Low-Temperature Electrocatalytic Conversion of CO2 to Liquid Fuels: Effect of the Cu Particle Size

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
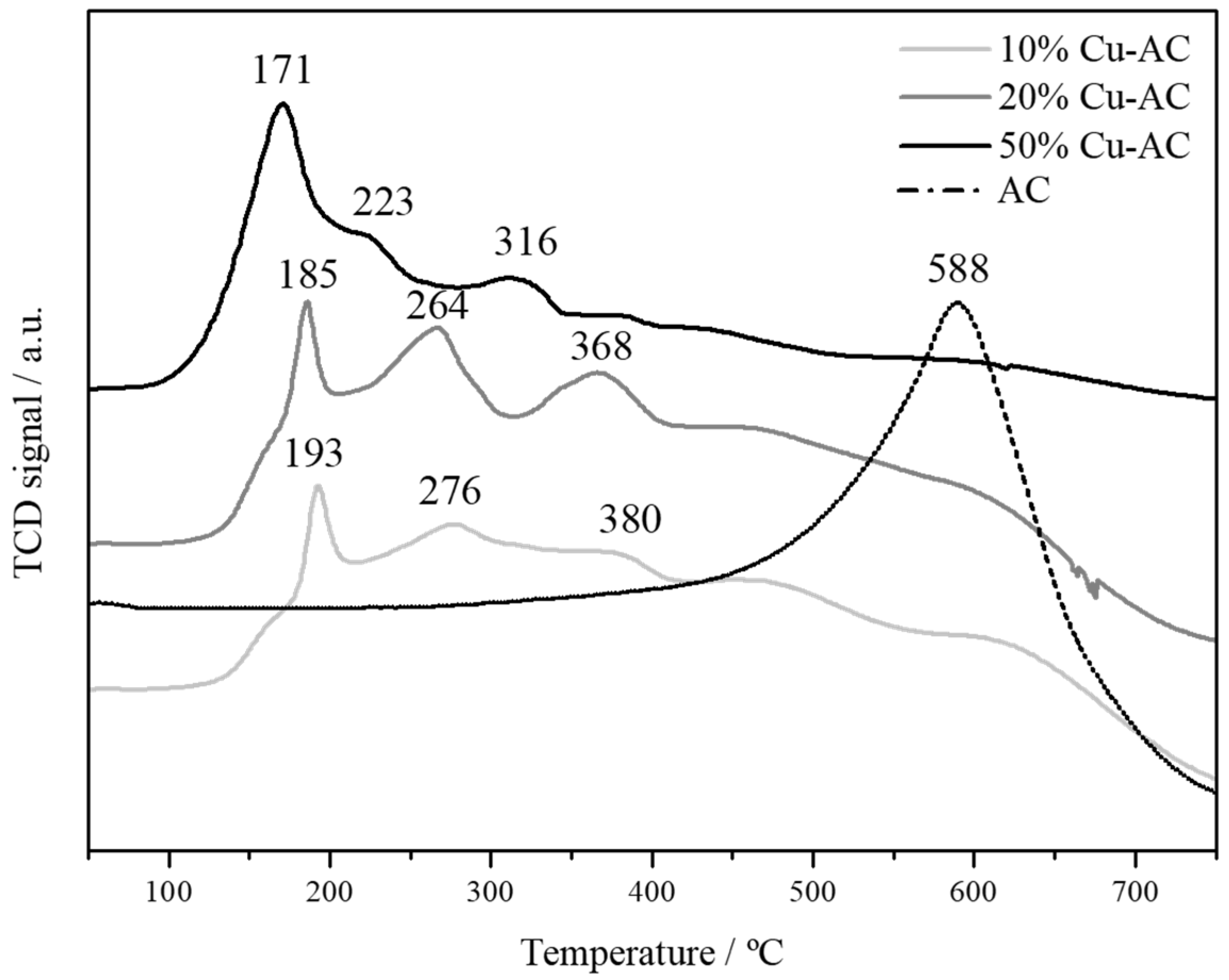
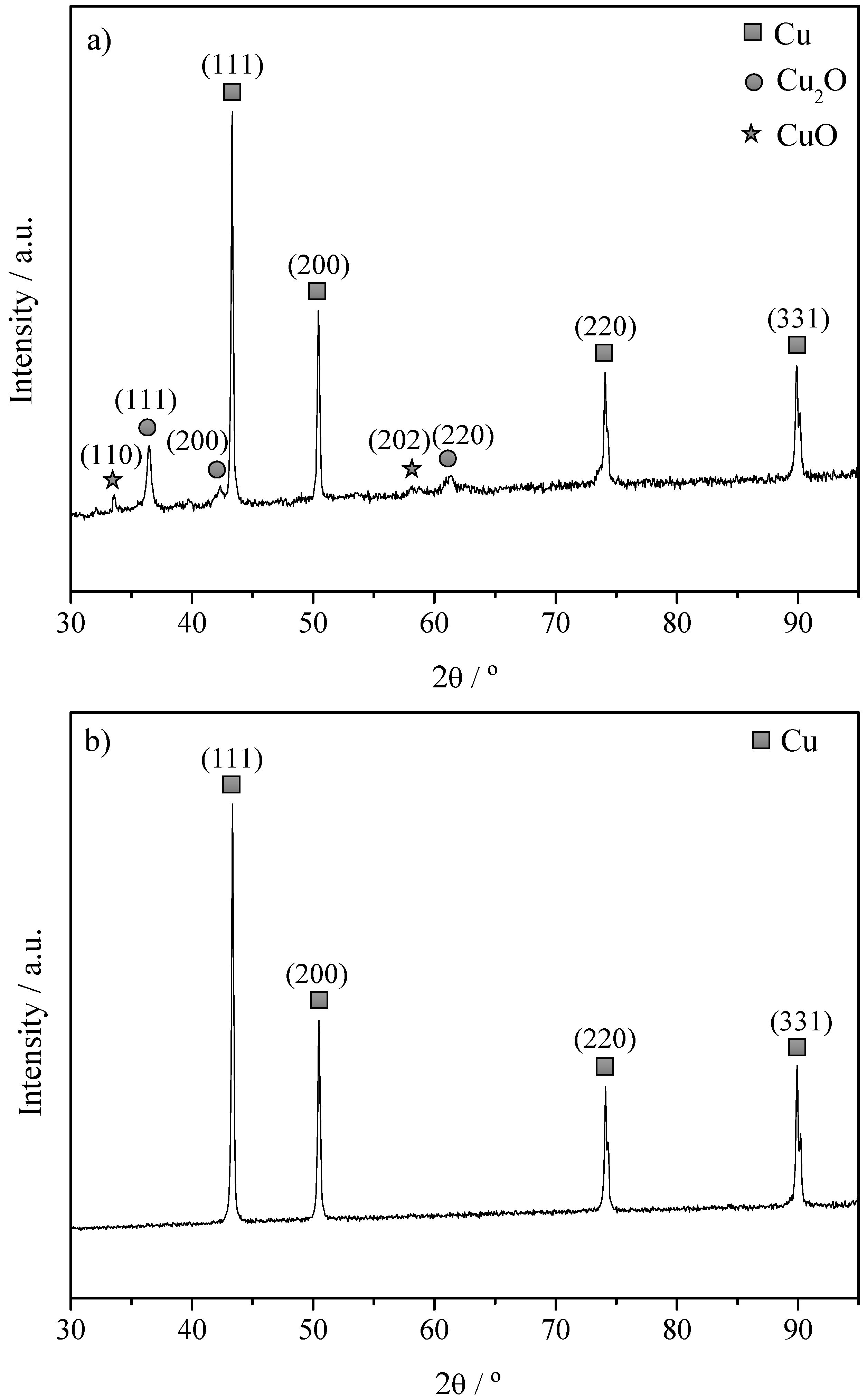
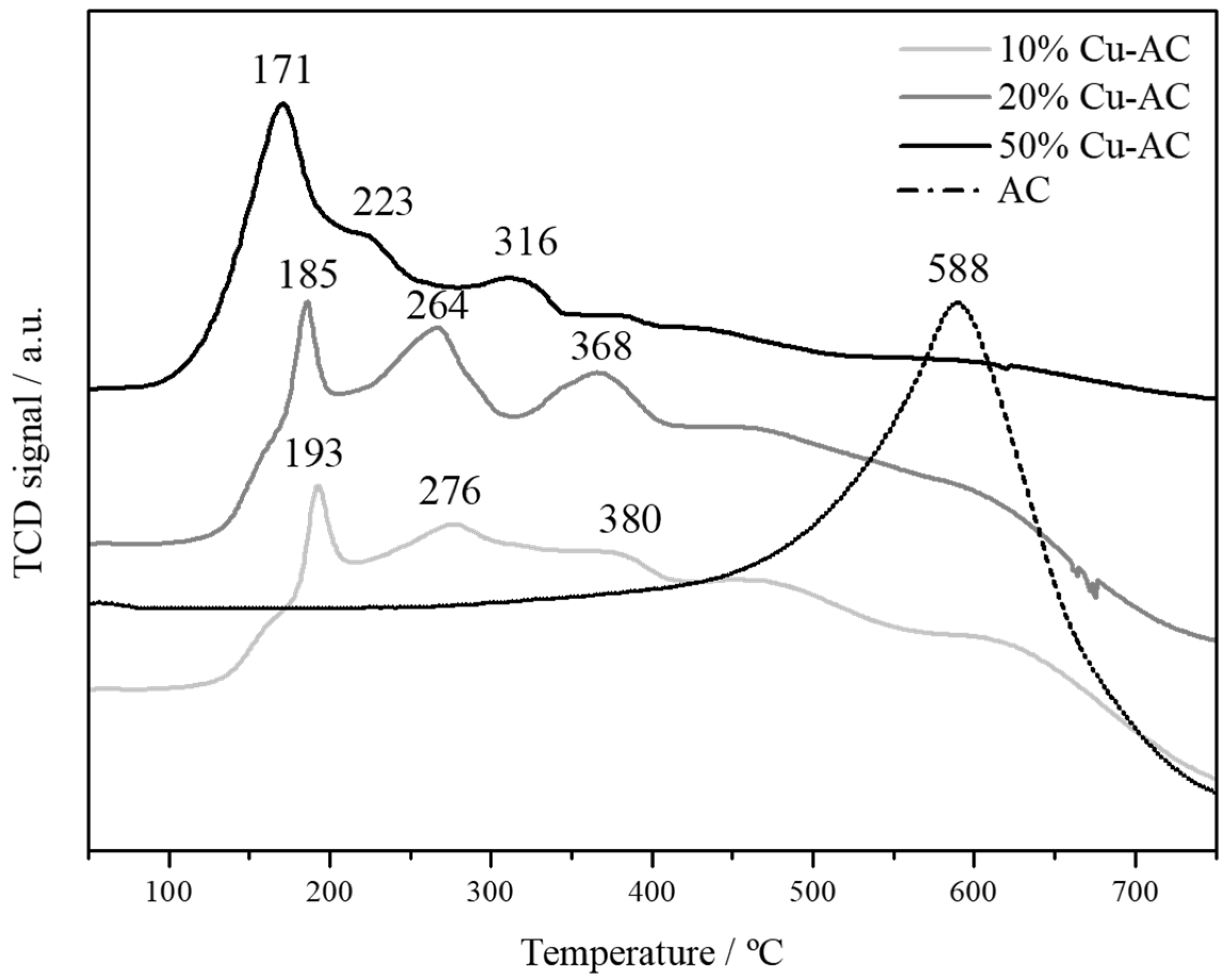
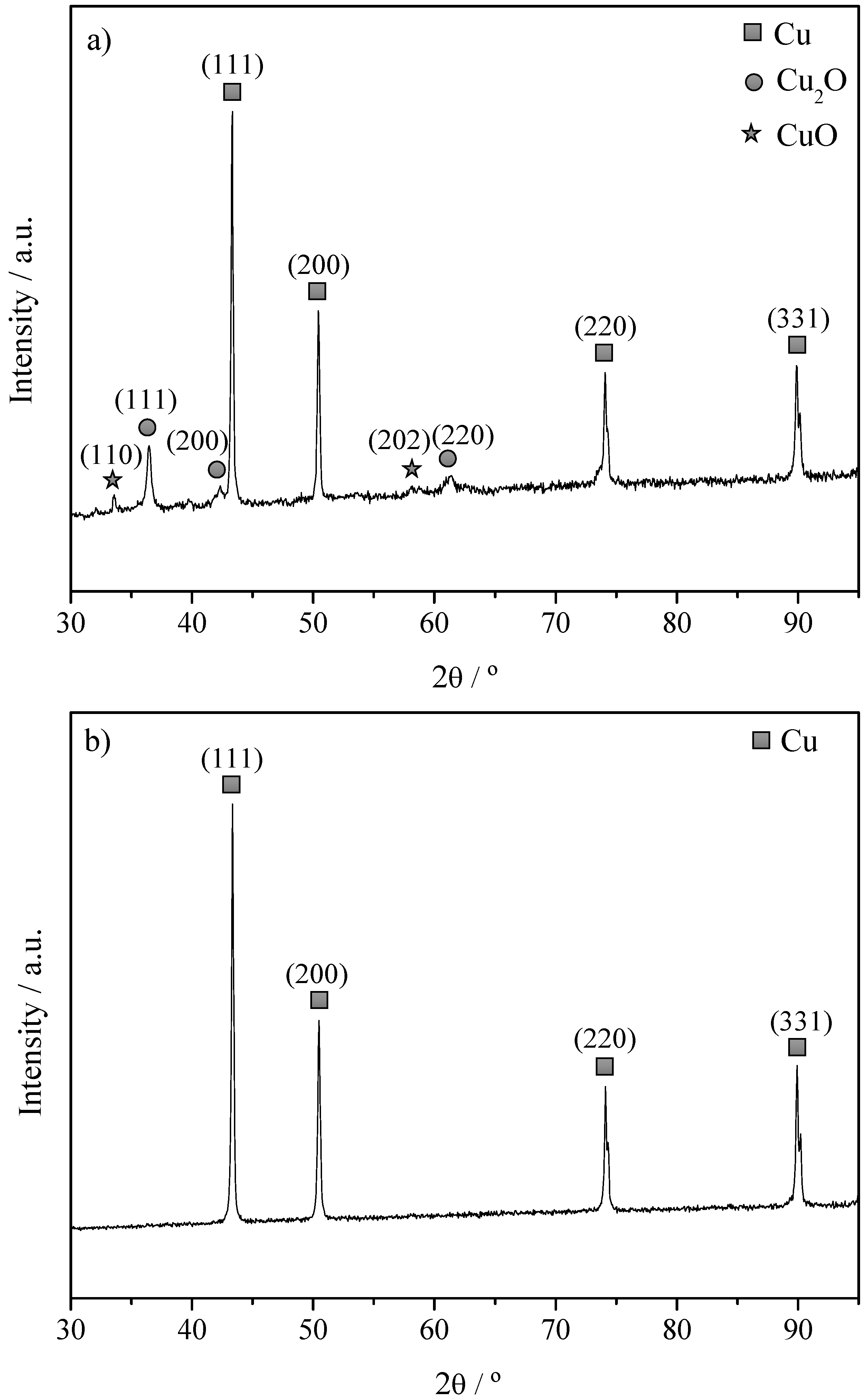
2.1. Characterization of The Cu Powder Catalysts and The Cu Electrodes
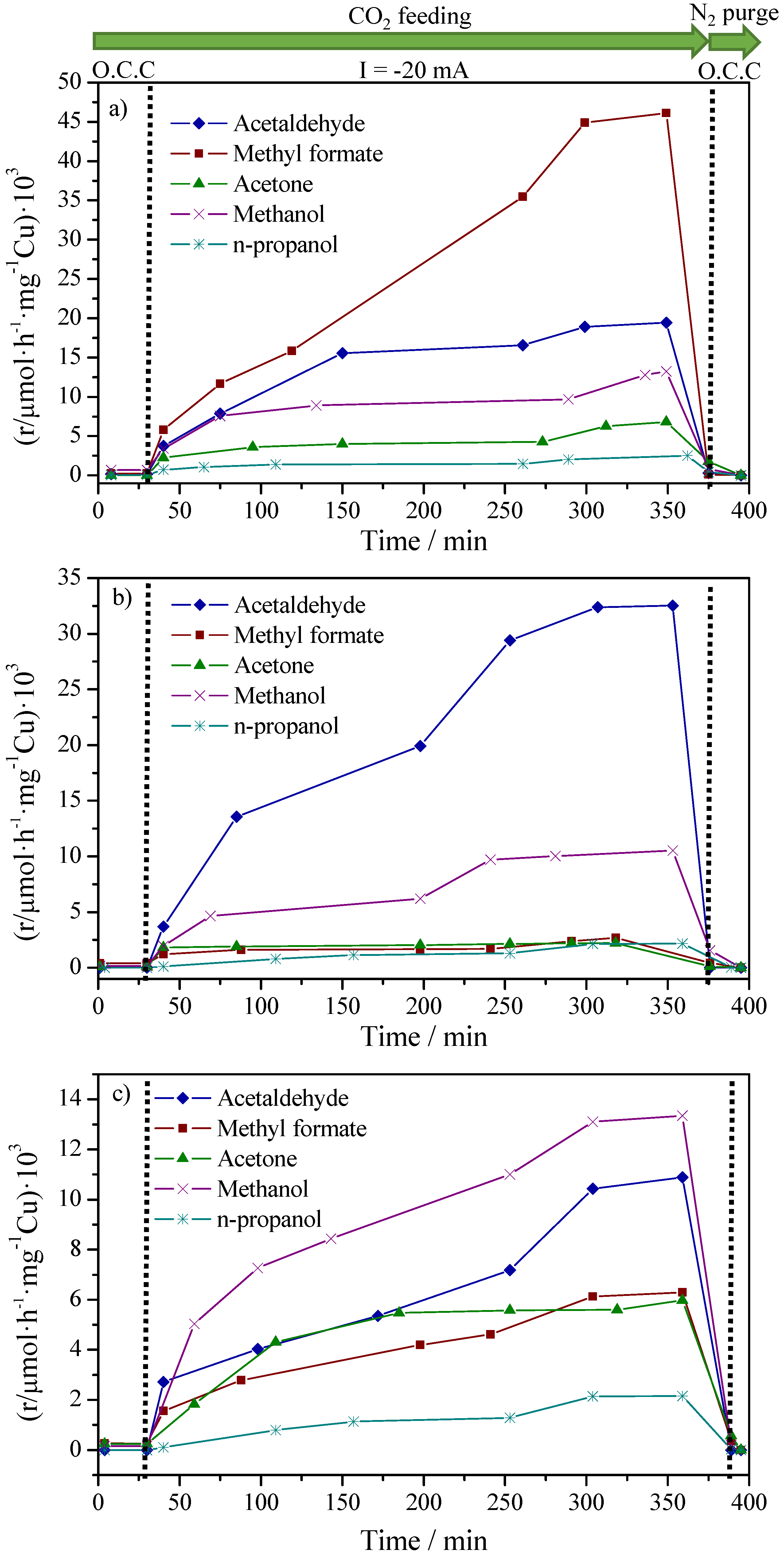
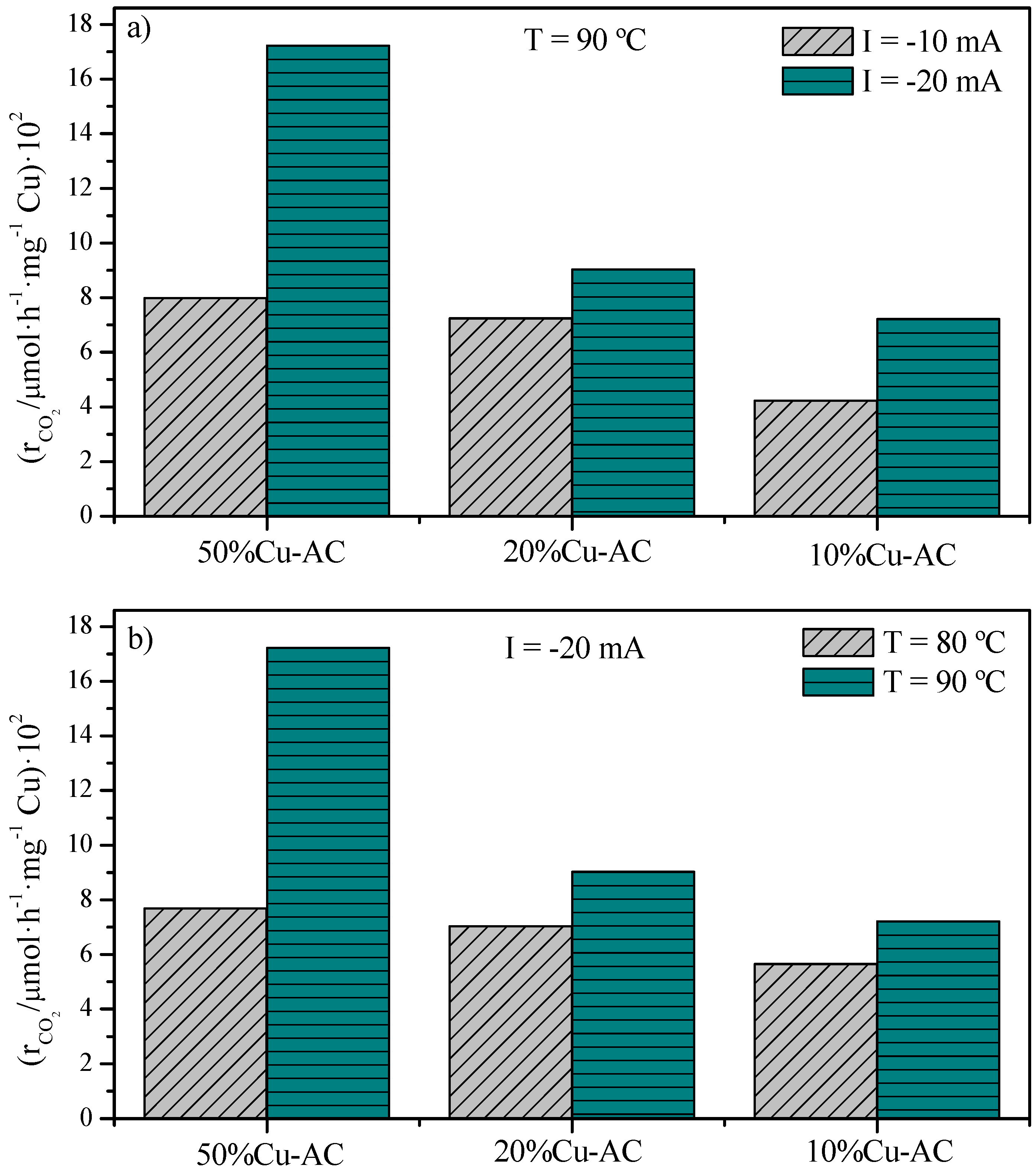
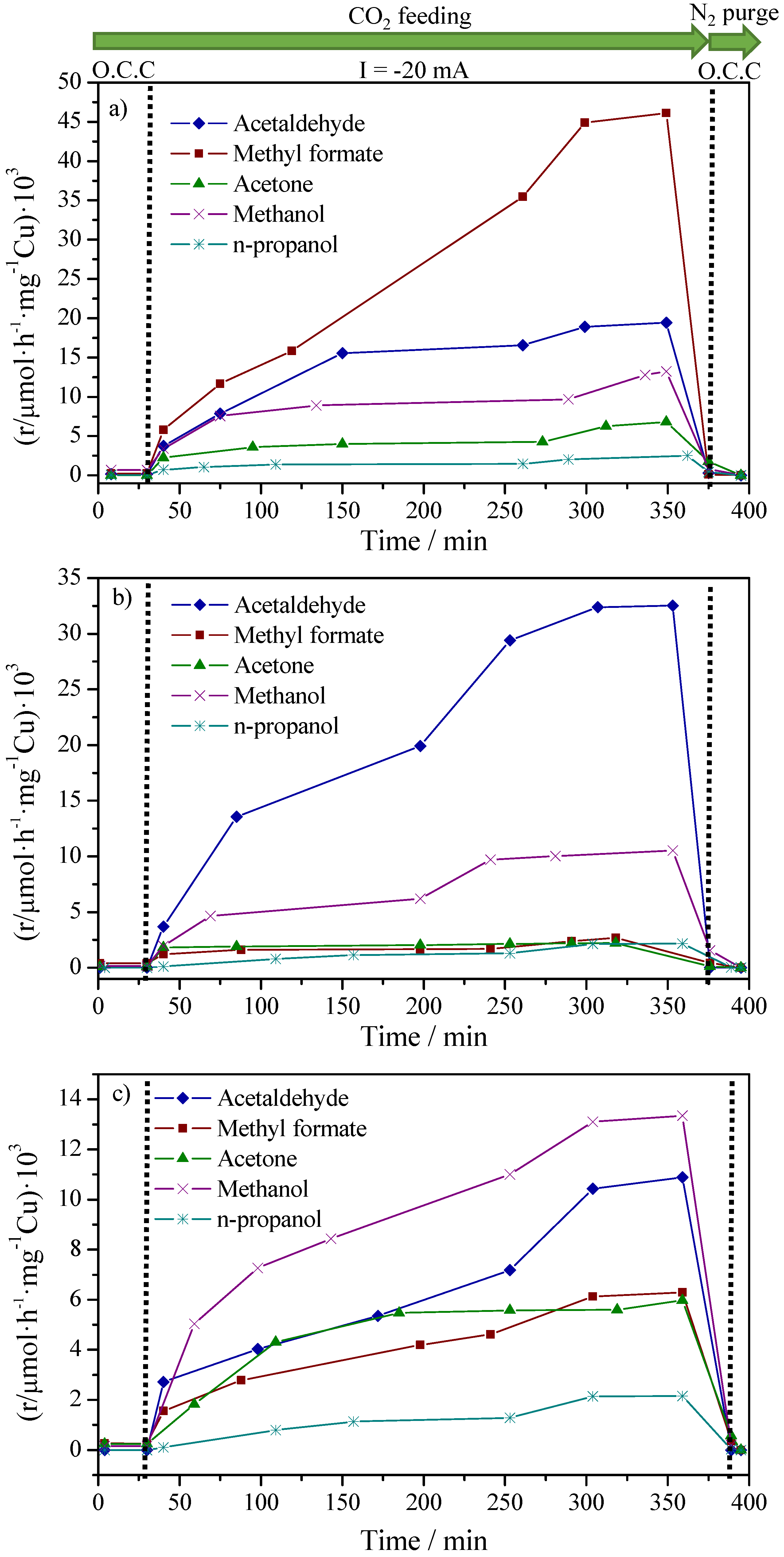
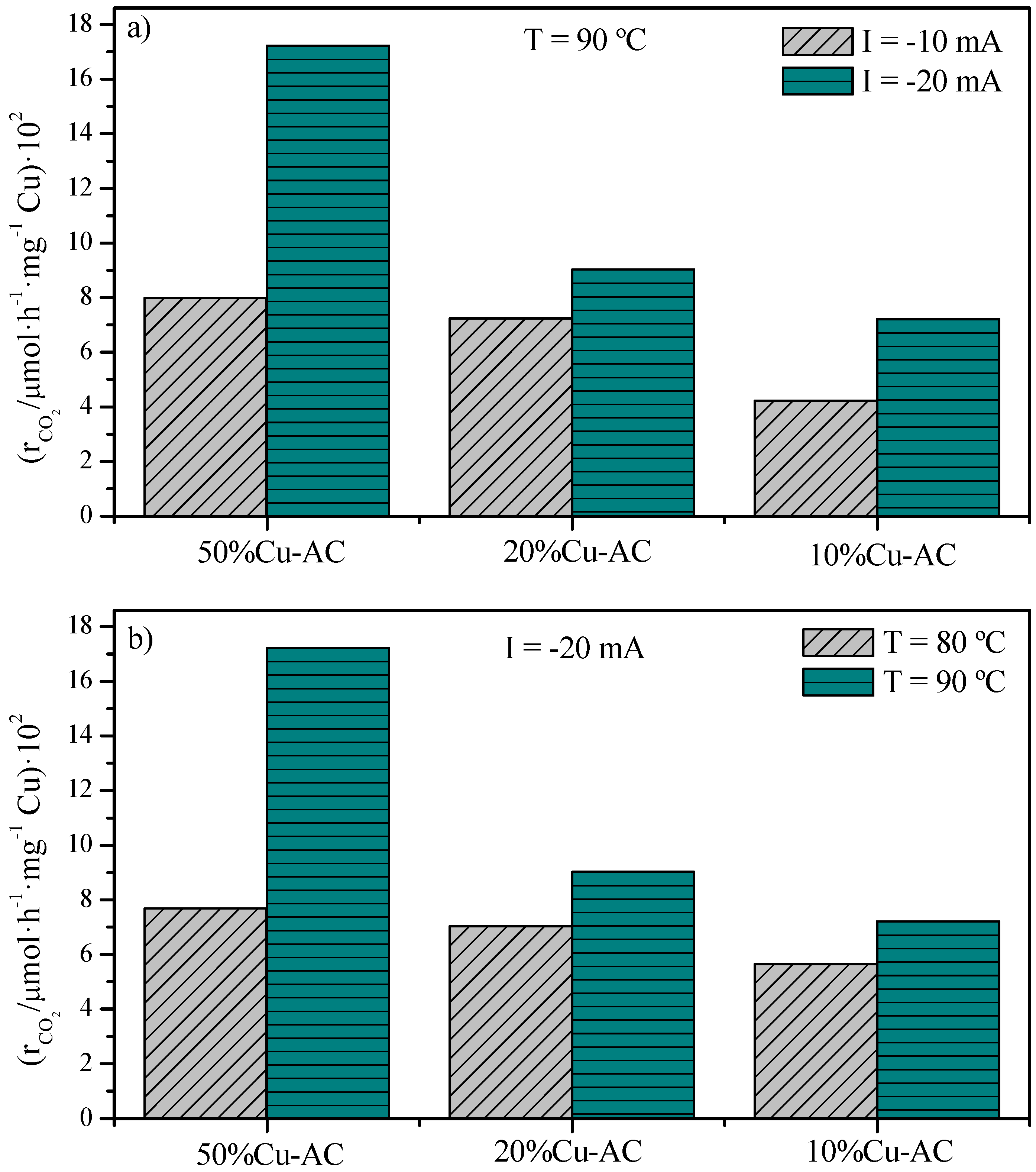
2.2. CO2 Conversion Electrocatalytic Experiments
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Umeyama, T.; Imahori, H. Carbon nanotube-modified electrodes for solar energy conversion. Energy Environ. Sci. 2008, 1, 120–133. [Google Scholar] [CrossRef]
- Bansode, A.; Urakawa, A. Towards full one-pass conversion of carbon dioxide to methanol and methanol-derived products. J. Catal. 2014, 309, 66–70. [Google Scholar] [CrossRef]
- Sekar, N.; Ramasamy, R.P. Recent advances in photosynthetic energy conversion. J. Photochem. Photobiol. C-Photochem. Rev. 2015, 22, 19–33. [Google Scholar] [CrossRef]
- Khiabani, N.H.; Yaghmaee, M.S.; Sarani, A.; Shokri, B. Synthesis-gas production from CH4-CO2-Ar via microwave plasma torch. Adv. Stud. Theor. Phys. 2012, 6, 1273–1287. [Google Scholar]
- Yu, Q.; Kong, M.; Liu, T.; Fei, J.; Zheng, X. Characteristics of the decomposition of CO2 in a dielectric packed-bed plasma reactor. Plasma Chem. Plasma Process. 2012, 32, 153–163. [Google Scholar] [CrossRef]
- Rahemi, N.; Haghighi, M.; Babaluo, A.A.; Jafari, M.F.; Allahyari, S. CO2 reforming of methane over Ni-Cu/Al2O3-ZrO2 nanocatalyst: The influence of plasma treatment and process conditions on catalytic properties and performance. Korean J. Chem. Eng. 2014, 31, 1553–1563. [Google Scholar] [CrossRef]
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Albo, J.; Alvarez-Guerra, M.; Castaño, P.; Irabien, A. Towards the electrochemical conversion of carbon dioxide into methanol. Green Chem. 2015, 17, 2304–2324. [Google Scholar] [CrossRef]
- Centi, G.; Quadrelli, E.A.; Perathoner, S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries. Energy Environ. Sci. 2013, 6, 1711–1731. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M. Co-electrolysis of CO2 and H2O in solid oxide cells: Performance and durability. Solid State Ionics 2011, 192, 398–403. [Google Scholar] [CrossRef]
- Blomen, E.; Hendriks, C.; Neele, F. Capture technologies: improvements and promising developments. Energy Procedia 2009, 1, 1505–1512. [Google Scholar] [CrossRef]
- Yamanaka, I.; Tabata, K.; Mino, W.; Furusawa, T. Electroreduction of Carbon Dioxide to Carbon Monoxide by Co-pthalocyanine Electrocatalyst under Ambient Conditions. ISIJ Int. 2015, 55, 399–403. [Google Scholar] [CrossRef] [Green Version]
- Ampelli, C.; Genovese, C.; Perathoner, S.; Centi, G.; Errahali, M.; Gatti, G.; Marchese, L. An electrochemical reactor for the CO2 reduction in gas phase by using conductive polymer based electrocatalysts. Chem. Eng. Trans. 2014, 41, 13–18. [Google Scholar]
- Lee, K.; Zhang, J.; Wang, H.; Wilkinson, D.P. Progress in the synthesis of carbon nanotube-and nanofiber-supported Pt electrocatalysts for PEM fuel cell catalysis. J. Appl. Electrochem. 2006, 36, 507–522. [Google Scholar] [CrossRef]
- Genovese, C.; Ampelli, C.; Perathoner, S.; Centi, G. Electrocatalytic conversion of CO2 to liquid fuels using nanocarbon-based electrodes. J. Energy Chem. 2013, 22, 202–213. [Google Scholar] [CrossRef]
- Genovese, C.; Ampelli, C.; Perathoner, S.; Centi, G. Electrocatalytic conversion of CO2 on carbon nanotube-based electrodes for producing solar fuels. J. Catal. 2013, 308, 237–249. [Google Scholar] [CrossRef]
- Gutiérrez-Guerra, N.; Moreno-López, L.; Serrano-Ruiz, J.C.; Valverde, J.L.; de Lucas-Consuegra, A. Gas phase electrocatalytic conversion of CO2 to syn-fuels on Cu based catalysts-electrodes. Appl. Catal. B-Environ. 2016, 188, 272–282. [Google Scholar]
- Rodriguez-Reinoso, F. The role of carbon materials in heterogeneous catalysis. Carbon 1998, 36, 159–175. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; Perez, V.; Rodriguez-Reinoso, F. Impregnation of activated carbon with chromium and copper salts: Effect of porosity and metal content. Carbon 1994, 32, 1259–1265. [Google Scholar] [CrossRef]
- Zhang, G.; Li, Z.; Zheng, H.; Fu, T.; Ju, Y.; Wang, Y. Influence of the surface oxygenated groups of activated carbon on preparation of a nano Cu/AC catalyst and heterogeneous catalysis in the oxidative carbonylation of methanol. Appl. Catal. B-Environ. 2015, 179, 95–105. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Sánchez, P.; Rodríguez-Gómez, A.; Valverde, J.L.; Dorado, F. Carbon nanofiber-based palladium/zinc catalysts for the hydrogenation of carbon dioxide to methanol at atmospheric pressure. Ind. Eng. Chem. Res. 2016, 55, 3556–3567. [Google Scholar] [CrossRef]
- Román-Martínez, M.C.; Cazorla-Amorós, D.; Linares-Solano, A.; De Lecea, C.S.M. TPD and TPR characterization of carbonaceous supports and Pt/C catalysts. Carbon 1993, 31, 895–902. [Google Scholar] [CrossRef]
- Gil, S.; Muñoz, L.; Sánchez-Silva, L.; Romero, A.; Valverde, J.L. Synthesis and characterization of Au supported on carbonaceous material-based catalysts for the selective oxidation of glycerol. Chem. Eng. J. 2011, 172, 418–429. [Google Scholar] [CrossRef]
- Trépanier, M.; Dalai, A.K.; Abatzoglou, N. Synthesis of CNT-supported cobalt nanoparticle catalysts using a microemulsion technique: Role of nanoparticle size on reducibility, activity and selectivity in Fischer–Tropsch reactions. Appl. Catal. A-Gen. 2010, 374, 79–86. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. The role of copper particle size in low pressure methanol synthesis via CO2 hydrogenation over Cu/ZnO catalysts. Catal. Sci. Technol. 2015, 5, 869–881. [Google Scholar] [CrossRef]
- Genovese, C.; Ampelli, C.; Perathoner, S.; Centi, G. A gas-phase electrochemical reactor for carbon dioxide reduction back to liquid fuels. Chem. Eng. Trans. 2013, 32, 289–294. [Google Scholar]
- Gangeri, M.; Perathoner, S.; Caudo, S.; Centi, G.; Amadou, J.; Begin, D.; Schlögl, R. Fe and Pt carbon nanotubes for the electrocatalytic conversion of carbon dioxide to oxygenates. Catal. Today 2009, 143, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Centi, G.; Perathoner, S.; Winè, G.; Gangeri, M. Electrocatalytic conversion of CO2 to long carbon-chain hydrocarbons. Green Chem. 2007, 9, 671–678. [Google Scholar] [CrossRef]
- Ahouari, H.; Soualah, A.; Le Valant, A.; Pinard, L.; Magnoux, P.; Pouilloux, Y. Methanol synthesis from CO2 hydrogenation over copper based catalysts. React. Kinet. Mech. Catal. 2013, 110, 131–145. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Valverde, J.L.; Sánchez, P.; Dorado, F. CO2 hydrogenation to methanol at atmospheric pressure: influence of the preparation method of Pd/ZnO catalysts. Catal. Lett. 2016, 146, 373–382. [Google Scholar] [CrossRef]
- Díez-Ramírez, J.; Dorado, F.; de la Osa, A.R.; Valverde, J.L.; Sánchez, P. Hydrogenation of CO2 to methanol at atmospheric pressure over Cu/ZnO catalysts: influence of the calcination, reduction, and metal loading. Ind. Eng. Chem. Res. 2017, 56, 1979–1987. [Google Scholar] [CrossRef]
- Reske, R.; Mistry, H.; Behafarid, F.; Roldan Cuenya, B.; Strasser, P. Particle size effects in the catalytic electroreduction of CO2 on Cu nanoparticles. J. Am. Chem. Soc. 2014, 136, 6978–6986. [Google Scholar] [CrossRef] [PubMed]
- Tonner, S.P.; Trimm, D.L.; Wainwright, M.S.; Cant, N.W. Dehydrogenation of methanol to methyl formate over copper catalysts. Ind. Eng. Chem. Prod. Res. Dev. 1984, 23, 384–388. [Google Scholar] [CrossRef]
- Caravaca, A.; Sapountzi, F.M.; de Lucas-Consuegra, A.; Molina-Mora, C.; Dorado, F.; Valverde, J.L. Electrochemical reforming of ethanol–water solutions for pure H2 production in a PEM electrolysis cell. Int. J. Hydrogen Energy 2012, 37, 9504–9513. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Powder Metal Loading/wt % | Electrode Metal Weight/mg·cm−2 | Surface Area/m2·g−1 | Total Pore Volume/cm3·g−1 | TPR-Tmax/°C | Mean Particle Size from XRD/nm |
|---|---|---|---|---|---|---|
| AC | - | - | 866 | 0.293 | - | - |
| 50% Cu-AC | 55 | 0.22 | 773 | 0.186 | 171 | 40 |
| 20% Cu-AC | 19 | 0.18 | 797 | 0.260 | 185 | 14 |
| 10% Cu-AC | 12 | 0.16 | 817 | 0.274 | 193 | 12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Lucas-Consuegra, A.; Serrano-Ruiz, J.C.; Gutiérrez-Guerra, N.; Valverde, J.L. Low-Temperature Electrocatalytic Conversion of CO2 to Liquid Fuels: Effect of the Cu Particle Size. Catalysts 2018, 8, 340. https://doi.org/10.3390/catal8080340
De Lucas-Consuegra A, Serrano-Ruiz JC, Gutiérrez-Guerra N, Valverde JL. Low-Temperature Electrocatalytic Conversion of CO2 to Liquid Fuels: Effect of the Cu Particle Size. Catalysts. 2018; 8(8):340. https://doi.org/10.3390/catal8080340
Chicago/Turabian StyleDe Lucas-Consuegra, Antonio, Juan Carlos Serrano-Ruiz, Nuria Gutiérrez-Guerra, and José Luis Valverde. 2018. "Low-Temperature Electrocatalytic Conversion of CO2 to Liquid Fuels: Effect of the Cu Particle Size" Catalysts 8, no. 8: 340. https://doi.org/10.3390/catal8080340
APA StyleDe Lucas-Consuegra, A., Serrano-Ruiz, J. C., Gutiérrez-Guerra, N., & Valverde, J. L. (2018). Low-Temperature Electrocatalytic Conversion of CO2 to Liquid Fuels: Effect of the Cu Particle Size. Catalysts, 8(8), 340. https://doi.org/10.3390/catal8080340