Electropositive Promotion by Alkalis or Alkaline Earths of Pt-Group Metals in Emissions Control Catalysis: A Status Report



Abstract
:| List of Contents | |
| 1. Introduction………………………………………………………………………………………………‥ | 3 |
| 2. Promotion Methodologies and Catalysts Formulations/Designs…………………………………… | 4 |
| 2.1. Electrochemical Catalysts Formulations. The Electrochemical Promotion of Catalysis | |
| (EPOC) Concept…………………………………………………………………………………………… | 4 |
| 2.1.1. Operation Modes of the Electrochemical Promotion of Catalysis (EPOC)………………‥ | 5 |
| 2.1.2. Certain Characteristics of EPOC Concept:…………………………………………………… | 7 |
| 2.2. Conventional Catalyst Formulations. The Conventional Catalysts Promotion (CCP) | |
| Method……………………………………………………………………………………………………… | 8 |
| 2.2.1. Estimation of the Promoter Coverage………………………………………………………… | 8 |
| 2.2.2. EPOC and CCP Comparison Issues…………………………………………………………… | 9 |
| 3. Results and Dissussion…………………………………………………………………………………… | 9 |
| 3.1. Promotion of Simple, “Model” Reactions………………………………………………………… | 9 |
| 3.1.1. CO Oxidation……………………………………………………………………………………‥ | 9 |
| 3.1.2. Light Hydrocarbons Oxidation………………………………………………………………… | 17 |
| 3.1.3. NO Reduction by CO…………………………………………………………………………… | 23 |
| 3.1.4. NO Reduction by Hydrocarbons or H2………………………………………………………‥ | 28 |
| 3.1.5. N2O Decomposition and/or Reduction………………………………………………………‥ | 37 |
| 3.2. Electropositive Promotion of PGMs Operated Under Simulated Practical Conditions……… | 42 |
| 3.2.1. Simulated TWC Conditions…………………………………………………………………‥‥ | 42 |
| 3.2.2. Oxygen Excess Conditions (Simulated Lean-Burn and Diesel Exhausts Gases)………… | 48 |
| 3.3. Mechanistic Implications: The mode of Action of Electropositive Promoters………………‥ | 54 |
| 3.3.1. Main Promotion Characteristics and Mechanistic Implications…………………………… | 54 |
| 3.3.2. Direct Spectroscopic and Other Analytical Technique Evidences………………………… | 60 |
| 4. Conclusions and Perspectives…………………………………………………………………………… | 65 |
| References……………………………………………………………………………………………………‥ | 66 |
1. Introduction
- Commercial TWCs use formulations based variously on two or three noble metals (Pt, Pd and Rh) where Rh is essential for an efficient control of NOx emissions (Rh is highly effective and therefore the key component in TWCs for the NO dissociation/reduction; Pt and Pd, although very active for CO and hydrocarbons oxidations, are almost ineffective for NOx reduction). However, Rh is rare, therefore its successful reduction or even replacement by another De-NOx efficient catalyst in TWC formulations is highly desirable.
- A small but significant portion of NO is still converted to the undesirable by-product N2O in commercial TWCs; Nitrous oxide emission control, due to N2O harmful impact on stratospheric ozone depletion and its outstanding global warming potential (the latter is about 310 and 21 times greater than that of CO2 and CH4, respectively [4,5]), is a current challenge in environmental catalysis technology.
- TWCs recycling for the noble metals recovery is nowadays marginally profitable, mainly due to the costly final separation of the recovered noble metals; simpler TWCs formulations (i.e., consisting of only one noble metal) are expected to substantially reduce both TWCs’ production and recycling costs.
- Commercial TWCs are no longer efficient in controlling NOx emissions from the advanced lean-burn and diesel engines that operate at net-oxidising conditions.
2. Promotion Methodologies and Catalysts Formulations/Designs
2.1. Electrochemical Catalysts Formulations. The Electrochemical Promotion of Catalysis (EPOC) Concept
2.1.1. Operation Modes of the Electrochemical Promotion of Catalysis (EPOC)
2.1.2. Certain Characteristics of EPOC Concept:
2.2. Conventional Catalyst Formulations. The Conventional Catalysts Promotion (CCP) Method
2.2.1. Estimation of the Promoter Coverage
2.2.2. EPOC and CCP Comparison Issues
3. Results and Discussion
3.1. Promotion of Simple, “Model” Reactions
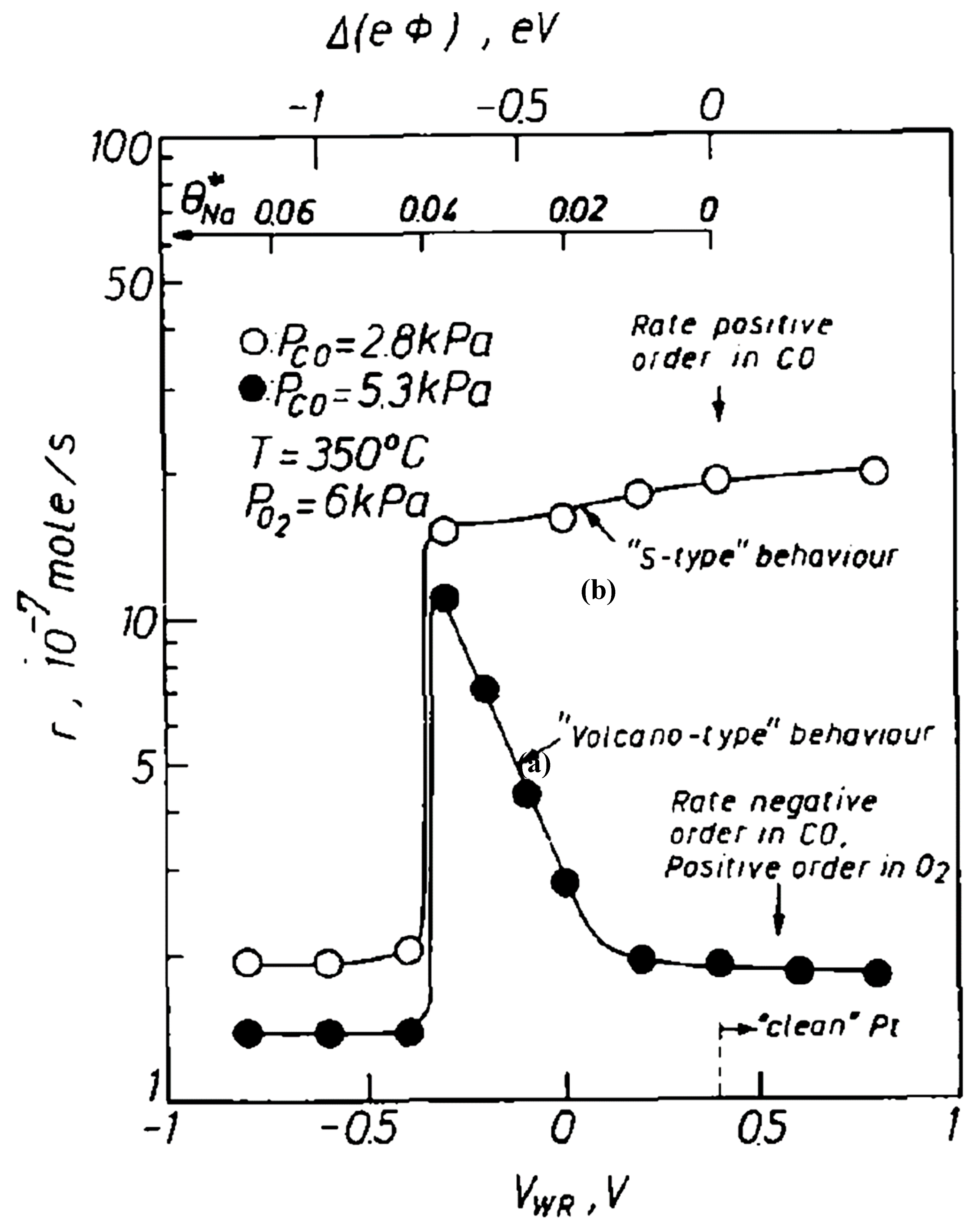
3.1.1. CO Oxidation
3.1.2. Light Hydrocarbons Oxidation
3.1.3. NO Reduction by CO
3.1.4. NO Reduction by Hydrocarbons or H2
- Alkanes oxidation, either using dioxygen (Section 3.1.2) or NO (this section) behaves similarly under electropositive promotion by alkalis: addition of alkali on PGM surfaces causes only poisoning. Interpretations given are similar in both cases, based on the low adsorption propensity of alkanes on PGM surfaces and on the O-poisoning of the surfaces resulted by the alkali-induced enhancement of NO dissociative chemisorption.
- On the opposite, electropositive promotion of the NO + alkenes reactions is very pronounced; apparently it is the greater observed, for emissions control catalysis model reactions reviewed in the present work, offering ρ values as high as = 420 and = 280.
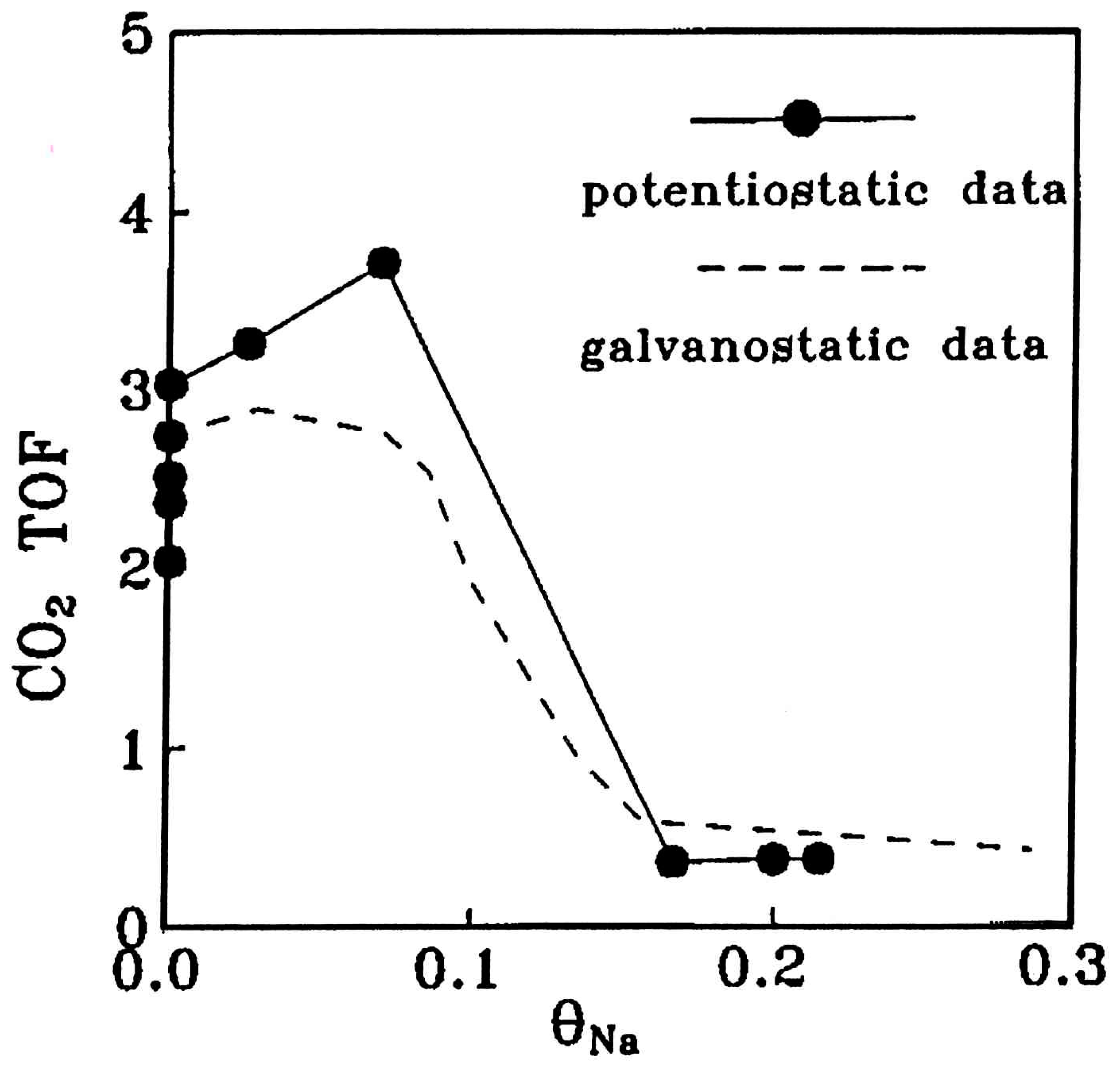
- Some studies performing one-by-one comparison of EPOC and CCP by means of Na promoted Pt/NO + C3H6 system have demonstrated the very close similarities on the promotion output characteristics of the two methods and have convincingly argued for the similar origin of promotion in both cases.
- Once again (see Section 3.1.3) the intensity of CCP was superior to that of EPOC, indicating that an intimate contact between active phase and promoter particles succeeded by nanostructured catalysts seems to play a significant role on the promotion intensity.
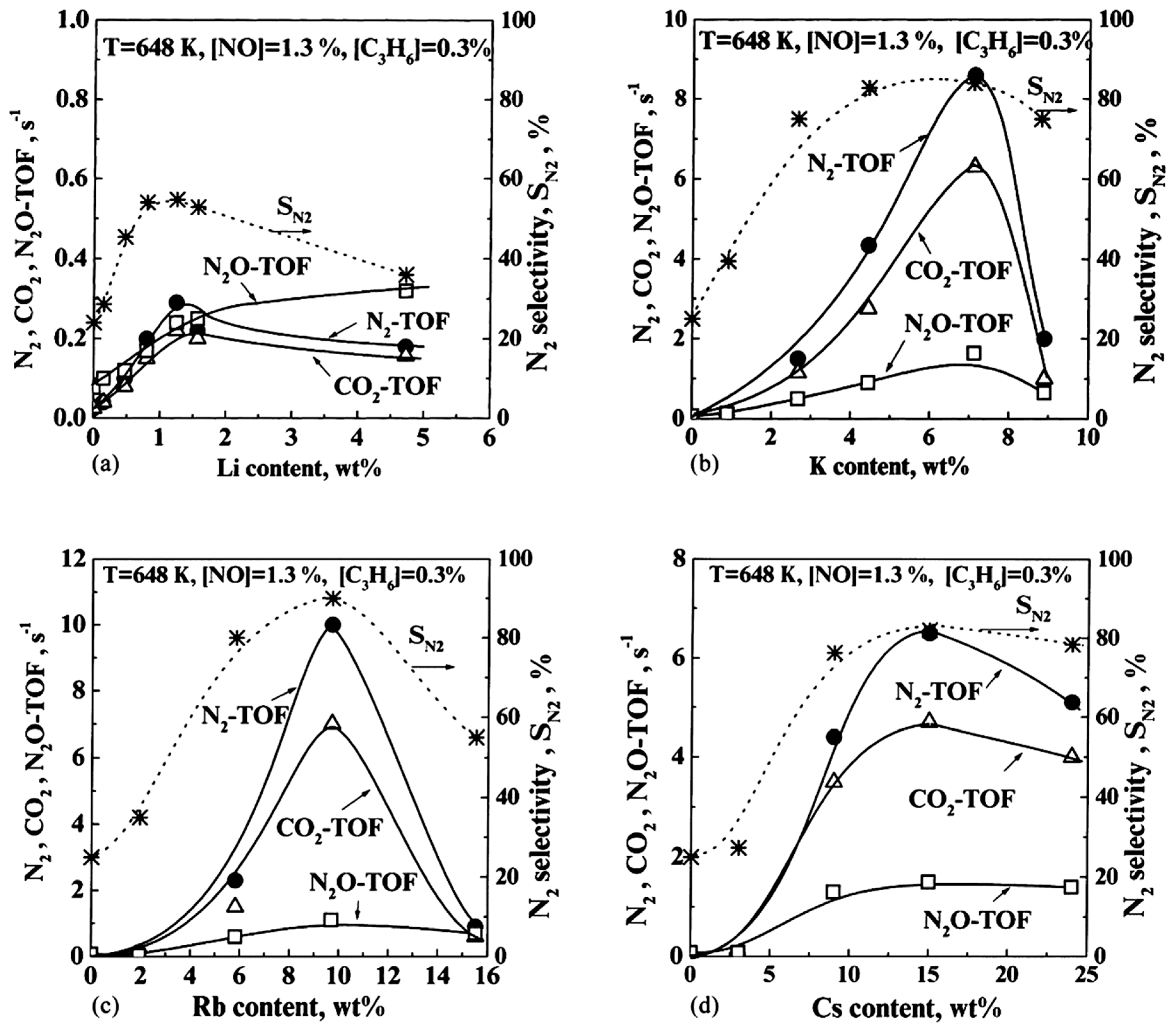
- Heavier alkalis were again (see Section 3.1.3) more effective than the lighter ones; Rb was the best promoter.
- Among Pt, Pd and Rh, alkali promotion was more effective on Pt, lower on Pd and even lower on Rh. As we shall see (Section 3.3) this is related to the initial, unpromoted propensity of each metal on the dissociative adsorption of NO.
- Promotion by alkalis was also found to be substantial on the NO reduction by H2 as well as on the direct NO decomposition reaction, although only few studies appeared in the open literature so far.
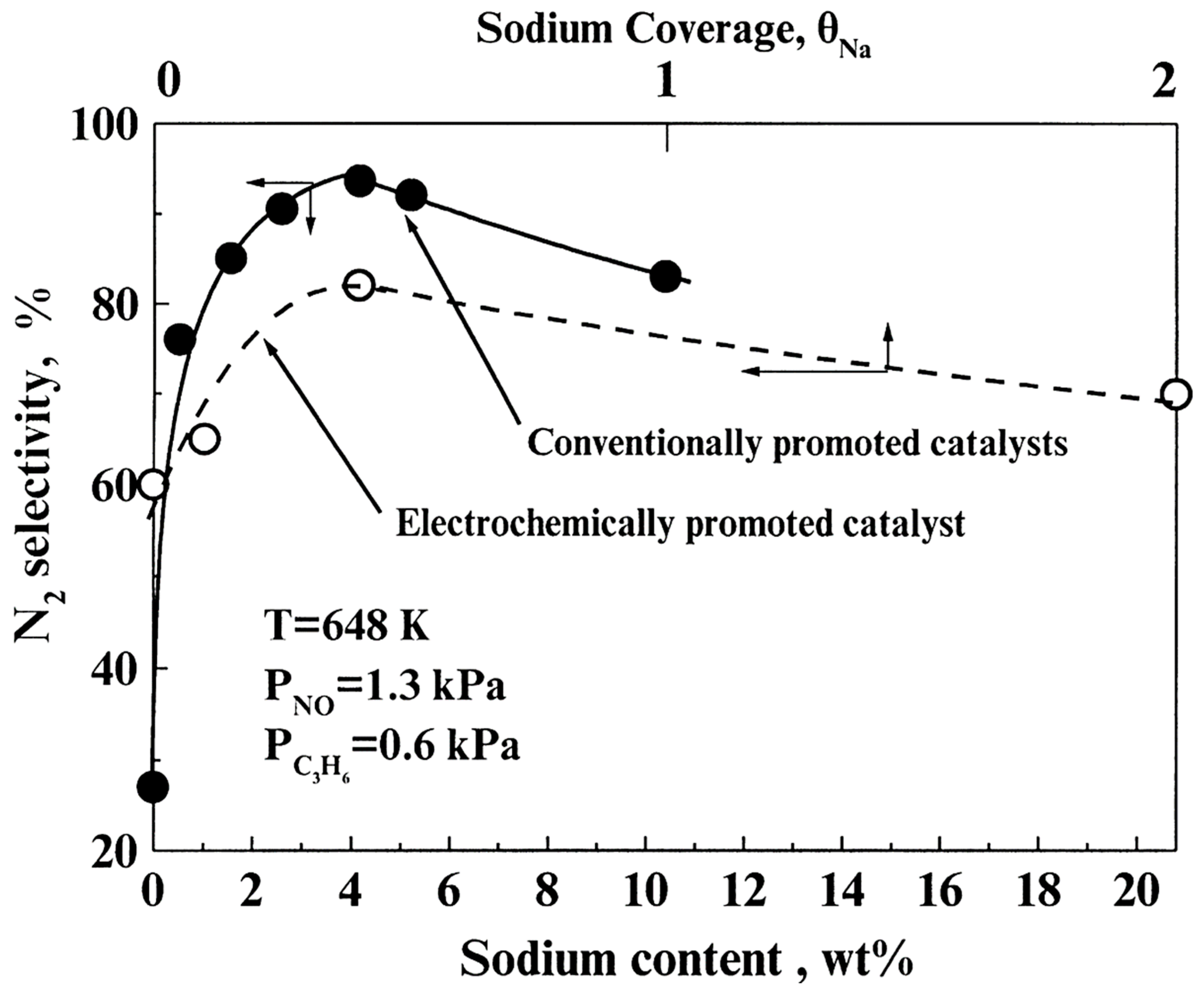
- Catalytic activity promotion was always accompanied by substantial increases on the system selectivity towards N2.
3.1.5. N2O Decomposition and/or Reduction
- Alkali addition on PGM catalysts typically promotes the direct N2O decomposition reaction (in the absence or presence of O2 in the feed stream) or its reduction by reducing agents.
- Among Pt, Rh and Ir catalysts, alkali-promotion of the N2O decomposition reaction was found to be very effective and superior on Pt, less effective on Rh and detrimental (poisoning) on Ir; in the case of Ir, alkali-induced promotion was obtained only under excess oxygen conditions, which was attributed to changes on the oxidation state of the metal.
- Comparative EPOC and CCP studies of alkali promotion on the N2O reduction by the use of several reducing agents (CH4, C3H8 and C3H6) demonstrated the great similarities on the output promotional characteristics of the two methods.
3.2. Electropositive Promotion of PGMs Operated Under Simulated Practical Conditions
3.2.1. Simulated TWC Conditions
3.2.2. Oxygen Excess Conditions (Simulated Lean-Burn and Diesel Exhausts Gases)
- Potassium ions electrochemically transferred to the Pt catalyst play a double role in the NSR process, as a promoter for the NO oxidation reaction and as storage sites through the formation of potassium nitrates.
- The maximum yield of Pt/(K)βAl2O3 to effectively reduce NOx to N2 was obtained at T = 300 °C (Figure 17). This temperature is suitable for both, gasoline and diesel engines working under lean burn conditions.
3.3. Mechanistic Implications: The mode of Action of Electropositive Promoters
3.3.1. Main Promotion Characteristics and Mechanistic Implications
3.3.2. Direct Spectroscopic and Other Analytical Technique Evidences
4. Conclusions and Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Kaspar, J.; Fornasiero, P.; Hickey, N. Automotive catalytic converters: Current status and some perspectives. Catal. Today 2003, 77, 419–449. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Heck, R.M. Catalytic converters: State of the art and perspectives. Catal. Today 1999, 51, 351–360. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M. Three-way Catalysis (Book Chapter). In Perovskites and Related Mixed Oxides: Concepts and Applications; Wiley-VCH, Vergal GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 559–586. Available online: https://onlinelibrary.wiley.com/doi/pdf/10.1002/9783527686605.ch25 (accessed on 12 November 2015).
- Wuebbles, D.J. Nitrous Oxide: No Laughing Matter. Science 2009, 326, 56–57. [Google Scholar] [CrossRef] [PubMed]
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Vayenas, C.G.; Bebelis, S.; Yentekakis, I.V.; Lintz, H.-G. Non-faradaic electrochemical modification of catalytic activity: A status report. Catal. Today 1992, 11, 303–442. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Pliangos, C.; Brosda, S.; Tsiplakides, D. Electrochemical Activation of Catalysis; Kluwer Academic/Plenum: New York, NY, USA, 2001. [Google Scholar]
- Stoukides, M.; Vayenas, C.G. The effect of electrochemical oxygen pumping on the rate and selectivity of ethylene oxidation on polycrystalline silver. J. Catal. 1981, 70, 137–146. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Vayenas, C.G. The effect of electrochemical oxygen pumping on the steady-state and oscillatory behaviour of CO oxidation on polycrystalline Pt. J. Catal. 1988, 111, 170–188. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Neophytides, S. Non-faradaic electrochemical modification of catalytic activity. J. Phys. Chem. 1988, 92, 5083–5085. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Ladas, S. Dependence of catalytic rates on catalyst work function. Nature 1990, 343, 625–627. Available online: https://www.nature.com/articles/343625a0 (accessed on 15 February 1990). [CrossRef]
- Yentekakis, I.V.; Bebelis, S. Study of the NEMCA effect in a single-pellet catalytic reactor. J. Catal. 1992, 137, 278–283. [Google Scholar] [CrossRef]
- Vernoux, P.; Lizarraga, L.; Tsampas, M.N.; Sapountzi, F.M.; De Lucas-Consuegra, A.; Valverde, J.-L.; Souentie, S.; Vayenas, C.G.; Tsiplakides, D.; Balomenou, S.; et al. Ionically Conducting Ceramics as Active Catalyst Supports. Chem. Rev. 2013, 113, 8192–8260. [Google Scholar] [CrossRef] [PubMed]
- Yentekakis, I.V.; Lambert, R.M.; Tikhov, M.; Konsolakis, M.; Kiousis, V. Promotion by sodium in emission control catalysis: A kinetic and spectroscopic study of the Pd-catalysed reduction of NO by propene. J. Catal. 1998, 176, 82–92. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M.; Lambert, R.M.; Macleod, N.; Nalbantian, L. Extraordinarily effective promotion by sodium in emission control catalysis: NO reduction by propene over Na-promoted Pt/γ-Al2O3. Appl. Catal. B 1999, 22, 123–133. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Moggridge, G.; Vayenas, C.G.; Lambert, R.M. In situ controlled promotion of catalyst surfaces via NEMCA: The effect of Na on the Pt-catalysed CO oxidation. J. Catal. 1994, 146, 292–305. [Google Scholar] [CrossRef]
- Palermo, A.; Lambert, R.M.; Harkness, I.R.; Yentekakis, I.V.; Marina, O.A.; Vayenas, C.G. Electrochemical promotion by Na of the platinum-catalysed reaction between CO and NO. J. Catal. 1996, 161, 471–479. [Google Scholar] [CrossRef]
- Harkness, I.R.; Hardacre, C.; Lambert, R.M.; Yentekakis, I.V.; Vayenas, C.G. Ethylene oxidation over platinum: In situ electrochemically controlled promotion using Na–β′′ alumina and studies with a Pt(111)/Na model catalyst. J. Catal. 1996, 160, 19–26. [Google Scholar] [CrossRef]
- Marina, O.A.; Yentekakis, I.V.; Vayenas, C.G.; Palermo, A.; Lambert, R.M. In situ controlled promotion of catalyst surfaces via NEMCA: The effect of Na on the Pt-catalysed NO reduction by H2. J. Catal. 1997, 166, 218–228. [Google Scholar] [CrossRef]
- Filkin, N.C.; Tikhov, M.S.; Palermo, A.; Lambert, R.M. A kinetic and spectroscopic study of the in situ electrochemical promotion by sodium of the platinum-catalysed combustion of propene. J. Phys. Chem. A 1999, 103, 2680–2687. [Google Scholar] [CrossRef]
- Konsolakis, M.; Palermo, A.; Tikhov, M.S.; Lambert, R.M.; Yentekakis, I.V. Electrochemical vs. Conventional promotion: A new tool to design effective, highly dispersed conventional catalysts. Ionics 1998, 4, 148–156. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M.; Lambert, R.M.; Palermo, A.; Tikhov, M. Successful application of electrochemical promotion to the design of effective conventional catalyst formulations. Solid State Ionics 2000, 136/137, 783–790. [Google Scholar] [CrossRef]
- Nicole, J.; Tsiplakides, D.; Pliangos, C.; Verykios, X.E.; Comninelis, C.; Vayenas, C.G. Electrochemical promotion and metal–support interactions. J. Catal. 2001, 204, 23–34. [Google Scholar] [CrossRef]
- Brosda, S.; Vayenas, C.G.; Wei, J. Rules of chemical promotion. Appl. Catal. B 2006, 68, 109–124. [Google Scholar] [CrossRef]
- Pekridis, G.; Kaklidis, N.; Konsolakis, M.; Athanasiou, C.; Yentekakis, I.V.; Marnellos, G.E. A comparison between electrochemical and conventional catalyst promotion: The case of N2O reduction by alkanes or alkenes over K-modified Pd catalysts. Solid State Ionics 2011, 192, 653–658. [Google Scholar] [CrossRef]
- Pliangos, C.; Yentekakis, I.V.; Verykios, X.E.; Vayenas, C.G. Non-Faradaic electrochemical modification of catalytic activity: VIII. Rh-catalysed C2H4 oxidation. J. Catal. 1995, 154, 124–136. [Google Scholar] [CrossRef]
- De Lucas-Consuegra, A.; Dorado, F.; Valverde, J.L.; Karoum, R.; Vernoux, P. Electrochemical activation of Pt catalyst by potassium for low temperature CO deep oxidation. Catal. Commun. 2008, 9, 17–20. [Google Scholar] [CrossRef]
- Mirkelamoglu, B.; Karakas, G. CO oxidation over palladium- and sodium-promoted tin dioxide: Catalyst characterization and temperature-programmed studies. Appl. Catal. A 2005, 281, 275–284. [Google Scholar] [CrossRef]
- Mirkelamoglu, B.; Karakas, G. The role of alkali-metal promotion on CO oxidation over PdO/SnO2 catalysts. Appl. Catal. A 2006, 299, 84–94. [Google Scholar] [CrossRef]
- Lee, C.-H.; Chen, Y.-W. Effect of basic additives on Pt/Al2O3 for CO and propylene oxidation under oxygen-deficient conditions. Ind. Eng. Chem. Res. 1997, 36, 1498–1506. [Google Scholar] [CrossRef]
- Tanikawa, K.; Egawa, C. Effect of barium addition on CO oxidation activity of palladium catalysts. Appl. Catal A 2011, 403, 12–17. [Google Scholar] [CrossRef]
- Minemura, Y.; Kuriyama, M.; Ito, S.; Tomishige, K.; Kunimori, K. Additive effect of alkali metal ions on preferential CO oxidation over Pt/Al2O3. Catal. Commun. 2006, 7, 623–626. [Google Scholar] [CrossRef]
- Minemura, Y.; Ito, S.; Miyao, T.; Naito, S.; Tomishige, K.; Kunimori, K. Preferential CO oxidation promoted by the presence of H2 over K–Pt/Al2O3. Chem. Commun. 2005, 11, 1429–1431. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kuriyama, M.; Ishida, Y.; Ito, S.; Tomishige, K.; Kunimori, K. Preferential CO oxidation in hydrogen-rich stream over Pt catalysts modified with alkali metals: Part I. Catalytic performance. Appl. Catal. A 2008, 343, 117–124. [Google Scholar] [CrossRef]
- Kuriyama, M.; Tanaka, H.; Ito, S.; Kubota, T.; Miyao, T.; Naito, S.; Timoshige, K.; Kunimori, K. Promoting mechanism of potassium in preferential CO oxidation on Pt/Al2O3. J. Catal. 2007, 252, 39–48. [Google Scholar] [CrossRef]
- Tanaka, H.; Kuriyama, M.; Ishida, Y.; Ito, S.; Kubota, T.; Miyao, T.; Naito, S.; Tomishige, K.; Kunimori, K. Preferential CO oxidation in hydrogen-rich stream over Pt catalysts modified with alkali metals: Part II. Catalyst characterization and role of alkali metals. Appl. Catal. A 2008, 343, 125–133. [Google Scholar] [CrossRef]
- Pedrero, C.; Waku, T.; Iglesia, E. Oxidation of CO in H2–CO mixtures catalysed by platinum: Alkali effects on rates and selectivity. J. Catal. 2005, 233, 242–255. [Google Scholar] [CrossRef]
- Kwak, C.; Park, T.-J.; Suh, D.J. Effects of sodium addition on the performance of PtCo/Al2O3 catalysts for preferential oxidation of carbon monoxide from hydrogen-rich fuels. Appl. Catal. A 2005, 278, 181–186. [Google Scholar] [CrossRef]
- Cho, S.-H.; Park, J.-S.; Choi, S.-H.; Kim, S.-H. Effect of magnesium on preferential oxidation of carbon monoxide on platinum catalyst in hydrogen-rich stream. J. Power Sources 2006, 156, 260–266. [Google Scholar] [CrossRef]
- De Lucas-Consuegra, A.; Princivalle, A.; Caravaca, A.; Dorado, F.; Guizard, C.; Valverde, J.L.; Vernoux, P. Preferential CO oxidation in hydrogen-rich stream over an electrochemically promoted Pt catalyst. Appl. Catal. B 2010, 94, 281–287. [Google Scholar] [CrossRef]
- Liu, K.; Wang, A.; Zhang, T. Recent advances in preferential oxidation of CO reaction over platinum group metal catalysts. ACS Catal. 2012, 2, 1165–1178. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Bebelis, S.; Despotopoulou, M. Non-faradaic electrochemical modification of catalytic activity 4. The use of β″-Al2O3 as the solid electrolyte. J. Catal. 1991, 128, 415–435. [Google Scholar] [CrossRef]
- Vernoux, P.; Gaillard, F.; Lopez, C.; Siebert, E. In-situ electrochemical control of the catalytic activity of platinum for the propene oxidation. Solid State Ionics 2004, 175, 609–613. [Google Scholar] [CrossRef]
- Billard, A.; Vernoux, P. Electrochemical catalysts for hydrocarbon combustion. Top. Catal. 2007, 44, 369–377. [Google Scholar] [CrossRef]
- De Lucas-Consuegra, A.; Dorado, F.; Valverde, J.L.; Karoum, R.; Vernoux, P. Low-temperature propene combustion over Pt/K-βAl2O3 electrochemical catalyst: Characterization, catalytic activity measurements and investigation of the NEMCA effect. J. Catal. 2007, 251, 474–484. [Google Scholar] [CrossRef]
- De Lucas-Consuegra, A.; Dorado, F.; Jimenez-Borja, C.; Caravaca, A.; Vernoux, P.; Valverde, J.L. Use of potassium conductors in the electrochemical promotion of environmental catalysis. Catal. Today 2009, 146, 293–298. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Tellou, V.; Bontzolaki, G.; Rapakousios, I.A. A comparative study of the C3H6 + NO + O2, C3H6 + O2 and NO + O2 reactions in excess oxygen over Na-modified Pt/γ-Al2O3 catalysts. Appl. Catal. B 2005, 56, 229–239. [Google Scholar] [CrossRef]
- Kotsionopoulos, N.; Bebelis, S. In situ electrochemical modification of catalytic activity for propane combustion of Pt/β′′-Al2O3 catalyst-electrodes. Top. Catal. 2007, 44, 379–389. [Google Scholar] [CrossRef]
- Kotsionopoulos, N.; Bebelis, S. Electrochemical characterization of the Pt/β′′–Al2O3 system under conditions of in situ electrochemical modification of catalytic activity for propane combustion. J. Appl. Electrochem. 2010, 40, 1883–1891. [Google Scholar] [CrossRef]
- Auvray, X.; Lindholm, A.; Milh, M.; Olsson, L. The addition of alkali and alkaline earths metals to Pd/Al2O3 to promote methane compustion. Effect of Pd and Ca loading. Catal. Today 2018, 299, 212–218. [Google Scholar] [CrossRef]
- Williams, F.J.; Palermo, A.; Tikhov, M.S.; Lambert, R.M. Electrochemical promotion by sodium of the rhodium-catalysed NO + CO reaction. J. Phys. Chem. B 2000, 104, 11883–11890. [Google Scholar] [CrossRef]
- Williams, F.J.; Palermo, A.; Tikhov, M.S.; Lambert, R.M. Mechanism of alkali promotion in heterogeneous catalysis under realistic conditions: Application of electron spectroscopy and electrochemical promotion to the reduction of NO by CO and by propene over rhodium. Surf. Sci. 2001, 482–485, 177–182. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V.; Palermo, A.; Lambert, R.M. Optimal promotion by rubidium of the CO + NO reaction over Pt/γ-Al2O3 catalysts. Appl. Catal. B 2001, 33, 293–302. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V. NO reduction by propene or CO over alkali-promoted Pd/YSZ catalysts. J. Hazard. Mater. 2007, 149, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, K.; Egawa, C. Effect of barium addition over palladium catalyst for CO–NO–O2 reaction. J. Mol. Catal. A 2011, 349, 94–99. [Google Scholar] [CrossRef]
- Lepage, M.; Visser, T.; Soulimani, F.; Inglesias-Juez, A.; Weckhuysen, B.M. Promotion Effects in the Reduction of NO by CO over Zeolite-Supported Rh Catalysts. J. Phys. Chem. C 2010, 114, 2282–2292. [Google Scholar] [CrossRef]
- Harkness, I.R.; Lambert, R.M. Electrochemical Promotion of the NO + Ethylene Reaction over Platinum. J. Catal. 1995, 152, 211–214. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Palermo, A.; Filkin, N.; Tikhov, M.S.; Lambert, R.M. In situ electrochemical promotion by sodium of the platinum-catalysed reduction of NO by propene. J. Phys. Chem. B 1997, 101, 3759–3768. [Google Scholar] [CrossRef]
- Williams, F.J.; Palermo, A.; Tikhov, M.S.; Lambert, R.M. Electrochemical promotion by sodium of the rhodium-catalysed reduction of NO by propene: Kinetics and spectroscopy. J. Phys. Chem. B 2001, 105, 1381–1388. [Google Scholar] [CrossRef]
- Williams, F.J.; Tikhov, M.S.; Palermo, A.; Macleod, N.; Lambert, R.M. Electrochemical promotion of rhodium-catalysed NO reduction by CO and by propene in the presence of oxygen. J. Phys. Chem. B 2001, 105, 2800–2808. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V. Strong promotional effects of Li, K, Rb and Cs on the Pt-catalysed reduction of NO by propene. Appl. Catal. B 2001, 29, 103–113. [Google Scholar] [CrossRef]
- Konsolakis, M.; Yentekakis, I.V. The reduction of NO by propene over Ba-promoted Pt/γ-Al2O3 catalysts. J. Catal. 2001, 198, 142–150. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Lambert, R.M.; Konsolakis, M.; Kallithrakas-Kontos, N. On the Effects of Residual Chloride and of Barium Promotion on Pt/γ-Al2O3 Catalysts in the Reduction of NO by Propene. Catal. Lett. 2002, 81, 181–185. [Google Scholar] [CrossRef]
- Macleod, N.; Isaac, J.; Lambert, R.M. Sodium promotion of the NO+C3H6 reaction over Rh/γ-Al2O3 catalysts. J. Catal. 2000, 193, 115–122. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Lambert, R.M.; Konsolakis, M.; Kiousis, V. The effect of sodium on the Pd-catalysed reduction of NO by methane. Appl. Catal. B 1998, 18, 293–305. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M.; Kiousis, V.; Lambert, R.M.; Tikhov, M. Promotion by sodium in emission control catalysis: The difference between alkanes and alkenes in the Pd-catalysed reduction of NO by hydrocarbons. G-NEST Int. J. 1999, 1, 121–130. [Google Scholar]
- Burch, R.; Watling, T.C. The difference between alkanes and alkenes in the reduction of NO by hydrocarbons over Pt catalysts under lean-burn conditions. Catal. Lett. 1997, 43, 19–23. [Google Scholar] [CrossRef]
- Burch, R.; Sullivan, J.A.; Watling, T.C. Mechanistic considerations for the reduction of NOx over Pt/Al2O3 and Al2O3 catalysts under lean-burn conditions. Catal. Today 1998, 42, 13–23. [Google Scholar] [CrossRef]
- Goula, M.A.; Charisiou, N.D.; Papageridis, K.N.; Delimitis, A.; Papista, E.; Pachatouridou, E.; Iliopoulou, E.F.; Marnellos, G.; Konsolakis, M.; Yentekakis, I.V. A comparative study of the H2-assisted selective catalytic reduction of nitric oxide by propene over noble metal (Pt, Pd, Ir)/γ-Al2O3 catalysts. J. Environ. Chem. Eng. 2016, 4, 1629–1641. [Google Scholar] [CrossRef]
- Machida, M.; Ikeda, S.; Kurogi, D.; Kijima, T. Low temperature catalytic NOx-H2 reactions over Pt/TiO2-ZrO2 in an excess oxygen. Appl. Catal. B 2001, 35, 107–116. [Google Scholar] [CrossRef]
- Costa, C.N.; Savva, P.G.; Andronikou, C.; Lambrou, P.S.; Polychronopoulou, K.; Belessi, V.C.; Stathopoulos, V.N.; Pomonis, P.J.; Efstathiou, A.M. An Investigation of the NO/H2/O2 (lean De-NOx) reaction on a highly active and selective Pt/La0.7Sr0.2Ce0.1FeO3 catalyst at low temperatures. J. Catal. 2002, 209, 456–471. [Google Scholar] [CrossRef]
- Polychronopoulou, K.; Efstathiou, A.M. NOx control via H2-selective catalytic reduction (H2-SCR) technology for stationary and mobile applications. Recent Pat. Mater. Sci. 2012, 5, 87–104. [Google Scholar] [CrossRef]
- Burch, R.; Coleman, M.D. An Investigation of Promoter Effects in the Reduction of NO by H2 under Lean-Burn Conditions. J. Catal. 2002, 208, 435–447. [Google Scholar] [CrossRef]
- Machida, M.; Watanabe, T. Effect of Na-addition on catalytic activity of Pt-ZSM-5 for low-temperature NO–H2–O2 reactions. Appl. Catal. B 2004, 52, 281–286. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. Low-temperature NOx reduction with H2+CO under oxygen-rich conditions over a Pd/TiO2/Al2O3 catalyst. Catal. Commun. 2002, 3, 61–65. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. Lean NOx reduction with CO + H2 mixtures over Pt/Al2O3 and Pd/Al2O3 catalysts. Appl. Catal. B 2002, 35, 269–279. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. In situ ammonia generation as a strategy for catalytic NOx reduction under oxygen rich conditions. Chem. Commun. 2003, 11, 1300–1301. [Google Scholar] [CrossRef]
- Macleod, N.; Lambert, R.M. An in situ DRIFTS study of efficient lean NOx reduction with H2 + CO over Pd/Al2O3: The key role of transient NCO formation in the subsequent generation of ammonia. Appl. Catal. B 2003, 46, 483–495. [Google Scholar] [CrossRef]
- Konsolakis, M.; Vrontaki, M.; Avgouropoulos, G.; Ioannides, T.; Yentekakis, I.V. Novel doubly-promoted catalysts for the lean NOx reduction by H2 + CO: Pd(K)/Al2O3–(TiO2). Appl. Catal. B 2006, 68, 59–67. [Google Scholar] [CrossRef]
- Wang, C.-B.; Chang, J.-G.; Wu, R.-C.; Yeh, C.-T. Promotion effect of coating alumina supported palladium with sodium hydroxide on the catalytic conversion of nitric oxide. Appl. Catal. B 1998, 17, 51–62. [Google Scholar] [CrossRef]
- Haber, J.; Nattich, M.; Machej, T. Alkali-metal promoted rhodium-on-alumina catalysts for nitrous oxide decomposition. Appl. Catal. B 2008, 77, 278–283. [Google Scholar] [CrossRef]
- Parres-Esclaped, S.; Lopez-Suerez, F.E.; Bueno-Lopez, A.; Illan-Gomez, M.J.; Ura, B.; Trawcczynski, J. Rh–Sr/Al2O3 catalyst for N2O decomposition in the presence of O2. Top. Catal. 2009, 52, 1832–1836. [Google Scholar] [CrossRef]
- Konsolakis, M.; Aligizou, F.; Goula, G.; Yentekakis, I.V. N2O decomposition over doubly-promoted Pt(K)/Al2O3-(CeO2-La2O3) structured catalysts: On the compined effects of promotion and feed composition. Chem. Eng. J. 2013, 230, 286–295. [Google Scholar] [CrossRef]
- Papista, E.; Pachatouridou, E.; Goula, M.A.; Marnellos, G.E.; Iliopoulou, E.; Konsolakis, M.; Yentekakis, I.V. Effect of alkali promoters (K) on nitrous oxide abatement over Ir/Al2O3 catalysts. Top. Catal. 2016, 59, 1020–1027. [Google Scholar] [CrossRef]
- Goncalves, F.; Figueiredo, J.L. Sinergistic effect between Pt and K in the catalytic reduction of NO and N2O. Appl. Catal. B 2006, 62, 181–192. [Google Scholar] [CrossRef]
- Pekridis, G.; Kaklidis, N.; Konsolakis, M.; Iliopoulou, E.F.; Yentekakis, I.V.; Marnellos, G. Correlation of surface characteristics with catalytic performance of potassium promoted Pd/Al2O3 catalysts: The case of N2O reduction by alkanes or alkenes. Top. Catal. 2011, 54, 1135–1142. [Google Scholar] [CrossRef]
- De Lucas-Consuegra, A.; Dorado, F.; Jimenez-Borja, C.; Valerde, J.L. Influence of the reaction conditions on the electrochemical promotion by potassium for the selective catalytic reduction of N2O by C3H6 on platinum. Appl. Catal. B 2008, 78, 222–231. [Google Scholar] [CrossRef]
- De Lucas Consuegra, A.; Dorado, F.; Jimenez-Borja, C.; Valverde, J.L. Electrochemical promotion of Pt impregnated catalyst for the treatment of automotive exhaust emissions. J. Appl. Electrochem. 2008, 38, 1151–1157. [Google Scholar] [CrossRef]
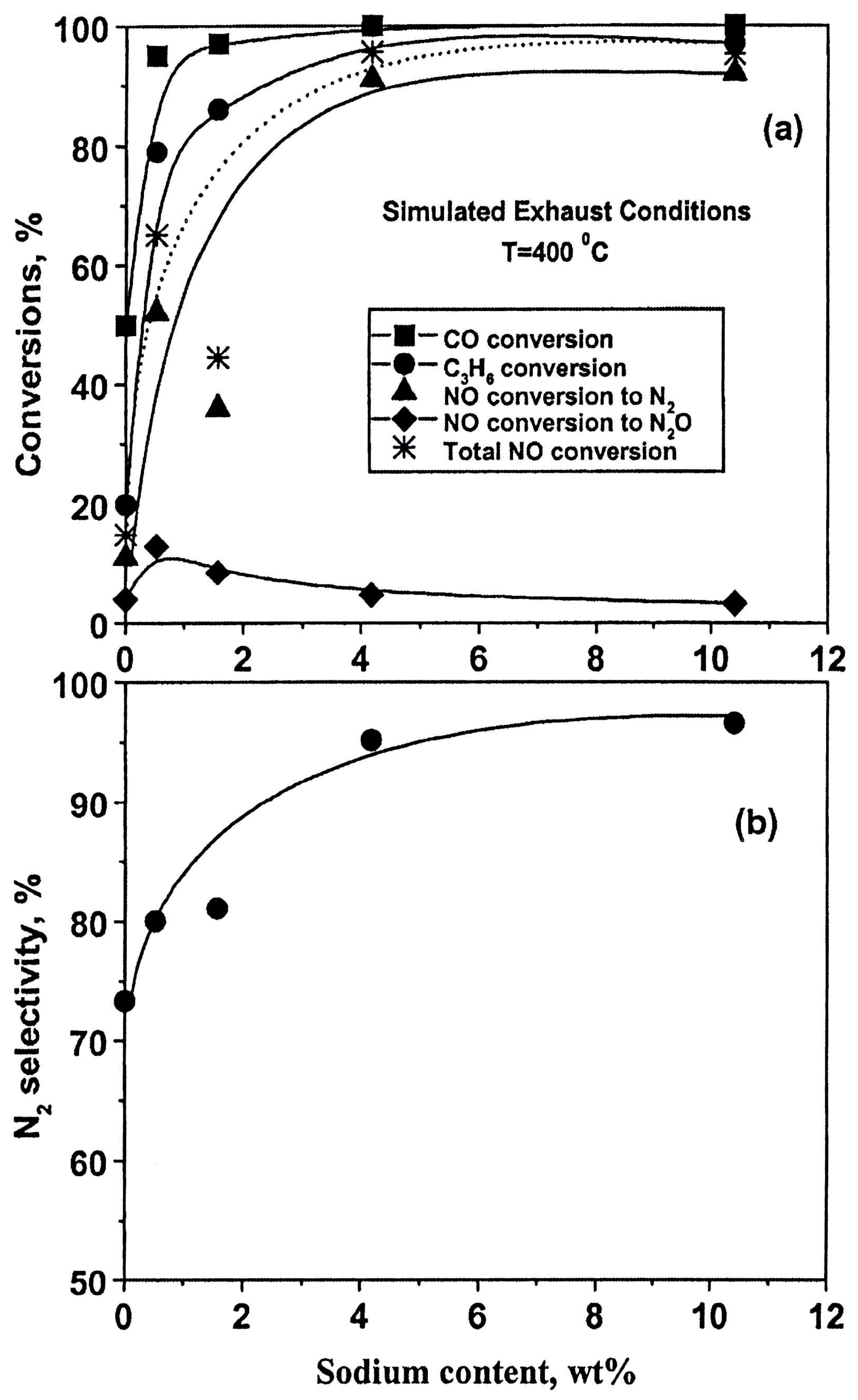
- Konsolakis, M.; Macleod, N.; Isaac, J.; Yentekakis, I.V.; Lambert, R.M. Strong promotion by Na of Pt/γ-Al2O3 catalysts operated under simulated exhaust conditions. J. Catal. 2000, 193, 330–337. [Google Scholar] [CrossRef]
- Macleod, N.; Isaac, J.; Lambert, R.M. Sodium promotion of Pd/γ-Al2O3 catalysts operated under simulated “three-way” conditions. J. Catal. 2001, 198, 128–135. [Google Scholar] [CrossRef]
- Macleod, N.; Isaac, J.; Lambert, R.M. A comparison of sodium-modified Rh/γ-Al2O3 and Pd/γ-Al2O3 catalysts operated under simulated TWC conditions. Appl. Catal. B 2001, 33, 335–343. [Google Scholar] [CrossRef]
- Tanaka, T.; Yokota, K.; Isomura, N.; Doi, H.; Sugiura, M. Effect of the addition of Mo and Na to Pt catalysts on the selective reduction of NO. Appl. Catal. B 1998, 16, 199–208. [Google Scholar] [CrossRef]
- Shinjoh, H.; Tanabe, T.; Sobukawa, H.; Sugiura, M. Effect of Ba addition on catalytic activity of Pt and Rh catalysts loaded on γ-alumina. Top. Catal. 2001, 16/17, 95–99. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yamada, T.; Kayano, K. Effect of basic metal additives on NOx reduction property of Pd-based three-way catalyst. Appl. Catal. B 2001, 30, 287–292. [Google Scholar] [CrossRef]
- Yentekakis, I.V.; Konsolakis, M.; Rapakousios, I.A.; Matsouka, V. Novel electropositively promoted monometallic (Pt-only) catalytic converters for automotive pollution control. Top. Catal. 2007, 42–43, 393–397. [Google Scholar] [CrossRef]
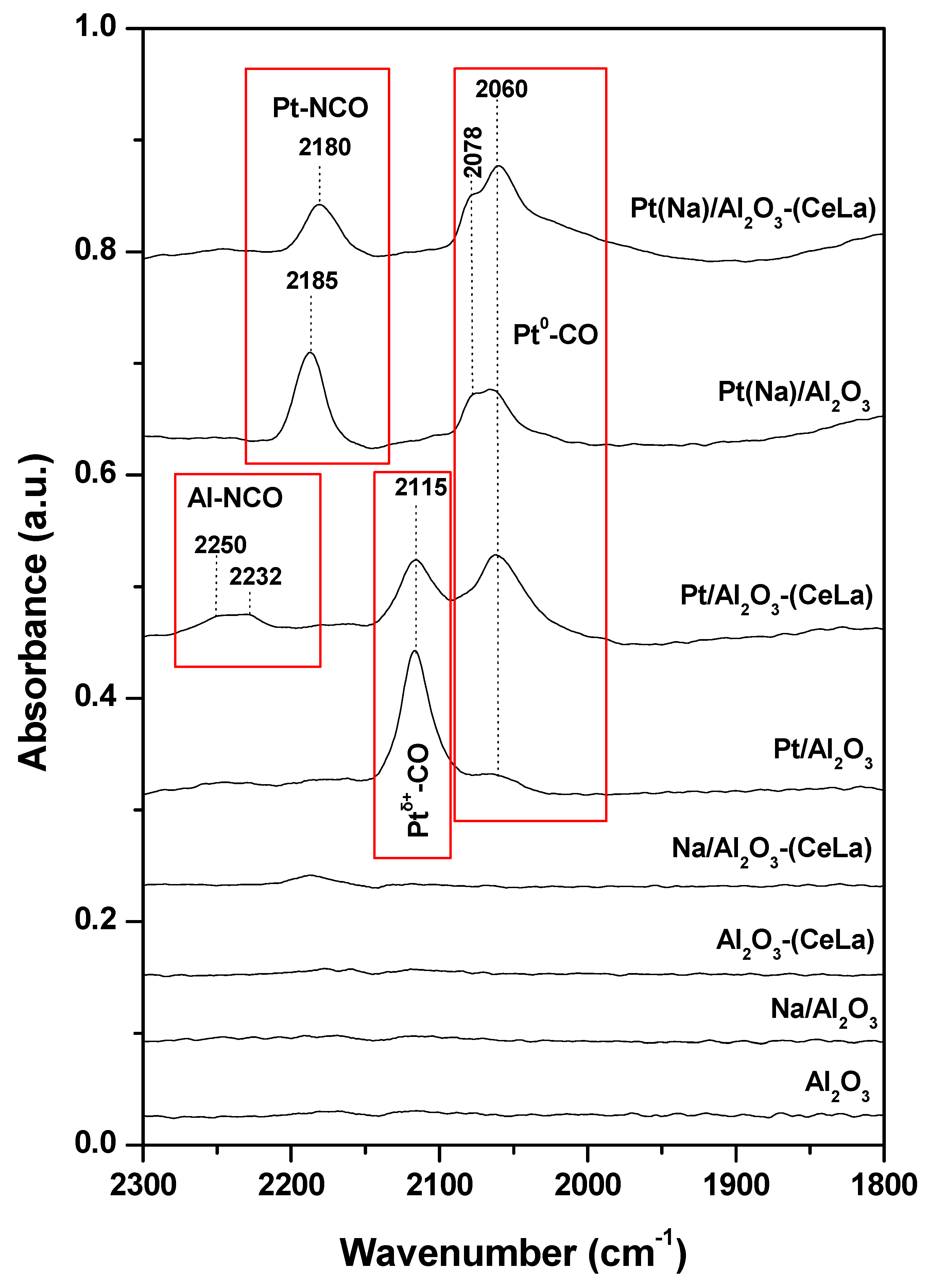
- Matsouka, V.; Konsolakis, M.; Lambert, R.M.; Yentekakis, I.V. In situ DRIFTS study of the effect of structure (CeO2-La2O3) and surface (Na) modifiers on the catalytic and surface behaviour of Pt/γ-Al2O3 catalyst under simulated exhaust conditions. Appl. Catal. B 2008, 84, 715–722. [Google Scholar] [CrossRef]
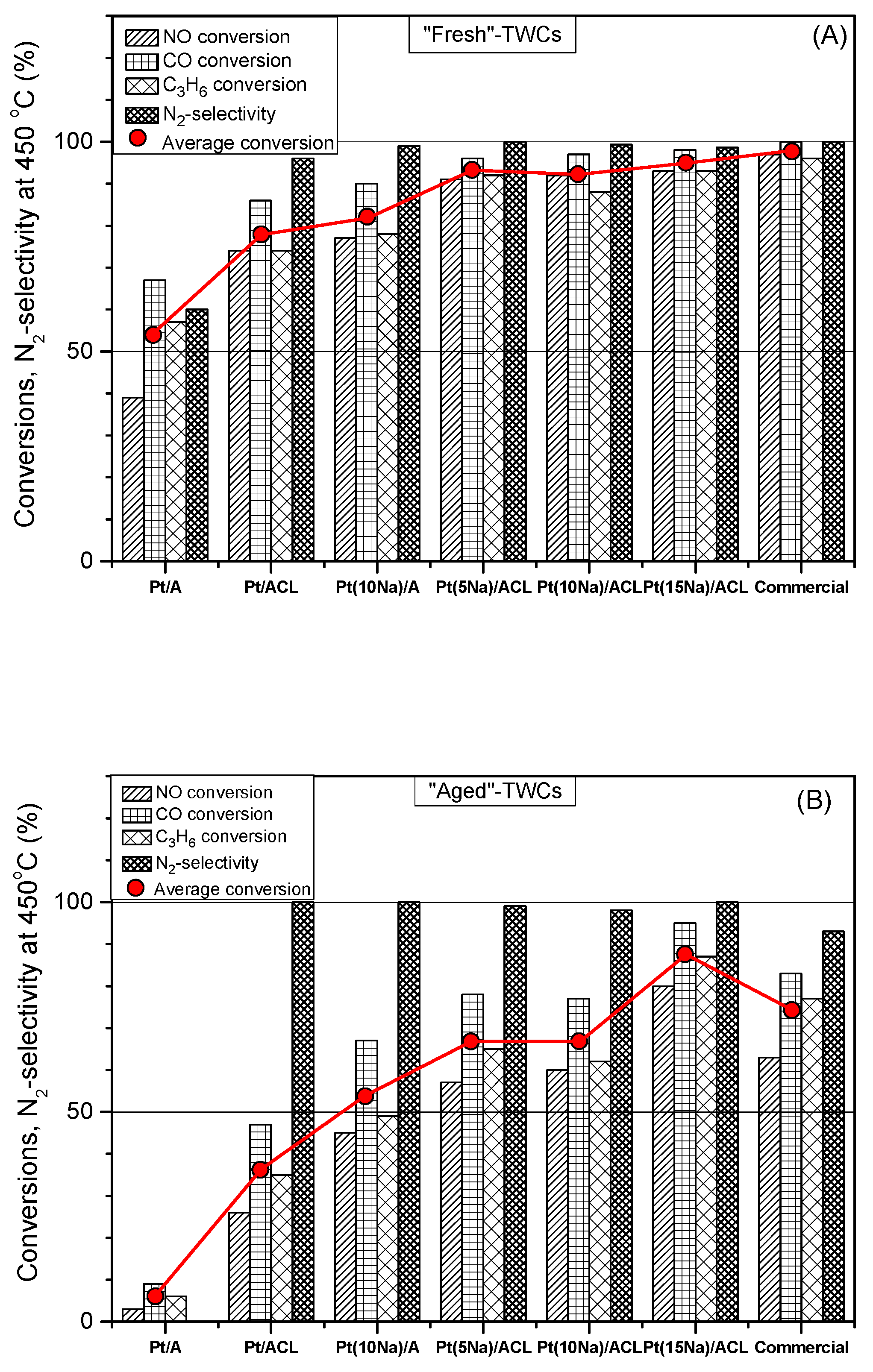
- Matsouka, V.; Konsolakis, M.; Yentekakis, I.V.; Papavasiliou, A.; Tsetsekou, A.; Boukos, N. Thermal aging behavior of Pt-only TWC converters under simulated exhaust conditions: Effect of rare earths (CeO2, La2O3) and alkali (Na) modifiers. Top. Catal. 2011, 54, 1124–1134. [Google Scholar] [CrossRef]
- Papavasiliou, A.; Tsetsekou, A.; Matsouka, V.; Konsolakis, M.; Yentekakis, I.V.; Boukos, N. Synergistic structural and surface promotion of monometallic (Pt) TWCs: Effectiveness and thermal aging tolerance. Appl. Catal. B 2011, 106, 228–241. [Google Scholar] [CrossRef]
- Vernoux, P.; Gaillard, F.; Lopez, C.; Siebert, E. Coupling catalysis to electrochemistry: A solution to selective reduction of nitrogen oxides in lean-burn engine exhausts? J. Catal. 2003, 217, 203–208. [Google Scholar] [CrossRef]
- Dorado, F.; de Lucas-Consuegra, A.; Jimenez, C.; Valverde, J.L. Influence of the reaction temperature on the elecrochemical promoted catalytic behaviour of platinum impregnated catalysts for the reduction of nitrogen oxides under lean burn conditions. Appl. Catal. A 2007, 321, 86–92. [Google Scholar] [CrossRef]
- Dorado, F.; de Lucas-Consuegra, A.; Vernoux, P.; Valverde, J.L. Electrochemical promotion of platinum impregnated catalyst for the selective catalytic reduction of NO by propene in presence of oxygen. Appl. Catal. B 2007, 73, 42–50. [Google Scholar] [CrossRef]
- De Lucas Consuegra, A.; Caravaca, A.; Dorado, F.; Valverde, J.L. Pt/K–βAl2O3 solid electrolyte cell as a “smart electrochemical catalyst” for the effective removal of NOx under wet reaction conditions. Catal. Today 2009, 146, 330–335. [Google Scholar] [CrossRef]
- De Lucas Consuegra, A.; Caravaca, A.; Sanchez, P.; Dorado, F.; Valverde, J.L. A new improvement of catalysis by solid-state electrochemistry: An electrochemically assisted NOx storage/reduction catalyst. J. Catal. 2008, 259, 54–65. [Google Scholar] [CrossRef]
- De Lucas Consuegra, A.; Caravaca, A.; Martin de Vidales, M.J.; Dorado, F.; Balomenou, S.; Tsiplakides, D.; Vernoux, P.; Valverde, J.L. An electrochemically assisted NOx storage/reduction catalyst operating under fixed lean burn conditions. Catal. Commun. 2009, 11, 247–251. [Google Scholar] [CrossRef]
- Burch, R.; Watling, T.C. The effect of promoters on Pt/Al2O3 catalysts for the reduction of NO by C3H6 under lean-burn conditions. Appl. Catal. B 1997, 11, 207–216. [Google Scholar] [CrossRef]
- Vernoux, P.; Leinekugel-Le-Cocq, A.-Y.; Gaillard, F. Effect of the addition of Na on Pt/Al2O3 catalysts for the reduction of NO by C3H8 and C3H6 under lean-burn conditions. J. Catal. 2003, 219, 247–257. [Google Scholar] [CrossRef]
- Wogerbauer, C.; Maciejewski, M.; Schubert, M.M.; Baiker, A. Effect of sodium on the catalytic properties of iridium black in the selective reduction of NOx by propene under lean-burn conditions. Catal. Lett. 2001, 74, 1–7. [Google Scholar] [CrossRef]
- Goula, G.; Katzourakis, P.; Vakakis, N.; Papadam, T.; Konsolakis, M.; Tikhov, M.; Yentekakis, I.V. The effect of potassium on the Ir/C3H6+NO+O2 catalytic system. Catal. Today 2007, 127, 199–206. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Koutsodontis, C.G. Non-Faradaic electrochemical activation of catalysis. J. Chem. Phys. 2008, 128, 182506–182513. [Google Scholar] [CrossRef]
- Lambert, R.M. Catalysis and Electrocatalysis at Nanoparticles; Wieckowski, A., Savinova, E., Vayenas, C.G., Eds.; Dekker: New York, NY, USA, 2003; p. 583. [Google Scholar]
- Lambert, R.M.; Palermo, A.; Williams, F.J.; Tikhov, M.S. Electrochemical promotion of catalytic reactions using alkali ion conductors. Solid State Ionics 2000, 136–137, 677–685. [Google Scholar] [CrossRef]
- Lang, N.D.; Holloway, S.; Norskov, J.K. Electrostatic adsorbate-adsorbate interactions: The poisoning and promotion of the molecular adsorption reaction. Surf. Sci. 1985, 150, 24–38. [Google Scholar] [CrossRef]
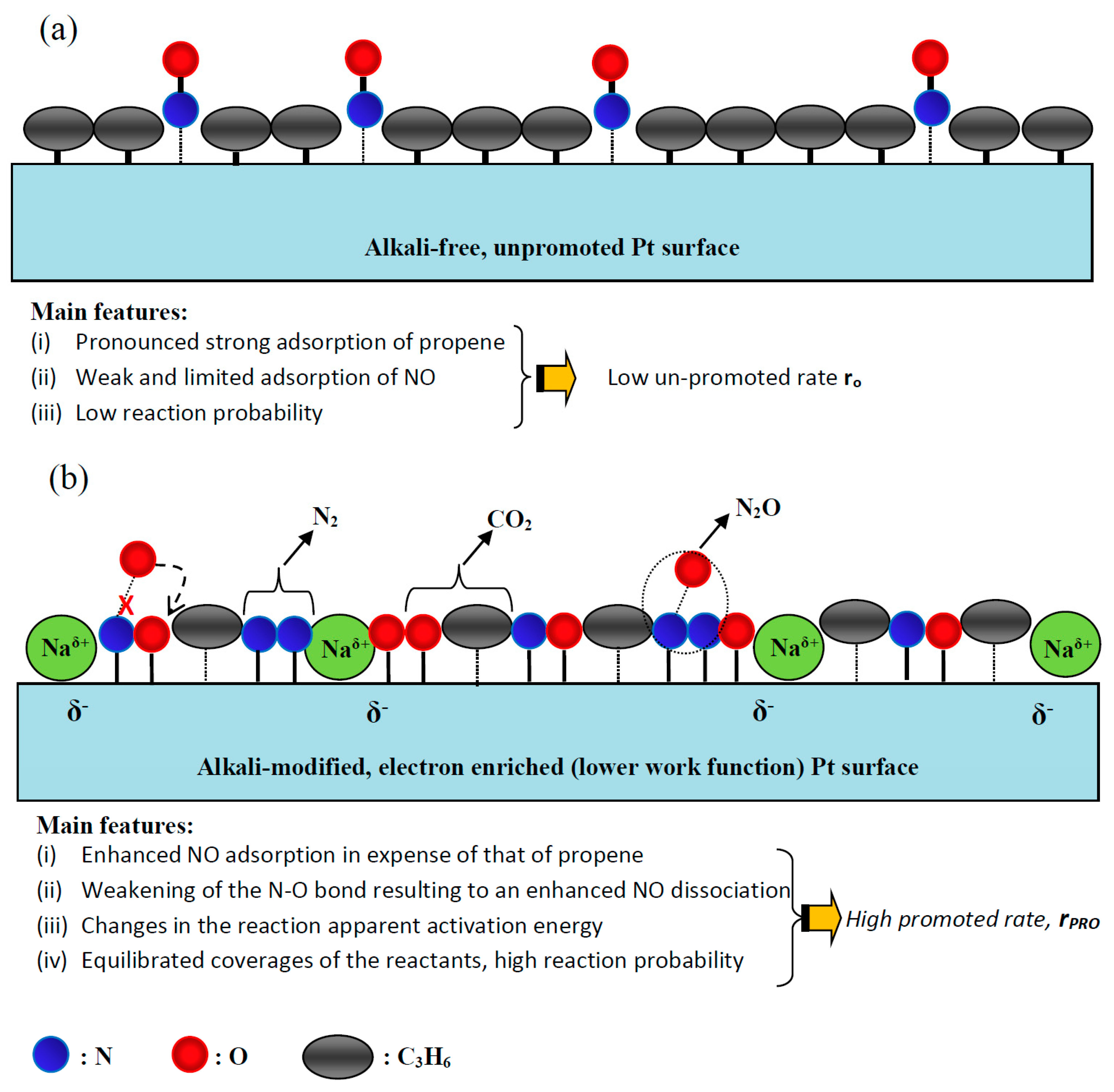
- Koukiou, S.; Konsolakis, M.; Lambert, R.M.; Yentekakis, I.V. Spectroscopic evidence for the mode of action of alkali promoters in Pt-catalyzed de-NOx chemistry. Appl. Catal. B 2007, 76, 101–106. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Brosda, S. Electron donation–backdonation and the rules of catalytic promotion. Top. Catal. 2014, 57, 1287–1301. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2778. [Google Scholar] [CrossRef]
- Harkness, I.R.; Lambert, R.M. Chemisorption and reactivity of nitric oxide on Na-dosed platinum{111}. J. Chem. Soc. Faraday Trans. 1997, 93, 1425–1429. [Google Scholar] [CrossRef]
- Garfunkel, E.L.; Maj, J.J.; Frost, J.C.; Farias, M.H.; Somorjai, G.A. Interaction of potassium with .pi.-electron orbital containing molecules on platinum(111). J. Phys. Chem. 1983, 87, 3629–3635. [Google Scholar] [CrossRef]
- Bertolini, J.C.; Derichele, P.; Massardier, J. The influence of potassium and the role of coadsorbed oxygen on the chemisorptive properties of Pt(100). Surf. Sci. 1985, 160, 531–541. [Google Scholar] [CrossRef]
- Pitchon, V.; Primet, M.; Praliaud, H. Alkali addition to silica-supported palladium: Infrared investigation of the carbon monoxide chemisorptions. Appl. Catal. 1990, 62, 317–334. [Google Scholar] [CrossRef]
- Liotta, F.L.; Martin, G.A.; Deganello, G. The influence of alkali metal ions in the chemisorption of CO and CO2 on supported palladium catalysts: A fourier transform infrared spectroscopic study. J. Catal. 1996, 164, 322–333. [Google Scholar] [CrossRef]
- Vayenas, C.G.; Archonta, D.; Tsiplakides, D. Scanning tunneling microscopy observation of the origin of electrochemical promotion and metal–support interactions. J. Electroanal. Chem. 2003, 554–555, 301–306. [Google Scholar] [CrossRef]
- Makri, M.; Vayenas, C.G.; Bebelis, S.; Besocke, K.H.; Gavalca, C. Atomic resolution STM imaging of electrochemically controlled reversible promoter dosing of catalysts. Surf. Sci. 1996, 369, 351–359. [Google Scholar] [CrossRef]
- Archonta, D.; Frantzis, A.; Tsiplakides, D.; Vayenas, C.G. STM observation of the origin of electrochemical promotion on metal catalyst-electrodes interfaced with YSZ and β″-Al2O3. Solid State Ionics 2006, 26–32, 2221–2225. [Google Scholar] [CrossRef]
- Gonzalez-Cobos, J.; de Lucas-Consuegra, A. A Review of surface analysis techniques for the investigation of the phenomenon of electrochemical promotion of catalysis with alkaline ionic conductors. Catalysts 2016, 6, 15. [Google Scholar] [CrossRef]

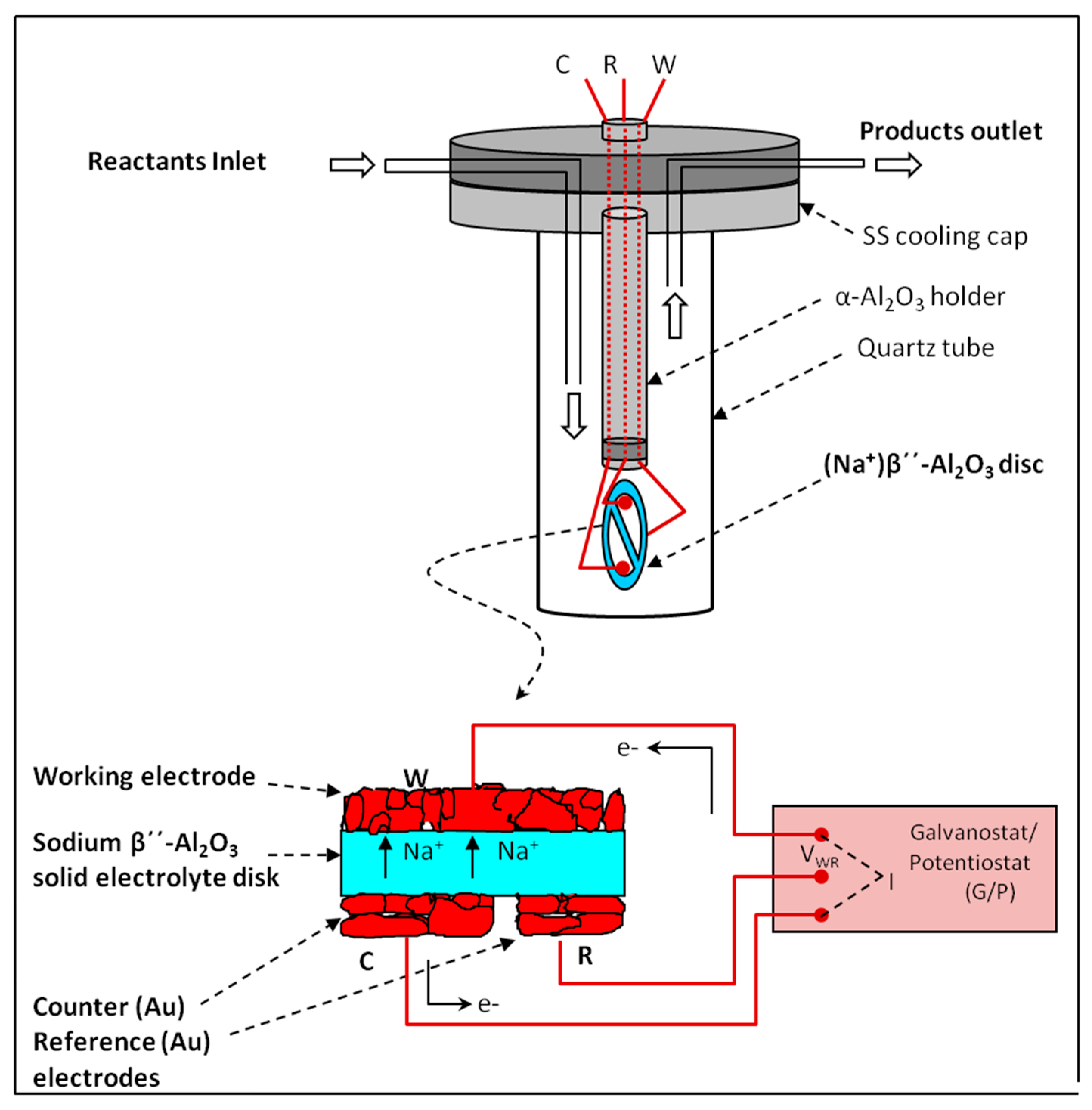











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactants | Catalyst, (promotion method applied) | Promoter | Reaction conditions | Promotion highlights and optimal achievements | Ref. |
|---|---|---|---|---|---|
| CO, O2 | Pt-film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 300–450 °C [CO] = 0–4% [O2] = 0–6% |
| [16] |
| CO, O2 | Pt-film over (K)β″Al2O3 (EPOC) | K | T = 200–350 °C [CO] = [O2] = 500 ppm |
| [27] |
| CO, O2 | 1 wt% PdO/SnO2 (CCP) | Na | T = 150 °C [CO]/[O] = 1.25 [CO]/[O] = 0.5 ([CO] = 2400 ppm) Na-loading: 0.1 wt% |
| [28] [29] |
| CO, O2 | 1 wt% Pd/γ-Al2O3 and 1 wt% Pd/CZ (CCP) | Ba | T = 50–250 °C [CO] = 2.2%, [O2] = 1.1% Ba-loadings: 0, 1, 5.5, 10, 15 wt% |
| [31] |
| CO, O2, H2 excess | 2 wt% Pt/(γ-Al2O3, SiO2, ZrO2, Nb2O5 or TiO2) (CCP) | Li, Na, K, Rb, Cs | T = 100–160 °C [CO] = [O2] = 0.2% Alkali loadings: A/Pt molar ratio = 0–20 |
| [32] [33] [34] [35] [36] |
| CO, O2, H2 excess | 1.6 wt% Pt/SiO2 (CCP) | Na, Rb, Cs | T = 110 °C [CO] = [O2] = 1%, [H2] = 70% Cs surface density = 0–6 atoms/nm2 |
| [37] |
| CO, O2, H2 excess | 1 wt% Pt/γ-Al2O3 1 wt% Pt-1.8wt%Co/γ-Al2O3 (CCP) | Na | T = 25–300 °C [CO] = 0.1%, [O2] = 0.1%, [H2] = 1% Na loading: 0.5–3 wt% Na |
| [38] |
| CO, O2, H2 excess | 2wt%Pt/γ-Al2O3 (CCP) | Mg | T = 100–250 °C [CO] = 1%, [O2] = 0.75%, [H2] = 65% [CO2] = 20%, [H2O] = 2% Mg loading = 3 wt% Mg |
| [39] |
| CO, O2, H2 excess | Pt film on (K)βAl2O3 solid electrolyte (EPOC) | K | T = 195 °C [CO] = 0.4%, [O2] = 0.2%, [H2] = 16% θK = 0–4% |
| [40] |
| Reactants | Catalyst and (Promotion method applied) | Promoter | Reaction conditions | Promotion highlights and optimal achievements | Ref. |
|---|---|---|---|---|---|
| Alkenes oxidation (Alkene + O2) | |||||
| C2H4, O2 | Pt-film over (Na)β″Al2O3 (EPOC) | Na | T = 291 °C; [C2H4] = 0.021%, [O2] = 5%; θNa = 0–3% |
| [42] |
| C2H4, O2 | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 300–470 °C; [C2H4] = 4.2%, [O2] = 8 and 16.2%; θNa = 0–35% |
| [18] |
| C3H6, O2 | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 340 °C [C3H6] = 0.62%, [O2] = 0–7% |
| [20] |
| C3H6, O2 | Pt film over NaSICon (Na3Zr2Si2PO12) solid electrolyte (EPOC) | Na | T = 300 °C, θNa = 0–6% - C3H6/O2 = 0.04%/0.2% (stoich.) - C3H6/O2 = 0.04%/8.3% (O2-rich) |
| [43] [44] |
| C3H6, O2 | Pt film over (K)βAl2O3 solid electrolyte (EPOC) | K | T=190–310 °C [C3H6] = 2000 ppm, [O2] = 1–7% |
| [45] [46] |
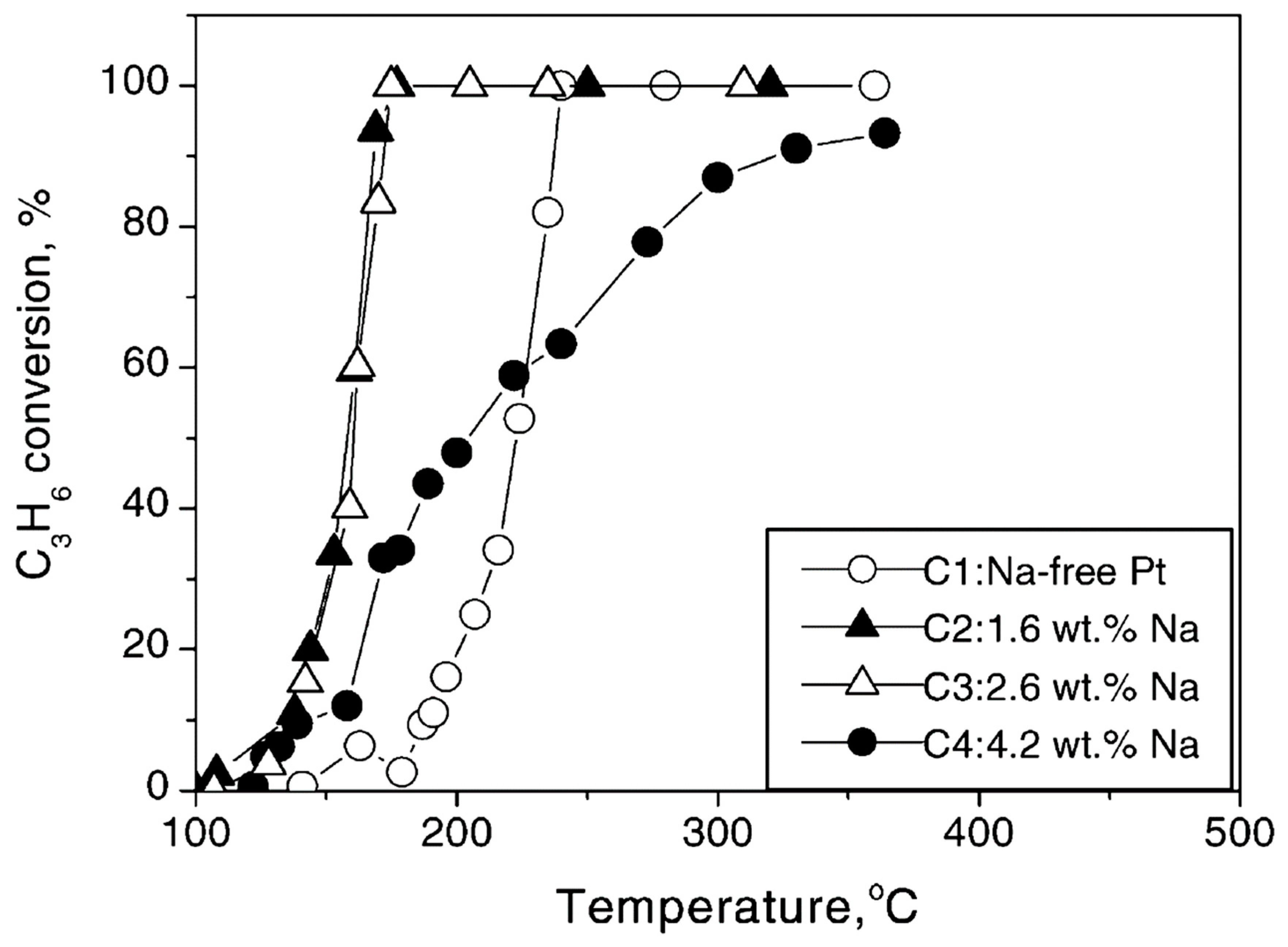
| C3H6, O2 | 0.5 wt% Pt/γ-Al2O3 (CCP) | Na | T = 100–500 °C [C3H6] = 1000 ppm, [O2] = 5% Na-loadings: 0, 1.6, 2.6, 4.2 wt% |
| [47] |
| Alkanes oxidation (alkane + O2) | |||||
| C3H8, O2 | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 320–440 °C [C3H8] = 0.2%, [O2] = 1% θNa = 0–3% |
| [48] [49] |
| CH4, O2 | 5 (or 2.5) wt% Pd/γ-Al2O3 (CCP) | Na, K, Ba, Ca | T = 250–600 °C; [CH4] = 500 ppm, [O2] = 8%, [H2O] = 5%, [CO2] = 5% Loadings: Ca: 0.38, 0.76, 1.5 wt%; Ba = 5 wt%; K = 1.48 wt%; Na = 0.87 wt% |
| [50] |
| Reactants | Catalyst and (Promotion method applied) | Promoter | Reaction conditions | Promotion highlights and maximum achievements | Ref. |
|---|---|---|---|---|---|
| NO, CO | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 300–400 °C [CO] = 0–1.5%, [NO] = 0–1.5% [NO]/[CO] = 0.75%/0.75% θNa = 3–20% |
| [17] |
| NO, CO | Rh film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 307 °C [CO] = [NO] = 1% |
| [51] [52] |
| NO, CO | 0.5 wt% Pt/γ-Al2O3 (CCP) | K, Rb, Cs | T = 150–500 °C [CO] = 0–3%, [NO] = 0–3% Promoter loadings used: wt% Rb: 1.9, 9.7, 15.5 wt% Cs: 9.0, 15.0, 24.0 wt% K: 2.7, 4.4, 8.8 |
| [53] |
| NO, CO | 0.5wt%Pd/YSZ (CCP) | Na | T = 352 °C [CO] = 0–4%, [NO] = 0–4% Na loadings: 0.017, 0.034, 0.068 and 0.102 wt% (or 6/1, 3/1, 1.5/1 and 1/1 Pd/Alkali) |
| [54] |
| NO, CO, O2 | 1 wt% Pd/(γ-Al2O3 or CZ) (CCP) | Ba | T = 50–300 °C [CO] = 2.2%, [NO] = 0.2% [O2] = 1% Ba-loadings: 0, 1, 5 and 15 wt% Ba |
| [55] |
| Reactants | Catalyst and (Promotion method applied) | Promoter | Reaction conditions | Promotion highlights and maximum achievements | Ref. |
|---|---|---|---|---|---|
| NO reduction by alkenes (NO + Alkene) | |||||
| NO, C2H4 | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 275–450 °C [NO] = 0.5–1%, [C2H4] = 1–3% θNa = 0–40% |
| [57] |
| NO, C3H6 | Pt film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 330–400 °C [NO] = 0–6%, [C3H6] = 0–0.4% θNa = 0–100% |
| [58] |
| NO, C3H6 | Rh film over (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 350 °C [NO] = 1–9%, [C3H6] = 1%, [O2] = 0–2% |
| [59] [60] |
| NO, C3H6 | 0.5 wt% Pd/YSZ (CCP) | Na, Li, K, Cs | T = 250–450 °C [NO] = 0–8%, [C3H6] = 0–8% Na loadings: 0–0.102 wt% (nominal θNa = 0–30%) |
| [14] [54] |
| NO, C3H6 | 0.5 wt% Pt/γ-Al2O3 (CCP) | Na, Li, K, Rb, Cs | T = 200–500 °C [NO] = 0–7%, [C3H6] = 0.3, 0.6% Alkali loadings: Li: 0–4.7 wt%; Na: 0–10.4 wt%; K: 0–8.8 wt%; Rb: 0–15.5 wt%; Cs: 0–24 wt%. |
| [15] [61] [21] [22] |
| NO, C3H6 | 0.5 wt% Pt/γ-Al2O3 (CCP) | Ba | T = 200–500 °C [NO] = 0–5%, [C3H6] = 0–2% Ba loadings: 0, 3.5, 9.7, 15.2, 22.3wt% |
| [62] [63] |
| NO, C3H6 | 0.5 wt% Rh/γ-Al2O3 (CCP) | Na | T = 200–550 °C [NO] = 0–1%, [C3H6] = 0.1% Na loadings: 0, 1.8, 3.0, 7.3, 11.3 wt% |
| [64] |
| NO reduction by alkanes (NO + Alkane) | |||||
| NO, CH4 | 0.5 wt% Pd/YSZ (CCP) | Na | T = 350–450 °C [NO] = 0–1.5%, [CH4] = 0–20% Na loadings: 0, 0.017, 0.034, 0.068, 0.102 wt% |
| [65] [66] |
| NO reduction by H2 and/or CO (NO + H2, NO + H2 + CO) in the absence or presence of O2 | |||||
| NO, H2 | Pt film on (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 300–450 °C [NO] = 0–1.6%, [H2] = 0–1% θNa = 0–12% |
| [19] |
| NO, H2, O2 | 1 wt% Pt/γ-Al2O3 1 wt% Pt/SiO2 (CCP) | Na (Mo) | T = 50–200 °C [NO] = 0.1%, [H2] = 0.4%, [O2] = 6% Na2O-laodings: 0, 0.27, 1, 5 and 10 wt% |
| [73] |
| NO, H2, O2 | 1 wt% Pt/ZSM-5 (CCP) | Na, K, Cs, Mg, Ca, Ba | T = 40–110 °C [NO] = 0.08%, [H2] = 0.08–0.56%, [O2] = 10% O2 Na loadings: 0, 3, 5, 10, 15 and 20 wt% |
| [74] |
| NO, H2, CO, O2 | 0.5 wt% Pd/γ-Al2O3 0.5 wt% Pd/γ-Al2O3-TiO2 (CCP) | K | T = 50–400 °C [NO] = 0.1%, [CO] = 0.25%, [H2] = 0.75%, [O2] = 6% O2 K loadings: 0, 0.1, 0.25, 0.5, 1.0 and 3.0 wt% K |
| [79] |
| Direct NO decomposition | |||||
| NO | (0.6–10 wt%)Pd/γ-Al2O3 (CCP) | Na | T = 550–900 °C [NO] = 4% in He Na loadings (as NaOH): 0, 1.3, 4.3, 6.5 and 13 wt% NaOH |
| [80] |
| Reactants | Catalyst and (Promotion method applied) | Promoter | Reaction conditions | Promotion highlights and maximum achievements | Ref. |
|---|---|---|---|---|---|
| N2O | 0.1 wt %Rh/γ-Al2O3 (CCP) | Li, Na, K, Cs | T = 250–450 °C [N2O] = 1% Alkali loadings (as alkali oxides): 0.033, 0.066, 0.078, 0.099 wt% |
| [81] |
| N2O, presence or absence of O2 | 0.5 wt% Rh/γ-Al2O3 (CCP) | Mg, Sr | T = 250–450 °C [N2O] = 0.1%; [N2O] = 0.1% + [O2] = 5%. Mg- or Sr-loading: 5 wt% |
| [82] |
| N2O, presence or absence of O2, CO, H2O | 0.5 wt% Pt/Al2O3-(CeO2-La2O3) (CCP) | K | T = 200–600 °C [N2O] = 0.1%; [N2O] = 0.1% + [O2] = 2%; [N2O] = 0.1% + [CO] = 1%; [N2O] = 0.1% + [CO] = 1% + [O2] = 2%. K-loadings: 0, 0.5, 1.0 and 2.0 wt% |
| [83] |
| N2O presence or absence of O2 | 0.5 wt% Ir/γ-Al2O3 (CCP) | K | T = 300–600 °C [N2O] = 0.1%; [N2O] = 0.1% + [O2] = 2%. K-loadings: 0, 0.25, 0.5 and 1.0wt% |
| [84] |
| N2O, NO | (0.1 and 0.5 wt%)Pt/(ROX 0.8) (CCP) | K | T = 50–500 °C [NO] = 0.1%; [N2O] = 0.05%; and [N2O] = 0.05% + [NO] = 0.1% K-loading: 3 and 5 wt% |
| [85] |
| N2O + CH4, N2O + C3H8, N2O + C3H6, presence or absence of O2 | 2 wt% Pd/γ-Al2O3,and Pd film on (K)β″Al2O3 solid electrolyte (CCP vs EPOC) | K | T=100–500 °C [N2O] = 0.15% [CH4] = 0.6% [C3H8] = 0.2% [C2H6] = 0.2% ([O2] = 3%) K-loadings: 0, 2.5, 4.5, 7.0, 9.0 wt% |
| [25] [86] |
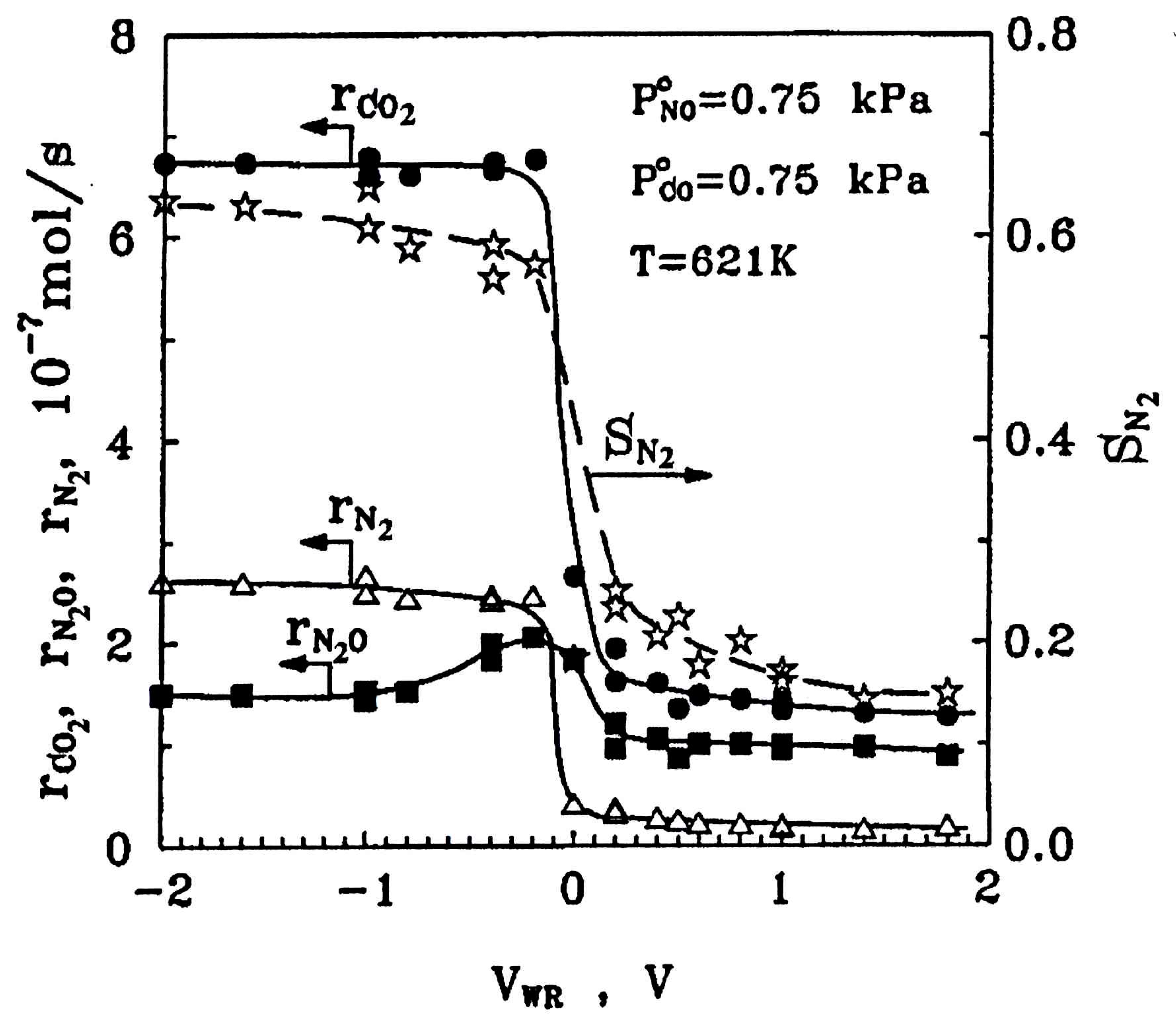
| N2O, C3H6, O2 | Pt film on (K)β″Al2O3 solid electrolyte (EPOC) | K | T = 180–580 °C [N2O] = 0.1%, [C2H6] = 0.2%, [O2] = 0.2–1.0% θK = 0–90% |
| [87] [88] |
| Reactants | Catalyst and (promotion method applied) | promoter | Conditions | Promotion highlights and achievements | Ref. |
|---|---|---|---|---|---|
| NO, C3H6, CO, O2 | 0.5wt%Pt/γ-Al2O3 (CCP) | Na | T = 200–500 °C (at simulated TWC conditions) [NO] = 1000 ppm, [CO] = 7000 ppm, [C3H6] = 1067 ppm, [O2] = 7800 ppm; (at S = SP = 1) w/F = 6 × 10−3 g∙s/cm3 Na loadings: 0, 0.52, 1.57, 4.18 and 10.4 wt% Na |
| [89] |
| NO, C3H6, CO, O2 | 0.5 wt% Pd/γ-Al2O3 and 0.5 wt% Rh/γ-Al2O3 (CCP) | Na | T = 100–340 °C (at simulated TWC conditions) [NO] = 1000 ppm, [CO] = 7000 ppm, [C3H6] = 1067 ppm, 6970 ≤ [O2] ≤ 8630 ppm (around SP: 0.9≤ S ≤1.1) w/F = 15 × 10−3 g∙s/cm3 Na loadings for Pd/Al2O3: 0, 1.8, 3.5, 7.0, 12.0 wt% Na loadings for Rh/Al2O3: 0, 3.0 and 7.3 wt%. |
| [90] [91] |
| NO, C3H6, CO, O2, H2, CO2, H2O | 1.67 wt% Pt/12.3wt% MoO3-SiO2 (CCP) | Na2O | T = 100–600 °C w/F = 9.91 × 10−3 g∙s/cm3 Na2O loadings: 0.05, 0.1, 1.0 wt%. Simulated oxidizing feed, S = 9.91:4.3% O2, 0.12% NO, 800ppm C3H6, 0.12% CO, 400ppm H2, 12.3% CO2, 3% H2O. Simulated exhaust feed, S-scan: 0.40–1.21%O2, 0.12% NO, 490–62 ppm C3H6, 0.45–1.50% CO, 0.15–0.50% H2, 10.0% CO2, 3.0% H2O. |
| [92] |
| NO, C3H6, CO, O2, H2, CO2, H2O | 1.67 wt% Pt/γ-Al2O3 0.9 wt% Rh/γ-Al2O3 (CCP) | Ba | T = 100–400 °C (at simulated TWC conditions) [NO] = 1000 ppm, [C3H6] = 334 ppm, [CO] = 1%, [H2] = 0.3%, [CO2] = 12%, [H2O] = 3%, [O2] = 0.75% GHSV = 200000 h−1 Ba-loading: 5.8, 11.5, 23 and 46 wt% Ba |
| [93] |
| Exhaust gas from an automotive engine | 5wt% Pd-commercial TWC catalyst supplied by N.E. ChemCat Corp. (CCP) | Ba, Sr, La | T=300–600 °C Real stoichiometric automotive engine exhaust gas GHSV = 68000 h−1 Ba, Sr od La-loading: 5wt% |
| [94] |
| C3H6, CO, H2, O2, CO2, H2O (NO-free) | 0.4 wt% Pt/γ-Al2O3 (CCP) | Na, K | T = 150–450 °C (simulated two-stroke motorcycle emissions). [CO] = 1–4.14%, [C3H6] = 0.08–0.7%, [O2] = 0.96–0.9%, [H2] = 0.2%, [CO2] = 10%, [H2O] = 0 or 10%. Stoichiometric numbers studied S = 1, 0.31, 0.17 Na-loading: 4.5 wt%; K-loading: 7.6 wt% |
| [30] |
| NO, C3H6, CO, O2, CO2, H2O | 0.1 wt% Pt/(γ-Al2O3-CeO2-La2O3-cordierite monolith) versus a Commercial 0.37Pt/0.08Rh-TWC(CCP) | Na | T = 150–500 °C (simulated TWC conditions) [NO] = 0.1%, [CO] = 0.7%, [C3H6] = 0.1067%, [O2] = 0.78%, [H2O] = 10%, [CO2] = 10% GHSV = 50500 h−1 Na-loading: 1 and 2 wt% Na (i.e., 5 and 10 wt% in the washcoat) |
| [95] [96] [97] [98] |
| reactants | Catalyst and (promotion method applied) | Promoter | Conditions | Promotion highlights and achievements | Ref. |
|---|---|---|---|---|---|
| NO, C3H6, O2 excess | Pt film on NASICON solid electrolyte (EPOC) | Na | T = 200–400 °C [NO] = 0.2%, [C3H6] = 0.2%, [O2] = 5% Na coverage: non estimated (catalyst potential VWR is given) |
| [99] |
| NO, C3H6, O2 excess | Pt impregnated on (Na)β″Al2O3 solid electrolyte (EPOC) | Na | T = 220–300 °C [NO] = 0.2%, [C3H6] = 0.2%, [O2] = 0.5, 1 and 5%. θNa = 0–8% |
| [100] [101] |
| NO, C3H6, O2 excess, H2O | Pt impregnated on (K)βAl2O3 solid electrolyte (EPOC) | K | T = 180–400 °C [NO] = 0.2%, [C3H6] = 0.2%, [O2] = 5%, [H2O] = 5% θK: varying, non-estimated |
| [102] |
| NO, C3H6, O2 excess | Pt film on (K)βAl2O3 solid electrolyte (EPOC) | K | T = 250–370 °C [NO] = 0.1%, [C3H6] = 0.1%, [O2] = 5% θK: varying, non-estimated |
| [103] [104] |
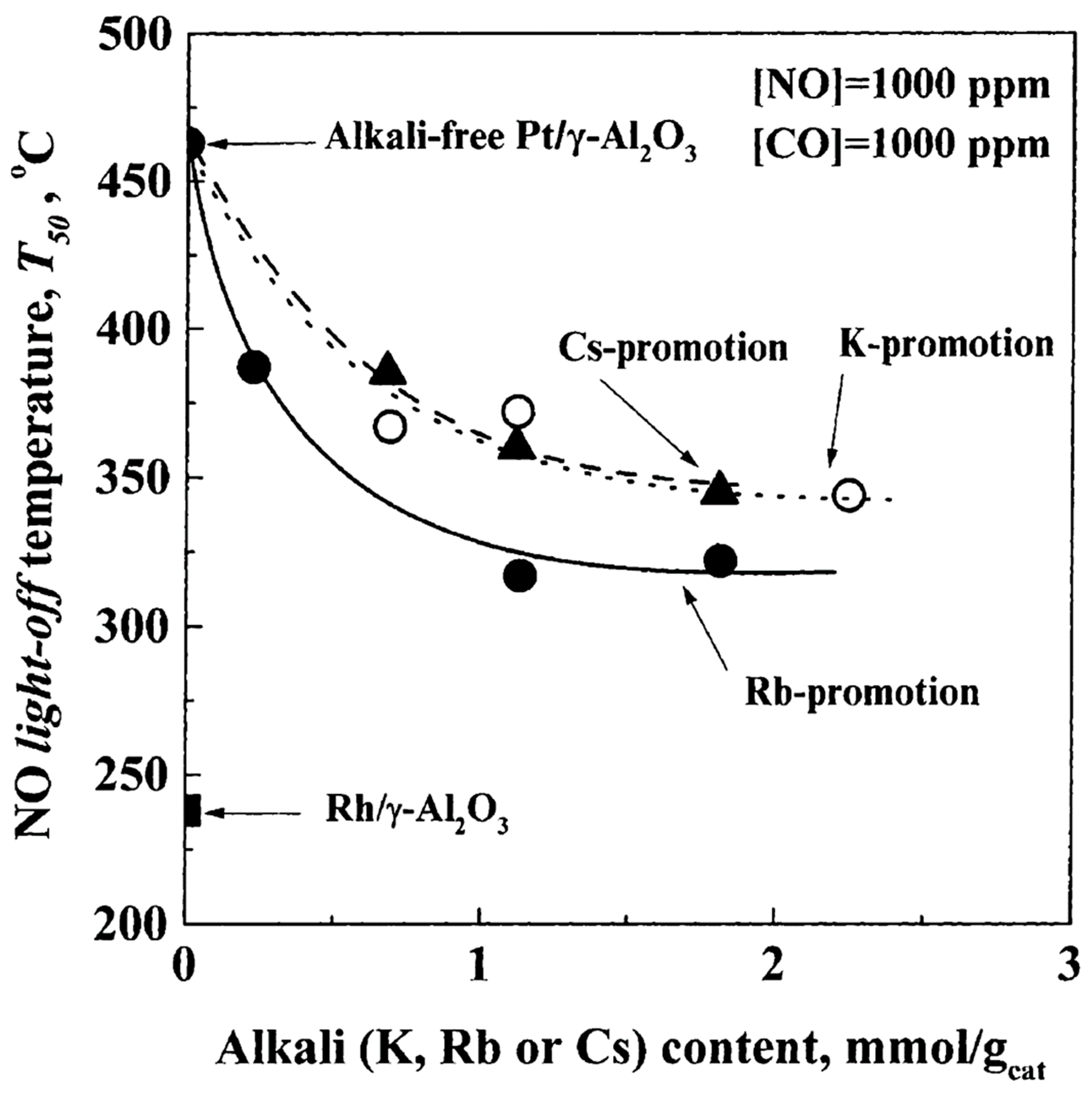
| NO, C3H6, O2 excess | 1 wt% Pt/γ-Al2O3 (CCP) | K, Cs, La, Mg, Ba | T = 200–500 °C [NO] = 0.1%, [C3H6] = 0.1%, [O2] = 5% Alk-loadings: A/Pt = 10/1 molar ratio |
| [105] |
| NO, C3H6 or C3H8, O2 excess | 0.9 wt% Pt/γ-Al2O3 (CCP) | Na | T = 200–650 °C [NO] = 0.2%, [HC] = 0.2% (HC: C3H6 or C3H8), [O2] = 5% GHSV = 20,000h−1 Na-loadings: 0.12, 1 and 5 wt% |
| [106] |
| NO, C3H6, O2 excess | 0.5 wt% Pt/γ-Al2O3 (CCP) | Na | T = 200–450 °C [NO] = 0.1%, [C3H6] = 0.1%, [O2] = 5% Na-loadings: 0, 1.6, 2.6 and 4.2 wt% |
| [47] |
| NO, C3H6, CO, H2O, CO2, O2 excess | Ir-black (CCP) | Na | T = 150–450 °C [NO] = 300 ppm, [C3H6] = 1800 ppm, [CO] = 450 ppm, [O2] = 8%, [H2O] = 10%, [CO2] = 10.7% Na-loadings: 0–10wt% |
| [107] |
| NO, C3H6, (O2) | Ir film on (K)β″Al2O3 solid electrolyte (EPOC) | K | T = 250–400 °C [NO] = 0.2%, [C3H6] = 0.2%, [O2] = 0–5% θK = 0–100% |
| [108] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yentekakis, I.V.; Vernoux, P.; Goula, G.; Caravaca, A. Electropositive Promotion by Alkalis or Alkaline Earths of Pt-Group Metals in Emissions Control Catalysis: A Status Report. Catalysts 2019, 9, 157. https://doi.org/10.3390/catal9020157
Yentekakis IV, Vernoux P, Goula G, Caravaca A. Electropositive Promotion by Alkalis or Alkaline Earths of Pt-Group Metals in Emissions Control Catalysis: A Status Report. Catalysts. 2019; 9(2):157. https://doi.org/10.3390/catal9020157
Chicago/Turabian StyleYentekakis, Ioannis V., Philippe Vernoux, Grammatiki Goula, and Angel Caravaca. 2019. "Electropositive Promotion by Alkalis or Alkaline Earths of Pt-Group Metals in Emissions Control Catalysis: A Status Report" Catalysts 9, no. 2: 157. https://doi.org/10.3390/catal9020157