Characteristics of Vanadium-Based Coal Gasification Slag and the NH3-Selective Catalytic Reduction of NO

Abstract
:1. Introduction
2. Results and Discussion
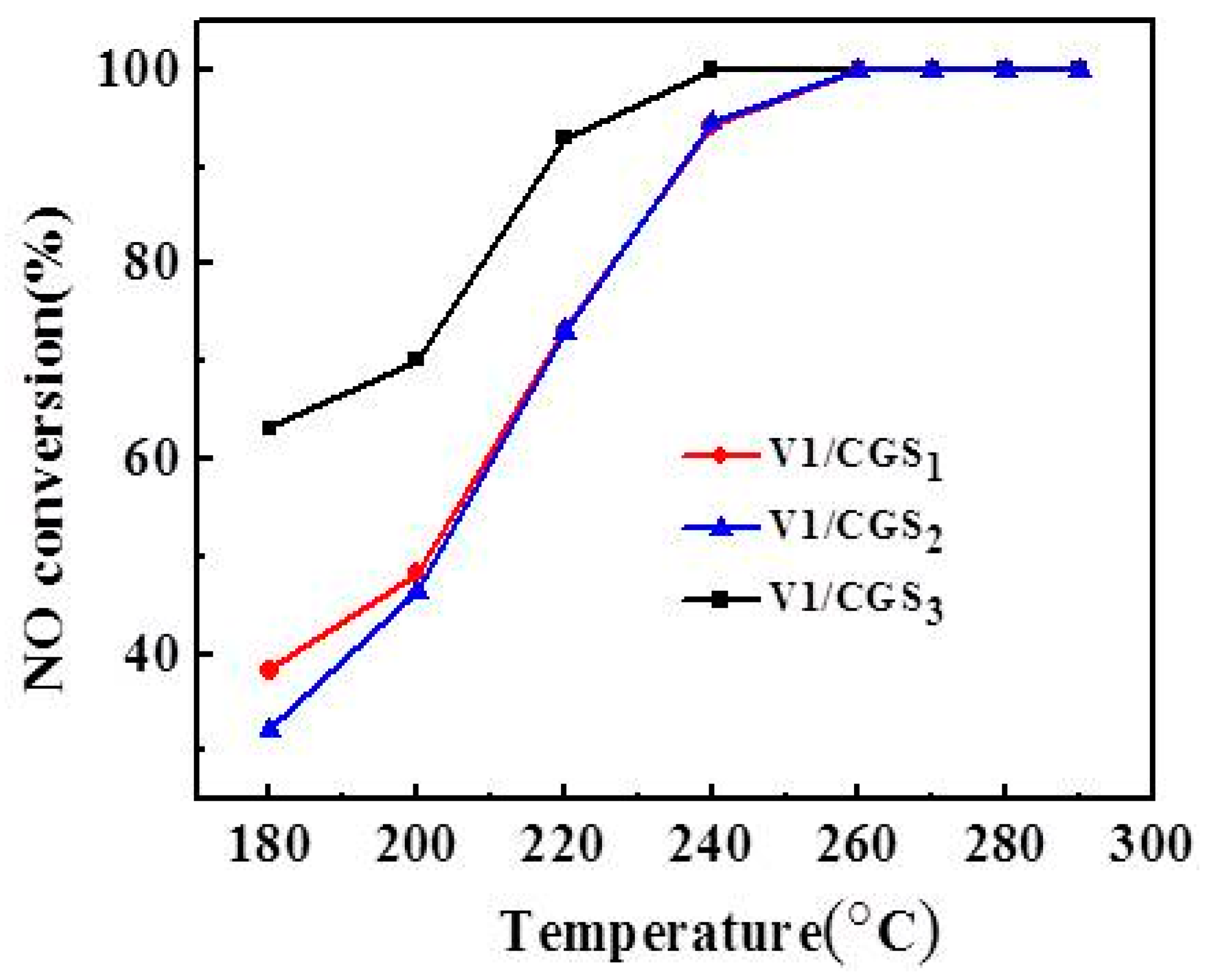
2.1. Effect of Carrier
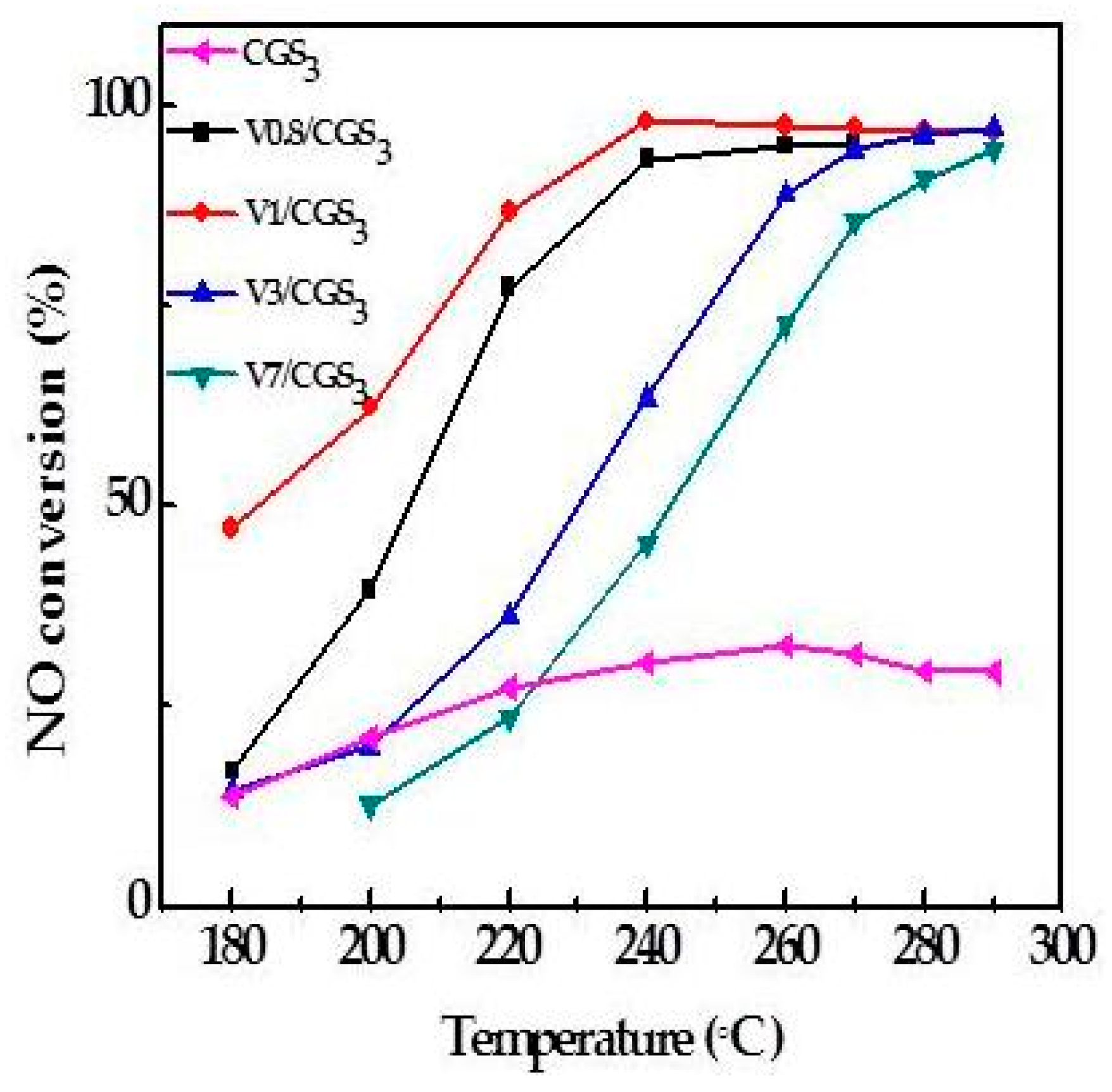
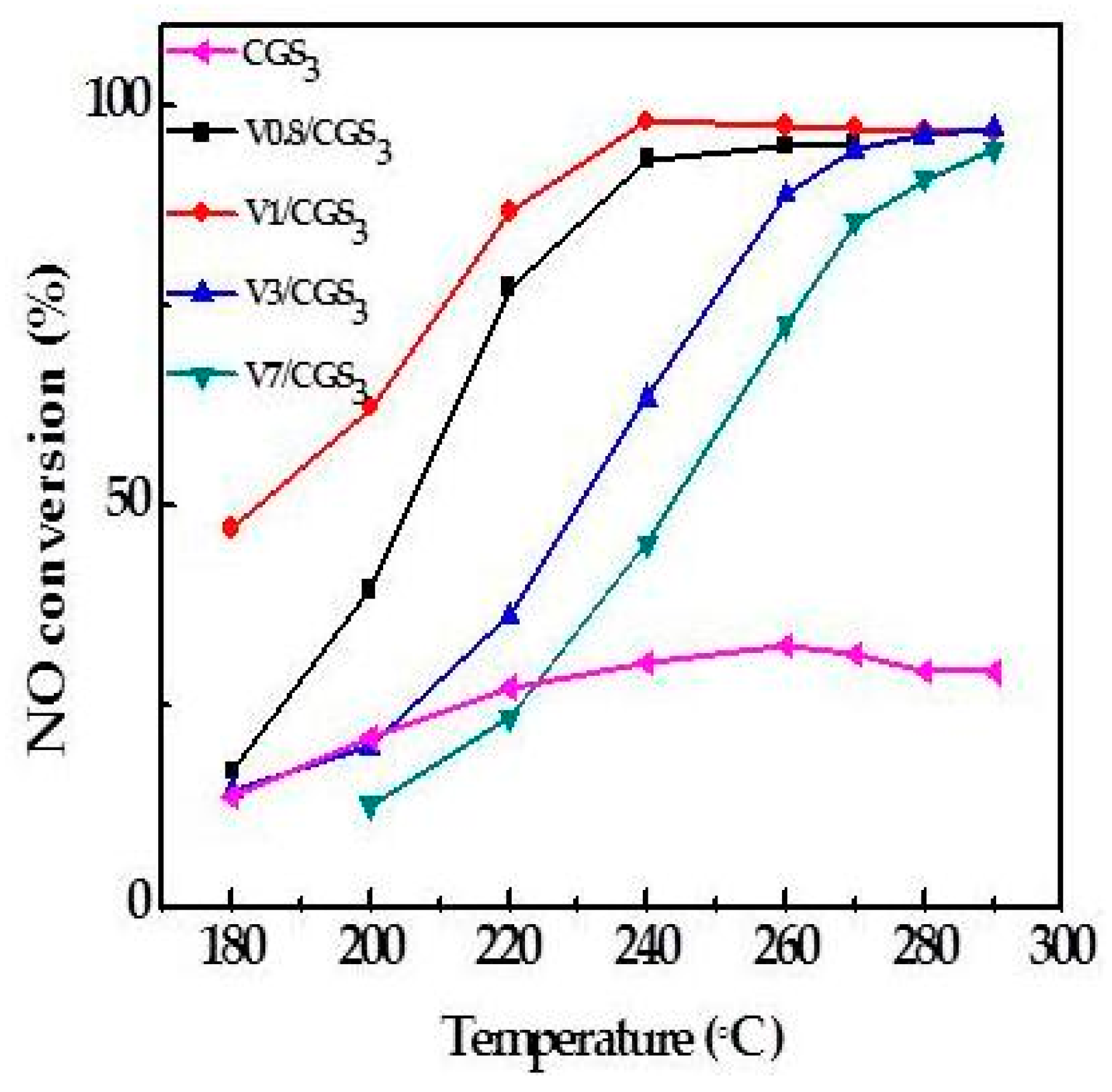
2.2. Effect of V Loading
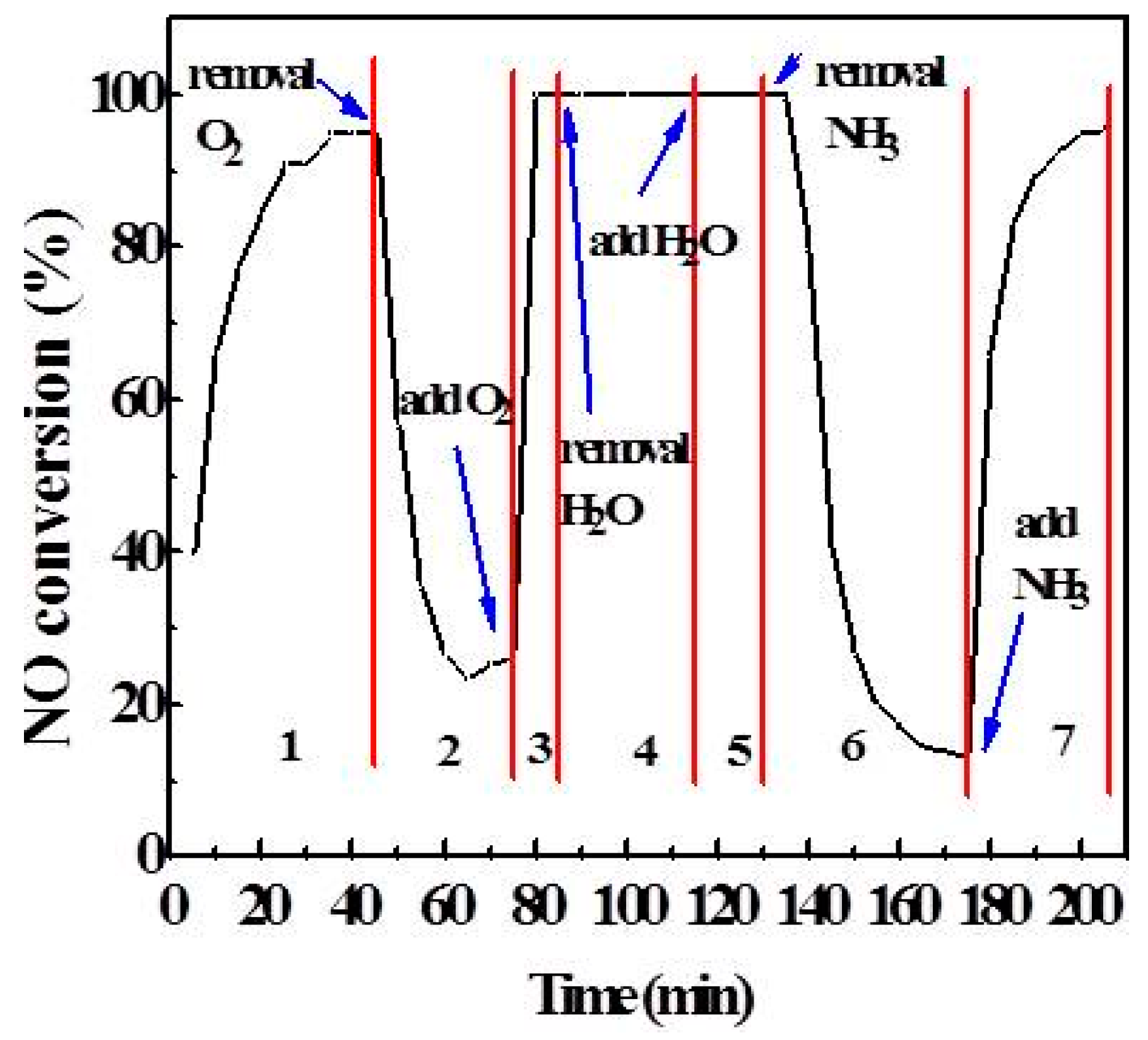
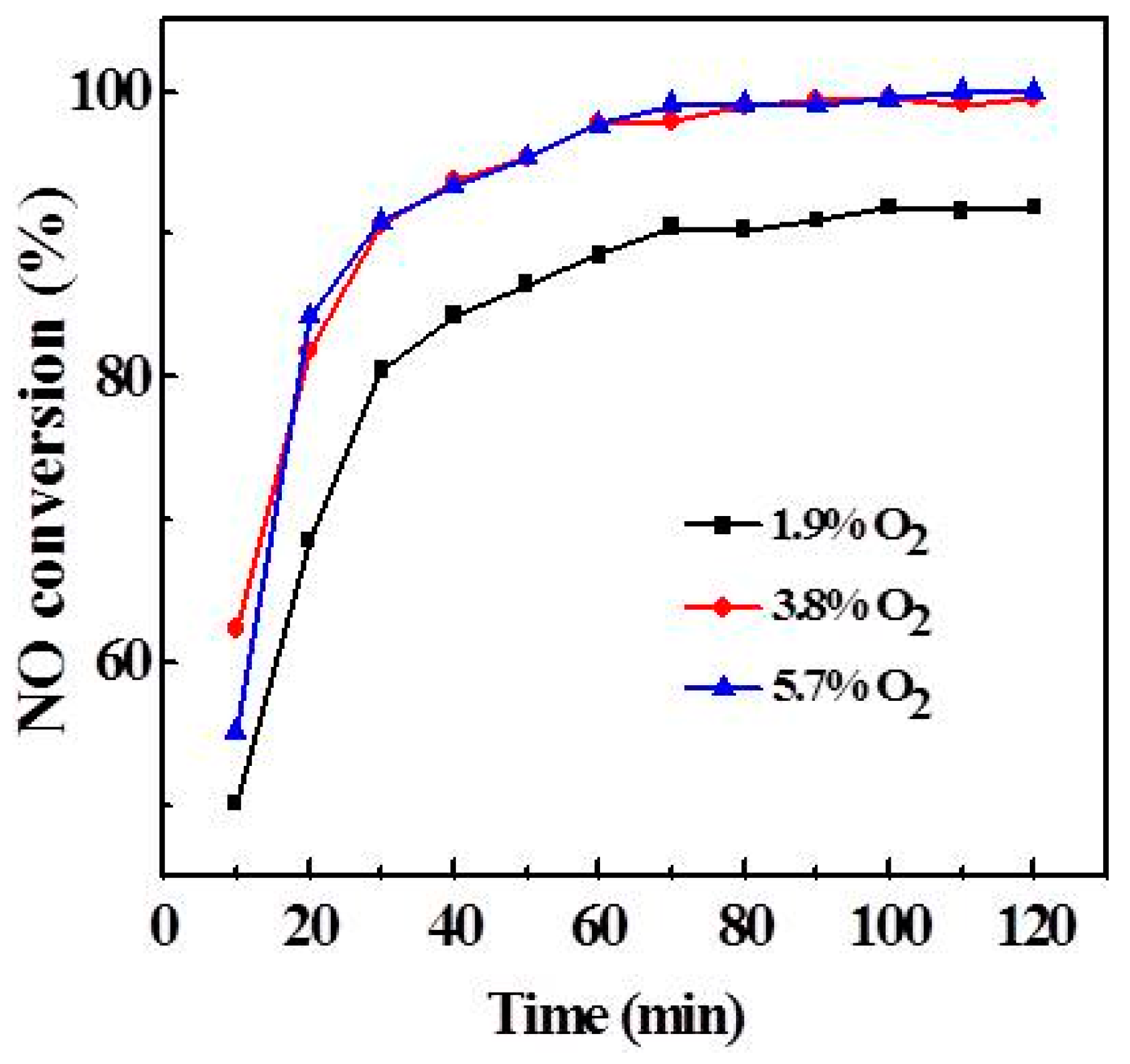
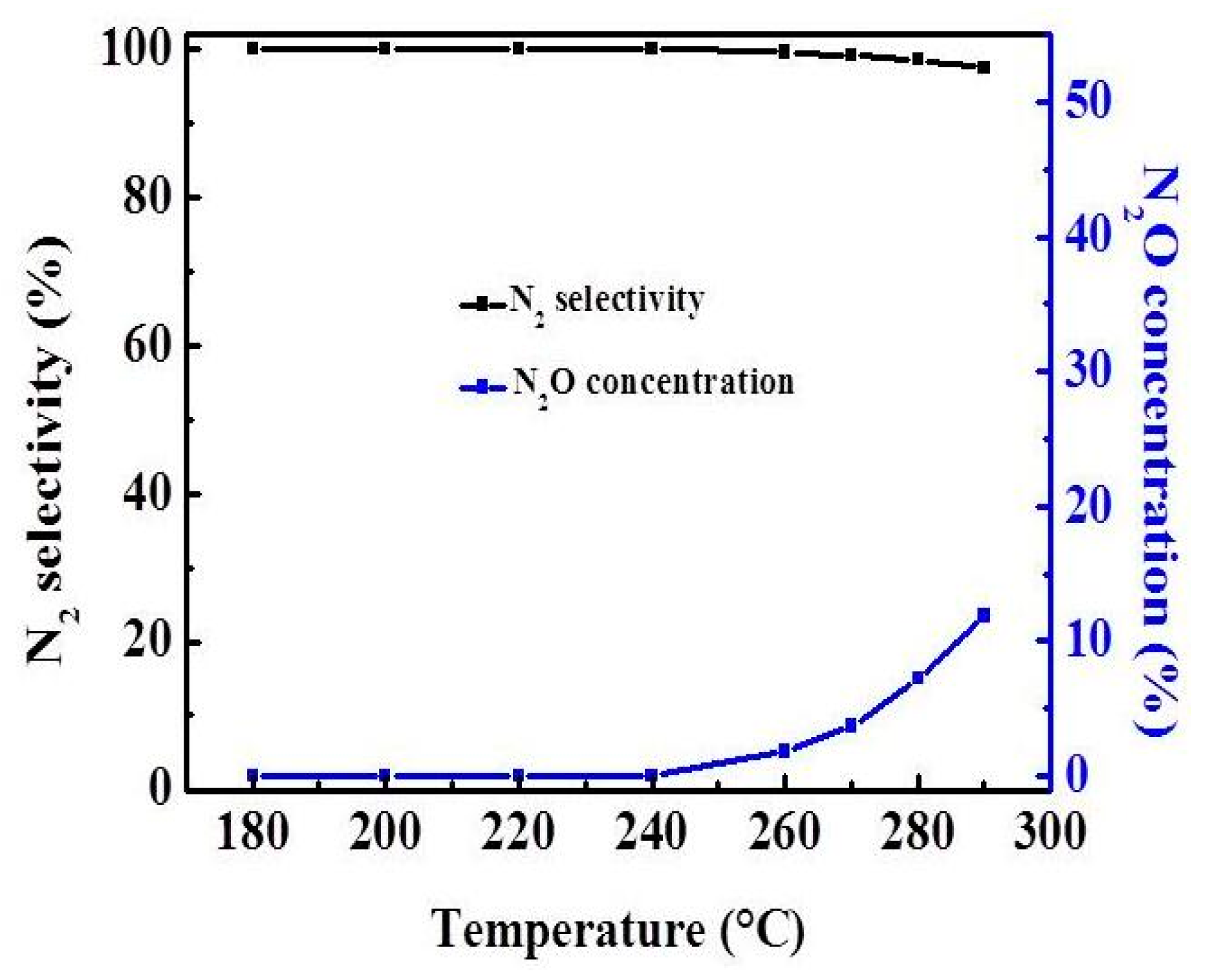
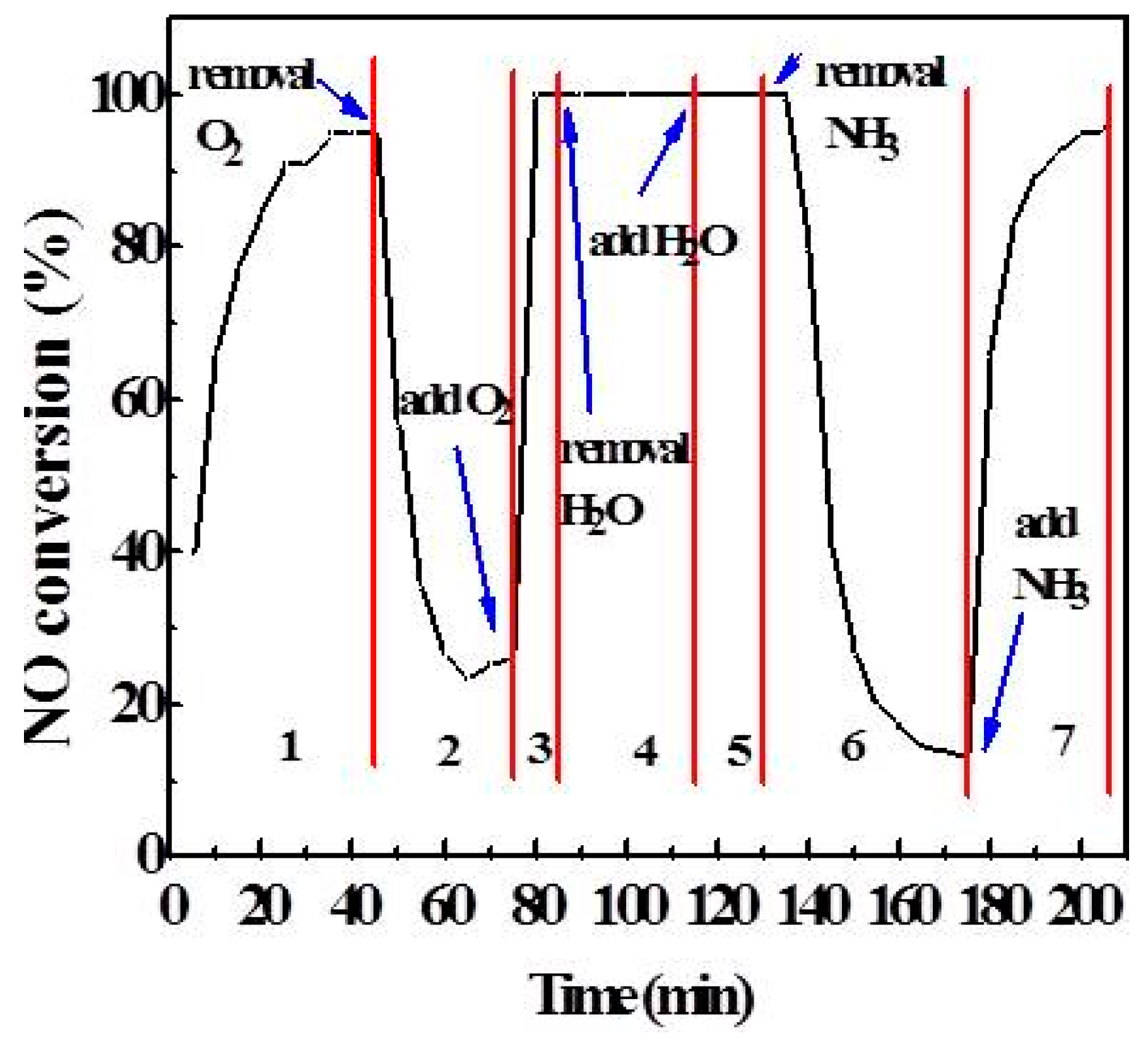
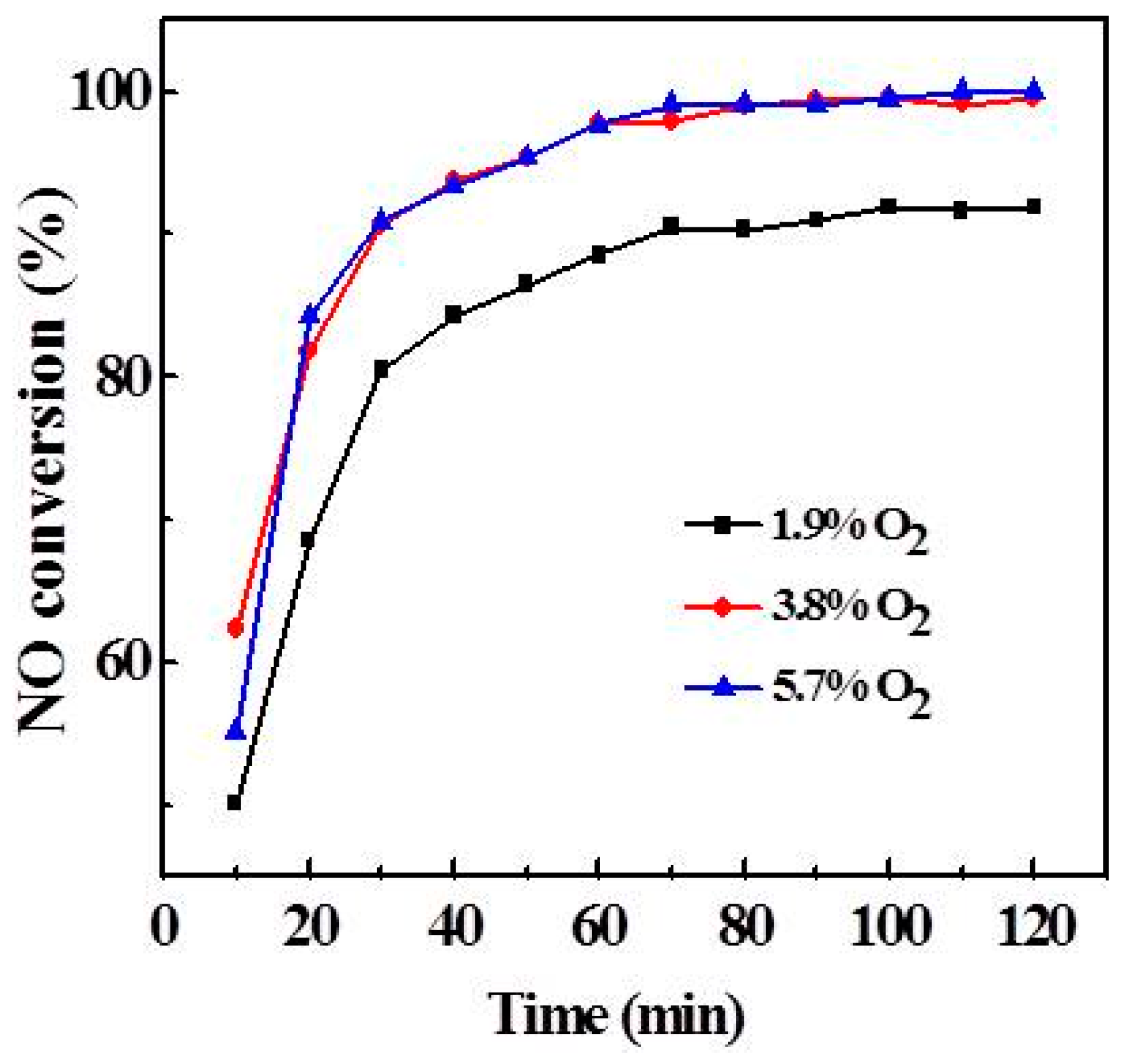
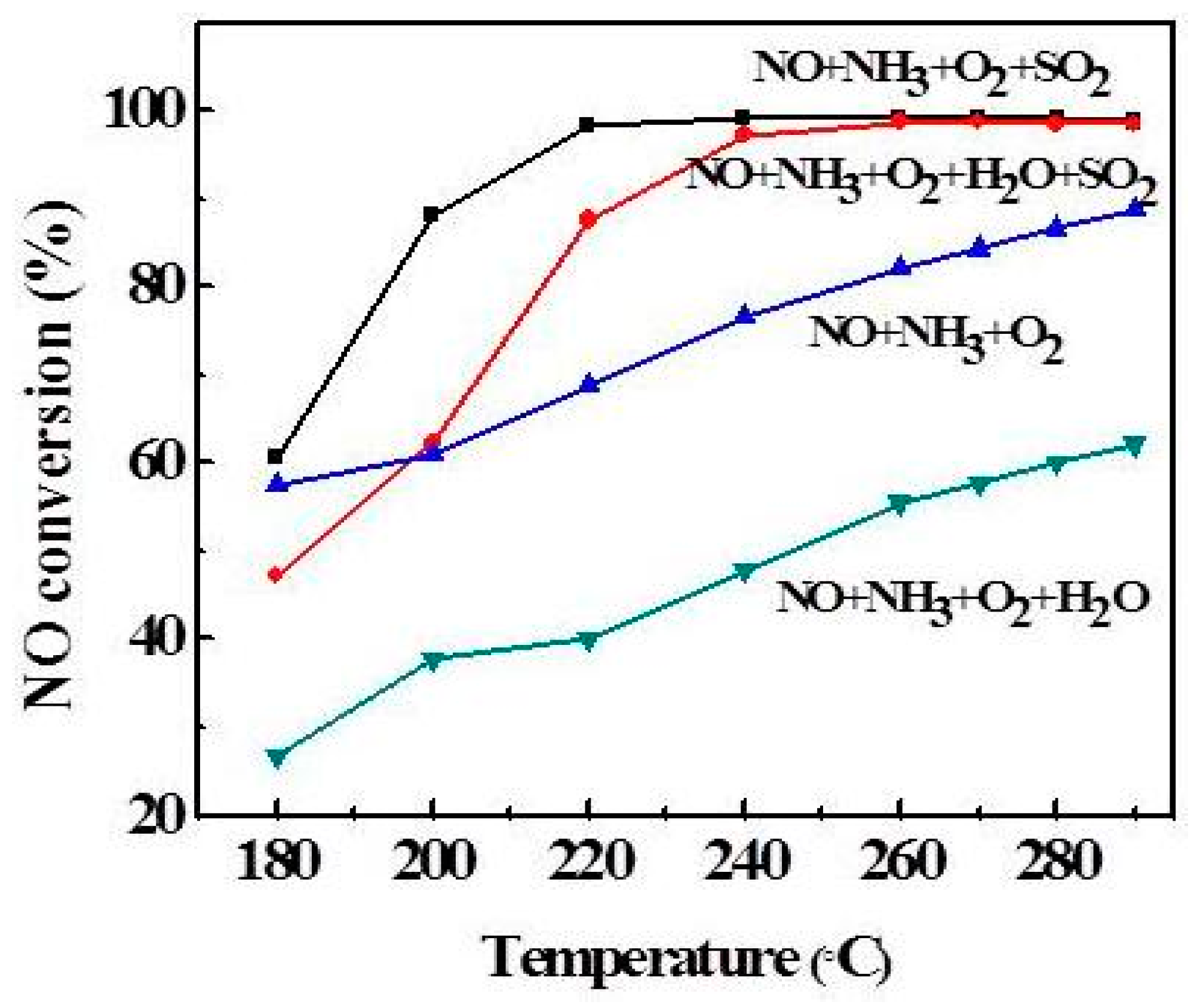
2.3. Effect of Atmosphere
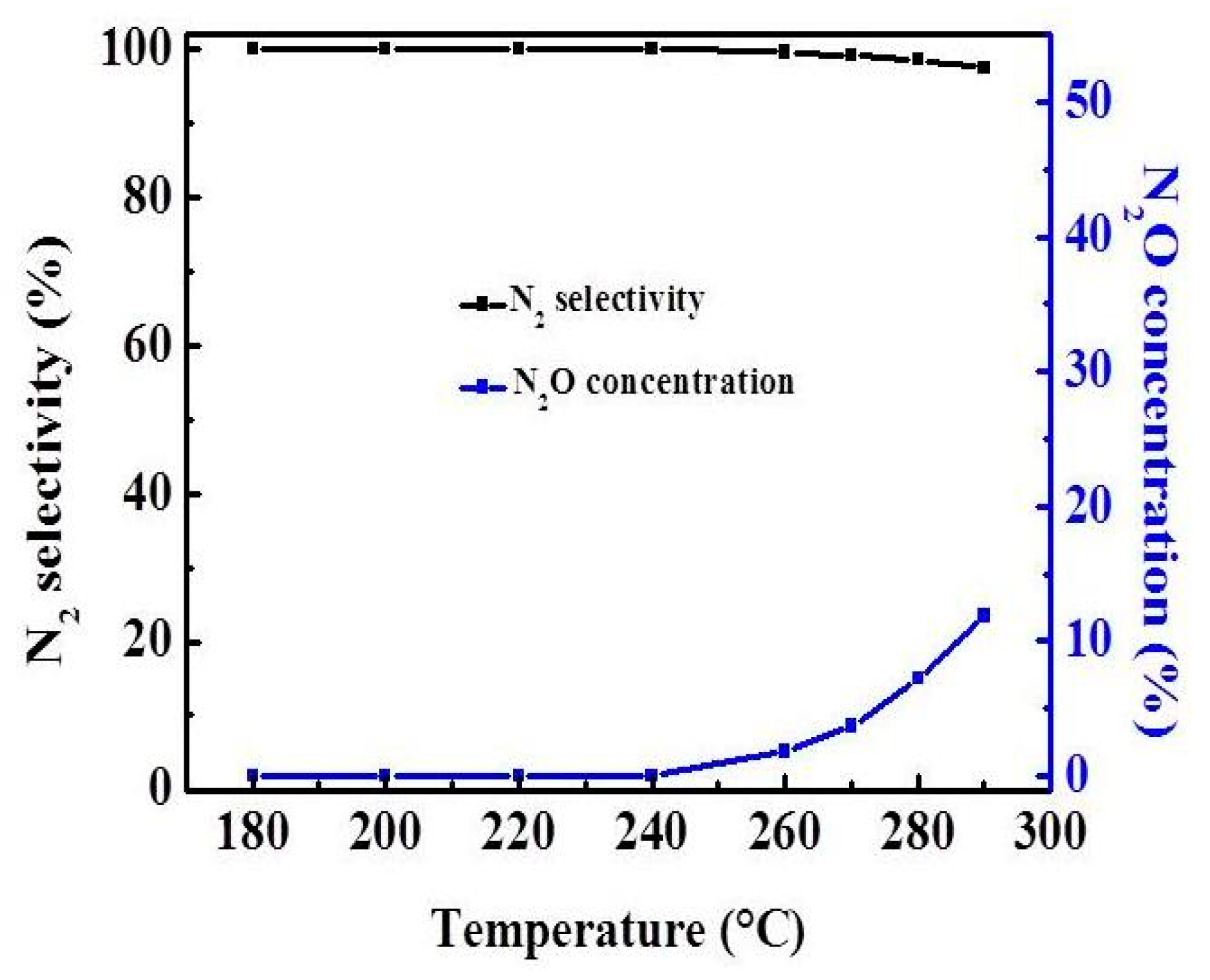
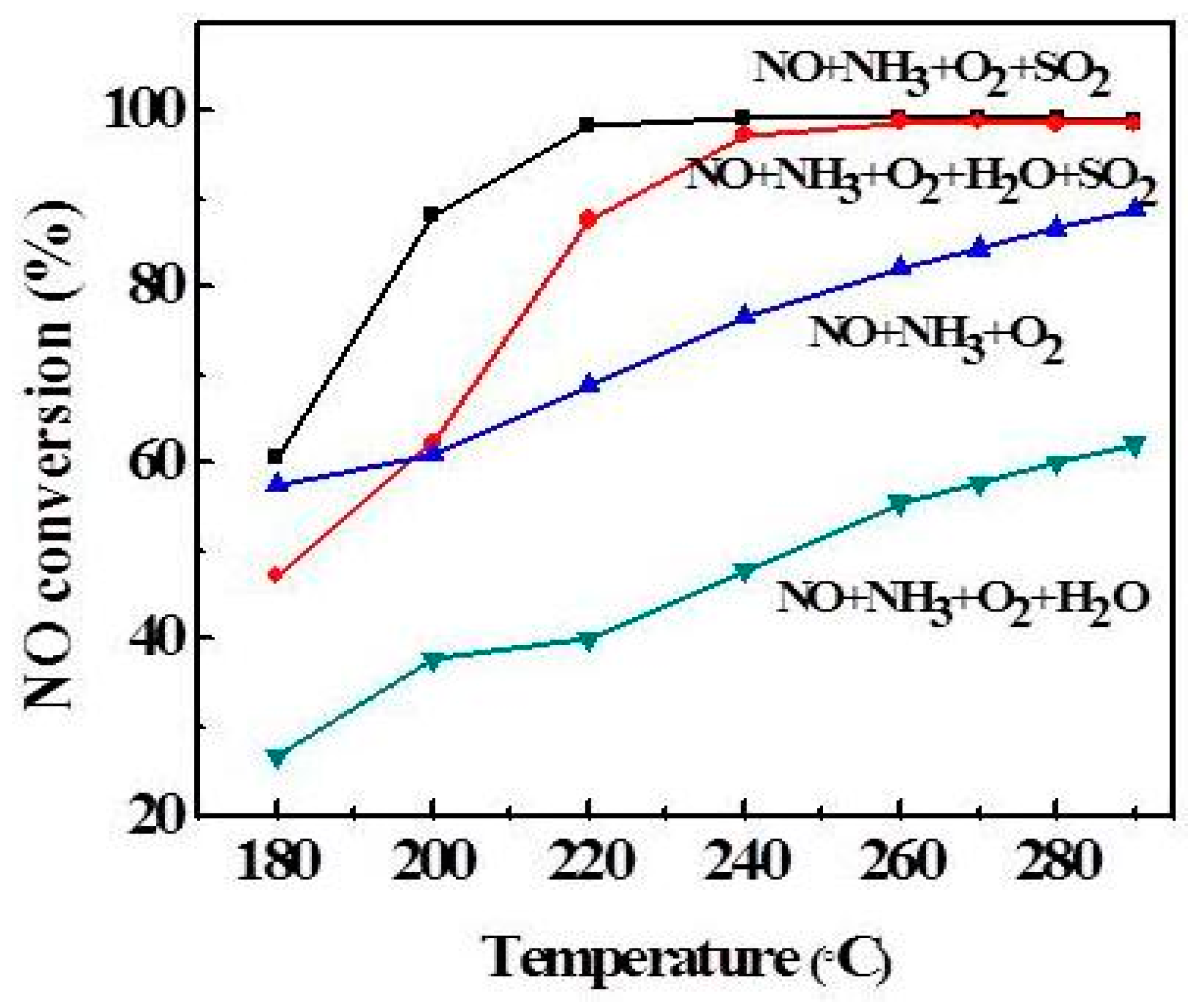
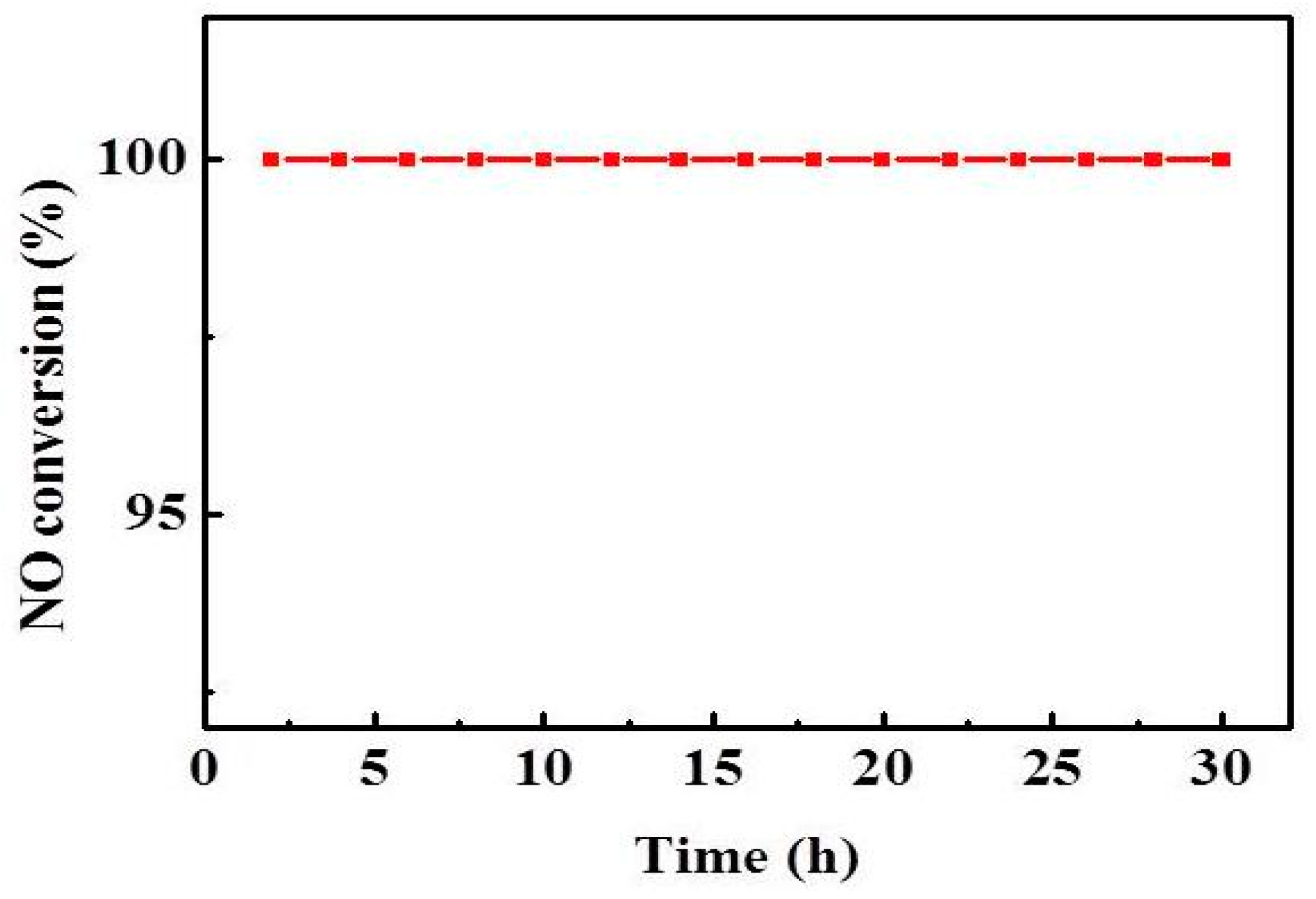
2.4. Effect of Reaction Temperature
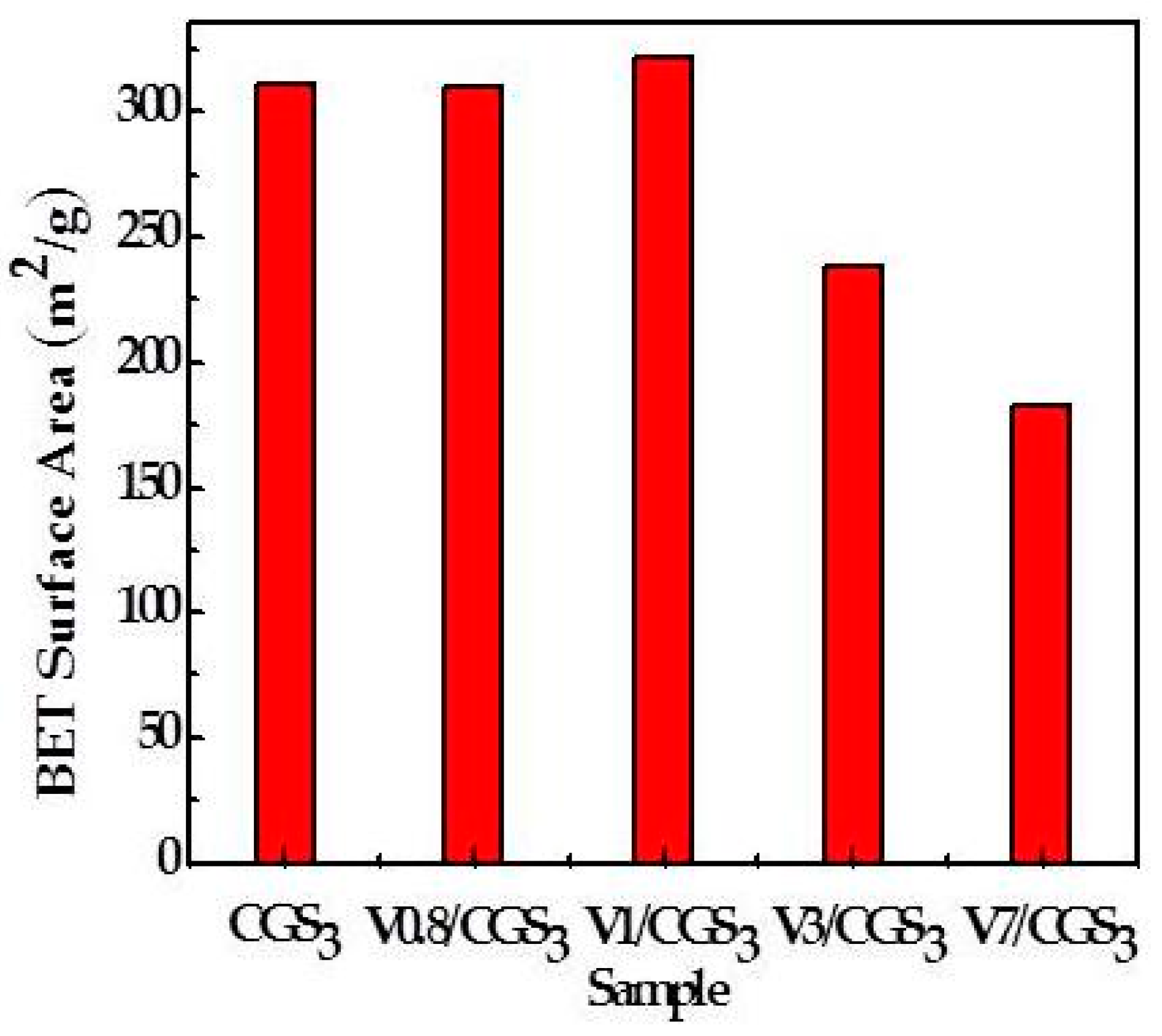
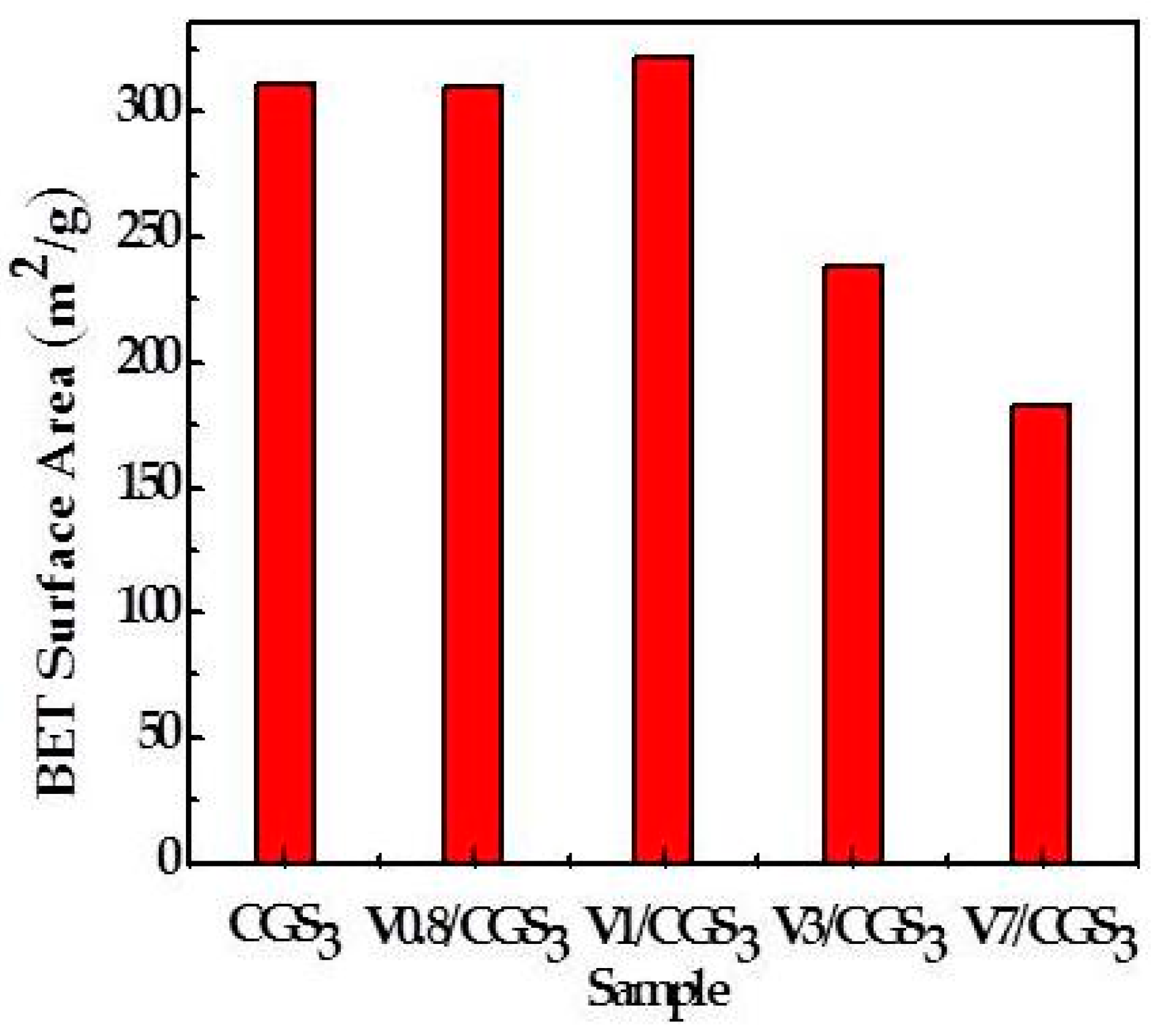
2.5. The Physical Properties of CGS
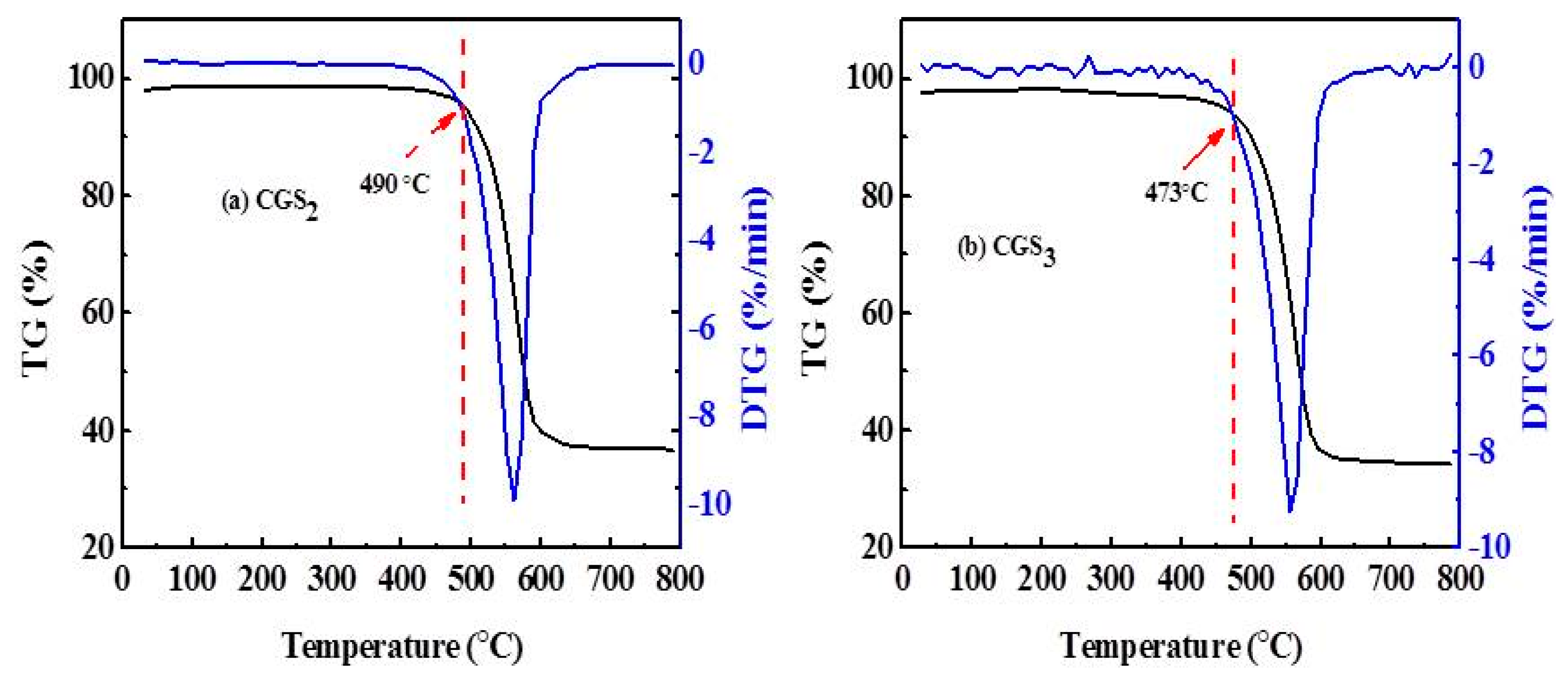
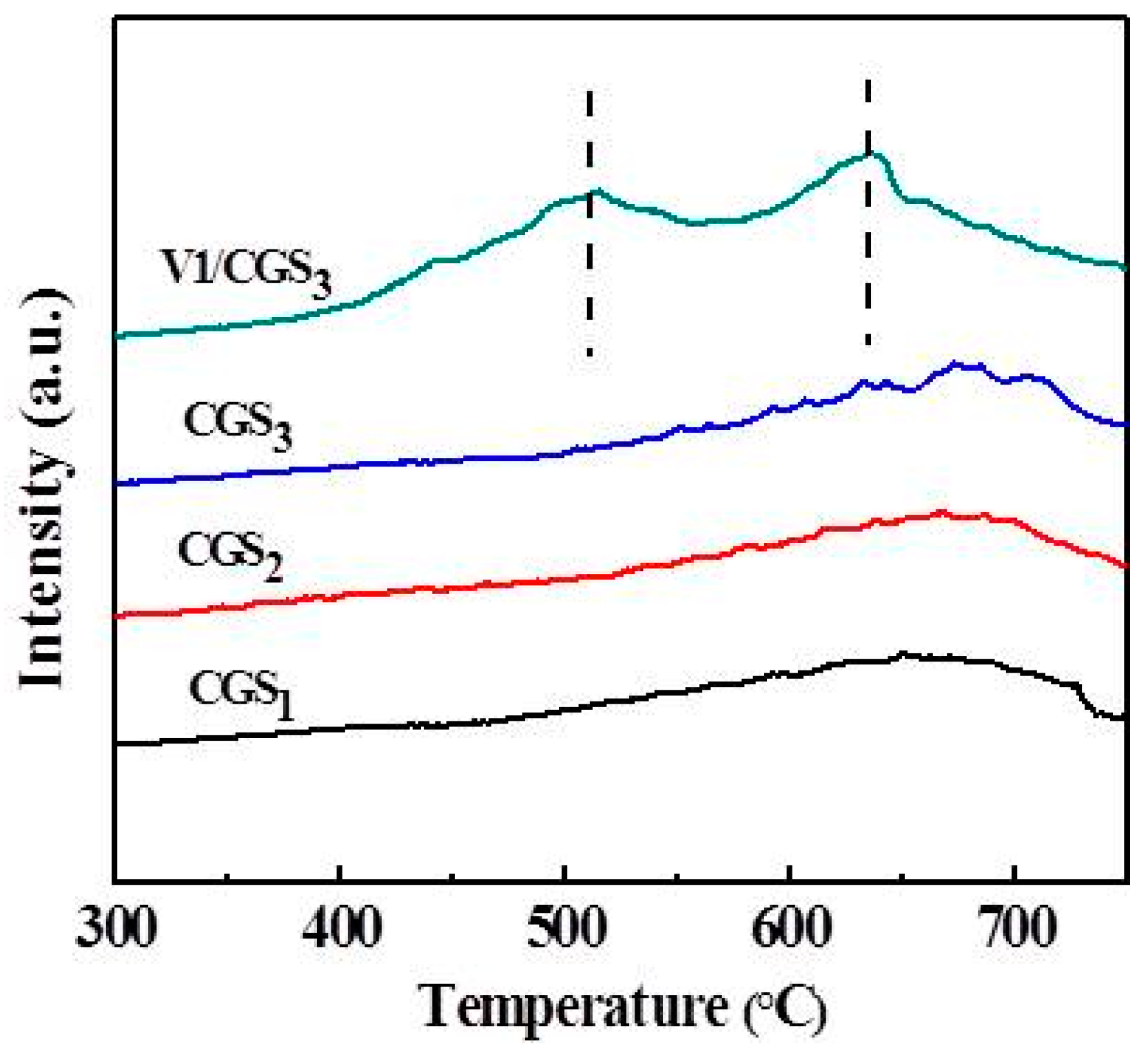
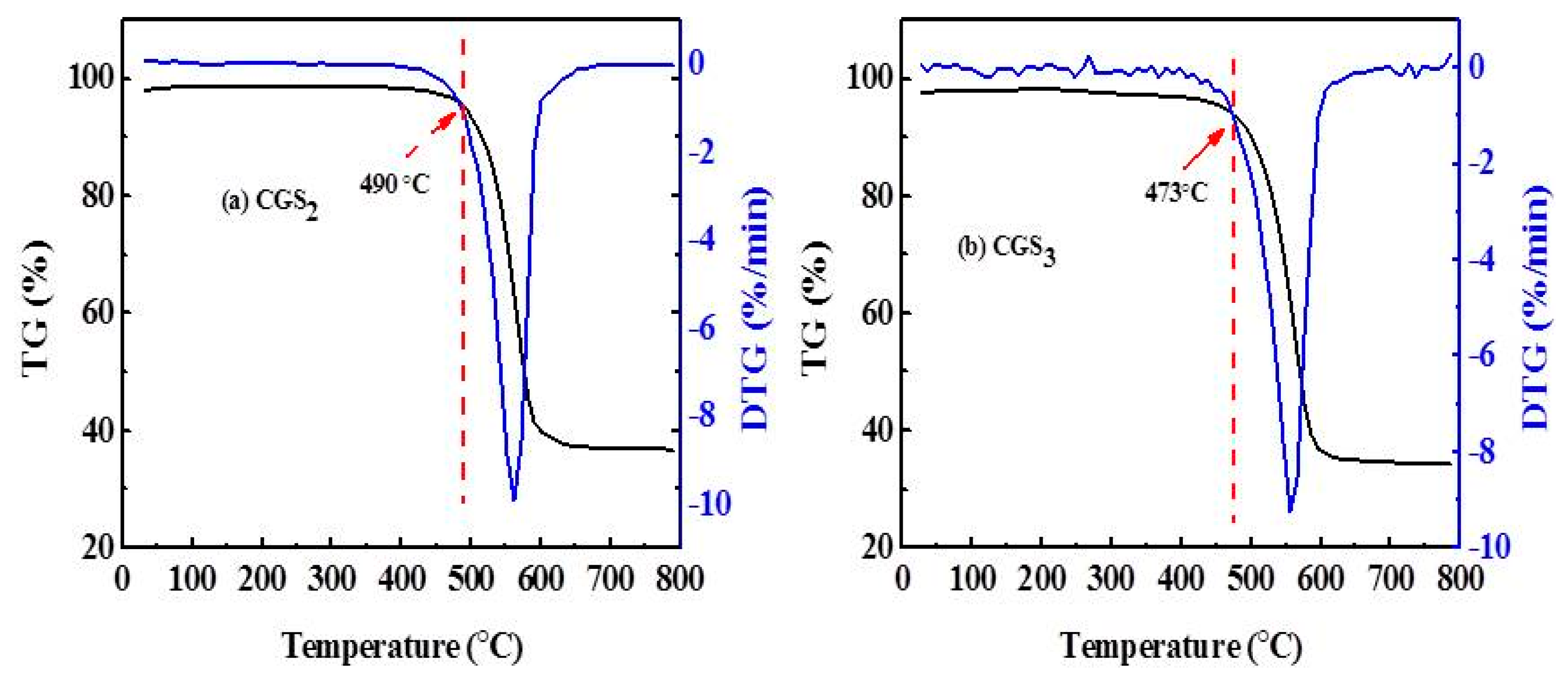
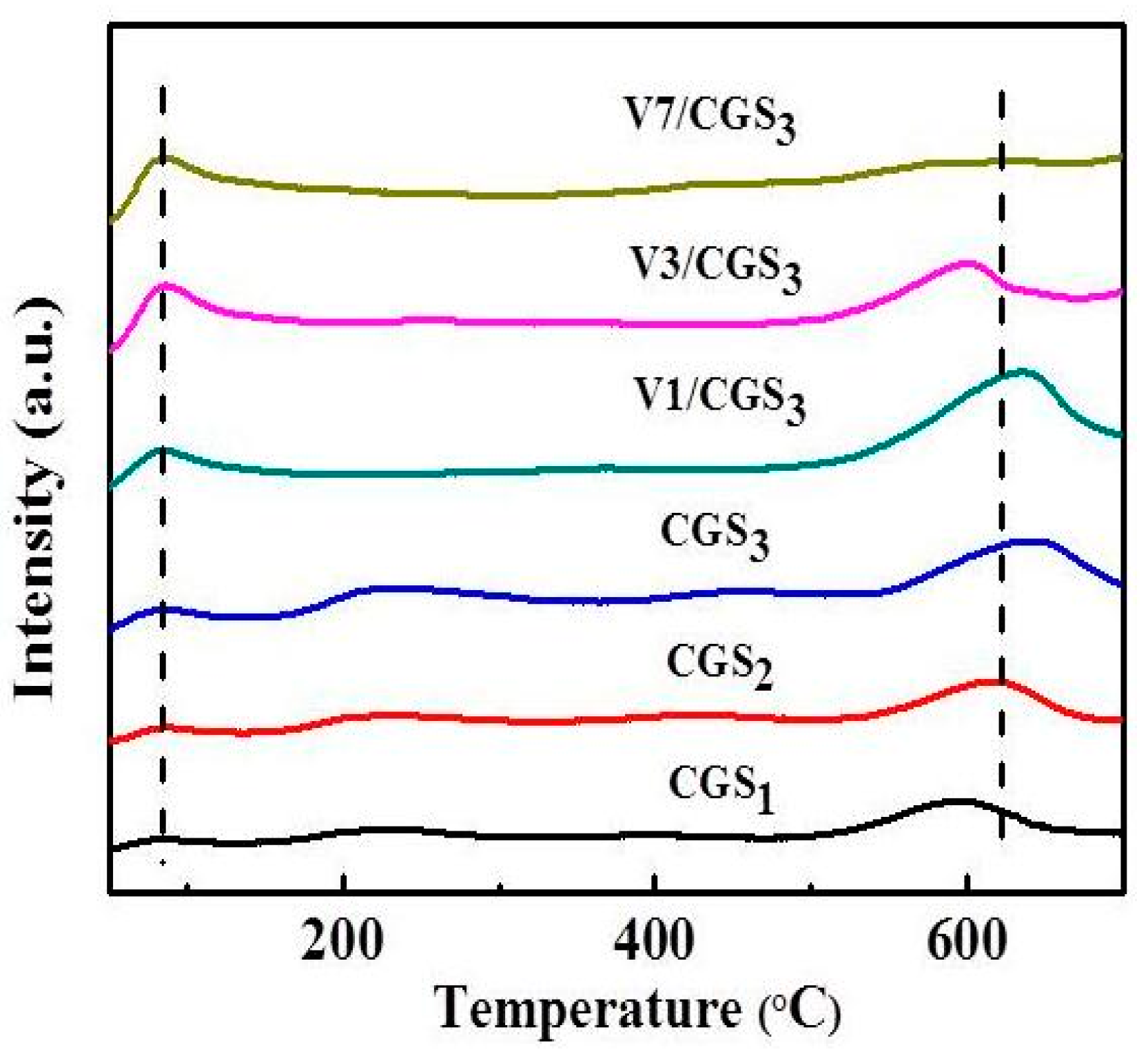
2.6. Thermo Gravimetric (TG) and Derivative Thermo Gravimetric (DTG
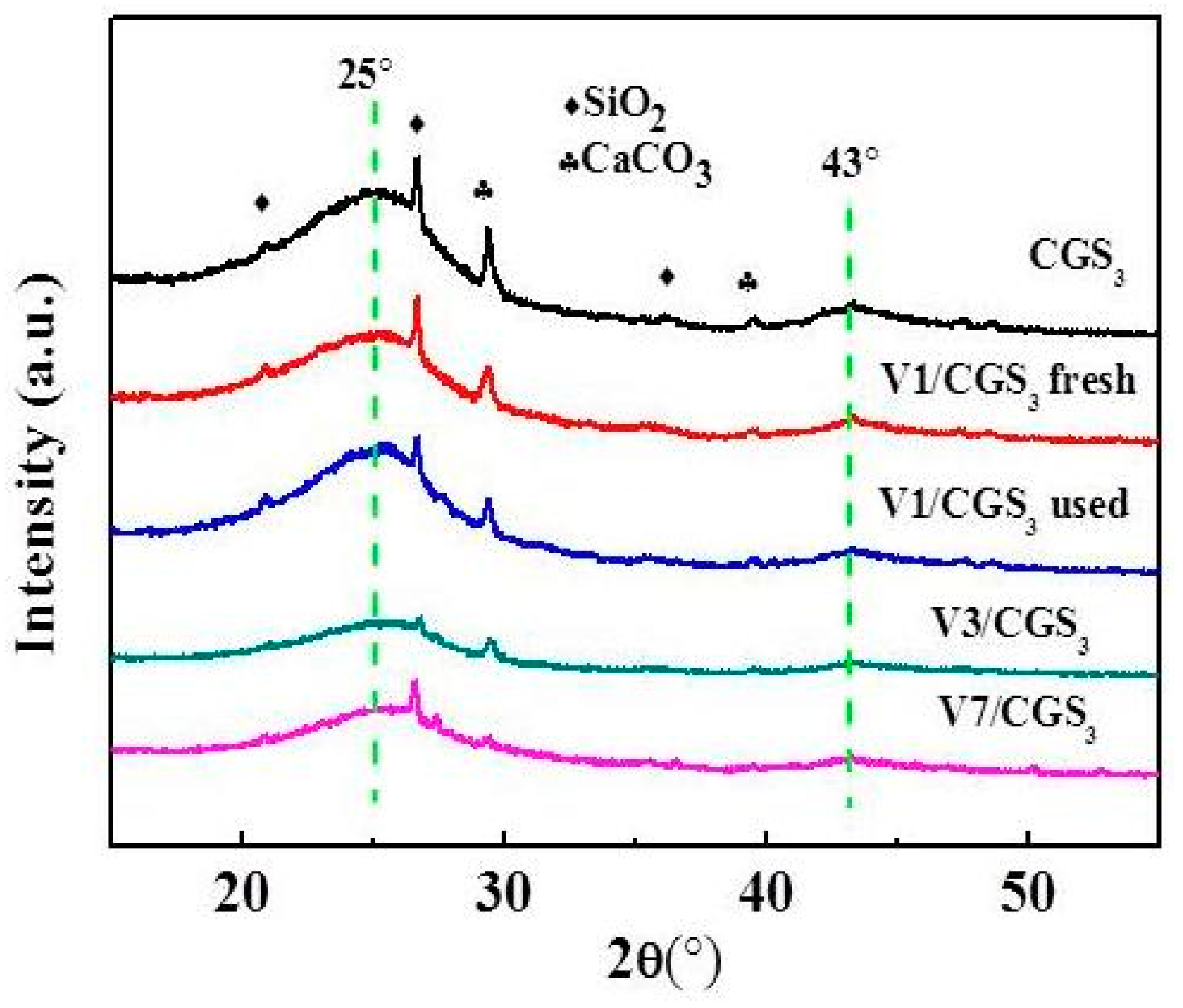
2.7. XRD
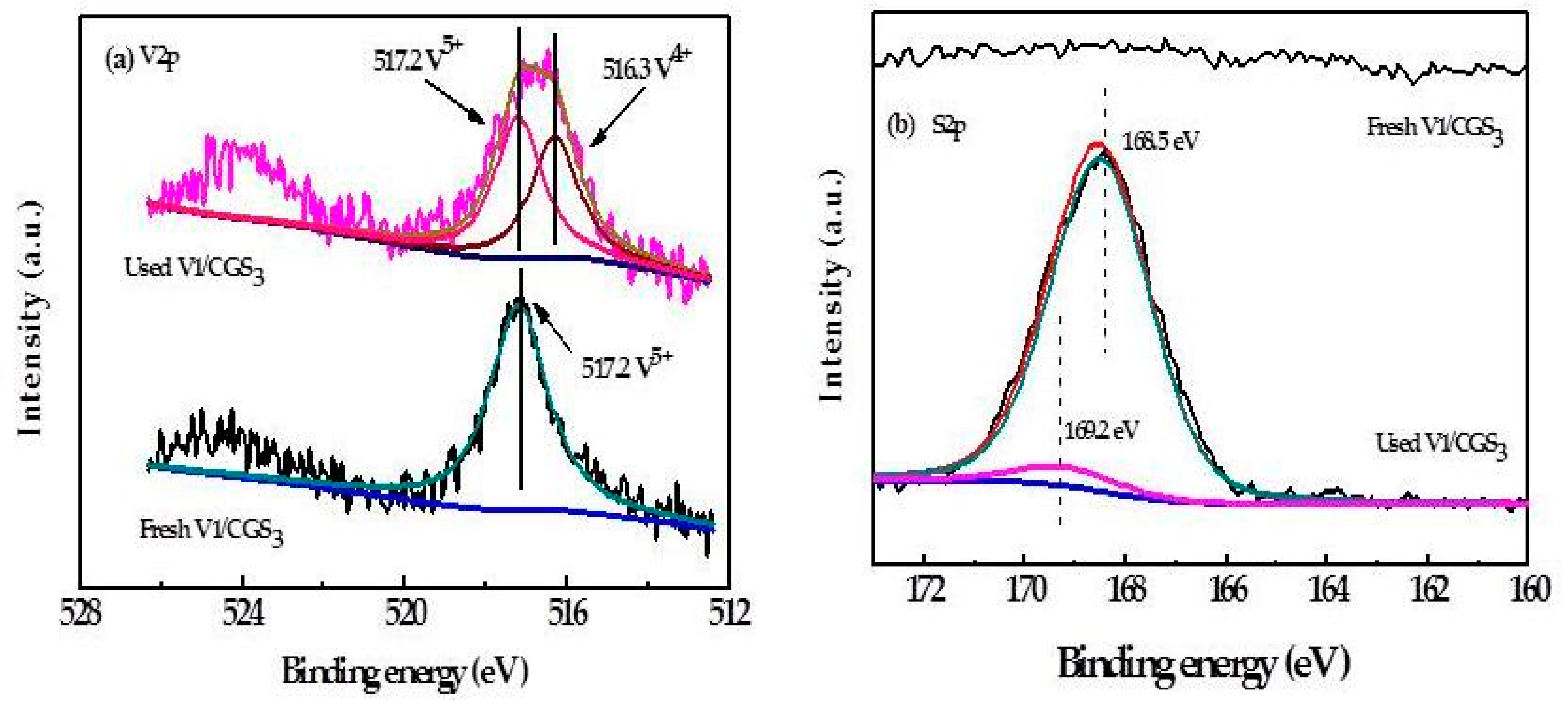
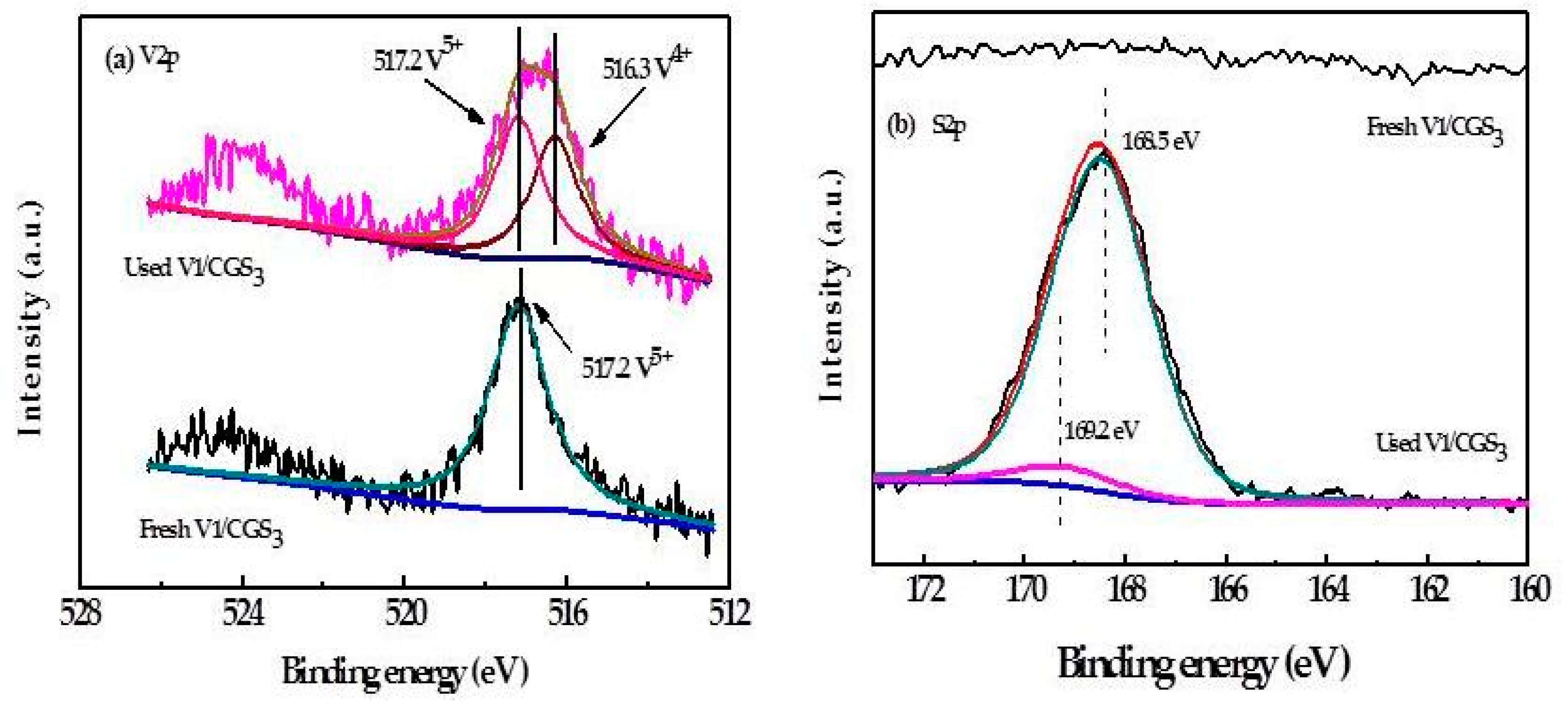
2.8. XPS
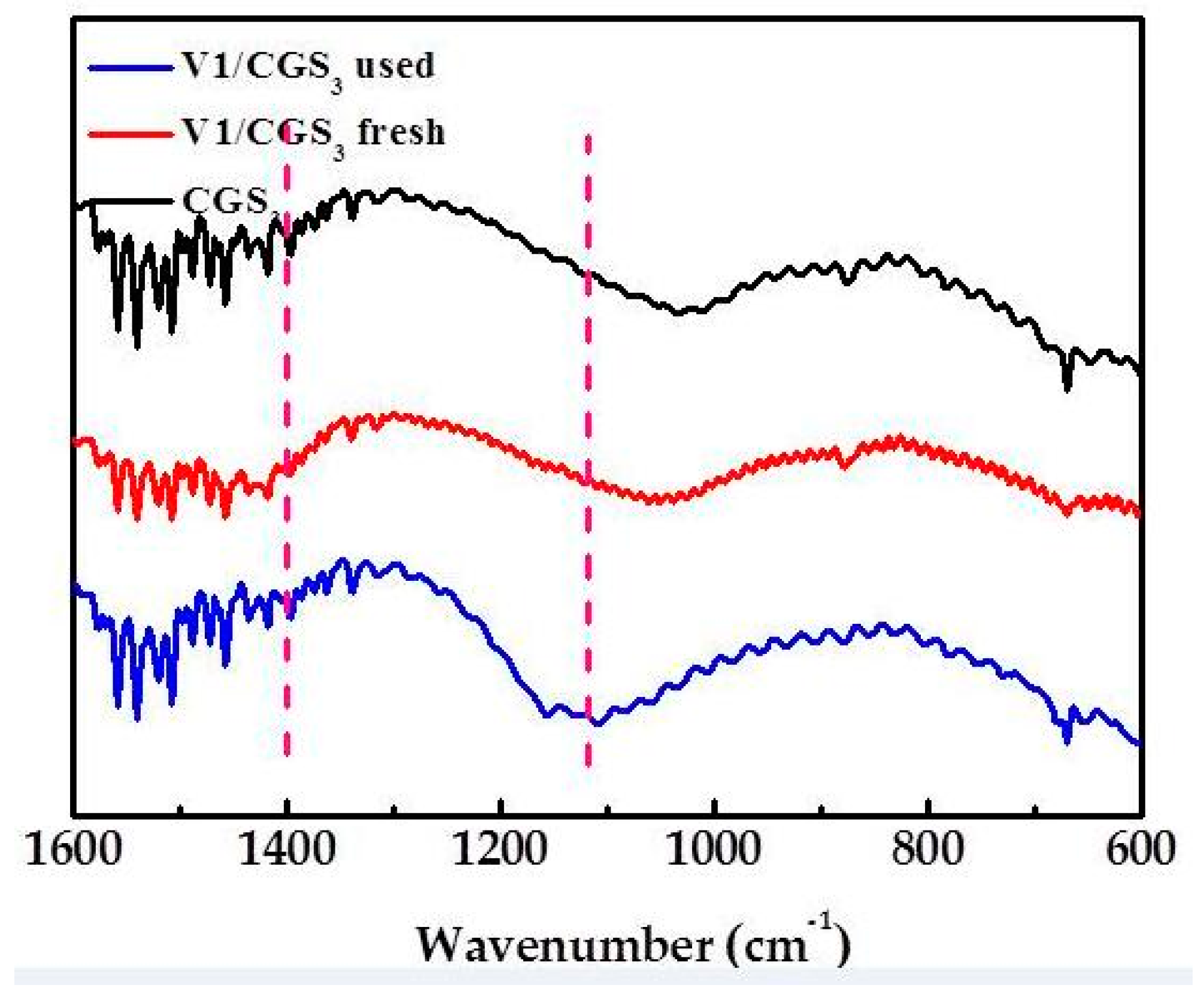
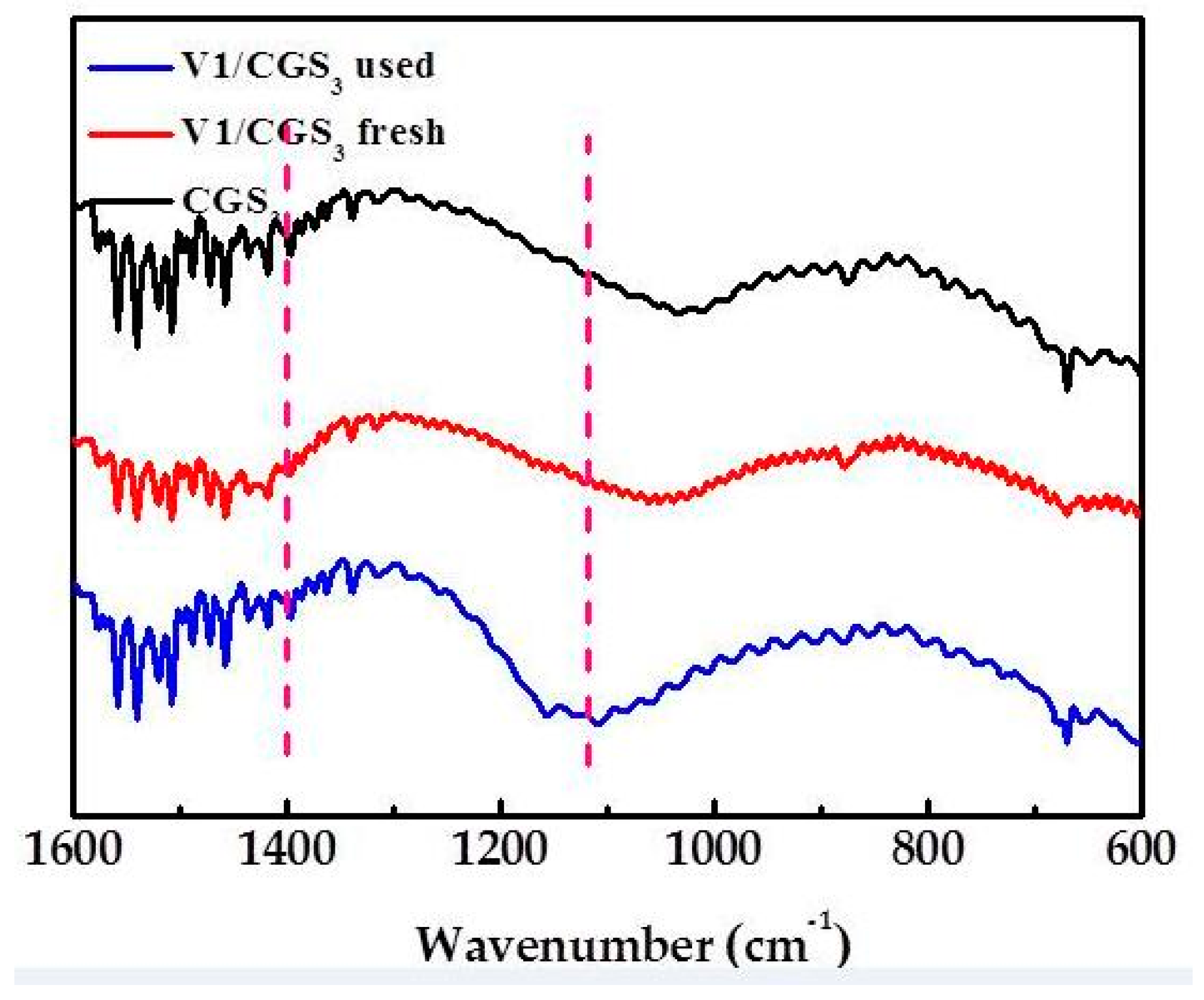
2.9. FTIR
2.10. SEM-EDS
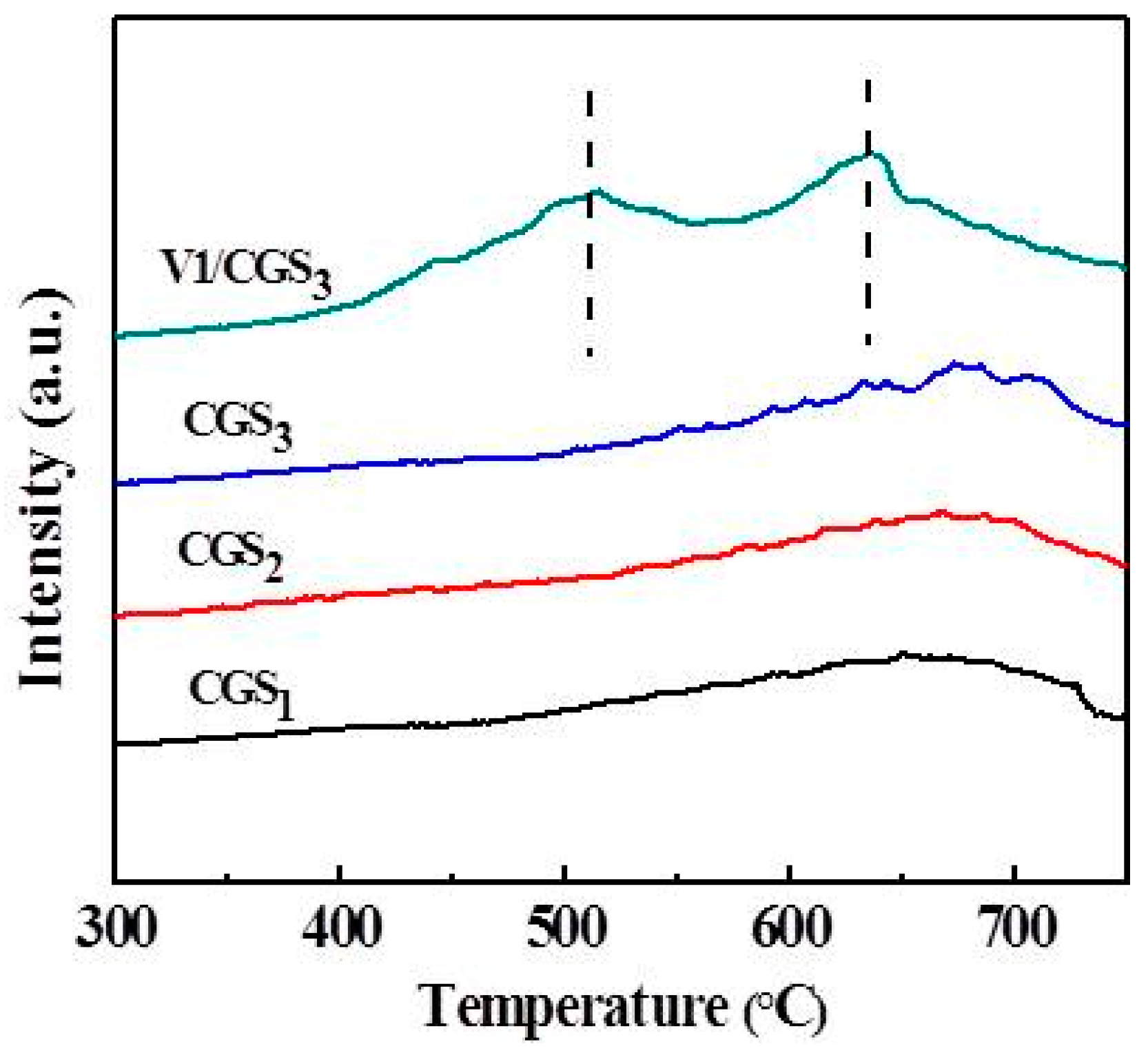
2.11. NH3-TPD
2.12. H2-TPR
3. Materials and Methods
3.1. Preparation of Supports and Catalysts
3.2. Catalytic Activity Test
3.3. Catalysts Characterization
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bosch, H.; Janssen, F. Formation and control of nitrogen oxides. Catal. Today 1988, 2, 369–379. [Google Scholar]
- Cho, S.M. Properly apply selective catalytic reduction for NOx removal. Chem. Eng. Prog. 1994, 90, 39–45. [Google Scholar]
- Chen, J.P.; Yang, R.T. Mechanism of poisoning of the V2O5/TiO2 catalyst for the reduction of NO by NH3. J. Catal. 1990, 125, 411–420. [Google Scholar] [CrossRef]
- Chen, J.P.; Yang, R.T. Selective catalytic reduction of NO with NH3 on SO42−/TiO2 superacid catalyst. J. Catal. 1993, 139, 277–288. [Google Scholar] [CrossRef]
- Huang, Z.; Zhu, Z.; Liu, Z. Combined effect of H2O and SO2 on V2O5/AC catalysts for NO reduction with ammonia at lower temperatures. Appl. Catal. B 2002, 39, 361–368. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, T. NO reduction with NH3 over an activated carbon—supported copper oxide catalysts at low temperatures. Appl. Catal. B 2000, 26, 25–35. [Google Scholar] [CrossRef]
- Singoredjo, L.; Korver, R.; Kapteijn, F.; Moulijn, J. Alumina supported manganese oxides for the low-temperature selective catalytic reduction of nitric oxide with ammonia. Appl. Catal. B 1992, 24, 297–316. [Google Scholar] [CrossRef]
- Grzybek, T.; Papp, H. Cheminform abstract: Selective catalytic reduction of nitric oxide by ammonia on Fe3+- promoted active carbon. Appl. Catal. B 1992, 24, 271–283. [Google Scholar] [CrossRef]
- Zhu, Z.; Niu, H.; Liu, Z.; Liu, S. Decomposition and reactivity of NH4HSO4 on V2O5/AC catalysts used for NO reduction with ammonia. J. Catal. 2000, 195, 268–278. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, Z.; Niu, H.; Liu, S. Promoting effect of SO2 on activated carbon-supported vanadia catalyst for NO reduction by NH3 at low temperatures. J. Catal. 1999, 187, 245–248. [Google Scholar] [CrossRef]
- Lorenc-Grabowska, E.; Gryglewicz, G. Adsorption characteristics of congo red on coal-based mesoporous activated carbon. Dyes Pigments 2007, 74, 34–40. [Google Scholar] [CrossRef]
- Rubio, B.; Izquierdo, M.T.; Mayoral, M.C.; Bona, M.T.; Andres, J.M. Unburnt carbon from coal fly ashes as a precursor of activated carbon for nitric oxide removal. J. Hazard. Mater. 2007, 143, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Acosta, A.; Iglesias, I.; Aineto, M.; Romero, M.; Rincón, J.M. Utilisation of igcc slag and clay steriles in soft mud bricks (by pressing) for use in building bricks manufacturing. Waste Manag. 2002, 22, 887–891. [Google Scholar] [CrossRef]
- Acosta, A.; Aineto, M.; Iglesias, I.; Romero, M.; Rincón, J.M. Physico-chemical characterization of slag waste coming from gicc thermal power plant. Mater. Lett. 2001, 50, 246–250. [Google Scholar] [CrossRef]
- Xu, Y.; Chai, X. Characterization of coal gasification slag based activated carbon and its potential application in lead removal. Environ. Technol. 2017, 41, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Hums, E. Is advanced scr technology at a standstill? A provocation for the academic community and catalyst manufacturers. Catal. Today 1998, 42, 25–35. [Google Scholar] [CrossRef]
- Huang, Z.; Niu, H. Effect of activated coke property on V2O5/AC activity for reduction of NO in flue gas at low temperature. Coal Convers. 2001, 24, 74–78. [Google Scholar]
- Davini, P. Adsorption and desorption of sulphur dioxide from simulated flue gas on active carbon: The effect of the ash content. Carbon 1993, 31, 47–51. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Superior Fe-ZSM-5 catalyst for selective catalytic reduction of nitric oxide by ammonia. J. Am. Chem. Soc. 1999, 121, 5595–5596. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Selective catalytic reduction of NO with ammonia over V2O5 doped TiO2 pillared clay catalysts. Appl. Catal. B 2000, 24, 13–21. [Google Scholar] [CrossRef]
- Long, R.Q.; Yang, R.T. Selective catalytic reduction of nitrogen oxides by ammonia over Fe3+-exchanged TiO2-pillared clay catalysts. J. Catal. 1999, 186, 254–268. [Google Scholar] [CrossRef]
- Pan, S.; Luo, H.; Li, L.; Wei, Z.; Huang, B. H2O and SO2 deactivation mechanism of MnOx/MWCNTs for low-temperature SCR of NOx with NH3. J. Mol. Catal. A-Chem. 2013, 377, 154–161. [Google Scholar] [CrossRef]
- Li, P.; Liu, Q.; Liu, Z. N2O and CO2 formation during selective catalytic reduction of NO with NH3 over V2O5/AC catalyst. Ind. Eng. Chem. Res. 2011, 50, 341–350. [Google Scholar]
- Boyano, A.; Gálvez, M.E.; Moliner, R.; Lázaro, M.J. Carbon-based catalytic briquettes for the reduction of NO: Effect of H2SO4 and HNO3 carbon support treatment. Fuel 2008, 87, 2058–2068. [Google Scholar] [CrossRef]
- Yang, W.; Liu, F.; Xie, L.; Lian, Z.; He, H. Effect of V2O5 additive on the SO2 resistance of a Fe2O3/AC catalyst for NH3-SCR of NOx at low temperatures. Ind. Eng. Chem. Res. 2016, 55, 2677–2685. [Google Scholar] [CrossRef]
- Potdar, H.S.; Jun, K.W.; Bae, J.W.; Kim, S.M.; Lee, Y.J. Synthesis of nano-sized porous γ-alumina powder via a precipitation/digestion route. Appl. Cataly. A-Gen. 2007, 321, 109–116. [Google Scholar] [CrossRef]
- Parida, K.M.; Pradhan, A.C.; Das, J.; Sahu, N. Synthesis and characterization of nano-sized porous gamma-alumina by control precipitation method. Mater. Chem. Phys. 2009, 113, 244–248. [Google Scholar] [CrossRef]
- Klimczak, M.; Kern, P.; Heinzelmann, T.; Lucas, M.; Claus, P. High-throughput study of the effects of inorganic additives and poisons on NH3-SCR catalysts—Part I: V2O5-WO3/TiO2 catalysts. Appl. Catal. B 2010, 95, 39–47. [Google Scholar] [CrossRef]
- Kröcher, O.; Elsener, M. Chemical deactivation of V2O5-WO3/TiO2 SCR catalysts by additives and impurities from fuels, lubrication oils, and urea solution: I. Catalytic studies. Appl. Catal. B 2008, 77, 215–227. [Google Scholar] [CrossRef]
- Jianrong, M.; Mengxue, A. A novel Ca/AC desulphurizer. Coal Convers. 2001, 24, 48–53. [Google Scholar]
- Yang, S.; Guo, Y.; Yan, N.; Qu, Z.; Xie, J.; Yang, C.; Jia, J. Capture of gaseous elemental mercury from flue gas using a magnetic and sulfur poisoning resistant sorbent Mn/γ-Fe2O3 at lower temperatures. J. Hazard. Mater. 2011, 186, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, Z.; Li, Q.; Wu, W.; Liu, Q. Multiple roles of SO2 in selective catalytic reduction of NO by NH3 over V2O5/AC catalyst. Ind. Eng. Chem. Res. 2014, 53, 7910–7916. [Google Scholar] [CrossRef]
- Liu, L.; Liu, Z.; Yang, J.; Huang, Z.; Liu, Z. Effect of preparation conditions on the properties of a coal-derived activated carbon honeycomb monolith. Carbon 2007, 45, 2836–2842. [Google Scholar] [CrossRef]
- Xiao, Y.; Liu, Z.; Liu, Q.; Wang, J.; Xing, X.; Huang, Z. Mechanism of SO2 influence on NO removal over V2O5/AC catalyst. Chin. J. Catal. 2008, 29, 81–85. [Google Scholar]
- Seo, P.W.; Lee, J.Y.; Shim, K.S.; Hong, S.H.; Hong, S.C.; Hong, S.I. The control of valence state: How V/TiO2 catalyst is hindering the deactivation using the mechanochemical method. J. Hazard. Mater. 2009, 165, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Wren, A.G.; Phillips, R.W.; Tolentino, L.U. Surface reactions of chlorine molecules and atoms with water and sulfuric acid at low temperatures. J. Colloid Interface Sci. 1979, 70, 544–557. [Google Scholar] [CrossRef]
- Zhu, Z.; Liu, Z.; Niu, H.; Liu, S.; Hu, T.; Liu, T. Mechanism of SO2 promotion for NO reduction with NH3 over activaed carbon—Supported vanadium oxide catalyst. J. Catal. 2001, 197, 6–16. [Google Scholar] [CrossRef]
- Pan, Y.; Zhao, W.; Zhong, Q.; Cai, W.; Li, H. Promotional effect of Si-doped V2O5/TiO2 for selective catalytic reduction of NOx by NH3. Acta Sci. Circumstantiae 2013, 25, 1703–1711. [Google Scholar]
- Frederickson, L.D.; Hausen, D.M. Infrared spectra-structure correlation study of vanadium-oxygen compounds. Anal. Chem 1963, 35, 818. [Google Scholar] [CrossRef]
- Khodayari, R.; Odenbrand, C.U.I. Regeneration of commercial SCR catalysts by washing and sulphation: Effect of sulphate groups on the activity. Appl. Catal. B 2001, 33, 277–291. [Google Scholar] [CrossRef]
- Zhuang, Q.; Kyotani, T.; Tomita, A. Drift and TK/TPD analyses of surface oxygen complexes formed during carbon gasification. Energy Fuels 1994, 8, 714–718. [Google Scholar] [CrossRef]
- Niwa, M.; Habuta, Y.; Okumura, K.; Katada, N. Solid acidity of metal oxide monolayer and its role in catalytic reactions. Catal. Today 2003, 87, 213–218. [Google Scholar] [CrossRef]
- Huang, B.; Huang, R.; Jin, D.; Ye, D. Low temperature scr of NO with NH3 over carbon nanotubes supported vanadium oxides. Catal. Today 2007, 126, 279–283. [Google Scholar] [CrossRef]
- Amiridis, M.D.; Wachs, I.E.; Deo, G.; Jehng, J.M.; Du, S.K. Reactivity of V2O5 catalysts for the selective catalytic reduction of NO by NH3: Influence of vanadia loading, H2O, and SO2. J. Catal. 1996, 161, 247–253. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | BET Surface Area (m2/g) | Micro-Pore Area (m2/g) | Mesopore Volume (cm3/g) | Micro-Pore Volume (cm3/g) |
|---|---|---|---|---|
| CGS1 | 212.8 | 94.5 | 0.13 | 0.04 |
| CGS2 | 318.7 | 93.1 | 0.27 | 0.04 |
| CGS3 | 311.4 | 111.0 | 0.18 | 0.05 |
| V1/CGS3 fresh | 321.5 | 115.6 | 0.19 | 0.05 |
| V1/CGS3 used | 203.5 | 61.2 | 0.15 | 0.03 |
| Samples | Composition (wt %) | ||||||
|---|---|---|---|---|---|---|---|
| SiO2 | Fe2O3 | CaO | Al2O3 | Na2O | TiO2 | V2O5 | |
| CGS1 | 14.5 | 20.0 | 7.9 | 3.9 | 0.8 | 0.2 | 0 |
| CGS2 | 12.8 | 6.4 | 5.1 | 5.1 | 0.8 | 0.2 | 0 |
| CGS3 | 11.9 | 3.0 | 2.9 | 3.1 | 0.5 | 0.1 | 0 |
| V1/CGS3 fresh | 8.9 | 2.9 | 2.6 | 2.6 | 0.4 | 0.1 | 1.3 |
| V1/CGS3 used | 7.4 | 2.5 | 3.0 | 2.3 | 0.3 | 0.1 | 1.0 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, F.; Gao, Y.; Huo, Q.; Han, L.; Wang, J.; Bao, W.; Chang, L. Characteristics of Vanadium-Based Coal Gasification Slag and the NH3-Selective Catalytic Reduction of NO. Catalysts 2018, 8, 327. https://doi.org/10.3390/catal8080327
Han F, Gao Y, Huo Q, Han L, Wang J, Bao W, Chang L. Characteristics of Vanadium-Based Coal Gasification Slag and the NH3-Selective Catalytic Reduction of NO. Catalysts. 2018; 8(8):327. https://doi.org/10.3390/catal8080327
Chicago/Turabian StyleHan, Fang, Yanchun Gao, Qihuang Huo, Lina Han, Jiancheng Wang, Weiren Bao, and Liping Chang. 2018. "Characteristics of Vanadium-Based Coal Gasification Slag and the NH3-Selective Catalytic Reduction of NO" Catalysts 8, no. 8: 327. https://doi.org/10.3390/catal8080327
APA StyleHan, F., Gao, Y., Huo, Q., Han, L., Wang, J., Bao, W., & Chang, L. (2018). Characteristics of Vanadium-Based Coal Gasification Slag and the NH3-Selective Catalytic Reduction of NO. Catalysts, 8(8), 327. https://doi.org/10.3390/catal8080327