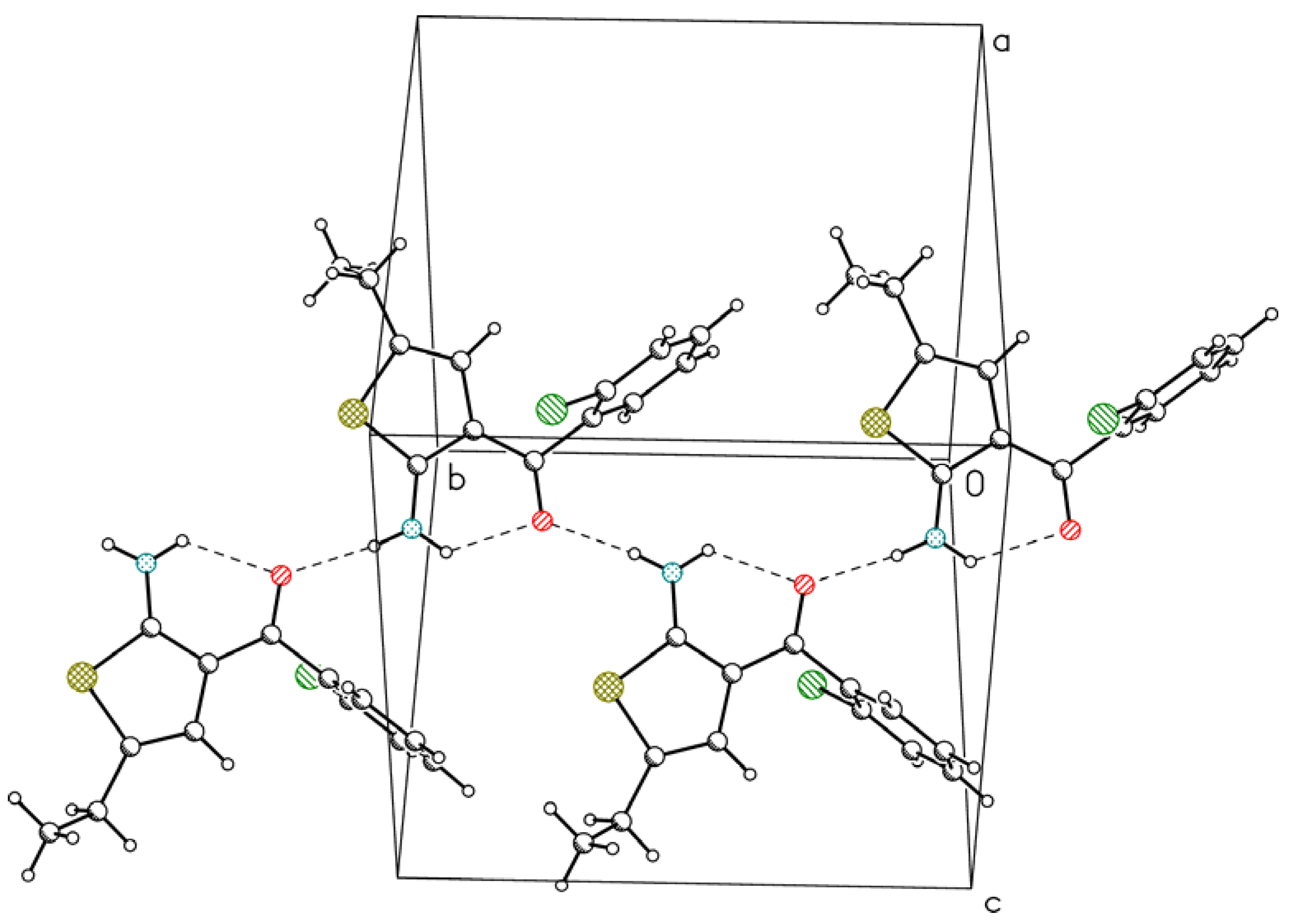{kind=link}
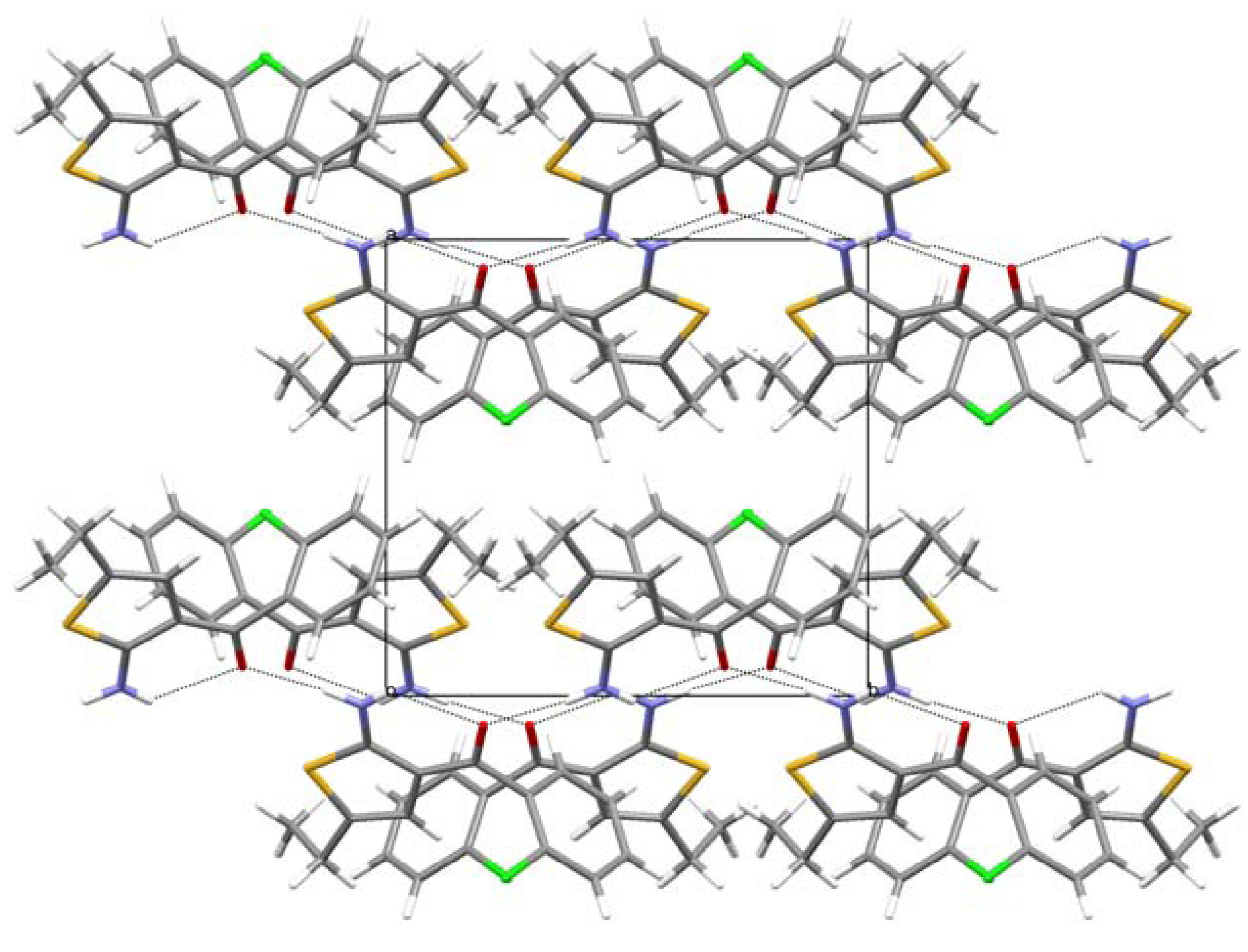{kind=link}
{kind=link}
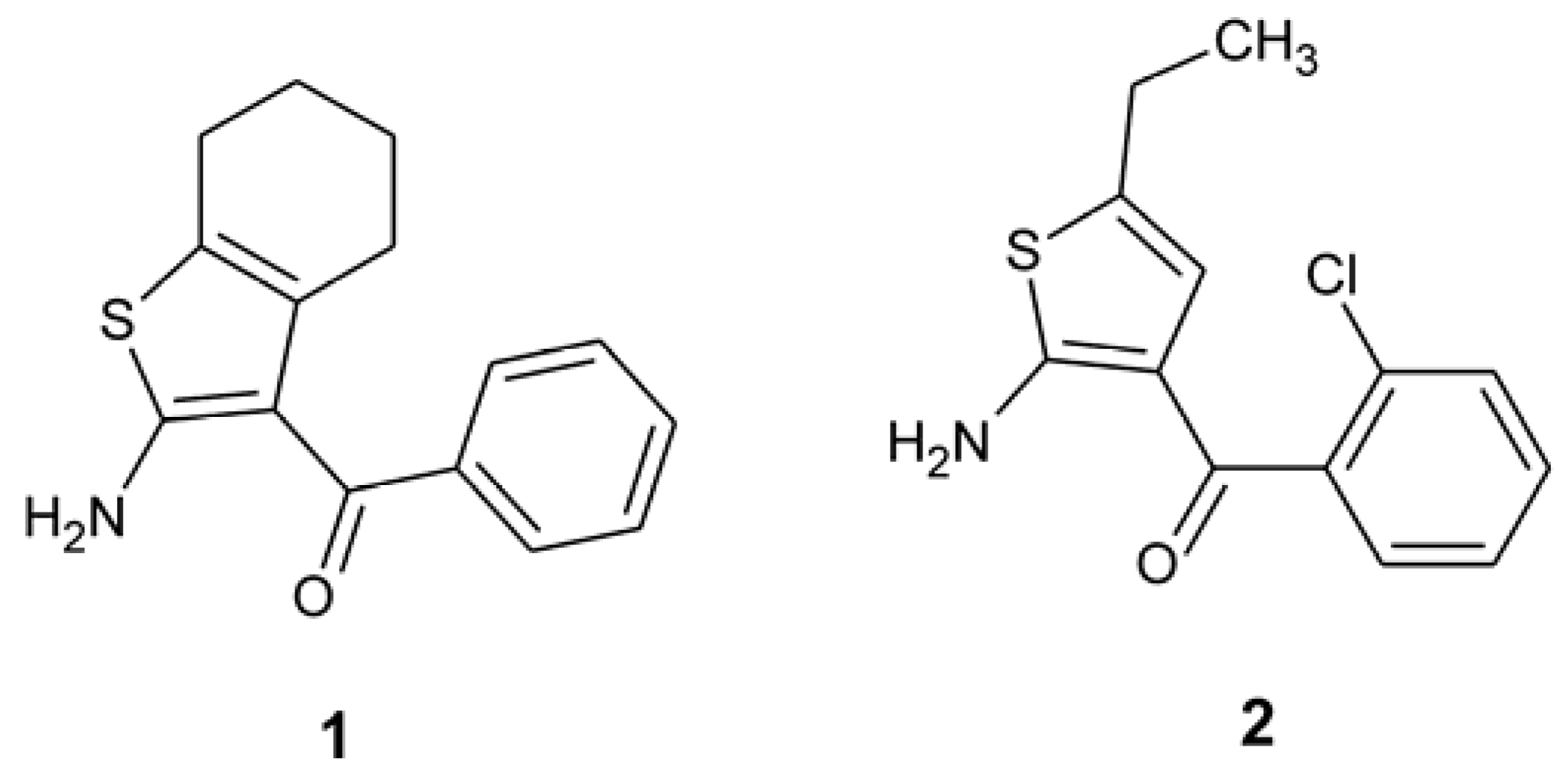{kind=link}
| 1 | 2 | |
|---|---|---|
| S1–C2 | 1.722(3) | 1.7311(15) |
| S1–C5 | 1.745(3) | 1.7611(16) |
| C2–N2 | 1.339(3) | 1.332(2) |
| C2–C3 | 1.405(3) | 1.402(2) |
| C3–C4 | 1.457(3) | 1.444(2) |
| C4–C5 | 1.342(3) | 1.343(2) |
| C10–O10 | 1.242(3) | 1.2485(19) |
| C3–C10 | 1.435(3) | 1.421(2) |
| C10–C11 | 1.499(4) | 1.509(2) |
| C2–S1–C5 | 91.76(12) | 92.30(7) |
| C3–C2–N2 | 127.5(2) | 127.10(14) |
| S1–C2–N2 | 120.9(2) | 121.98(12) |
| C2–C3–C10 | 119.0(2) | 122.28(14) |
| C4–C3–C10 | 129.9(2) | 126.24(14) |
| C3–C10–C11 | 121.2(2) | 117.33(13) |
| C3–C10–O10 | 121.3(3) | 124.13(13) |
| C11–C10–O10 | 117.4(2) | 118.51(13) |
| C12–C11–C16 | 118.9(3) | 118.41(15) |
| S1–C2–C3–C10 | −176.6(2) | −178.96(12) |
| C2–C3–C10–O10 | −17.5(4) | −0.5(3) |
| C4–C3–C10–O10 | 162.3(3) | 179.09(16) |
| C2–C3–C10–C11 | 159.4(2) | 177.67(14) |
| C4–C3–C10–C11 | −20.8(4) | −2.7(2) |
| C3–C10–C11–C12 | 137.6(3) | 82.15(19) |
| C3–C10–C11–C16 | −46.9(4) | −96.86(18) |
| D | H | A | D–H | H···A | D···A | D–H···A |
|---|---|---|---|---|---|---|
| 1 | ||||||
| N2 | H2A | O10 | 0.85(3) | 2.03(3) | 2.667(4) | 131(3) |
| N2 | H2B | O10 i | 0.85(3) | 1.99(3) | 2.832(3) | 173(3) |
| 2 | ||||||
| N2 | H2A | O10 | 0.87(2) | 2.16(2) | 2.7785(19) | 127.8(17) |
| N2 | H2B | O10 ii | 0.87(3) | 2.02(3) | 2.8750(19) | 169(2) |
| C4 | H4 | Cl12 iii | 0.98(2) | 2.97(2) | 3.8384(17) | 149.0(16) |
| C6 | H6A | Cl12 iv | 1.01(3) | 2.89(2) | 3.8187(19) | 153.0(18) |
| C7 | H7B | Cl12 v | 0.99(3) | 2.95(3) | 3.8454(19) | 152(2) |

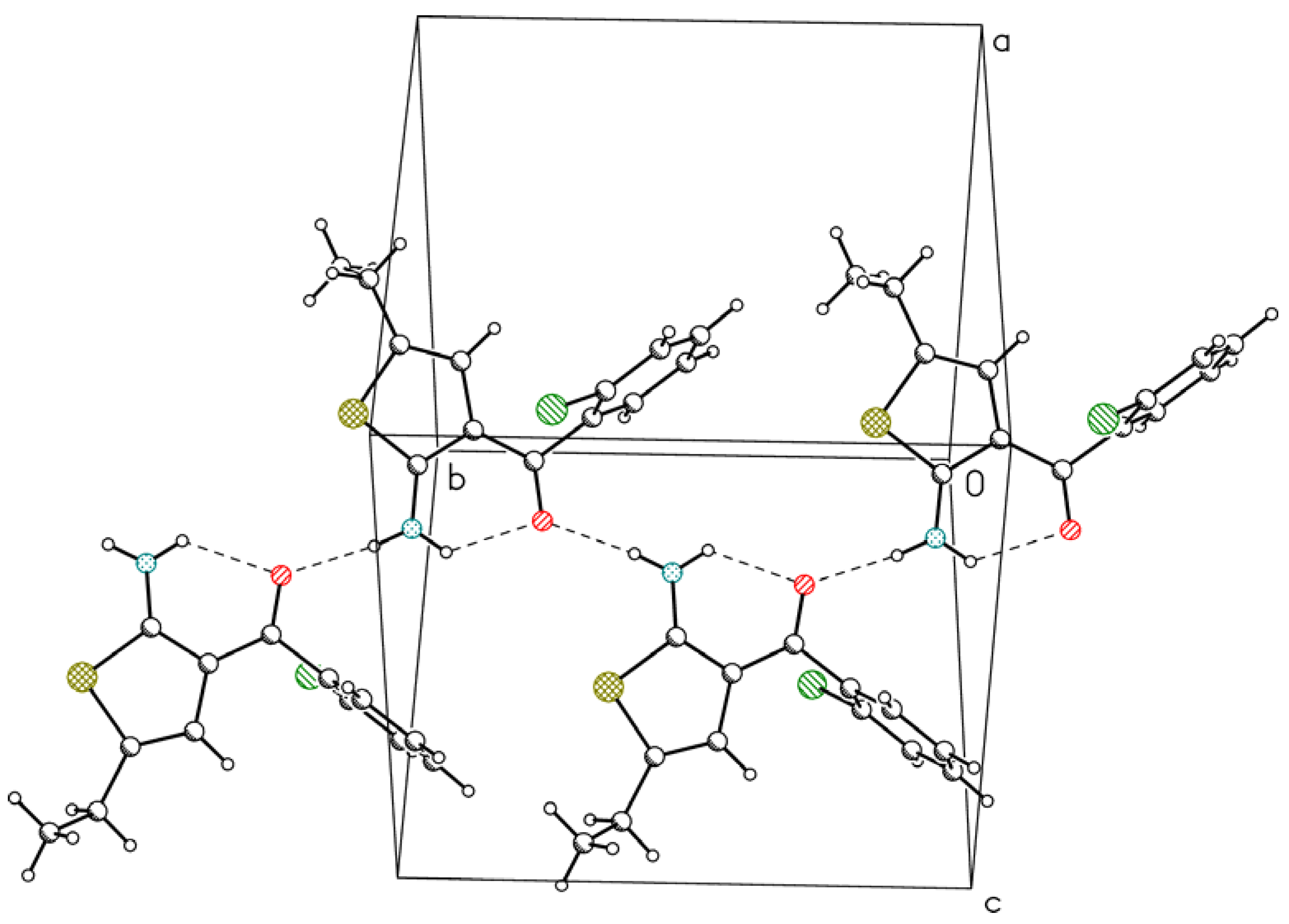
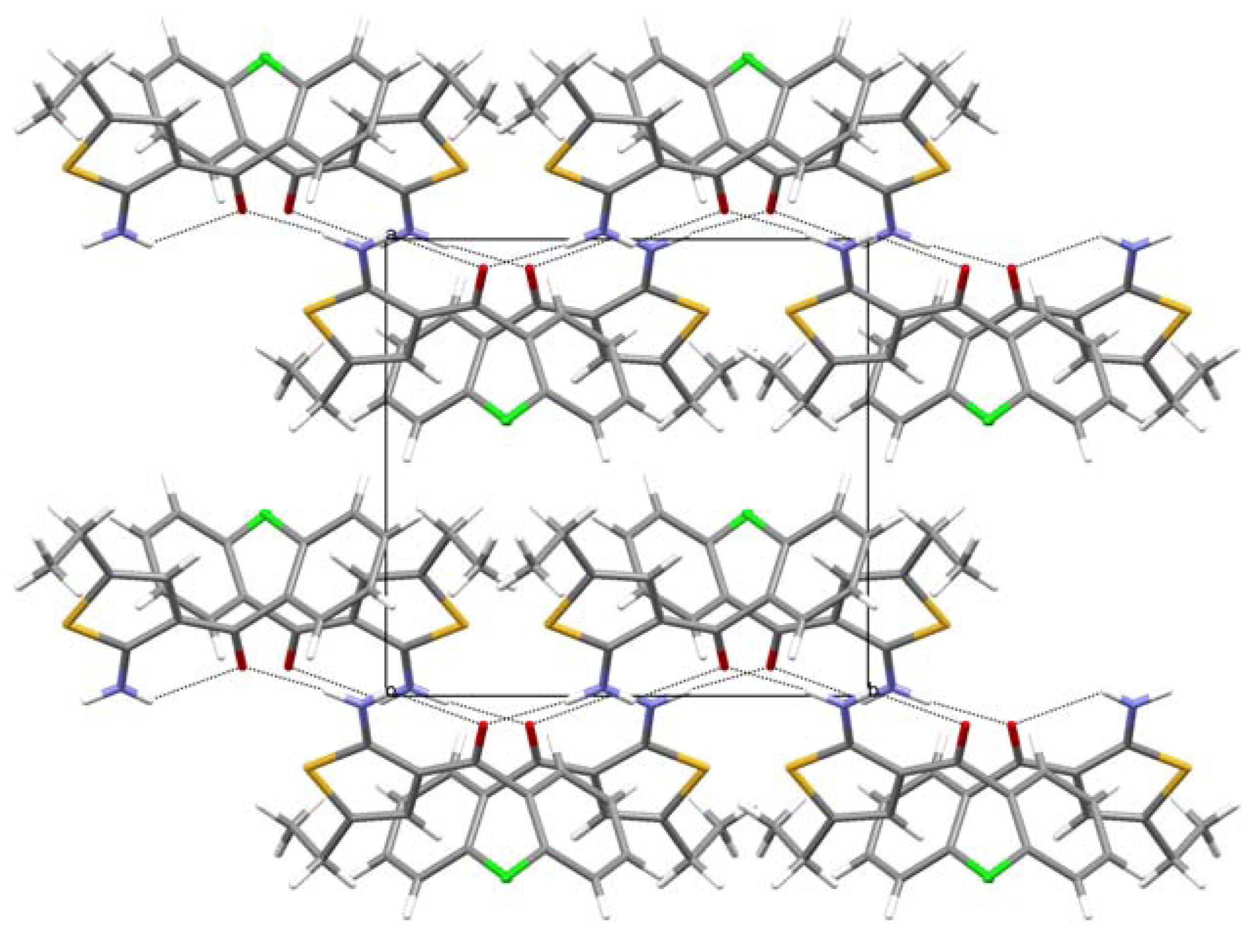
3. Experimental Section
| Compound | 1 | 2 |
|---|---|---|
| Formula | C15H15NOS | C13H12ClNOS |
| Formula weight | 257.34 | 265.75 |
| Crystal system | orthorhombic | monoclinic |
| Space group | Pna21 | P21/c |
| a (Å) | 9.2080(4) | 10.6092(8) |
| b (Å) | 14.0485(7) | 10.8355(8) |
| c (Å) | 10.3826(6) | 11.1346(9) |
| β (º) | 90 | 98.643(6) |
| V (Å3) | 1343.08(12) | 1265.45(17) |
| Z | 4 | 4 |
| Dx (g cm−3) | 1.72 | 1.40 |
| F(000) | 544 | 552 |
| μ (mm−1) | 0.23 | 4.07 |
| Crystal size (mm) | 0.3 × 0.15 × 0.15 | 0.35 × 0.2 × 0.1 |
| Θ range (°) | 3.29-28.18 | 4.21-73.59 |
| hkl range | −11 ± h ± 11 | −11 ± h ± 13 |
| −7 ± k ± 17 | −10 ± k ± 13 | |
| −8 ± l ± 13 | −13 ± l ± 13 | |
| Reflections: | ||
| collected | 3556 | 4715 |
| unique (Rint) | 1984 (0.021) | 2476 (0.0145) |
| with I > 2σ(I) | 1720 | 2400 |
| Number of parameters | 171 | 202 |
| Weighting scheme: | ||
| A | 0.0481 | 0.0624 |
| B | 0.1426 | 0.396 |
| R(F) [I > 2σ(I)] | 0.037 | 0.035 |
| wR(F2) [I > 2σ(I)] | 0.087 | 0.099 |
| R(F) [all data] | 0.045 | 0.036 |
| wR(F2) [all data] | 0.092 | 0.099 |
| Goodness of fit | 1.04 | 1.07 |
| max/min Δρ (e Å−3) | 0.14/−0.24 | 0.35/−0.31 |
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Sabnis, R.W.; Rangnekar, D.W.; Sonawane, N.D. 2-Aminothiophenes by the Gewald Reaction. J. Heterocycl. Chem. 1999, 36, 333–345. [Google Scholar] [CrossRef]
- Puterová, Z.; Krutošiková, A.; Végh, D. Gewald reaction: Synthesis, properties and applications of substituted 2-aminothiophenes. Arkivoc 2010, 209–246. [Google Scholar]
- Cannito, A.; Perrisin, M.; Luu-Duc, C.; Huguer, F.; Gaultier, C.; Narcisse, G. Synthèse et propriétés pharmacologiques de quelques thiéno[2,3-d]pyrimidin-4-one 2-thiones. Eur. J. Med. Chem. 1990, 25, 635–639. [Google Scholar]
- Nikolakopoulos, G.; Figler, H.; Linden, J.; Scammells, P. 2-Aminothiophene-3-carboxylates and carboxamides as adenosine A1 receptor allosteric enhancers. Bioorg. Med. Chem. 2006, 14, 2358–2365. [Google Scholar]
- Lütjens, H.; Zickgraf, A.; Figler, H.; Linden, J.; Olsson, R.A.; Scammells, P.J. 2-Amino-3-benzoylthiophene allosteric enhancers of A1 adenosine agonist binding: new 3-, 4-, and 5-modifications. J. Med. Chem. 2005, 46, 1870–1877. [Google Scholar]
- Allen, F.H. The Cambridge Structural Database: A quarter of a million crystal structures and rising. ActaCrystallogr. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Aurelio, L.; Valant, C.; Flynn, B.L.; Sexton, P.M.; White, J.M.; Christopoulos, A.; Scammells, P.J. Effects of conformation restriction of 2-amino-3-benzoylthiophenes on A1 adenosine receptor modulation. J. Med. Chem. 2010, 53, 6550–6559. [Google Scholar]
- Bruns, R.F.; Fergus, J.H.; Coughenour, L.L.; Courtland, G.G.; Pugsley, T.A.; Dodd, J.H.; Tinney, F.J. Structure-activity relationships for enhancement of adenosine A1 receptor binding by 2-amino-3-benzoylthiophenes. Mol. Pharmacol. 1990, 38, 950–958. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Duax, W.L.; Norton, D.A. Atlas of Steroid Structure; Plenum: NewYork, NY, USA, 1975; Volume 1, pp. 16–22. [Google Scholar]
- Bolduc, A.; Dufresne, S.; Skene, W.G. EDOT-containing azomethine: An easily prepared electrochromically active material with tuneable colours. J. Mater. Chem. 2010, 20, 4820–4826. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar]
- CrysAlisPro Setup, Version 1.171.35.15 (release 3 August 2011 CrysAlis171.NET); Oxford Diffraction: Palo Alto, CA, USA, 2010.
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr. 1993, 26, 343–350. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kubicki, M.; Dutkiewicz, G.; Yathirajan, H.S.; Dawar, P.; Ramesha, A.R.; Dayananda, A.S. Crystal and Molecular Structures of Two 2-Aminothiophene Derivatives. Crystals 2012, 2, 1058-1066. https://doi.org/10.3390/cryst2031058
Kubicki M, Dutkiewicz G, Yathirajan HS, Dawar P, Ramesha AR, Dayananda AS. Crystal and Molecular Structures of Two 2-Aminothiophene Derivatives. Crystals. 2012; 2(3):1058-1066. https://doi.org/10.3390/cryst2031058
Chicago/Turabian StyleKubicki, Maciej, Grzegorz Dutkiewicz, Hemmige S. Yathirajan, Pankaj Dawar, Andagar R. Ramesha, and Alaloor S. Dayananda. 2012. "Crystal and Molecular Structures of Two 2-Aminothiophene Derivatives" Crystals 2, no. 3: 1058-1066. https://doi.org/10.3390/cryst2031058