
Synthesis, X-ray Diffraction, NMR and Thermolysis Studies of Cadmium Tin Sulfido Complexes

Fakultät Für Chemie und Mineralogie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany
*
Author to whom correspondence should be addressed.
Crystals 2023, 13(5), 721; https://doi.org/10.3390/cryst13050721
Submission received: 29 March 2023
/
Revised: 13 April 2023
/
Accepted: 21 April 2023
/
Published: 25 April 2023
Abstract
:Three molecular sulfido complexes containing a Cd-S-Sn structure motive, [{(tmeda)Cd}3(SnPh)2S6] (1), [{(tmeda)Cd}2(SnPh2)2S4] (2) and [{(iPr3P)Cd}3Cd4(SnPh2)6S13] (3), have been synthesized and characterized by single-crystal X-ray diffraction. So far, 2 is the first cadmium tin sulfido complex soluble in common organic solvents. Analysis by 113Cd and 119Sn NMR spectroscopy shows a slow rearrangement in solution, while spin coupling between the nuclei can be used to identify the species in solution. Thermolysis of 2 results in CdS and SnS as solid thermolysis products.
1. Introduction
Compounds featuring a Cd-S-Sn structure motif are rare in the literature. To the best of our knowledge, there are only the purely inorganic, supertetrahedral cluster Cs10[Cd4Sn4S17] [1] and [{(C6F5)3Ge}2Cd{S(SnEt3)2}] [2]. Suitable starting materials for the solution-based synthesis of cadmium tin sulfido complexes are cadmium chalcogenolate complexes which are known in the literature with different ligands coordinating the Cd2+ ions, e.g., tmeda-stabilized chalcogenolates [3,4,5,6,7]. Moreover, tertiary phosphine ligands have been used to synthesize complexes such as [Cd4(SPh)6(PPh3)4](ClO4)2 [8], [Cd(SEt)(S2CSEt)(P{c-hex}3)]2 [9] or [Cd17S4(SH)2(SPh)24(PPh3)2] [10] (Et = C2H5, Ph = C6H5, c-hex = cyclo-C6H11) and also a mixed zinc-cadmium thiophenolate complex [11].
Cadmium tin sulfido complexes are promising materials as precursors for functional inorganic materials. One example is Cu2CdSnS4 (CCTS): a direct p-type semi-conductor with a band gap of 1.37 eV [12,13] which has already been used in lab-scale thin film solar cells [14]. A high conversion efficiency above 10% has been reported [15]. Even if CCTS is not considered for commercial solar cells due to the toxicity of cadmium compounds, it is expected to provide insights into the relationship between occupation of cation positions and physical properties. This is challenging for a related well-known material for solar cell application: Cu2ZnSnS4 (CZTS). Due to the same electron number of Cu+ and Zn2+, even for a known elemental composition it is hardly possible to distinguish these elements by routine X-ray diffraction. Two structures for CZTS have been reported: the kesterite [16] structure with space group Iand the stannite structure [17] with space group I2m. According to theoretical calculations, both structures are close in energy with a slightly higher stability (only 3 meV per atom) for the kesterite structure. The different structures result in different electrical properties [18,19,20,21,22]. Analyzing the crystal structure, including disorder of the Zn and Cu positions, is experimentally possible by special techniques such as neutron diffraction [23], X-ray absorption spectroscopy [24] or high resolution transmission electron microscopy [25], which are generally not available in a routine lab.
Thin films of CZTS and CCTS are accessible by spray-aerosol-assisted chemical vapor deposition (AACVD) [26,27,28]. This method starts from four separate sources for the four elements in CCTS. Obtaining a stoichiometrically precise product is difficult. For the synthesis of CZTS, the reduction of such a four component system to a three component system has already been achieved by using thiourea [29], xanthate [30] or dithiocarbamate [31] complexes of copper, zinc and tin. A further reduction to a two-component system has been suggested [30] and it was already possible to obtain CZTS(e) in thermolysis reactions of a two component mixture of zinc tin chalcogenido complexes of the general formula [(tmeda)Zn(SnR2)2S3] (R = Me, Ph, tBu) [32] with a copper chalcogenolate compound. As mentioned before, structural analyses of the thermolysis products are difficult, whereas the different electron densities of Cu+ and Cd2+ allow for their assignment by X-ray diffraction. Thus, thermolysis reactions of cadmium tin chalcogenido complexes can support the analyses to get a better understanding for the thermolysis reaction.
Herein we report our first results on the synthesis and characterization of cadmium tin sulfido complexes. The synthesis pathway is related to the synthesis of zinc tin chalcogenido complexes [32] which are accessible by reaction of the zinc complex [(tmeda)Zn(SSiMe3)2] [33] with a diorganotin acetate. Here, we use the cadmium analogue [(tmeda)Cd(SSiMe3)2] as well as similar tmeda-stabilized cadmium silylsulfido complexes [34,35] to generate a Cd–S–Sn structural motif.
2. Materials and Methods
All syntheses were performed applying Schlenk technique or in a MBRAUN UniLab glove box with N2 as inert gas. All solvents were dried according to common methods, distilled prior to use and stored over molecular sieve 4Å. Cd(OAc)2 was dried by refluxing in acetic anhydride. iPr3P was obtained by the reaction of iPrMgBr with PCl3 [36]. R2Sn(OAc)2 (R = Me, Ph, Vin; Vin = C2H3) were prepared by the reaction of (R2SnO)3 with a 2:1 mixture of acetic acid and Ac2O [37,38,39] and recrystallized from n-heptane [40]. While (Ph2SnO)3 is commercially available, (Me2SnO)3 and (Vin2SnO)3 were synthesized from Me2SnCl2 and Vin2SnCl2 (obtained by metathesis reaction of SnVin4 and SnCl4 [41]) in aqueous NH3 [42,43]. Ph3SnOAc was prepared by the reaction of Ph3SnCl and AgOAc [44]. (Me3Si)2S was prepared by reaction of Na2S (obtained by the reaction of the elements in liquid NH3 [45]) and Me3SiCl [46].
NMR spectra were measured using a Bruker Avance III HD 400 spectrometer with 400 MHz proton frequency in dried and distilled C6D6 or THF-d8 as solvents. 1H NMR spectra were referenced to the protonated solvent residue signal (7.16 ppm for C6D5H) or TMS as internal standard for THF-d8. Heteronuclei NMR spectra were referenced using the Ξ-scale [47]. Elemental analyses (C, H, N) were measured with a Heraeus Vario EL, TG/DTA analyses with a Netzsch STA 449 F1 coupled to an Aëolus QMS 403 D mass spectrometer. XRF analyses were measured with a Bruker S2 PICOFOX TXRF spectrometer. Suspended samples in water were dropped on a polished quartz glass disc and dried at 80 °C.
2.1. Syntheses of [{(tmeda)Cd}3(SnPh)2S6] (1) and [{(tmeda)Cd}2(SnPh2)2S4] · DME (2)
A quantity of 461 mg (2.0 mmol) Cd(OAc)2 was suspended in 20 mL DME. Then, 0.60 mL (4.0 mmol) tmeda was added under stirring. The suspension was cooled to –30 °C, and 0.84 mL (4.0 mmol) (Me3Si)2S was added dropwise to obtain a colorless solution. Then, 782 mg (2.0 mmol) Ph2Sn(OAc)2 was dissolved in 15 mL DME, and the solution was added to [(tmeda)Cd(SSiMe3)2]. After stirring at −30 °C for 3 h, small amounts of a colorless solid formed which was removed by filtration at −30 °C. The colorless solution was stored at −70 °C for two weeks. Finally, 60 mg (0.05 mmol, 7%) colorless blocks of 1 were obtained and separated by filtration. Elemental analysis for CHN found (calculated for C30H58Cd3N6S6Sn2): C 27.9 (28.4), H 4.9 (4.6), N 6.9 (6.6).
The volume of the solution cooled to −30 °C was reduced in vacuo to approx. 8 mL and stored at −25 °C for three weeks. Finally, 450 mg (0.37 mmol, 37%) colorless blocks of 2 were obtained and separated by filtration. 1H NMR (C6D6, 400 MHz, 26 °C): δ/ppm = 1.72 (br, 4H, tmeda-CH2); 2.00 (br, 24H, tmeda-CH3); 2.02 (br, 4H, tmeda-CH2); 3.12 (s, 6H, DME-CH3); 3.32 (s, 4H, DME-CH2); 7.00–7.38 (m, 12H, m+p-H SnPh2); 7.91 (d, 3J1H–117/119Sn = 69 Hz, 3J1H–1H = 6.8 Hz, 4H, o-H SnPh2); 8.32 (d, 3J1H–117/119Sn = 65 Hz, 3J1H–1H = 7.3 Hz, 4H, o-H SnPh2). 13C{1H} NMR (C6D6, 101 MHz, 26 °C): δ/ppm = 45.9 (br, tmeda-CH3); 46.3 (tmeda-CH3); 56.3 (tmeda-CH2); 57.3 (br, tmeda-CH2); 58.4 (DME-CH3), 71.9 (DME-CH2); 128.3 (SnPh2); 128.6 (SnPh2); 128.8 (SnPh2); 128.9 (SnPh2); 129.7 (J13C–117/119Sn = 15 Hz, SnPh2); 135.5 (J13C–117/119Sn = 53 Hz, SnPh2); 135.7 (J13C–117/119Sn = 50 Hz, SnPh2); 136.4 (J13C–117/119Sn = 49 Hz, SnPh2); 140.7 (SnPh2); 144.2 (SnPh2); 146.1 (SnPh2). 113Cd{1H} NMR (C6D6, 88.8 MHz, 26 °C): δ/ppm = −167 (2J113Cd–117/119Sn = 183 Hz); −67 (br, 2J113Cd–117/119Sn = 200 Hz). 119Sn{1H} NMR (C6D6, 149 MHz, 26 °C): δ/ppm = 18 (2J119Sn–117Sn = 195 Hz, (Ph2SnS)3); 30 (1J119Sn–13C = 361 Hz, 2J119Sn–113Cd = 186 Hz, 2J119Sn–111Cd = 180 Hz, J119Sn–13C = 62 Hz, J119Sn–13C = 52 Hz); 33 (br, 2J119Sn–111/113Cd = 201 Hz). Elemental analysis CHN found (calculated for C40H62Cd2N4O2S4Sn2): C 39.5 (39.3), H 4.9 (5.1), N 5.0 (4.6).
2.2. Synthesis of [{(iPr3P)Cd}3Cd4(SnPh2)6S13] (3)
A quantity of 461 mg (2.0 mmol) Cd(OAc)2 was suspended in 20 mL DME. Then, 0.76 mL (4.0 mmol) iPr3P was added under stirring. Within 30 min, a clear and colorless solution was obtained which was cooled to −25 °C. Then, 0.84 mL (4.0 mmol) (Me3Si)2S was added dropwise. Subsequently, 782 mg (2.0 mmol) Ph2Sn(OAc)2 was dissolved in 15 mL DME, cooled to −30 °C and added to the first solution. The solution was warmed up to 0 °C within 1 h and stored at 5 °C for five days. Finally, 80 mg (0.03 mmol, 8%) colorless cubes of 3 were obtained and separated by filtration. Elemental analysis for CHN found (calculated for C99H123Cd7P3S13Sn6): C 35.5 (35.8), H 3.4 (3.7).
2.3. X-ray Crystal Structure Analyses
Crystallographic data are given in Table 1. Measurements were performed using a STOE IPDS 2T image plate diffractometer system equipped with a sealed Mo X-ray tube and a graphite monochromator crystal (λ(Mo-Kα) = 71.073 pm) or a STOE STADIVARI, equipped with an X-ray micro-source (Cu-Kα, λ = 154.186 pm) and a DECTRIS Pilatus 300k detector. Data reduction and numerical absorption correction were carried out with STOE X-AREA software [48]. All structures were solved by direct methods using SHELXS-2018 and refined with SHELXL-2018 [49] using WinGX [50] as graphical frontend. In the structure analysis of 2, all non-hydrogen atoms (besides disordered C atoms) were refined with anisotropic thermal parameters; hydrogen atoms were included on idealized positions applying the riding model. Due to weak diffraction and limited quality of the dataset of 3, the C atoms were refined with isotropic thermal parameters. Diamond 4.6.8 was used for visualization of the crystal structures [51]. CCDC 2252002 and 2252037 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: [email protected]).
The PXRD measurements were carried out on a STOE STADI-P diffractometer equipped with a sealed Cu X-ray tube and a germanium (111) monochromator crystal (λ(Cu-Kα1) = 154.060 pm). Samples of the air-sensitive complexes were prepared in a glove box under N2 atmosphere, filled in glass capillaries (Hilgenberg, outer diameter 0.3 mm or 0.5 mm) and measured in Debye–Scherrer mode. Rietveld analyses of the diffraction patterns were performed with Bruker TOPAS 5 software [52] using the fundamental parameter approach. Crystal structure data for CdS (sphalerite) [53], CdS (wurtzite) [53] and SnS [54] were taken from the literature.
3. Results & Discussion
3.1. Syntheses
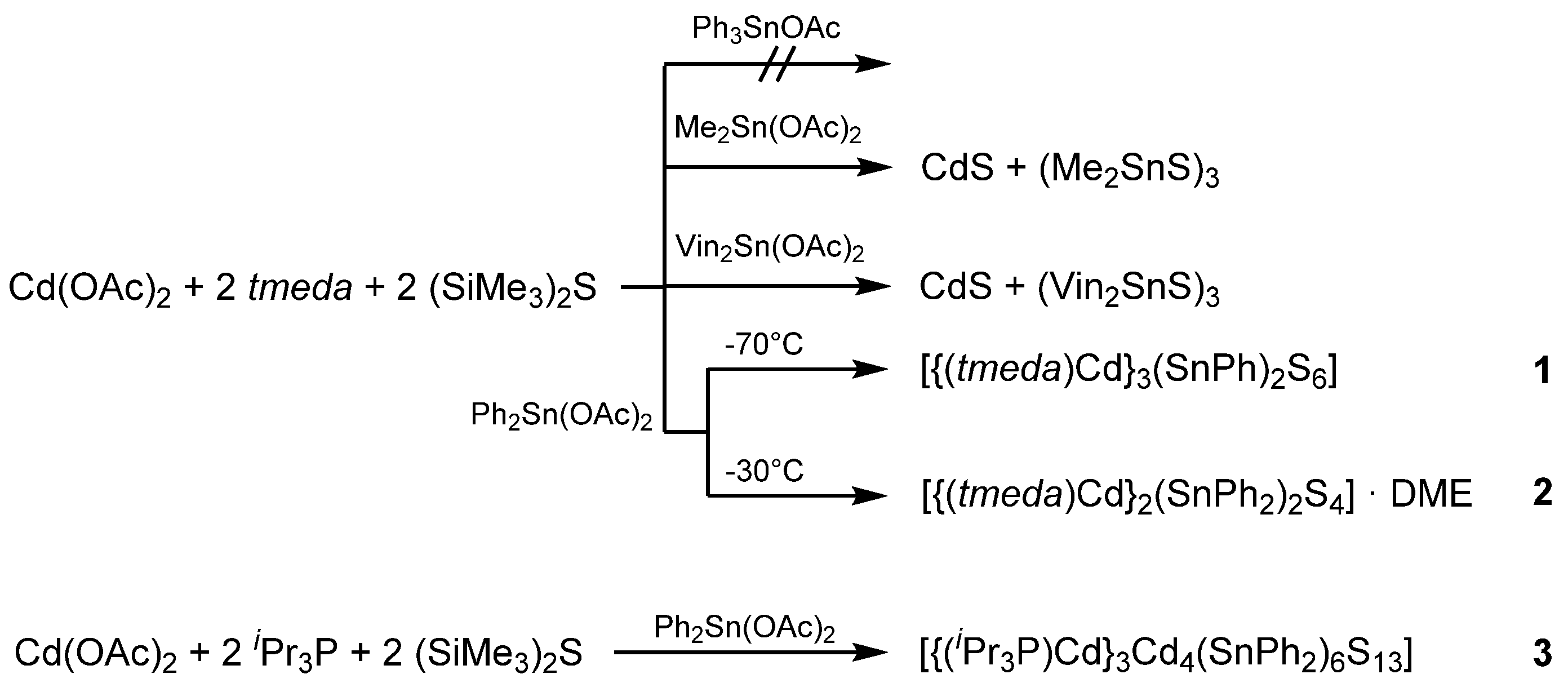
The reactions carried out are summarized in Scheme 1. No reaction was observed using Ph3SnOAc as reactant. Only Ph3SnOAc could be crystallized from the reaction solution. The reaction with Me2Sn(OAc)2 led to slow decomposition with formation of a yellowish solid even at −70 °C. The precipitate was analyzed by X-ray fluorescence spectroscopy and showed the presence of Cd and S, as expected for decomposition to CdS. Crystals obtained from the reaction solution were identified as (Me2SnS)3 [55]. NMR experiments performed directly after the synthesis showed the 119Sn NMR signal for (Me2SnS)3 and no soluble 113Cd species. Direct decomposition into CdS and organotin sulfides was also observed for the reaction of Vin2Sn(OAc)2 with [(tmeda)Cd(SSiMe3)2].
With sterically more demanding phenyl groups, the formation of two cadmium tin sulfido complexes was possible. With a low yield of 7% the complex [{(tmeda)Cd}3(SnPh)2S6] (1) with monophenylstannyl groups was isolated. It is known from the literature that tin compounds with organic groups can undergo metathesis reactions as shown in Scheme 2.
A second cadmium tin sulfido complex, [{(tmeda)Cd}2(SnPh2)2S4] (2), could be isolated from the reaction solution with a moderate yield of 37%. While the hard Lewis base tmeda works well for Zn2+ complexes which are relatively stable, the complexes with the softer Lewis acid Cd2+ decompose readily or undergo exchange reactions. Therefore, tmeda was replaced by the soft Lewis base iPr3P, which has already been applied to successfully stabilize copper tin chalcogenido complexes [56]. With low yields, a third cadmium tin sulfido complex [{(iPr3P)Cd}3Cd4(SnPh2)6S13] (3) was obtained.
3.2. Single-Crystal Structure Analyses
The crystals of 1 and 3 are very small and, due to the low yield and their high sensitivity, recrystallization was not possible. In case of 1, only the heavy atom framework could be identified, and only rough positions of the carbon atoms could be assigned by single-crystal structure analysis. Therefore, a detailed discussion of geometric parameters of 1 is not possible.
Complex 1 crystallizes in the triclinic space group P with one molecule of 1 forming the asymmetric unit (Figure 1). The structure contains two PhSnS33– groups which are arranged on opposite sides of the molecule. The tin atoms are tetrahedrally coordinated. Three cadmium atoms are situated between the PhSnS33– groups with bonds to two sulfur atoms and coordination of one chelating tmeda molecule for each cadmium atom. Due to the large radius of Cd2+, a tmeda molecule is too small for ideal chelating of the Cd2+ ion, which may explain the low stability of the complex.
Compound 2 (Figure 2a, Table 2) crystallizes with one formula unit and one molecule DME as asymmetric unit in the monoclinic space group P21/n. The heavy atoms form an eight-membered ring of alternating tin and cadmium atoms bridged by sulfur atoms in a boat-like conformation (Figure 2b). Tin atoms arranged in an eight membered ring are a rare structure motif which has only been reported for a few organotin sulfides [57], an antimony compound with a {Sb-S-Sn-S}2 ring [58] and in a copper tin sulfido complex [56]. Both tin atoms are coordinated by two sulfur atoms and two phenyl groups. The Sn–S distances are similar (236.38(9)-239.17(9) pm) and in good agreement with Sn–S distances in copper tin sulfido complexes [56,59,60]. S–Sn–S bond angles (103.55(3)° for Sn1 and 107.79(3)° for Sn2) are smaller than the ideal tetrahedral angle. While sulfur atoms S2, S3 and S4 bridge one tin and one cadmium atom, S1 reveals, in addition, a longer interaction to Cd1 (Cd1···S1 300.01(9) pm). Thus, Cd1 exhibits a [4+1] coordination with a distorted trigonal bipyramidal coordination, S1 and N2 occupying the axial positions (S1···Cd1–N2 162.00(8)°). The distance Cd1–N2 (253.3(4) pm) is significantly longer than Cd1–N1 (237.7(4) pm) due to the interaction Cd1···S1. For Cd2 a weaker additional interaction to S3 (322.51(9) pm) can be assumed, in agreement with a smaller difference in Cd2–N distances (236.2(3) and 243.8(3) pm). The other Cd–S bonds are similar with 247.82(9)-249.34(8) pm. These geometric parameters are in agreement with literature examples such as the Cd10S4-core in the cluster [NEt4]4[Cd10S4(p-SC6H4Me)12Br4] [61] for Cd–S bonds or [(tmeda)Cd(SSiPh3)2] [35] for the tmeda coordination to cadmium atoms.
Complex 3 (Table 3) crystallizes in the cubic space group Pa with eight formula units per unit cell. A third of the molecule forms the asymmetric unit, and a three-fold axis through atoms S5 and Cd3 generates the molecule crystallographically (Figure 3). Four cadmium atoms (Cd3, Cd2 and symmetry equivalent atoms) bind to sulfur atoms only, while three cadmium atoms (Cd1) bind to three sulfur atoms and one iPr3P ligand. The Cd–P distance (262(2) pm) is in the same range as observed for phosphine stabilized cadmium thiolate complexes [9,10,62]. Six tin atoms are located in the outer sphere of the CdS core which are coordinated by two sulfur atoms and two phenyl groups. The Sn–S distances vary between 237.8(9) pm and 245.9(9) pm, i.e., they are longer than observed in 1 and 2. The core of the molecule is formed by Cd3 and S5, both occupying positions on the axis. Cd3 is tetrahedrally coordinated by four sulfur atoms (S5 and S1), S5 binds to four {CdS3}4– units. This is the typical structural motive for cadmium sulfide thiolate cluster molecules [63]. Bond distances in the core are similar to literature values of that motif varying from 247.5(9) pm to 252.8(9) pm and are also similar to Cd–S distances in both CdS modifications with distances of 252.5 pm to 254.0 pm. For the peripheral cadmium atoms Cd1 and Cd2 the distances vary from 243(2) pm to 262.2(9) pm, probably due to steric interactions of the phosphine ligands and the SnPh2 groups. A total number of 13 sulfur atoms has been observed in cadmium sulfide thiolate clusters [64].
3.3. Characterization of the Complexes by PXRD and NMR Spectroscopy
After separation of crystals of 1 from the mother liqueur and drying in vacuo, the X-ray powder diffraction pattern shows only a very broad reflection, i.e., the crystals loose crystallinity during the preparation process for powder diffraction. Crystals of 1 are hardly soluble in common organic solvents. NMR spectra of 1 in THF-d8 show only two weak 119Sn signals (Figure S2) (see supplementary materials). The signal at 13 ppm can be assigned to (Ph2SnS)3. A 113Cd signal could not be detected, probably due to decomposition. Based on these observations, formation of CdS and phenyltin sulfides is very likely. NMR analysis of 3 (Figure S6) resulted only in a 31P signal for uncoordinated iPr3P. Neither 113Cd nor 119Sn signals could be observed. Due to the low yield and the low stability further characterization was not feasible.
X-ray diffraction of a freshly prepared powder sample of 2 showed mainly broad reflections matching the simulated pattern based on single-crystal structure data in reflection positions and intensities (Figure 4). Repeated measurements of the same sample resulted in a continuous broadening of the reflections. After approx. 4 h, the reflections became sharp again, and the diffraction pattern did not change any more. The origin of this behavior might be the loss of DME solvent molecules.
Crystals of 2 are soluble in C6D6 and THF-d8. Since the signals are significantly sharper in THF-d8, the NMR spectra of a solution of 2 in THF-d8 are presented in Figure 5. The chemical shifts obtained in C6D6 are given in the experimental part, and the spectra can be found in the SI (Figure S3). The proton NMR spectrum reveals dynamic effects as broad signals for the tmeda ligands. In the aromatic region, several signals can be found, i.e., the presence of several phenyltin species is likely. The intensity ratio of tmeda to phenyl-groups matches the expected 32:20 ratio known from the crystal structure. DME shows up in the NMR spectra with an intensity corresponding to one molecule DME per molecule of 2.
The presence of several tin species is confirmed by the 119Sn spectrum where three signals are visible. A signal at 13 ppm can be assigned to (Ph2SnS)3 with a 2J coupling to the magnetically nonequivalent 117Sn isotope (7.7% natural abundance). Both signals at 29 ppm and 33 pm show satellite peaks due to spin–spin coupling with the cadmium nuclei 111Cd and 113Cd (12.8% and 12.2% natural abundance of 111Cd and 113Cd, respectively; both with I = 1/2). The 119Sn satellites of the signal at 29 ppm show a splitting of 180 Hz (2J119Sn–111Cd) and 186 Hz (2J119Sn–113Cd). The latter coupling is larger due to the higher gyromagnetic value of 113Cd. The signal at 33 ppm is slightly broadened and splitting of the satellite peaks is not resolved. Determination of the satellite intensities helps to ascertain the number of two coupled cadmium nuclei for both tin signals. The coupling to 13C of the phenyl groups is also visible and the coupling constants are given in the experimental part. Two signals can be found in the 113Cd spectrum, at –166 ppm and –63 ppm. Satellite peaks are also present; coupling to the respective tin spins is indicated by the differently colored arrows in Figure 5. Again, the satellite intensities allow the verification of the number of coupled nuclei. The signal at –63 ppm clearly indicates coupling with two 117/119Sn nuclei. For the weaker signal at –166 ppm, the satellite intensity is approx. 2% lower, and the satellite signal is broadened which may indicate a coupling to a third nucleus.
The above findings allow the conclusion that an exchange reaction (Scheme 3) may occur in solution. The presence of 2 is most likely and assigned by the signals marked in red in Figure 5, as each tin atom is in the neighborhood of two cadmium atoms and vice versa. Due to the clear presence of (Ph2SnS)3, another cadmium-rich species must exist. The formation of a species such as [{(tmeda)Cd}2(SnPh2)S3] is possible and is in agreement with the NMR spectroscopy results. The lower satellite intensity of the 113Cd signal at −166 pm can be explained with a coupling of the 113Cd nucleus with one 117/119Sn nucleus and one magnetically nonequivalent 111Cd. The 119Sn signal clearly shows coupling with two 111/113Cd nuclei. Unfortunately, even at low temperature the three couplings with similar coupling constants cannot be resolved.
An 119Sn-119Sn EXSY spectrum (Figure S4) did not show any correlation between the tin signals, i.e., it is probably not an equilibrium reaction. After having kept the sample in solution within the NMR tube for a week, the amount of (Ph2SnS)3 has increased and a yellowish solid has developed (Figure S5). The latter may be due to the formation of cadmium sulfide. As observed for 1 and 3, 2 also slowly decomposes in solution.
3.4. Thermal Behavior and Thermolysis Study
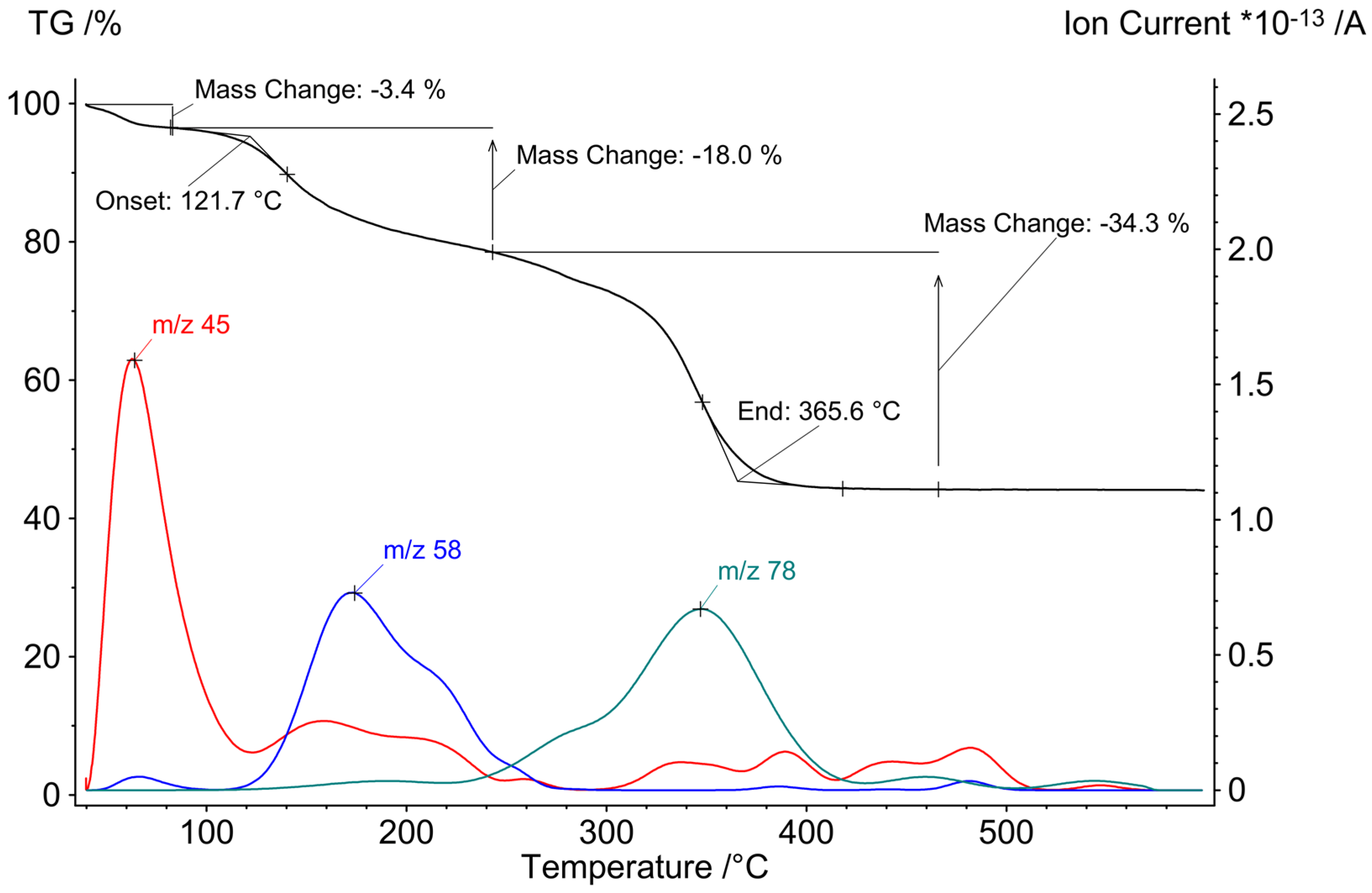
Complex 2 is a potential source of Cd, Sn and S in thermolysis reactions. The results of a simultaneous thermogravimetric experiment (TG) up to 600 °C are shown in Figure 6. The observed weight loss of 55.7% is higher than expected according to the schematic expected thermolysis reaction equation as shown in Scheme 4.
DME is evaporating from crystals of 2 at temperatures below 100 °C as detected by the mass spectrometry signal at m/z = 45 (MeOCH2+). According to only 3.4% mass loss, approx. 0.43 equivalents of DME were present in the sample of 2 after evacuation prior to the TG measurement. Thermolysis of the complex begins at 122 °C. The increase of the mass signal at m/z = 58 (Me2NCH2+) indicates the elimination of the tmeda ligands (calc. mass loss 19.8%, measured 18%). In the last thermolysis step ending at 366 °C, mass signals of phenyl groups (m/z = 78 C6H6+) can be detected. The mass loss of 34.3% is 8% higher than expected. The partial elimination of tin as volatile phenylstannanes is likely, this has already been observed in the thermolysis of Zn-Sn-S complexes [32].
The X-ray powder diffraction pattern of the solid thermolysis product (Figure S7) shows reflections of a mixture of CdS in wurtzite and sphalerite structure as well as reflections of SnS. Due to broad and overlapping reflections for both CdS phases, the exact ratio could not be determined. Rietveld analysis provides a mass ratio CdS:SnS of 56.6(4) %: 43.4(5)%, in quite good agreement with the loss of volatile tin compounds during thermolysis as confirmed by the TG experiment results. According to temperature-dependent powder X-ray diffraction (Figure S8) 2 is thermally stable up to 90 °C. Starting at 270 °C, reflections of CdS and SnS are observed.
4. Conclusions
We successfully synthesized three cadmium tin sulfido complexes and characterized them by single-crystal X-ray structure analysis. In addition to S2− ligands, the Cd2+ ions are coordinated by tmeda as a chelating N-donating ligand (1, 2) and iPr3P as P-donating ligand (3), respectively. Different organotin (R = Me, Vin, Ph) acetates were tested in the synthesis. Only the reaction with diphenyltin acetate led to the formation of crystalline cadmium tin sulfido complexes. The complexes [{(tmeda)Cd}3(SnPh)2S6] (1) and [{(iPr3P)Cd}3Cd4(SnPh2)6S13] (3) were obtained in low yields and are quite unstable showing rapid decomposition to CdS and phenyltin sulfides. [{(tmeda)Cd}2(SnPh2)2S4] (2) was isolated in a moderate yield of 37%. Crystals of 2 are more stable; however, NMR studies in solution revealed a slow rearrangement reaction with formation of (Ph2SnS)3 and [{(tmeda)Cd}2(SnPh2)S3]. Thermolysis studies of 2 show the formation of a solid mixture of 57 mass% CdS and 43 mass% SnS due to formation of volatile phenylstannanes during the thermolysis reaction. While the formation of the binary sulfides and a 1:1 stoichiometric ratio of Cd:Sn in the complex appear suitable for co-thermolysis reactions with a copper sulfide source to form CCTS, the loss of volatile phenylstannanes reduces the amount of tin in the thermolysis product. Moreover, the reduction of SnIV to SnII, which is a common side reaction [65] during thermolysis, must be avoided for the formation of CCTS. Taking advantage of the findings in the thermolytic formation of CZTS [32] behavior of 2 in such co-thermolysis experiments will be studied in future works.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cryst13050721/s1, Figure S1: 1H, 113Cd{1H} and 119Sn{1H} NMR spectra of the reaction product of [(tmeda)Cd(SSiMe3)2] and Me2Sn(OAc)2 in C6D6; Figure S2: 1H and 119Sn{1H} NMR spectra of the crystals of 1 dissolved in THF-d8. The solubility of 1 in THF is very low; only decomposition products could be detected; Figure S3: 1H, 113Cd{1H}, 119Sn{1H} and 13C{1H} NMR spectra of 2 in C6D6; Figure S4: A 119Sn-119Sn-EXSY spectrum of 2 in C6D6 does not show a correlation signal, i.e., there is probably no equilibrium reaction between the tin containing species; Figure S5: 119Sn{1H} NMR spectra of 2 in C6D6 (left) and THF-d8 (right) after 1 week show the slow decomposition with increasing signal intensity for (Ph2SnS)3. In C6D6 an addition signal for (Ph3Sn)2S at –52 ppm was detected; Figure S6: 1H, 31P{1H} and 119Sn{1H} NMR spectra of crystals of 3 dissolved in THF-d8. The solubility of 3 in THF is very low; only decomposition products could be detected; Figure S7: X-Ray powder diffraction pattern (λ = 154.060 pm) obtained from the solid residue of thermolysis of 2 up to 600 °C and results of the Rietveld refinement (wRp = 6.52%, Rp = 4.58%, Rexp = 1.69%). Observed, calculated intensities, difference curve as well as reflection positions of wurtzite CdS (first row), sphalerite CdS (second row) and SnS (third row). (Due to overlapping reflection positions of the sphalerite and wurtzite phase of CdS and broadening of reflections due to low crystallinity, there are some differences in the calculated and observed reflection intensities.); Figure S8: Temperature dependent X-ray powder diffraction pattern (λ = 154.060 pm) of 2 up to 500 °C. The crystal structure of 2 remains stable up to 90 °C. Starting at 270 °C, reflections of CdS and SnS are observed.
Author Contributions
Writing—original draft preparation, D.F.; investigation, N.H.B.; formal analysis, M.I.; project administration, H.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Universität Leipzig and the Operational Program of the European Regional Development Fund 2014–2020, project “In situ investigations of energy related materials” (Project 100357551). This measure is cofinanced by tax money on the basis of the budget adopted by the members of the Saxon Landtag.
Data Availability Statement
CCDC 2252002 and 2252037 contain the crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Powder diffraction data, NMR data and thermogravimetry analysis are included in the article or supporting information file.
Acknowledgments
We thank Katrin Hoffmann and Katrin Steinke for measuring NMR spectra and Manuela Roßberg for CHN analyses.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Palchik, O.; Iyer, R.G.; Canlas, C.G.; Weliky, D.P.; Kanatzidis, M.G. K10M4M′4S17 (M = Mn, Fe, Co, Zn; M′ = Sn, Ge) and Cs10Cd4Sn4S17: Compounds with a Discrete Supertetrahedral Cluster. Z. Anorg. Allg. Chem. 2004, 630, 2237–2247. [Google Scholar] [CrossRef]
- Bochkarev, M.N.; Andreevichev, V.S.; Vyazankin, N.S. Bis[tri(pentafluorophenyl)germyl]cadmium complexing reactions. Izv. Akad. Nauk. Ser. Khim. 1973, 1973, 702–703. [Google Scholar] [CrossRef]
- Eichhöfer, A.; Buth, G. Synthesis and Structure of the Group 12 Pyrimidinethiolate Complexes ∞3[Zn(S-2-N2C4H3)2], ∞2[Cd(S-2-N2C4H3)2], [Hg(S-2-N2C4H3)2] and [Cd(S-2-N2C4H3)2(tmeda)]. Eur. J. Inorg. Chem. 2005, 2005, 4160–4167. [Google Scholar] [CrossRef]
- Edwards, A.J.; Fallaize, A.; Raithby, P.R.; Rennie, M.-A.; Steiner, A.; Verhorevoort, K.L.; Wright, D.S. A halidefree route to Groups 12 and 13 organometallic and metalloorganic complexes. J. Chem. Soc. Dalton Trans. 1996, 1996, 133–137. [Google Scholar] [CrossRef]
- Yosef, M.; Schaper, A.K.; Fröba, M.; Schlecht, S. Stabilization of the thermodynamically favored polymorph of cadmium chalcogenide nanoparticles CdX (X = S, Se, Te) in the polar mesopores of SBA-15 silica. Inorg. Chem. 2005, 44, 5890–5896. [Google Scholar] [CrossRef]
- Denny, J.A.; Foley, W.S.; Almaraz, E.; Reibenspies, J.H.; Bhuvanesh, N.; Darensbourg, M.Y. Comparisons of zinc with cadmium in N2S2 coordination and as S-bonded adducts to tungsten carbonyls. Dalton Trans. 2012, 41, 143–148. [Google Scholar] [CrossRef]
- Jill Black, S.; Einstein, F.W.B.; Hayes, P.C.; Kumar, R.; Tuck, D.G. Reactions of cadmium thiolate (Cd(SR)2, R = n-butyl, phenyl) and the molecular structure of the 2,2’-bipyridine adduct of bis(n-butyl thioxanthato)cadmium(II). Inorg. Chem. 1986, 25, 4181–4184. [Google Scholar] [CrossRef]
- Philip, A.W.D.; Payne, N.C.; Vittal, J.J.; Yu, Y. Synthesis and NMR spectra (phosphorus-31, cadmium-111/113, selenium-77) of adamantane-like phosphine complexes with the (µ-ER)6Cd4 core and the crystal and molecular structure of [(µ-SPri)6(CdPPh3)2(CdOClO3)2] · EtOH. Inorg. Chem. 1993, 32, 4632–4639. [Google Scholar] [CrossRef]
- Dean, P.A.W.; Vittal, J.J. SYNTHESIS AND STRUCTURE OF cis-syn-[Cd(μ-SEt)(S2CSEt)(P{c-C6H11}3)]2. Main Group Met. Chem. 2002, 25, 695–696. [Google Scholar] [CrossRef]
- Gruber, F. Synthesis and Structure Determination of Two Neutral Cadmium Thiophenolate Clusters. Z. Anorg. Allg. Chem. 2012, 638, 2467–2469. [Google Scholar] [CrossRef]
- DeGroot, M.W.; Taylor, N.J.; Corrigan, J.F. Controlled Synthesis of Ternary II−II′−VI Nanoclusters and the Effects of Metal Ion Distribution on Their Spectral Properties. Inorg. Chem. 2005, 44, 5447–5458. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Nakazawa, T. Electrical and Optical Properties of Stannite-Type Quarternary Semiconductor Thin Films. Jpn. J. Appl. Phys. Part 1 1988, 27, 2094–2097. [Google Scholar] [CrossRef]
- Matsushita, H.; Ichikawa, T.; Katsui, A. Structural, thermodynamical and optical properties of Cu2-II-IV-VI4 quaternary compounds. J. Mater. Sci. 2005, 40, 2003–2005. [Google Scholar] [CrossRef]
- Hadke, S.; Levcenko, S.; Sai Gautam, G.; Hages, C.J.; Márquez, J.A.; Izquierdo-Roca, V.; Carter, E.A.; Unold, T.; Wong, L.H. Suppressed Deep Traps and Bandgap Fluctuations in Cu2CdSnS4 Solar Cells with ≈8% Efficiency. Adv. Energy Mater. 2019, 9, 1902509. [Google Scholar] [CrossRef]
- Fan, P.; Lin, J.; Hu, J.; Yu, Z.; Zhao, Y.; Chen, S.; Zheng, Z.; Luo, J.; Liang, G.; Su, Z. Over 10% Efficient Cu2CdSnS4 Solar Cells Fabricated from Optimized Sulfurization. Adv. Funct. Mater. 2022, 32, 2207470. [Google Scholar] [CrossRef]
- Gao, F.; Yamazoe, S.; Maeda, T.; Nakanishi, K.; Wada, T. Structural and Optical Properties of In-Free Cu2ZnSn(S,Se)4 Solar Cell Materials. Jpn. J. Appl. Phys. 2012, 51, 10NC29. [Google Scholar] [CrossRef]
- Nitsche, R.; Sargent, D.F.; Wild, P. Crystal growth of quaternary 122464 chalcogenides by iodine vapor transport. J. Cryst. Growth 1967, 1, 52–53. [Google Scholar] [CrossRef]
- Chen, S.; Gong, X.G.; Walsh, A.; Wei, S.-H. Crystal and electronic band structure of Cu2ZnSnX4 (X=S and Se) photovoltaic absorbers: First-principles insights. Appl. Phys. Lett. 2009, 94, 41903. [Google Scholar] [CrossRef]
- Chen, S.; Gong, X.G.; Walsh, A.; Wei, S.-H. Electronic structure and stability of quaternary chalcogenide semiconductors derived from cation cross-substitution of II-VI and I-III-VI2 compounds. Phys. Rev. B 2009, 79, 165211. [Google Scholar] [CrossRef]
- Persson, C. Electronic and optical properties of Cu2ZnSnS4 and Cu2ZnSnSe4. J. Appl. Phys. 2010, 107, 53710. [Google Scholar] [CrossRef]
- Paier, J.; Asahi, R.; Nagoya, A.; Kresse, G. Cu2ZnSnS4 as a potential photovoltaic material: A hybrid Hartree-Fock density functional theory study. Phys. Rev. B 2009, 79, 115126. [Google Scholar] [CrossRef]
- Nakamura, S.; Maeda, T.; Wada, T. Phase Stability and Electronic Structure of In-Free Photovoltaic Materials: Cu2ZnSiSe4, Cu2ZnGeSe4 and Cu2ZnSnSe4. Jpn. J. Appl. Phys. 2010, 49, 121203. [Google Scholar] [CrossRef]
- Schorr, S. The crystal structure of kesterite type compounds: A neutron and X-ray diffraction study. Sol. Energy Mater. Sol. Cells 2011, 95, 1482–1488. [Google Scholar] [CrossRef]
- Just, J.; Lützenkirchen-Hecht, D.; Frahm, R.; Schorr, S.; Unold, T. Determination of secondary phases in kesterite Cu2ZnSnS4 thin films by X-ray absorption near edge structure analysis. Appl. Phys. Lett. 2011, 99, 262105. [Google Scholar] [CrossRef]
- Mendis, B.G.; Shannon, M.D.; Goodman, M.C.J.; Major, J.D.; Claridge, R.; Halliday, D.P.; Durose, K. Direct observation of Cu, Zn cation disorder in Cu2ZnSnS4 solar cell absorber material using aberration corrected scanning transmission electron microscopy. Prog. Photovolt: Res. Appl. 2014, 22, 24–34. [Google Scholar] [CrossRef]
- Tombak, A.; Kilicoglu, T.; Ocak, Y.S. Solar cells fabricated by spray pyrolysis deposited Cu2CdSnS4 thin films. Renew. Energy 2020, 146, 1465–1470. [Google Scholar] [CrossRef]
- Nakayama, N.; Ito, K. Sprayed films of stannite Cu2ZnSnS4. Appl. Surf. Sci. 1996, 92, 171–175. [Google Scholar] [CrossRef]
- Kamoun, N.; Bouzouita, H.; Rezig, B. Fabrication and characterization of Cu2ZnSnS4 thin films deposited by spray pyrolysis technique. Thin Solid Films 2007, 515, 5949–5952. [Google Scholar] [CrossRef]
- Madarásza, J.; Bombiczb, P.; Okuyaa, M.; Kanekoa, S. Thermal decomposition of thiourea complexes of Cu(I), Zn(II), and Sn(II) chlorides as precursors for the spray pyrolysis deposition of sulfide thin films. Solid State Ionics 2001, 141–142, 439–446. [Google Scholar] [CrossRef]
- Kociok-Köhn, G.; Molloy, K.C.; Sudlow, A.L. Molecular routes to Cu2ZnSnS4: A comparison of approaches to bulk and thin-film materials. Can. J. Chem. 2014, 92, 514–524. [Google Scholar] [CrossRef]
- Ramasamy, K.; Malik, M.A.; O’Brien, P. The chemical vapor deposition of Cu2ZnSnS4 thin films. Chem. Sci. 2011, 2, 1170–1172. [Google Scholar] [CrossRef]
- Fuhrmann, D.; Dietrich, S.; Krautscheid, H. Zinc Tin Chalcogenide Complexes and Their Evaluation as Molecular Precursors for Cu2ZnSnS4 (CZTS) and Cu2ZnSnSe4 (CZTSe). Inorg. Chem. 2017, 56, 13123–13131. [Google Scholar] [CrossRef] [PubMed]
- DeGroot, M.W.; Corrigan, J.F. Coordination Complexes of Zinc with Reactive ESiMe3 (E = S, Se, Te) Ligands. Organometallics 2005, 24, 3378–3385. [Google Scholar] [CrossRef]
- Babcock, J.R.; Zehner, R.W.; Sita, L.R.; Babcock, J.R.; Zehner, R.W.; Sita, L.R. A Heterocumulene Metathesis Route to Cd[ESiMe3]2 and Passivated CdE (E = S and Se) Nanocrystals // A Heterocumulene Metathesis Route to Cd[ESiMe3]2 and Passivated CdE (E = S and Se) Nanocrystals. Chem. Mater. 1998, 10, 2027–2029. [Google Scholar] [CrossRef]
- Foody, M.J.; Weimer, M.S.; Bhandari, H.; Hock, A.S. Comparison of Ligand Architecture on Vapor Deposition Precursors: Synthesis, Characterization, and Reactivity of Volatile Cadmium Bis-Amidinate Complexes. Inorg. Chem. 2021, 60, 6191–6200. [Google Scholar] [CrossRef] [PubMed]
- Davies, W.C. Tertiary phosphines containing secondary alkyl radicals // 247. Tertiary phosphines containing secondary alkyl radicals. J. Chem. Soc. 1933, 1933, 1043–1044. [Google Scholar] [CrossRef]
- Maeda, Y.; Okawara, R. Studies on dialkyltin diacetate derivatives. J. Organomet. Chem. 1967, 10, 247–256. [Google Scholar] [CrossRef]
- Dietzel, S.; Jurkschat, K.; Tzschach, A.; Zschunke, A. Synthese und spektroskopische Untersuchungen von Di-t-butylzinn(IV)-dicarboxylaten. Z. Anorg. Allg. Chem. 1986, 537, 163–168. [Google Scholar] [CrossRef]
- Mokal, V.B.; Jain, V.K. Steric effects on the formation of isolable products in the reactions of dibutyltin oxides with carboxylic acids. J. Organomet. Chem. 1992, 441, 215–226. [Google Scholar] [CrossRef]
- Lockhart, T.P.; Manders, W.F.; Holt, E.M. Solution and Solid-state Molecular Structures of Me2Sn(OAc)2 (I) and Its Hydrolyzate, ([Me2Sn(OAc)]2O)2 (II), by Solution and Solid-state 13C NMR. X-ray Diffraction Study of II. J. Am. Chem. Soc. 1986, 108, 6611–6616. [Google Scholar] [CrossRef]
- Seyferth, D.; Stone, F.G.A. Vinyl Derivatives of the Metals. I. Synthesis of Vinyltin Compounds. J. Am. Chem. Soc. 1957, 79, 515–517. [Google Scholar] [CrossRef]
- Brown, M.P.; Okawara, R.; Rochow, E.G. Infrared spectra of some methyl derivatives of germanium and tin. Spectrochim. Acta 1960, 16, 595–601. [Google Scholar] [CrossRef]
- Puff, H.; Schuh, W.; Sievers, R.; Wald, W.; Zimmer, R. Niedermolekulare Diorganozinn-Sauerstoff-Verbindungen: Di-t-butyl- und Di-t-amylzinnoxid. J. Organomet. Chem. 1984, 260, 271–280. [Google Scholar] [CrossRef]
- Anderson, H.H. Volatile n-Butyltin and Phenyltin Tricarboxylates. Organotin Oxymonocarboxylates. Inorg. Chem. 1964, 3, 912–914. [Google Scholar] [CrossRef]
- Brauer, G. Handbuch der Präparativen Anorganischen Chemie; Ferdinand Enke Verlag: Stuttgart, Germany, 1978. [Google Scholar]
- Schmidt, M.; Ruf, H. Über die Umsetzung von Organohalogensilanen mit Natriumselenid. Z. Anorg. Allg. Chem. 1963, 321, 270–273. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Grange, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts (IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- STOE & Cie GmbH. X-Area Version 1.70; STOE & Cie GmbH: Darmstadt, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. Diamond Version 3.2k; CRYSTAL IMPACT: Bonn, Germany, 2014. [Google Scholar]
- Bruker AXS. TOPAS Version 5; Bruker AXS: Karlsruhe, Germany, 2014. [Google Scholar]
- Ulrich, F.; Zachariasen, W. XIV. Über die Kristallstruktur des α- und ß-CdS, sowie des Wurtzits. Z. Kristallogr. 1925, 62, 260–273. [Google Scholar] [CrossRef]
- Wiedemeier, H.; von Schnering, H.G. Refinement of the structures of GeS, GeSe, SnS and SnSe. Z. Kristallogr. 1978, 148, 295–303. [Google Scholar] [CrossRef]
- Menzebach, B.; Bleckmann, P. Die Kristallstruktur von Hexamethylcyclotristannathian. J. Organomet. Chem. 1975, 91, 291–294. [Google Scholar] [CrossRef]
- Fuhrmann, D. Synthese, Charakterisierung und Thermolyse von potentiellen molekularen Vorläufern für Cu2ZnSnE4 und Ag2ZnSnE4 (E = S, Se). Ph.D. Thesis, Universität Leipzig, Leipzig, Germany, 2017. [Google Scholar]
- Dakternieks, D.; Jurkschat, K.; Schollmeyer, D.; Wu, H. Synthesis, NMR studies and molecular structures of [(PhSSn)2(CH2)3]2 and [PhSn(S2CNEt2)]2(S)(CH2)3. J. Organomet. Chem. 1995, 492, 145–150. [Google Scholar] [CrossRef]
- Feng, M.-L.; Ye, D.; Huang, X.-Y. Quaternary tin(IV) antimony(III) sulfide decorated with lanthanum(III) ethylenediamine complexes: [La(en)4SbSnS5]2 x 0.5 H2O. Inorg. Chem. 2009, 48, 8060–8062. [Google Scholar] [CrossRef] [PubMed]
- Hauser, R.; Merzweiler, K. [(PhSnS3)2(CuPPhMe2)6], ein sechskerniger Kupfer(I)-Komplex mit PhSnS3-Liganden. Z. Anorg. Allg. Chem. 2002, 628, 905–906. [Google Scholar] [CrossRef]
- Eußner, J.P.; Dehnen, S. Bronze, silver and gold: Functionalized group 11 organotin sulfide clusters. Chem. Commun. 2014, 50, 11385–11388. [Google Scholar] [CrossRef]
- Adams, R.D.; Zhang, B.; Murphy, C.J.; Yeung, L.K. Halide enhancement of the luminescence of Cd10S4 thiolate clusters. Chem. Commun. 1999, 1999, 383–384. [Google Scholar] [CrossRef]
- Wallbank, A.I.; Lebold, T.P.; Borecki, A.; Polgar, A.M.; Waters, B.M.; Workentin, M.S.; Corrigan, J.F. ZnII and CdII Ferrocenechalcogenolate Complexes. Eur. J. Inorg. Chem. 2017, 2017, 372–377. [Google Scholar] [CrossRef]
- Bendova, M.; Puchberger, M.; Schubert, U. Characterization of “Cd10S4(SPh)12”, the Thermal Decomposition Product of (NMe4)4[Cd10S4(SPh)16]: Synthesis of a Neutral Cd54 Sulfide Cluster and of a Polymeric Chain of Thiolate-Bridged Cd17 Sulfide Clusters. Eur. J. Inorg. Chem. 2010, 2010, 3299–3306. [Google Scholar] [CrossRef]
- Ka-Luo, T.; Xiang-Lin, J.; Shao-Juan, J.; You-Qi, T. Cadmium Thiolates obtained from Cambridge Structural Database. Jiegou Huaxue 1995, 14, 399. [Google Scholar]
- Weber, A.; Krauth, H.; Perlt, S.; Schubert, B.; Kötschau, I.; Schorr, S.; Schock, H.W. Multi-stage evaporation of Cu2ZnSnS4 thin films. Thin Solid Film. 2009, 517, 2524–2526. [Google Scholar] [CrossRef]
Scheme 1.
Syntheses of the cadmium tin sulfido complexes 1–3.
Scheme 2.
Metathesis reaction of diphenyltin compounds.

Figure 1.
Molecular structure of 1. Heavy atoms are drawn in ball and stick representation, bonds to carbon atoms shown as sticks. Hydrogen atoms are omitted for clarity.
Figure 1.
Molecular structure of 1. Heavy atoms are drawn in ball and stick representation, bonds to carbon atoms shown as sticks. Hydrogen atoms are omitted for clarity.

Figure 2.
(a) Molecular structure of 2; atoms are drawn as 50% probability ellipsoids. (b) heavy atom core of 2 with tmeda ligands as sticks and without phenyl groups. Hydrogen atoms and DME molecules are omitted for clarity.
Figure 2.
(a) Molecular structure of 2; atoms are drawn as 50% probability ellipsoids. (b) heavy atom core of 2 with tmeda ligands as sticks and without phenyl groups. Hydrogen atoms and DME molecules are omitted for clarity.
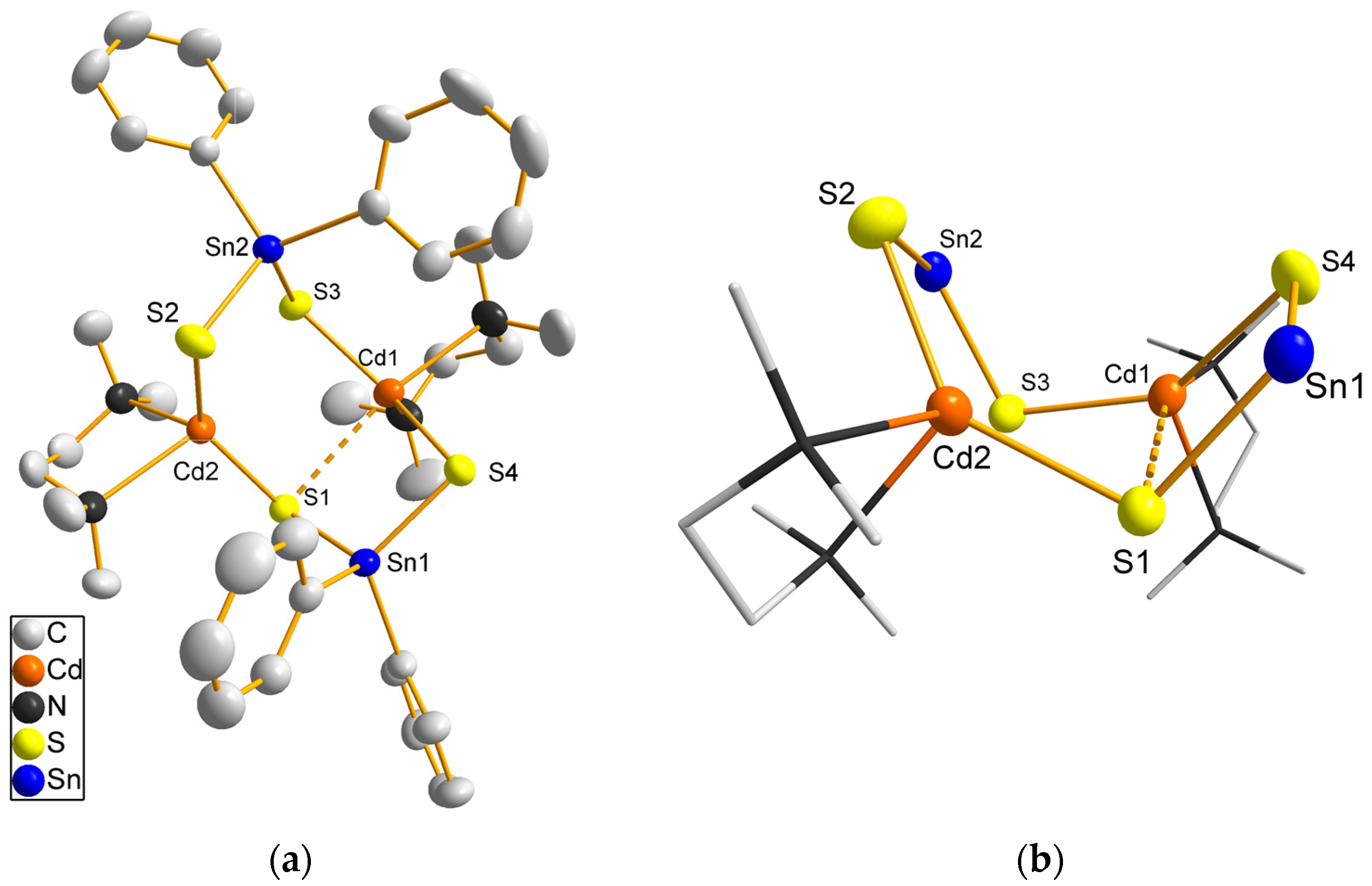
Figure 3.
Molecular structure of 3. Heavy atoms are shown as 50% probability ellipsoids, carbon atoms as sticks. Hydrogen atoms are omitted for clarity.
Figure 3.
Molecular structure of 3. Heavy atoms are shown as 50% probability ellipsoids, carbon atoms as sticks. Hydrogen atoms are omitted for clarity.
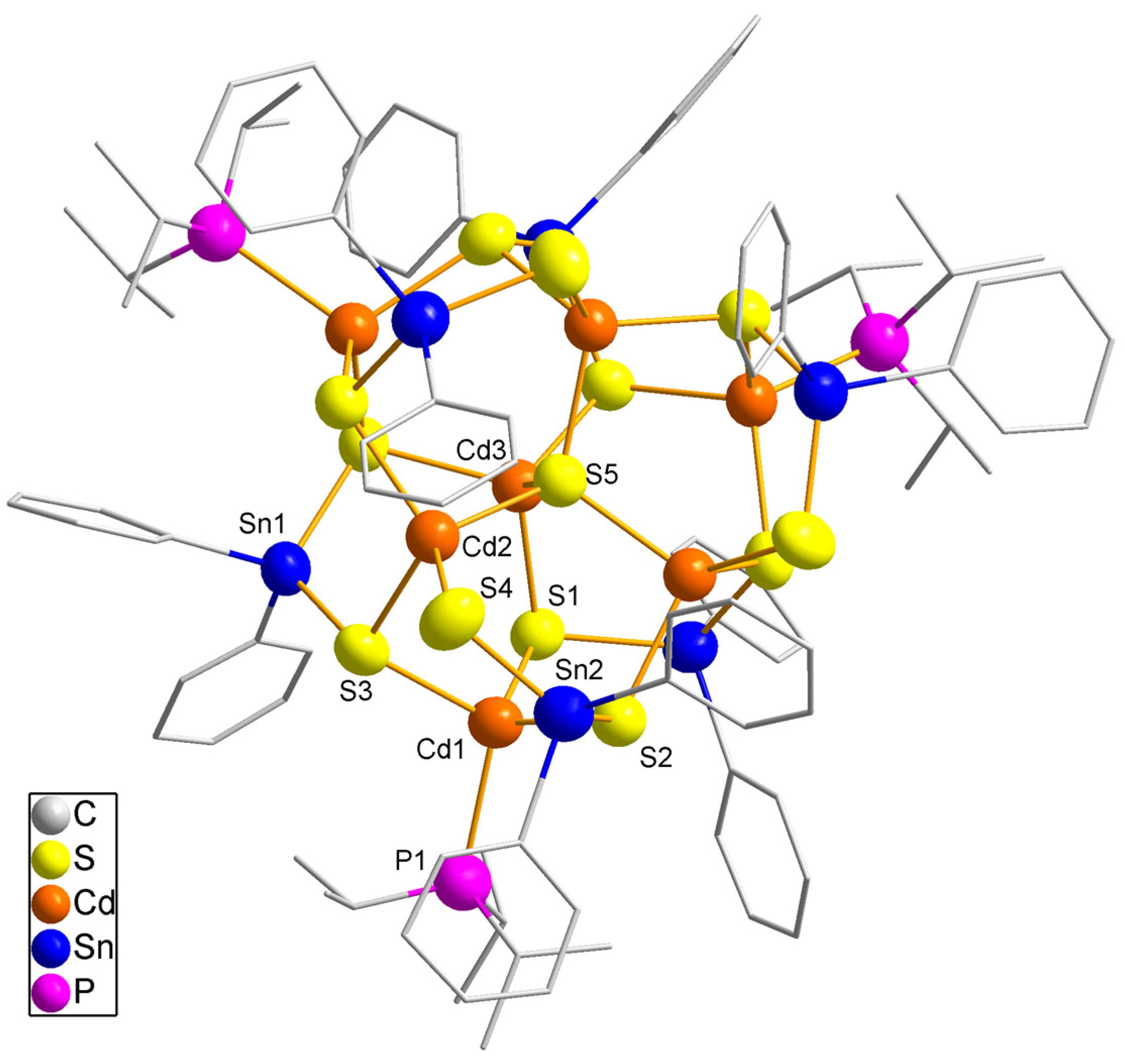
Figure 4.
Simulated and measured X-ray (λ(Cu-Kα1 = 154.060 pm) powder diffraction patterns of 2.
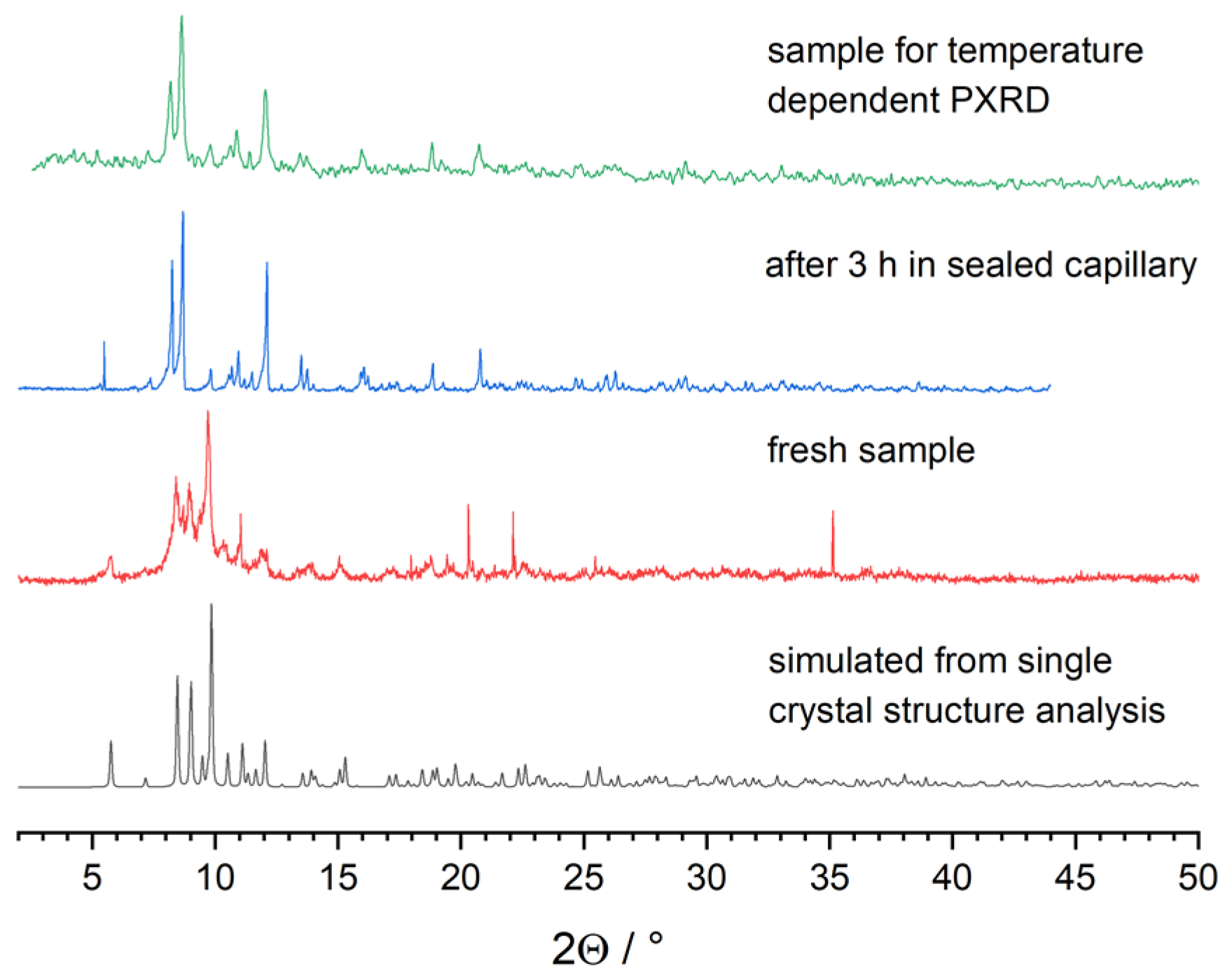
Figure 5.
1H, 119Sn{1H} and 113Cd{1H} NMR spectra of 2 recorded at 26 °C in THF-d8. The intensity ratios of the main and satellite signals in the 119Sn and 113Cd spectra are given below the spectra.
Figure 5.
1H, 119Sn{1H} and 113Cd{1H} NMR spectra of 2 recorded at 26 °C in THF-d8. The intensity ratios of the main and satellite signals in the 119Sn and 113Cd spectra are given below the spectra.

Scheme 3.
Rearrangement reaction in solution confirmed by NMR spectroscopy.
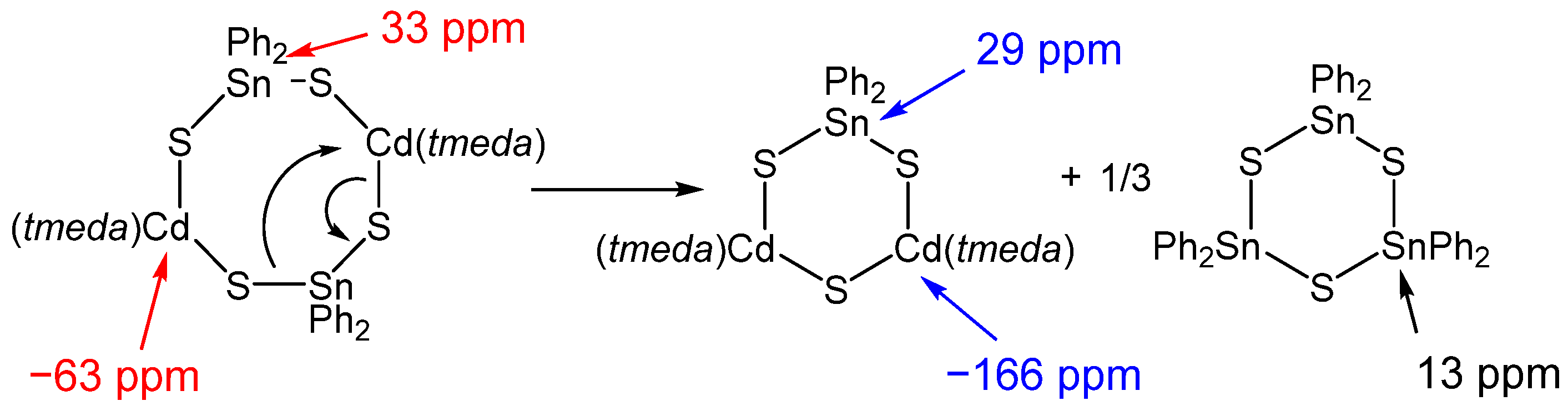
Scheme 4.
Proposed thermolysis reaction of 2 forming CdS, SnS and volatile thermolysis products (red). The amount of DME in the TG sample of 2 was calculated based on the first mass loss in the TG curve attributed to DME. The percentage numbers correspond to the expected mass loss according to the given overall reaction equation.
Scheme 4.
Proposed thermolysis reaction of 2 forming CdS, SnS and volatile thermolysis products (red). The amount of DME in the TG sample of 2 was calculated based on the first mass loss in the TG curve attributed to DME. The percentage numbers correspond to the expected mass loss according to the given overall reaction equation.
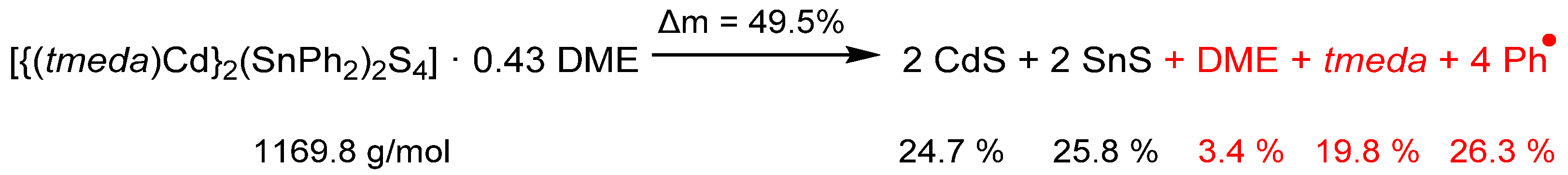
Figure 6.
TG curve and selected ion current curves for the thermolysis of 2 at temperatures up to 600 °C (* means “multiplied with”).
Figure 6.
TG curve and selected ion current curves for the thermolysis of 2 at temperatures up to 600 °C (* means “multiplied with”).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal structure data of 1–3.
| 1 | 2 | 3 | |
|---|---|---|---|
| Formula | C30H58Cd3N6S6Sn2 | C40H62Cd2N4O2S4Sn2 | C99H123Cd7P3S13Sn6 |
| molar mass M/g mol−1 | 1269.84 | 1221.35 | 3321.6 |
| temperature/K | 180(2) | 200(2) | 180(2) |
| Wavelength | 154.186 | 71.073 | 154.186 pm |
| crystal color and shape | colorless block | colorless block | colorless cube |
| crystal size/mm | 0.01·0.02·0.02 | 0.17·0.24·0.35 | 0.02·0.02·0.02 |
| crystal system | triclinic | monoclinic | cubic |
| space group | P | P21/c | Pa |
| a/pm | 1279.0(4) | 1029.73(5) | 2889.9(2) |
| b/pm | 1473.1(4) | 2470.59(8) | 2889.9(2) |
| c/pm | 1647.3(4) | 1995.41(9) | 2889.9(2) |
| α/° | 114.98(3) | 90 | 90 |
| β/° | 92.85(3) | 99.724(4) | 90 |
| γ/° | 111.21(3) | 90 | 90 |
| V/106 pm3 | 2545.7(2) | 5003.5(4) | 24,134(5) |
| Z | 2 | 4 | 8 |
| calc. density/g cm−3 | 1.8 | 1.62 | 1.83 |
| absorption coefficient µ/mm−1 | 20.31 | 2.03 | 22.12 |
| θ range/° | 2.6–26.7 | 3.4–40 | |
| measured reflections | 29,186 | 9608 | |
| independent reflections (Rint) | 10,534 (0.0263) | 2369 (0.1115) | |
| observed reflections (I > 2σ(I)) | 7717 | 1229 | |
| Parameters | 494 | 220 | |
| R1 (observed reflections) | 0.0278 | 0.0736 | |
| wR2 (all data) | 0.0634 | 0.1686 | |
| max./min. residual e- density/10−6 pm−3 | 1.1 and −0.6 | 0.3 and −0.8 |
Table 2.
Bond lengths/pm and angles/° for the molecular structure of 2.
| Bond Lengths | Bond Angles | ||
|---|---|---|---|
| Sn1-S1 | 239.17(9) | S1-Sn1-S4 | 103.55(3) |
| Sn1-S4 | 236.86(9) | S2-Sn2-S3 | 107.79(3) |
| Sn2-S2 | 236.38(9) | S1-Cd1-S3 | 91.23(3) |
| Sn2-S3 | 238.15(9) | S1-Cd1-S4 | 85.39(3) |
| Cd1-S1 | 300.01(9) | S3-Cd1-S4 | 141.72(3) |
| Cd1-S3 | 248.04(9) | S1-Cd2-S2 | 139.06(3) |
| Cd1-S4 | 248.7(1) | N-Cd1-N | 75.9(1) |
| Cd2-S1 | 249.34(8) | N-Cd2-N | 77.9(1) |
| Cd2-S2 | 247.82(9) | ||
Table 3.
Bond lengths/pm and angles/° for the molecular structure of 3.
| Bond Lengths | Bond Angles | ||
|---|---|---|---|
| Sn1-S1 | 241.5(9) | S1-Sn1-S3 | 113.0(3) |
| Sn1-S3 | 236.8(9) | S2-Sn2-S4 | 122.0(4) |
| Sn2-S4 | 237(1) | S1-Cd1-S2 | 108.6(3) |
| Sn2-S2 | 245.9(9) | S1-Cd1-S3 | 105.1(3) |
| Cd1-S1 | 257.7(9) | S2-Cd1-S3 | 112.7(3) |
| Cd1-S2 | 252.4(9) | S2-Cd2-S3 | 110.7(3) |
| Cd1-S3 | 255.7(9) | S2-Cd2-S4 | 101.8(4) |
| Cd1-P1 | 261.5(9) | S3-Cd2-S4 | 101.7(3) |
| Cd2-S2 | 254.2(9) | S2-Cd2-S5 | 100.3(2) |
| Cd2-S3 | 262.2(9) | S3-Cd2-S5 | 105.8(4) |
| Cd2-S4 | 244(1) | S4-Cd2-S5 | 120.1(4) |
| Cd2-S5 | 251.6(9) | S1-Cd3-S5 | 111.3(2) |
| Cd3-S1 | 252.8(9) | S1-Cd3-S1 | 107.6(2) |
| Cd3-S5 | 247.5(9) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fuhrmann, D.; Bollfraß, N.H.; Icker, M.; Krautscheid, H. Synthesis, X-ray Diffraction, NMR and Thermolysis Studies of Cadmium Tin Sulfido Complexes. Crystals 2023, 13, 721. https://doi.org/10.3390/cryst13050721
AMA Style
Fuhrmann D, Bollfraß NH, Icker M, Krautscheid H. Synthesis, X-ray Diffraction, NMR and Thermolysis Studies of Cadmium Tin Sulfido Complexes. Crystals. 2023; 13(5):721. https://doi.org/10.3390/cryst13050721
Chicago/Turabian StyleFuhrmann, Daniel, Nick Hermann Bollfraß, Maik Icker, and Harald Krautscheid. 2023. "Synthesis, X-ray Diffraction, NMR and Thermolysis Studies of Cadmium Tin Sulfido Complexes" Crystals 13, no. 5: 721. https://doi.org/10.3390/cryst13050721
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.