Photoelectrocatalytic H2 and H2O2 Production Using Visible-Light-Absorbing Photoanodes


 and
and
Abstract
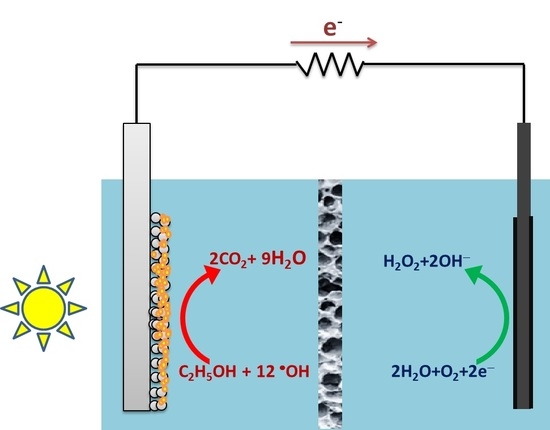
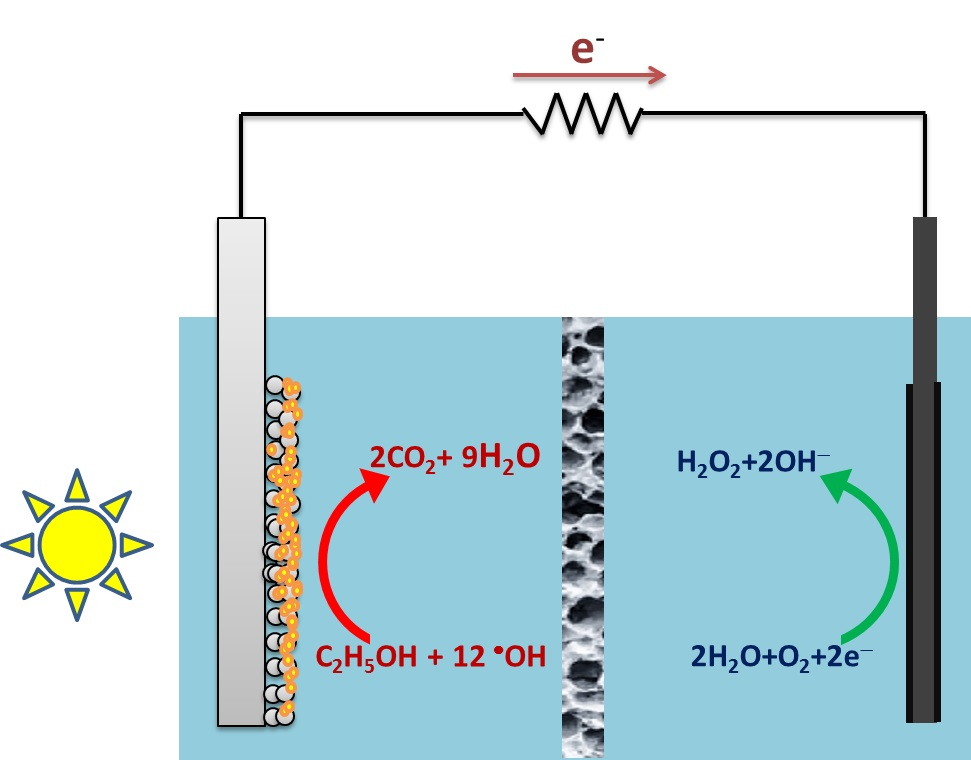
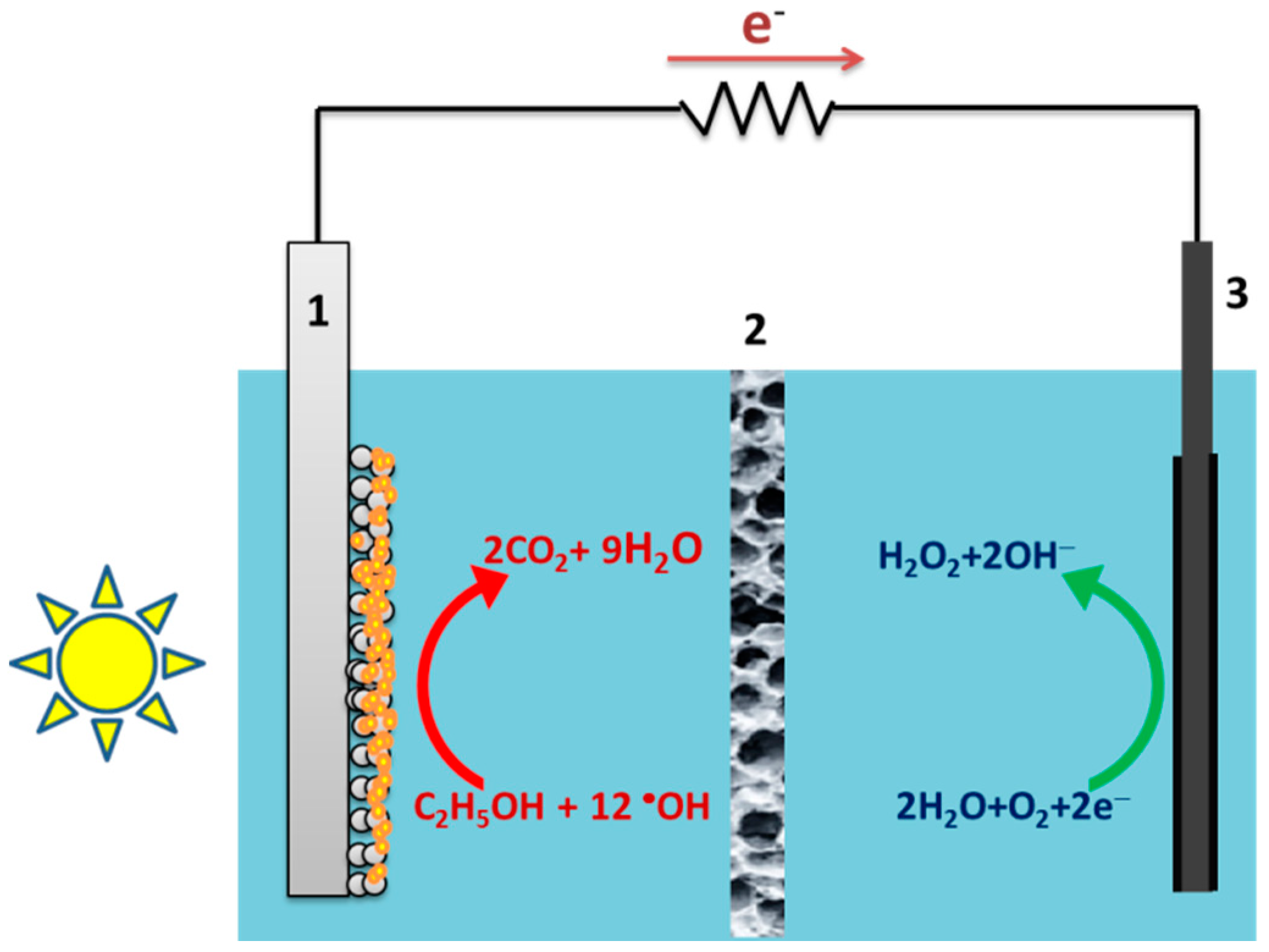
:
1. Introduction
2. Results and Discussion
2.1. Design of the Reactor
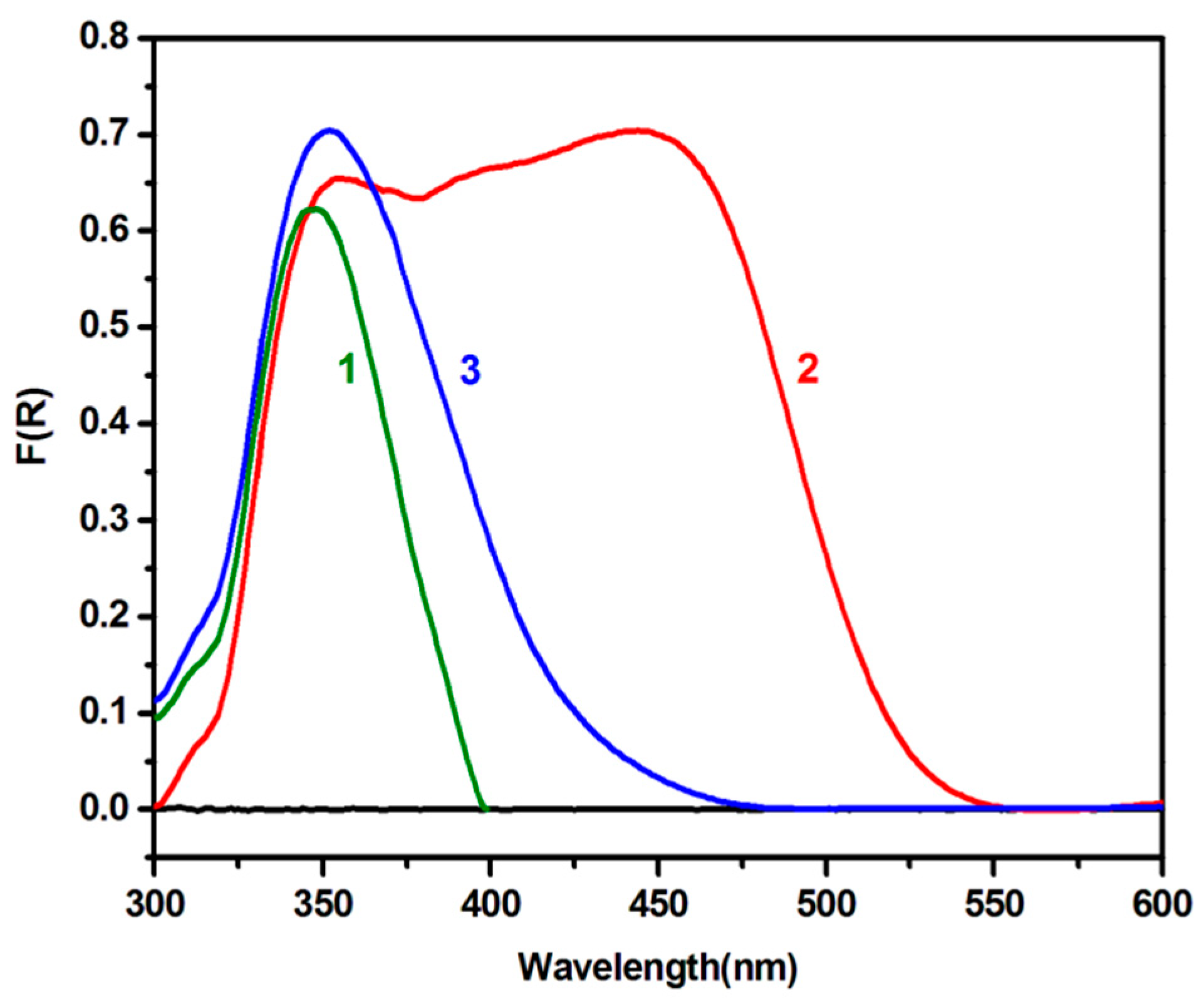
2.2. Characterization of the Photoanode and the Counter-Electrode
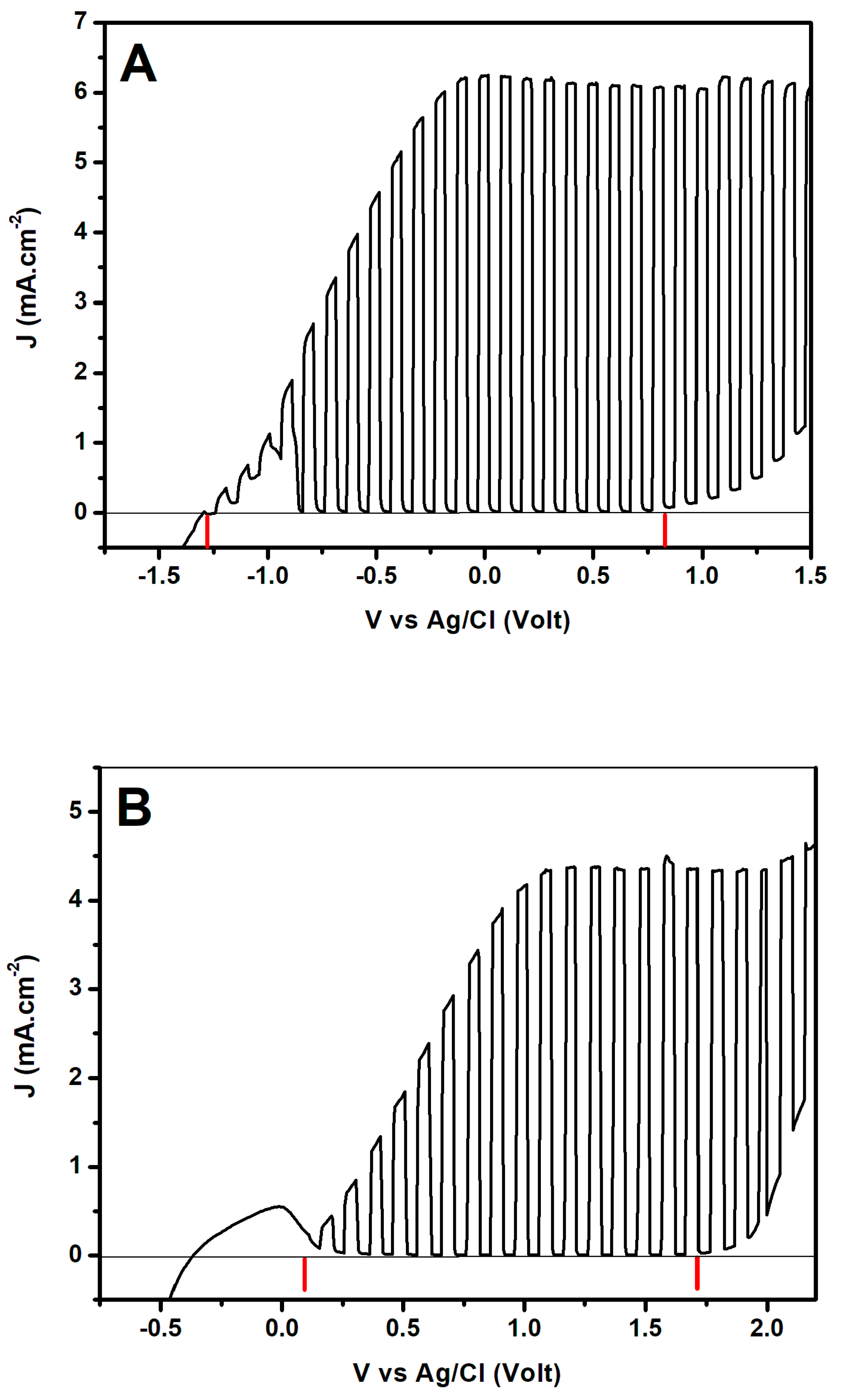
2.3. Photocurrents Generated by the Presently Used Photoanodes and Counter-Electrodes
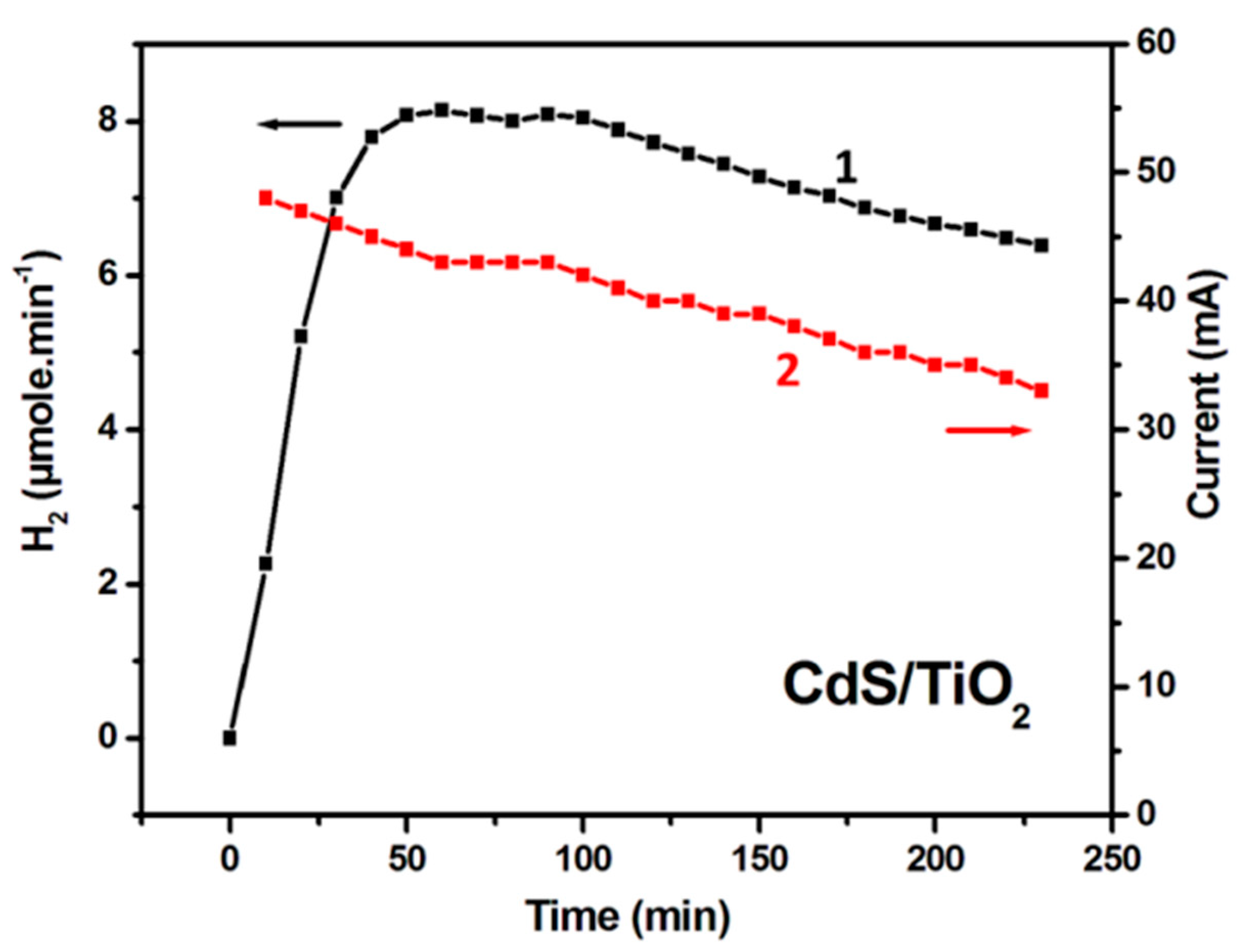
2.4. Examples of Photoelectrocatalytic Hydrogen Production by Using CdS/TiO2 or WO3 Photoanodes
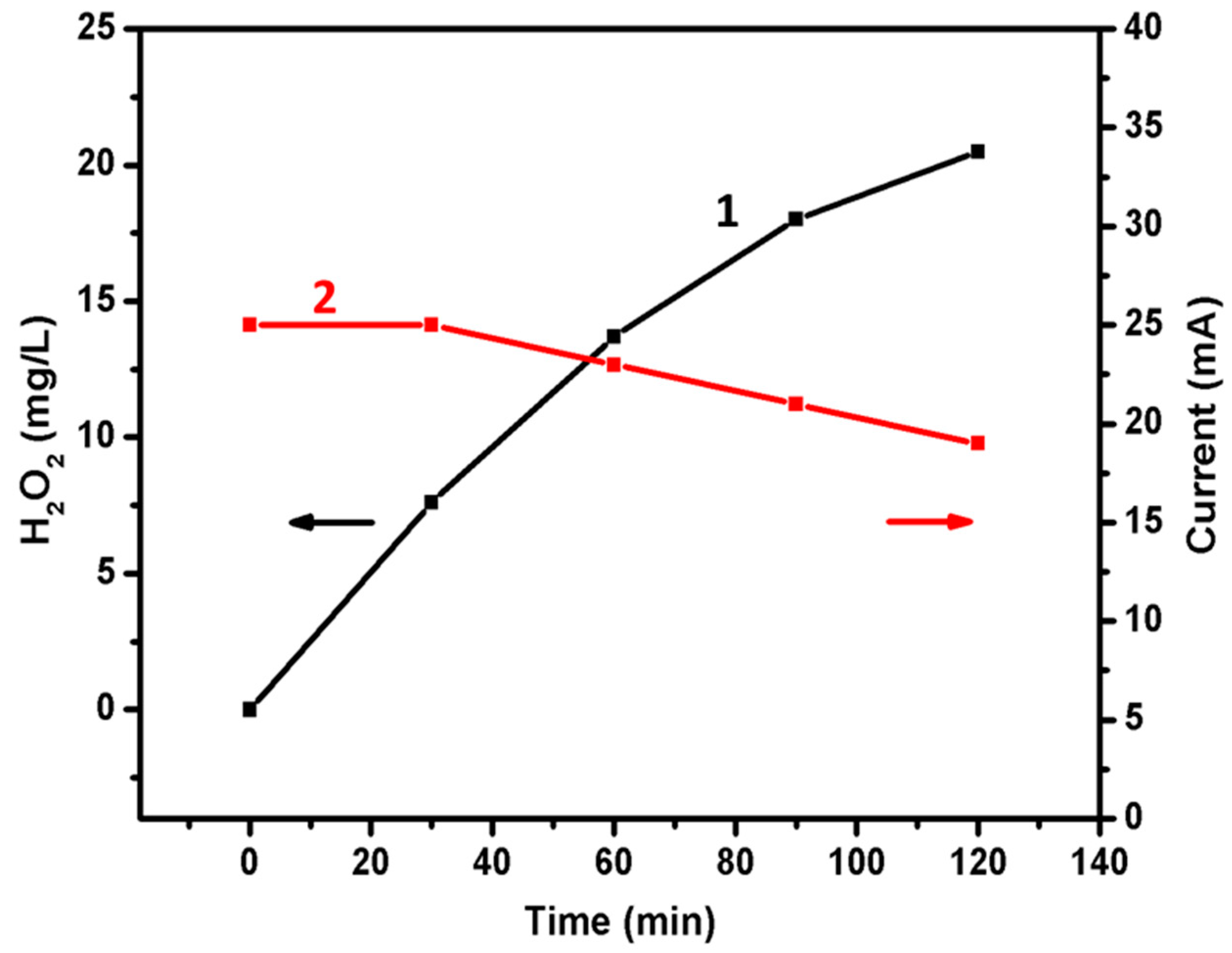
2.5. Photoelectrocatalytic Hydrogen Peroxide Production
3. Materials and Methods
3.1. Materials
3.2. Preparation of Photoanode Electrodes
3.3. Construction of the Counter-Electrode
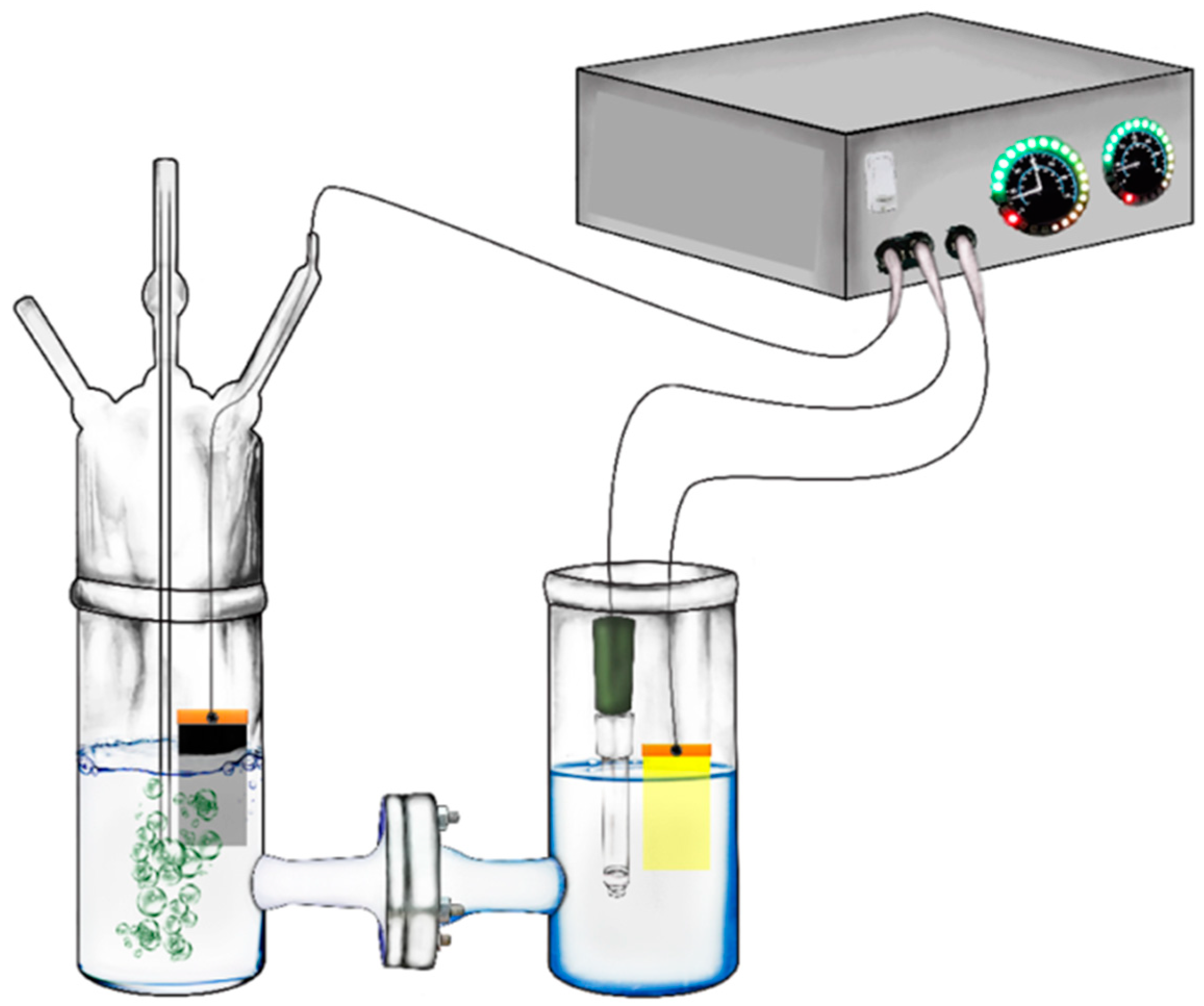
3.4. Description of the Reactor
3.5. Measurements and Characterizations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shi, X.; Zhang, Y.; Siahrostami, S.; Zheng, X. Light-Driven BiVO4–C Fuel Cell with Simultaneous Production of H2O2. Adv. Energy Mater. 2018, 8, 1801158. [Google Scholar] [CrossRef]
- McDonnell-Worth, C.J.; MacFarlane, D.R. Progress Towards Direct Hydrogen Peroxide Fuel Cells (DHPFCs) as an Energy Storage Concept. Aust. J. Chem. 2018, 71, 781–788. [Google Scholar] [CrossRef]
- Disselkamp, R.S. Can aqueous hydrogen peroxide be used as a stand-alone energy source? Int. J. Hydrogen Energy 2010, 35, 1049–1053. [Google Scholar] [CrossRef]
- Prater, K.B. Polymer electrolyte fuel cells: A review of recent developments. J. Power Sources 1994, 51, 129–144. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.Z.; Hin, J.N.C.; Wang, H.; Friedrich, K.A.; Schulze, M. A review of platinum-based catalyst layer degradation in proton exchange membrane fuel cells. J. Power Sources 2009, 194, 588–600. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Doukas, E.; Balta, P.; Raptis, D.; Avgouropoulos, G.; Lianos, P. A Realistic Approach for Photoelectrochemical Hydrogen Production. Materials 2018, 11, 1269. [Google Scholar] [CrossRef] [PubMed]
- Lianos, P. Review of recent trends in photoelectrocatalytic conversion of solar energy to electricity and hydrogen. Appl. Catal. Environ. 2017, 210, 235–254. [Google Scholar] [CrossRef]
- Raptis, D.; Dracopoulos, V.; Lianos, P. Renewable energy production by photoelectrochemical oxidation of organic wastes using WO3 photoanodes. J. Hazard. Mater. 2017, 333, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, E.A.; Kozhevnikova, N.S.; Cherepanova, S.V.; Lyubina, T.P.; Gerasimov, E.Y.; Kaichev, V.V.; Vorontsov, A.V.; Tsybulya, S.V.; Rempel, A.A.; Parmon, V.N. Photocatalytic oxidation of ethanol vapors under visible light on CdS-TiO2 nanocatalyst. J. Photochem. Photobiol. A 2012, 250, 103–109. [Google Scholar] [CrossRef]
- Liao, Q.; Li, L.; Chen, R.; Zhu, X.; Wang, H.; Ye, D.; Cheng, X.; Zhang, M.; Zhou, Y. Respective electrode potential characteristics of photocatalytic fuel cell with visible-light responsive photoanode and air-breathing cathode. Int. J. Hydrogen Energy 2015, 40, 16547–16555. [Google Scholar] [CrossRef]
- Li, L.; Xue, S.; Chen, R.; Liao, Q.; Zhu, X.; Wang, Z.; He, X.; Feng, H.; Cheng, X. Performance characteristics of a membraneless solar responsive photocatalytic fuel cell with an air-breathing cathode under different fuels and electrolytes and air conditions. Electrochim. Acta 2015, 182, 280–288. [Google Scholar] [CrossRef]
- Mase, K.; Yoneda, M.; Yamada, Y.; Fukuzumi, S. Efficient Photocatalytic Production of Hydrogen Peroxide from Water and Dioxygen with Bismuth Vanadate and a Cobalt(II) Chlorin Complex. ACS Energy Lett. 2016, 1, 913–919. [Google Scholar] [CrossRef]
- Lianos, P. Production of electricity and hydrogen by photocatalytic degradation of organic wastes in a photoelectrochemical cell: The concept of the Photofuelcell: A review of a re-emerging research field. J. Hazard. Mater. 2011, 185, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Leng, W.H.; Zhu, W.C.; Ni, J.; Zhang, Z.; Zhang, J.Q.; Cao, C.N. Photoelectrocatalytic destruction of organics using TiO2 as photoanode with simultaneous production of H2O2 at the cathode. Appl. Catal. A Gen. 2006, 300, 24–35. [Google Scholar] [CrossRef]
- Ueno, H.; Nemoto, J.; Ohnuki, K.; Horikawa, M.; Hoshino, M.; Kaneko, M. Photoelectrochemical reaction of biomass-related compounds in a biophotochemical cell comprising a nanoporous TiO2 film photoanode and an O2-reducing cathode. J. Appl. Electrochem. 2009, 39, 1897–1905. [Google Scholar] [CrossRef]
- Grätzel, M. Photoelectrochemical cells. Nature 2001, 414, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Sfaelou, S.; Syggelou, L.; Dracopoulos, V.; Travlos, A.; Lianos, P. Effect of the Nature of Cadmium Salts on the Effectiveness of CdS SILAR Deposition and Its Consequences on the Performance of Sensitized Solar Cells. J. Phys. Chem. 2014, 118, 22873–22880. [Google Scholar] [CrossRef]
- Nicolau, Y.F. Solution deposition of thin solid compound films by a successive ionic-layer adsorption and reaction process. Appl. Surf. Sci. 1985, 22/23, 1061–1074. [Google Scholar] [CrossRef]
- Li, Z.; Luo, W.; Zhang, M.; Feng, J.; Zou, Z. Photoelectrochemical cells for solar hydrogen production: Current state of promising photoelectrodes, methods to improve their properties, and outlook. Energy Environ. Sci. 2013, 6, 347–370. [Google Scholar] [CrossRef]
- Sfaelou, S.; Pop, L.C.; Monfort, O.; Dracopoulos, V.; Lianos, P. Mesoporous WO3 photoanodes for hydrogen production by water splitting and PhotoFuelCell operation. Int. J. Hydrogen Energy 2016, 41, 5902–5907. [Google Scholar] [CrossRef]
- Hodes, G.; Cahen, D.; Manassen, J. Tungsten trioxide as a photoanode for a photoelectrochemical cell (PEC). Nature 1976, 260, 312–313. [Google Scholar] [CrossRef]
- Soltani, T.; Tayyebi, T.; Hong, H.; Mirfasih, M.H.; Lee, B.K. A novel growth control of nanoplates WO3 photoanodes with dual oxygen and tungsten vacancies for efficient photoelectrochemical water splitting performance. Sol. Energy Mater. Sol. Cells 2019, 191, 39–49. [Google Scholar] [CrossRef]
- Kalamaras, E.; Lianos, P. Current Doubling effect revisited: Current multiplication in a PhotoFuelCell. J. Electroanal. Chem. 2015, 751, 37–42. [Google Scholar] [CrossRef]
- Murau, P.C. Dissolution of Tungsten by Hydrogen Peroxide. Anal. Chem. 1961, 33, 1125–1126. [Google Scholar] [CrossRef]
- Orel, B.; Krasovec, U.O.; Groselj, N.; Kosec, M.; Drazi, G.; Reisfeld, R. Gasochromic Behavior of Sol-Gel Derived Pd Doped Peroxopolytungstic Acid (W-PTA) Nano-Composite Films. J. Sol. Gel. Sci. Technol. 1999, 14, 291–308. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Photoanode | Photocurrent (mA) | Fuel Production Rate (μmole min−1) | Faradaic Efficiency (%) | |
|---|---|---|---|---|
| H2 | H2O2 | |||
| CdS/TiO2 | 42 | 7.8 | 60 | - |
| WO3 | 18 | 3.4 | 59 | - |
| WO3 | 22 | 5.0 | - | 73 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papagiannis, I.; Doukas, E.; Kalarakis, A.; Avgouropoulos, G.; Lianos, P. Photoelectrocatalytic H2 and H2O2 Production Using Visible-Light-Absorbing Photoanodes. Catalysts 2019, 9, 243. https://doi.org/10.3390/catal9030243
Papagiannis I, Doukas E, Kalarakis A, Avgouropoulos G, Lianos P. Photoelectrocatalytic H2 and H2O2 Production Using Visible-Light-Absorbing Photoanodes. Catalysts. 2019; 9(3):243. https://doi.org/10.3390/catal9030243
Chicago/Turabian StylePapagiannis, Ioannis, Elias Doukas, Alexandros Kalarakis, George Avgouropoulos, and Panagiotis Lianos. 2019. "Photoelectrocatalytic H2 and H2O2 Production Using Visible-Light-Absorbing Photoanodes" Catalysts 9, no. 3: 243. https://doi.org/10.3390/catal9030243
APA StylePapagiannis, I., Doukas, E., Kalarakis, A., Avgouropoulos, G., & Lianos, P. (2019). Photoelectrocatalytic H2 and H2O2 Production Using Visible-Light-Absorbing Photoanodes. Catalysts, 9(3), 243. https://doi.org/10.3390/catal9030243